
Abstract
To investigate the effects of ELK1 on hippocampal cells and its related mechanisms in the progression of vascular dementia (VD). Twenty-seven SD rats were divided into three groups: sham group (n = 9), Model group (n = 9), and Model+ELK1-OE group (n = 9). The VD animal model was established in the Model group by bilateral common carotid artery occlusion (BCCAO). The Morris water maze test was performed to observe the learning and memory abilities of the rats in the three groups. HE staining was used to examine the morphological changes in the hippocampal region, and immunohistochemical staining was conducted to observe the expression levels of ELK1. In the in vitro experiments, hippocampal tissues were isolated from rats in the sham group to prepare hippocampal single-cell suspensions. After different stimuli, Western blot was performed to observe protein expression, flow cytometry for apoptosis was used to assess hippocampal cell apoptosis, and a colorimetric assay kit was employed to detect the Fe2 + levels in the hippocampal cells of each group. In the in vivo experiments, the levels of ELK1 in VD rats were significantly lower than those in the sham group. ELK1 improved the learning and memory abilities of VD rats. After ELK1 intervention, the arrangement of neural cells in the hippocampal region of the rats was more regular, the nucleoli were clearer, the number of glial cells decreased, and the number of inflammatory cells was reduced. In the in vitro experiments, ELK1 promoted the expression of P-mTOR, P-CREB, and nuclear YAP, inhibited the expression of NOX4 and nuclear TFEB, and reduced hippocampal cell apoptosis and Fe2 + content. ELK1 inhibits the progression of vascular dementia by regulating mTOR/CREB/YAP/TFEB signaling-induced ferroptosis in hippocampal cells.
Similar content being viewed by others

Introduction
Vascular dementia (VD), a prevalent cognitive impairment disorder in the elderly, is associated with several causes, including neurodegeneration and cerebrovascular disease1,2. The second most common kind of dementia after Alzheimer’s disease (AD), VD is a cognitive impairment caused by cerebrovascular diseases3. Cerebrovascular illnesses that impact the blood flow to the brain, such ischemia, infarction, or hemorrhage, are typically linked to VD. Because of this, brain tissue is damaged and neurons die4. People with VD often have executive dysfunction, trouble paying attention, memory loss, and other symptoms that make their social and personal lives much worse. As the population becomes older, more and more people are getting VD. It is assumed that VD causes around 20% of dementia cases across the globe. This number may be significantly higher in certain locations5. VD has a big impact on the people who have it, but it also puts a lot of stress on families and society as a whole. Consequently, a comprehensive knowledge of the pathophysiology of VD and the development of effective preventive and treatment strategies have considerable clinical and practical significance.
Issues in the hippocampus area, which also helps with managing stress and controlling emotions, may be linked to emotional issues in patients with VD6. In individuals with VD, the hippocampus area is often profoundly impacted, exhibiting pathological alterations including neuronal death, diminished synaptic connections, and compromised neurogenesis. These alterations immediately cause memory and learning capacities to become worse. Studies indicate that individuals with vascular dementia have substantial anatomical and functional anomalies in the hippocampus area. Imaging studies indicate that individuals with vascular dementia have a reduced hippocampus, less gray matter, and decreased synaptic plasticity7. People with VD may have emotional difficulties that are linked to issues with the hippocampus area, which also helps manage emotions and deal with stress8. So, it’s very important to preserve hippocampus cells and keep them working normally to stop and cure VD9,10. At now, strategies to safeguard the hippocampus area mostly concentrate on enhancing cerebral blood flow, diminishing oxidative stress, and suppressing inflammatory responses11,12. These therapies have limited effectiveness, and more research is needed to identify the precise mechanisms of VD development and develop more potent intervention techniques.
ELK1 is a transcription factor that controls how genes are expressed in many kinds of cells. It is a member of the ETS family. ELK1 is involved in controlling how neural cells grow and function in the nervous system13,14. Research indicates that ELK1 is expressed in neural stem cells and has a role in controlling neuronal production and differentiation15. ELK1 also helps the body deal with stress and protects the brain. Some research have shown that ELK1 being expressed in an unusual way is linked to disorders including Huntington’s chorea and Parkinson’s disease16. Nonetheless, there is little knowledge on ELK1 in vascular dementia, and its precise role in hippocampus cells remains unidentified. This research seeks to investigate the role of ELK1 in hippocampus cells and its impact on vascular dementia (VD) to identify novel targets and treatment approaches.
To elucidate the specific molecular mechanisms underlying ELK1’s protective role, this study will integrate in vivo and in vitro models, focusing on the potential signaling axis of mTOR/CREB/YAP/TFEB. At the mechanism validation level, we will employ a stepwise gain-of-function and loss-of-function experimental strategy: First, we will confirm the overall protective effect of ELK1 through overexpression; Next, we will employ a CREB-specific inhibitor (666 − 15) to validate whether CREB is an indispensable downstream effector of ELK1/mTOR; Building upon this, we will co-treat with a YAP activator (PY-60) to test whether YAP activation can bypass inhibited CREB to exert protective effects, thereby confirming YAP’s downstream position in this pathway; Finally, apply ferroptosis activator (Erastin) to confirm ferroptosis as the ultimate execution mechanism of cell death in this model, establishing a direct causal link between upstream signaling alterations and this terminal phenotype. This systematic approach aims to decipher the causal chain by which ELK1 suppresses hippocampal cell ferroptosis through regulation of the mTOR/CREB/YAP/TFEB signaling cascade.
Materials and methods
Bioinformatics analysis
The GEO database was used to extract the vascular dementia dataset GSE122063, which included gene expression data from 36 patients with vascular dementia and 44 healthy people. First, the Robust Multi-array Average technique was used to normalize, background-correct, and multi-probe average the chip data. Subsequently, the Combat algorithm was used to adjust for batch effects in the processed data. The limma package’s differential expression analysis required a |log2 Fold Change| of more than 0.58 and a p-value of less than 0.05. Additionally, the VennDiagram program was used to find overlapping genes, and the FerrDb database was used to find iron-related genes. The ClusterProfiler software (Version 4.6.0, https://bioconductor.org/packages/ClusterProfiler/) for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways was used to do enrichment analysis on these genes that are the same in both groups. Finally, the ggplot2 tool was used to illustrate the results of the differential analysis in the form of heatmaps, bar plots, and scatter plots. We utilized the Spearman correlation method to look at the associations between genes that were differentially expressed17,18,19.
Experimental animals
Henan Scibeis Biotechnology Co., Ltd. provided 27 SPF-grade male SD rats (body weight 250–300 g, age 6–8 weeks) (Permit No.: SYXK (Yu) 2020-005). Each rat was housed in a sophisticated climate-controlled habitat that allowed them to eat and drink whenever they wanted. The temperature was 22 ± 2 °C, the relative humidity was 60% ± 5%, and the light and dark cycles were 12 h long. A week of getting used to things was needed before the experiment could start. All animal experiments were approved by the Hebei General Hospital Committee and conducted according to the guidelines of the General Hospital. All methods in this experiment were performed in accordance with relevant guidelines and regulations and conformed to the 3R and ARRIVE guidelines.
ELK1 overexpression lentivirus injection
The rats were placed on the experimental table and gently fixed. The SD rats were placed in the anesthesia box to induce anesthesia using 3% isoflurane for 2 ~ 3 min. The anesthetized rats were transferred to the animal surgery table, and anesthesia was maintained using 2% isoflurane, and the respiration and response of the rats were observed to ensure that they were completely anesthetized. The anesthetized rats were fixed on the stereotaxic apparatus to ensure that the head was flat. The upper and lower incisors of the rat were secured using pliers to ensure that the head was stable. Sterilize the rat’s head with iodine or alcohol. Make a small incision in the middle of the head to expose the skull. Using the coordinate system of a stereotaxic device, determine the location of the lateral ventricles. Typically, the lateral ventricles are located 1.0 mm anterior to the fontanelle, 1.5 mm paracentral to the median line, and 3.5 mm below the surface of the skull. Using a surgical drill to drill a small hole at the determined coordinates, approximately 1 mm in diameter. Inserting a syringe needle to a predetermined depth and slowly inject lentiviral solution (2–5 µL). Keep the needle stable during injection and keep the needle in the original position for 1–2 min after injection to prevent fluid reflux. Remove the syringe needle slowly and gently press the drilled hole with sterile gauze. The skin incision was closed with surgical thread. The rats were given medication after surgery, which comprised two subcutaneous injections of ketoprofen (10 mg/kg) a day for two days to reduce pain and swelling. After being placed back in their cages, the rats were watched to make sure they were eating and doing the same things they usually do. The number, place, and time of the injection were only a few of the numerous details about the surgery that were written down. After surgery, watch the rats closely to make sure they are recovering properly. On the seventh day after injection, rats were randomly selected from each group to confirm the efficacy of transfection and the distribution of lentivirus in brain tissue. Following cardiac perfusion, brains were harvested and frozen sections were prepared. To label the nuclei, slices were fixed with 4% paraformaldehyde, washed with PBS, and stained with DAPI (Biyun Tian, C1002). Using a fluorescent microscope (Olympus, Japan), we saw and recorded green fluorescence signals from GFP in the hippocampus region. We used ImageJ software (Version 1.53c, National Institutes of Health, https://imagej.nih.gov/ij/) to put the pictures together and look at them.
Animal model construction and grouping
To simulate the chronic cerebral ischemia pathology of vascular dementia, this study employed a bilateral common carotid artery permanent ligation (BCCAO/2VO) model. This paradigm reliably induces a sustained reduction in cerebral blood flow, ultimately leading to neuronal damage and cognitive deficits reliant on the hippocampus. It serves as a conventional model for investigating the pathophysiological mechanisms and treatment strategies of vascular dementia20. Before surgery, rats were restricted from food intake for 12 h and water intake for 4 h. Briefly, rats were anesthetized using 3% isoflurane. The common carotid artery was isolated from the vagus nerve through a ventral neck incision using a vitreous nerve separator. The common carotid artery was tied up twice with silk thread (4 − 0), with an incision created in between. The artery was also exposed, but it wasn’t tied up in the group that underwent fake surgery. The rats returned to normal survival conditions after surgery. 27 rats were divided into three groups (n = 9 per group): (1) sham group: sham surgery and saline administration via intragastric administration; (2) Model: BCCAO surgery and saline administration via intragastric administration; and (3) Model+ELK1-OE group: BCCAO surgery and administration of ELK1-OE lentivirus by injection via the lateral ventricle.
Morris water maze experiment
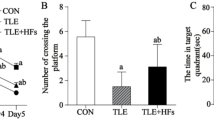
The rats in each group were subjected to the place navigation test and the spatial probe test 2 weeks after surgery. The place navigation test was conducted over 5 days, with 4 trials per day and an interval of 1 h between trials. The trials started at the points where the bisectors of each quadrant crossed the pool wall. Each time, a different entry point was selected. The rats were placed in the pool with their backs to the wall, and the time it took them to climb onto the platform was measured. The rats were sent to the platform and told to stay there for 10 s if they couldn’t find it and climb up it in 120 s. After then, the escape latency was recorded at 120 s. The average escape latency on the fifth day was used to figure out the score for the locati0n navigation test. The spatial probe test was conducted 24 h after the place navigation test, with the platform removed. The rats were placed into the pool at the same entry points and the number of times they crossed the original platform location within 120 s was recorded as the spatial probe test score.
HE staining
After completing the behavioral tests, nine rats were randomly selected from each group and anesthetized with 3% isoflurane for 2–3 min. The anesthetized rats were transferred to an animal operating table and anesthesia was maintained with 2% isoflurane. The brain was perfused and preserved with 40 g/L paraformaldehyde, then embedded in regular paraffin wax and sliced to a thickness of 5 μm. The sections were treated with xylene to remove the paraffin, ethanol to add water, hematoxylin and eosin to stain them, and water to remove the water. The sections were transparent in xylene and sealed with conventional neutral gum and coverslips. The sections were stained with HE staining, and the morphological changes in the hippocampal region were observed under a light microscope.
Immunohistochemical staining
After xylene removed the paraffin, graded ethanol solutions were used to rehydrate pieces of brain tissue collected at the same time. Then, citrate buffer was used to get the antigen back. After the sections were treated with 30 g/L H2O2 and goat serum, BDNF antibody (dilution ratio 1:200), biotinylated goat anti-rabbit IgG, and horseradish peroxidase-labeled streptavidin were added. After DAB staining, the slices were dried, cleared, and sealed with neutral gum. Negative controls were performed by substituting PBS for the primary antibody. Photographs were taken under a 400x field of view in the hippocampal region. Each section randomly selected 3 non-overlapping regions in the hippocampus and measured the average absorbance value of ELK1-positive granules per high-power field using the Image-J image analysis system.
Preparation of single-cell suspensions from hippocampus
The hippocampal tissue from “1.6” was rinsed 3–4 times with D-Hank’s solution, then the tissue was minced and digested with approximately 5 times the volume of trypsin (2.5 g/L) at 37 °C with slow shaking for 30 min. The digested tissue was then treated with pre-chilled DMEM containing 100 mL/L calf serum. To get the final volume of 2 mL, the mixture was filtered through a 200-mesh sieve, spun in a centrifuge for five minutes at 1000 rpm, rinsed with D-Hank’s solution, and then put in serum-free DMEM. The single-cell suspensions in the centrifuge tubes had a cell concentration of around 1 × 107/mL. We utilized Trypan Blue to see how many cells were alive, and cells that were more than 95% alive were OK for the experiment.
Cell culture and grouping
The hippocampal cells from “1.8” were subjected to various treatments, including untreated hippocampal cells; cells cultured in 1% O2 and 3% low serum medium with 100 µM FeCl3; cells cultured in 1% O2 and 3% low serum medium with 100 µM FeCl3 and ELK1-OE lentivirus; cells treated with 1% O2, 3% low serum medium, 100 µM FeCl3, ELK1-OE lentivirus, and 1µM CREB inhibitor 666 − 159 (Shanghai ZeYe Biotechnology Co.); cells treated with 1% O2, 3% low serum medium, 100 µM FeCl3, ELK1-OE lentivirus, CREB inhibitor 666 − 159, and YAP activator PY-60 (SELLECK) 1 µM; and cells treated with 1% O2, 3% low serum medium, 100 µM FeCl3, ELK1-OE lentivirus, CREB inhibitor 666 − 159, YAP activator PY-60, and ferroptosis activator Erastin (MCE) 10 µM. Groups: (-) group, (-) + hypoxia + FeCl₃ group, (-) + hypoxia + FeCl₃ + ELK1-OE group, (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 group, (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 group, (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 + Erastin group.
This study employs a stepwise inhibitor/activator treatment to sequentially intervene in the mTOR/CREB/YAP/TFEB signaling axis, aiming to validate its upstream-downstream relationships and causal roles in ferroptosis. Specifically: (1) Determining if CREB is an essential downstream node of ELK1/mTOR in this pathway by using a CREB inhibitor in conjunction with ELK1 overexpression (ELK1-OE); (2) To verify that YAP is an essential downstream effector of CREB and that it may partially counteract phenotypes induced by CREB inhibition, both CREB inhibition and YAP activation are conducted concurrently; (3) The application of ferroptosis activators aims to corroborate ferroptosis as the primary mechanism of cell death in this model and demonstrate that the protective effects of the aforementioned signaling pathways ultimately culminate in the inhibition of ferroptosis. This “rescue assay” strategy facilitates the transition from inferring correlations to elucidating causal mechanisms.
To independently assess the potential cytotoxicity of key compounds (666 − 15, PY-60, Erastin) used in this study at specified concentrations on hippocampal cells, we established additional cytotoxicity validation subgroups. Cells were cultured under standard conditions (normal oxygen, complete medium) and divided into two groups: (1) Normal control group (NC group): no compound treatment; (2) 666 − 159+PY-60 + Erastin group: Simultaneously treated with CREB inhibitor 666 − 15 (1 µM), YAP activator PY-60 (1 µM), and ferroptosis activator Erastin (10 µM) in the medium. Cells were harvested 48 h post-treatment for subsequent flow cytometric apoptosis analysis to assess cell viability.
Western blot
Total protein was extracted from cells according to the protocol of the total protein extraction kit. For the nuclear proteins YAP and TFEB, we used a nuclear/cytoplasmic protein separation kit to isolate and extract the nuclear protein fraction for subsequent detection. The BCA method was employed to measure the protein, and boiling it in loading buffer for five minutes changed its shape. After putting 40 µg of protein into each sample, the electrophoresis data was transferred to PVDF membranes. The membranes were blocked for an hour with 5% skim milk, and then the primary antibodies were put in the fridge for the full night. The membranes were treated with secondary antibodies for an hour at room temperature after being washed. The membranes were made using ECL chemiluminescent reagent, and a gel imaging device was utilized to take pictures of them. Total protein was normalized using GAPDH (Proteintech, 1:5000) as an internal control, while nuclear protein samples were standardized with the nuclear marker Lamin B1 (Proteintech, 1:1000). The gray value ratio of each target protein band to the internal control band was calculated.
Detection of MDA, GSH, and GPX levels in hippocampal cells
After the cells were treated as described in “1.9”, they were digested with trypsin, and the cell suspension was collected and centrifuged (1000 rpm, 5 min) to discard the supernatant and retain the cell sediment. The levels of the corresponding indicators were calculated by successively adding the reaction reagents and colorimetric reagents of the kit and measuring the absorbance values.
DCFH-DA staining
The hippocampal cells were plated at 8 × 104/dish in confocal dishes and treated 24 h later. After treatment, the cells were washed with PBS three times for five minutes each time. They were then put in a cell incubator for twenty minutes in the dark with 1.5 mL of serum-free medium that had 10 µmol/L DCFH-DA in it. After three 5-minute PBS rinses, the cells were quickly looked at and photographed using a confocal microscope.
AnnexinV/PI flow cytometry apoptosis assay
We used Annexin V-FITC/PI double-staining flow cytometry to quantify ferroptosis-associated apoptosis levels and evaluate the impact of various treatment conditions on cell viability. The hippocampal cells were sown in 6-well plates at a density of 1 × 102 cells/well and treated according to the technique described in section “CellcCulture and grouping”. In addition to the groups stated in section “CellcCulture and grouping”, there was also a negative control group (NC group) that was not treated. After 48 h of treatment, trypsin was used to breakdown the cells, which were then washed three times with cold PBS and spun at 1200 rpm for three minutes. Resuspend cells in 1× binding buffer for counting. Put 100 µL of cell suspension (1 × 102 cells/mL) and 5 µL of Annexin V-FITC and PI antibodies into a flow cytometry tube. After mixing everything well, let it rest in the dark for 15 min at room temperature. Finally, filter the cells, put them back in 400 µL of 1× binding buffer, and utilize flow cytometry to look at them.
Data Analysis: To get the total apoptosis rate, we included the percentages of early apoptotic cells (Annexin V+/PI-) and late apoptotic/necrotic cells (Annexin V+/PI+). To evaluate how cytotoxic the treatment conditions were, the total apoptotic rates of each treatment group were compared to those of the NC group.
Statistical analysis
We utilized SPSS 22.0 software (IBM Corp., Armonk, NY, USA, https://www.ibm.com/analytics/spss-statistics-software) to do a statistical analysis of the data, and we showed the experimental results as mean ± standard deviation. We used one-way ANOVA to look at the quantitative data from each group and the LSD test to compare groups with similar variances in pairs. A P value lower than 0.05 was considered statistically significant.
Results
Bioinformatics analysis results
The analysis removed 2,446 highly differentially expressed genes (DEGs), 1,010 of which were upregulated and 1,436 of which were downregulated. In the GO analysis, DEGs were shown to be significantly enriched in molecular activities including oxidoreductase activity, transcription factor activity, and transferrin receptor binding. This suggests that these genes may play a role in cellular signal transmission and metabolic processes. The inflammatory response and cellular damage mechanisms in vascular dementia may be associated with the substantial enrichment of DEGs in biological processes, such as responses to external stimuli, cellular reactions to chemical stress, and the control of DNA-binding transcription factor activity. The KEGG pathway analysis also demonstrated that DEGs were mostly found in important pathways that are important for cell survival, growth, and death, such as the MAPK signaling pathway, chemical carcinogenesis-reactive oxygen species, and ferroptosis. This indicates that these genes may play a role in the pathogenesis of vascular dementia via regulating inflammatory responses and cellular death. The heatmap analysis revealed a substantial correlation between SP1 and DNMT1, suggesting that these two genes may collaboratively influence the genesis of vascular dementia. SP1 may serve as a transcription factor that regulates DNA methylation by modifying DNMT1 expression, hence influencing the functional regulation of neurons. Moreover, the inverse relationship between DNMT1 and AP-2α may indicate that the overexpression of AP-2α might impede the function of DNMT1, hence affecting cell proliferation and differentiation. The correlation bar chart analysis showed that CREB and YAP were positively related, whereas YAP and NCOA4 were negatively related. Additionally, the positive correlation between YAP and SLC7A11, GPX4 suggested that these genes may play an important role in maintaining the redox balance within cells. The negative correlation between mTOR and TFEB suggests the importance of autophagy in vascular dementia. Meanwhile, the correlation scatter plot analysis showed a negative correlation between GPX4 and ROS, which shows how important antioxidant defense is for protecting neurons. The positive correlation between NCOA4 and HIF-1α illustrated the complexity of cellular adaptation mechanisms in hypoxic environments, while the positive correlation between TFEB and the autophagy marker protein LC3 further validated the essential function of autophagy in preserving intracellular homeostasis (Fig. 1).

Bioinformatics analysis. A: Differential gene volcano plot; B: Wayne diagram of differential and iron death-related genes; C: MF enrichment analysis bubble plot; D: BP enrichment analysis histogram; E: KEGG enrichment analysis bubble plot; F: KEGG enrichment analysis clustering plot; G: correlation pie chart; H: correlation bar chart; I: correlation scatter plot.
ELK1 improves vascular dementia in rats
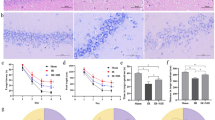
To determine whether bilateral common carotid artery occlusion (BCCAO) successfully induced a vascular dementia model, we assessed spatial learning and memory abilities in rats using the Morris water maze test and examined hippocampal pathological morphology via HE staining. Behavioral test results showed that compared with the sham-operated group, rats in the model group exhibited significantly prolonged escape latency. Additionally, rats in the model group demonstrated significantly fewer crossings in the quadrant containing the target platform compared with the sham-operated group (Fig. 1A). Histomorphological analysis revealed a marked reduction in pyramidal cell numbers in the hippocampal region of the model group compared to the sham-operated group. Additionally, pyramidal cells in the model group exhibited disorganized arrangement, morphological abnormalities, and visible necrotic neurons compared to the sham-operated group (Fig. 2B). Collectively, these findings demonstrate that BCCAO surgery successfully induced rat vascular dementia-like cognitive impairment and hippocampal pathological damage, confirming the validity of the established model.

ELK1 improves vascular dementia in rats. A: Morris water maze test to assess learning and memory abilities in sham-operated rats, model rats, and model rats overexpressing ELK1. B: Histological changes in the hippocampal region of the three groups were observed via HE staining. C: Expression levels of ELK1 were assessed through immunohistochemical staining. Groups: Sham surgery group, model group, model + ELK1-OE group. D: Expression and distribution of ELK1-OE lentivirus in the hippocampal region. Scale bar: 100 μm. Data are expressed as mean ± standard deviation. N = 9, *P < 0.05, **P < 0.01, nsP>0.05: indicates no statistically significant difference.
To evaluate the intervention effect of ELK1 on vascular dementia, we further examined relevant behavioral, histomorphological, and protein expression indicators. Results from the Morris water maze experiment showed that compared with the model group, rats in the model + ELK1 overexpression group exhibited significantly shorter escape latency. Additionally, rats in the model + ELK1 overexpression group demonstrated significantly increased crossings in the target quadrant compared with the model group (Fig. 1A), indicating that ELK1 overexpression improves cognitive function in rats with vascular dementia. Histomorphological analysis revealed that compared with the model group, the model + ELK1-OE group exhibited a marked increase in hippocampal pyramidal cell numbers and more regular cell arrangement. Compared with the model group, the model + ELK1-OE group showed clear nuclear membrane and nucleolus structures, abundant cytoplasm, and morphology more closely resembling that of the sham-operated group (Fig. 2B). Immunohistochemical staining results indicated that compared with the sham group, ELK1 expression levels in the hippocampal region were significantly reduced in the model group. Compared with the model group, ELK1 expression was significantly elevated in the model + ELK1-OE group (Fig. 2C).
To confirm successful delivery and expression of ELK1-OE lentivirus in the hippocampus following lateral ventricle injection, we performed GFP fluorescence detection. The results showed obvious green fluorescence signals in both the cell bodies and processes of hippocampal neurons in rats, with the signals mainly concentrated in the hippocampal CA1 region. This indicates that ELK1-OE lentivirus delivered via lateral ventricle injection can be effectively transported to the hippocampus and achieve stable expression, providing reliable viral expression validation for subsequent functional studies (Fig. 2D).
ELK1 regulates mTOR/CREB/YAP signaling
To elucidate the effects of ELK1 on hippocampal cell signaling pathways under hypoxic and iron-loaded conditions, we detected the expression of relevant proteins via Western blotting. Compared with the (-) group, the (-) + hypoxia + FeCl₃ group exhibited significantly decreased expression of ELK1, total mTOR, phosphorylated mTOR, phosphorylated CREB, and nuclear YAP protein, while NOX4 expression was significantly elevated. Compared with the (-) + hypoxia + FeCl₃ group, the (-) + hypoxia + FeCl₃ + ELK1-OE group showed significantly increased expression of ELK1, total mTOR, phosphorylated mTOR, phosphorylated CREB, and nuclear YAP protein, while NOX4 expression was significantly downregulated. Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE group, the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 group showed significantly decreased phosphorylation of mTOR, phosphorylation of CREB, and nuclear YAP protein expression, significantly increased NOX4 expression, while ELK1 and total mTOR expression showed no significant difference; Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 group, the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 group showed significantly increased phosphorylated mTOR and nuclear YAP protein expression, significantly decreased NOX4 expression, while ELK1 and total mTOR, and phosphorylated CREB expression showed no significant differences; Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 group, the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 + Erastin group exhibited significantly decreased phosphorylated mTOR expression, NOX4 expression was significantly increased, while ELK1, total mTOR, phosphorylated CREB, and nuclear YAP protein expression showed no significant differences (Fig. 3A).

ELK1 acts by regulating mTOR/CREB/YAP signaling. A: Western blot detected ELK1, total mTOR, P-mTOR, P-CREB, nuclear YAP, and NOX4 protein expression in hippocampal cells after different stimulation treatments. B: Levels of MDA, GSH, and GPX were detected according to the kit. C: DCFH-DA staining to detect ROS levels. Groups: (-) group, (-) + hypoxia + FeCl₃ group, (-) + hypoxia + FeCl₃ + ELK1-OE group, (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 group, (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 group, (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 + Erastin group. Data are expressed as mean ± standard deviation. GAPDH served as the internal control protein, and Lamin B1 as the nuclear protein internal control. N = 3. *P < 0.05, **P < 0.01, nsP>0.05: indicates no statistically significant difference.
Simultaneously, we employed colorimetric assays to detect cellular oxidative stress and antioxidant markers. Compared with the (-) group, the (-) + hypoxia + FeCl₃ group exhibited significantly elevated MDA levels and markedly reduced GSH and GPX levels. Compared with the (-) + hypoxia + FeCl₃ group, the (-) + hypoxia + FeCl₃ + ELK1-OE group showed significantly decreased MDA levels and markedly increased GSH and GPX levels. Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE group, the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 group showed significantly elevated MDA levels and significantly reduced GSH and GPX levels; Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 group, the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 group showed significantly reduced MDA levels and significantly increased GSH and GPX levels; Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 group, the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 + Erastin group exhibited significantly elevated MDA levels and markedly reduced GSH and GPX levels (Fig. 3B).
Additionally, intracellular reactive oxygen species (ROS) levels were detected using DCFH-DA staining. Compared with the (-) group, ROS levels were significantly elevated in the (-) + hypoxia + FeCl₃ group; Compared with the (-) + hypoxia + FeCl₃ group, ROS levels were significantly reduced in the (-) + hypoxia + FeCl₃ + ELK1-OE group; Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE group, ROS levels were significantly elevated in the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 group; Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 group, ROS levels were significantly reduced in the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 group; Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 group, ROS levels were significantly elevated in the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 + Erastin group (Fig. 3C).
ELK1 induces ferroptosis in hippocampal cells by regulating mTOR/CREB/YAP/TFEB signaling
To elucidate the effects of ELK1 on the expression of ferroptosis-related proteins in hippocampal cells, we detected relevant markers via Western blotting. Compared with the (-) group, the (-) + hypoxia + FeCl3 group exhibited significantly decreased expression of SLC7A11 and GPX4 proteins, while NCOA4, nuclear TFEB, Beclin, and SQSTM1 protein expression was significantly elevated. Compared with the (-) + hypoxia + FeCl3 group, the (-) + hypoxia + FeCl3+ ELK1-OE group showed significantly increased SLC7A11 and GPX4 protein expression, while NCOA4, nuclear TFEB, Beclin, and SQSTM1 protein expression was significantly decreased; Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE group, the (-) + hypoxia + FeCl3+ ELK1-OE + 666 − 159 group showed significantly decreased SLC7A11 and GPX4 protein expression, while NCOA4, nuclear TFEB, Beclin, and SQSTM1 protein expression was significantly increased; Compared with the (-) + hypoxia + FeCl3 + ELK1-OE + 666 − 159 group, the (-) + hypoxia + FeCl3+ ELK1-OE + 666 − 159 + PY-60 group showed significantly increased SLC7A11 and GPX4 protein expression, while NCOA4, nuclear TFEB, Beclin, and SQSTM1 protein expression was significantly decreased; Compared with the (-) + hypoxia + FeCl3+ ELK1-OE + 666 − 159 + PY-60 group, the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 + Erastin group showed significantly decreased SLC7A11 and GPX4 protein expression, while NCOA4, nuclear TFEB, Beclin, and SQSTM1 protein expression were significantly increased (Fig. 4A).

ELK1 induces iron death in hippocampal cells by regulating mTOR/CREB/YAP/TFEB signaling. A: Western blot analysis of SLC7A11, GPX4, NCOA4, nuclear TFEB, Beclin, and SQSTM1 protein expression in hippocampal cells under different stimulation treatments. B: Colorimetric assay of Fe²⁺ levels in hippocampal cells under different treatment conditions. Groups: (-) control group, (-) + hypoxia + FeCl3 group, (-) + hypoxia + FeCl₃ + ELK1-OE group, (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 group, (-) + hypoxia + FeCl3+ ELK1-OE + 666 − 159 + PY-60 group, (-) + hypoxia + FeCl3+ ELK1-OE + 666 − 159 + PY-60 + Erastin group. C: Flow cytometry analysis of hippocampal cell apoptosis rates under different treatment conditions. Groups: Normal control group, 666 − 159+PY-60 + Erastin group. Data are expressed as mean ± standard deviation. GAPDH served as the housekeeping protein, and Lamin B1 as the nuclear protein marker. N = 3. *P < 0.05, **P < 0.01, nsP>0.05: indicates no statistically significant difference.
Simultaneously, intracellular Fe2+ levels were measured using a colorimetric assay. Compared to the (-) group, Fe2+ levels were significantly elevated in the (-) + hypoxia + FeCl₃ group; Compared with the (-) + hypoxia + FeCl₃ group, Fe2+ levels were significantly reduced in the (-) + hypoxia + FeCl₃ + ELK1-OE group; Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE group, Fe2+ levels were significantly increased in the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 group; Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 group, Fe2+ levels were significantly reduced in the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 group; Compared with the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 group, Fe2+ levels were significantly elevated in the (-) + hypoxia + FeCl₃ + ELK1-OE + 666 − 159 + PY-60 + Erastin group (Fig. 4B).
To verify the potential cytotoxicity of the various inhibitor and agonist compounds used in this study on hippocampal cells, we assessed apoptosis rates via flow cytometry. As shown in Fig. 4C, compared to the NC group, the apoptosis rates in the 666 − 159+PY-60 + Erastin groups did not significantly increase (P > 0.05) and remained at low levels (< 15%). These results indicate that the key compounds used in this study did not induce additional cytotoxicity in hippocampal cells at the concentrations employed. Consequently, the effects observed in subsequent analyses can be attributed to biological regulation within specific signaling pathways (Fig. 4C).
Mechanistic studies confirm that ELK1 alleviates the progression of vascular dementia by regulating the mTOR/CREB/YAP/TFEB signaling cascade in hippocampal neurons, thereby suppressing ferroptosis-related protein expression, apoptosis, and iron overload (Fig. 5). The original membranes and raw data for this study were supplemented in Supplementary File 1 and Supplementary File 2, respectively.

ELK1 inhibits the progression of vascular dementia by regulating mTOR/CREB/YAP/TFEB signaling-induced iron death in hippocampal cells.
Discussion
Neuronal cell loss and a significant decline in brain function characterize Vascular Dementia (VD), a cognitive impairment resulting from cerebrovascular diseases21. Ferroptosis, a kind of cell death reliant on iron, has garnered significant attention in vascular disease research22. This study focuses on analyzing the roles and interconnections among several stages of the ferroptosis process, aiming to elucidate the specific pathways and regulatory mechanisms of ferroptosis in hippocampal cells in vascular dementia.
Vascular dementia causes the blood flow to the brain to slow down, which leads to ischemia and hypoxia in brain cells. This, in turn, creates metabolic issues and more oxidative stress in the cells. A lot of cells are dying in the hippocampus, which is a part of the brain that is extremely sensitive to ischemia and hypoxia23. Research indicates that a substantial amount of Fe3+ is produced with the death of hippocampal cells. This Fe3+ may then go into other hippocampal cells via the transferrin receptor (TfR) and link with ferritin within the cells24. Iron is stored in the protein ferritin. Its major jobs are to keep Fe3 + safe and protect cells from harm caused by too much free iron25.
Fe3+ reacts with ferritin and then with the protein NCOA4 to make autophagosomes. The NCOA4 protein, which is involved in selective autophagy, may be able to identify and bind to ferritin, which lets it enter autophagosomes26. When ferritin is broken down to make autophagosomes, a lot of Fe2+ is released. The accumulation of Fe2+ within the cell makes it much more likely that reactive oxygen species (ROS) will form. When there are too many ROS, they may induce lipid peroxidation of the cell membrane, which leads to ferroptosis27.
The accumulation of ROS not only directly induces ferroptosis but also influences cellular physiological processes via many communication pathways. One key stage in the process is the activation of p38 mitogen-activated protein kinase (p38 MAPK)28. P38 phosphorylation may start downstream signaling cascades, including the phosphorylation of the transcription factor SP1. SP1 phosphorylation activates the transcription of DNA methyltransferase 1 (DNMT1)29. When DNMT1 expression goes up, it stops the action of the transcription factor AP-2α, which in turn stops the production of other genes. This leads to higher levels of DNA methylation. When DNMT1 stops AP-2α from working, it also stops AP-2α from promoting the transcription of the ELK1 gene. The transcription factor ELK1, which is part of several cellular signaling pathways, increases the expression and transcription of the mechanistic target of rapamycin (mTOR). mTOR activation is a key part of signaling pathways. It helps cells grow and metabolize, but it is also significantly associated to the development of many diseases30. The active mTOR may add more phosphate groups to the cAMP response element-binding protein (CREB)31. Phosphorylated CREB starts the transcription of YAP (Yes-Associated Protein), which is an important part of the Hippo signaling cascade. YAP activation inhibits the expression of glutathione peroxidase 4 (GPX4) and SLC7A11 (Solute Carrier Family 7 Member 11). Lower levels of two important antioxidant proteins, GPX4 and SLC7A11, weaken the cellular antioxidant system, which makes ROS levels rise and speeds up ferroptosis32. Additionally, via phosphorylating the transcription factor EB (TFEB), phosphorylated mTOR may inhibit the expression of genes associated with autophagy33. TFEB, an important regulator of autophagy, is phosphorylated and remains in the cytoplasm, where it can’t enter the nucleus and start transcription. Thus, inhibiting TFEB prevents ferroptosis by lowering the expression of genes that are related to autophagy34. Autophagy usually keeps cells healthy by getting rid of damaged proteins and organelles35. On the other hand, autophagy exacerbates ferroptosis by causing ROS buildup and Fe2+ emissions36.
In short, the ferroptosis mechanism causes a lot of cell death in hippocampus cells in the vascular dementia model. This process starts with the release of Fe3 + and the buildup of ferritin. Then, NCOA4 starts the autophagy process, which releases a lot of Fe2 + and ROS, which causes ferroptosis. The buildup of ROS leads to the phosphorylation of SP1 via the phosphorylation of p38. This increases the expression of DNMT1 and stops AP-2α. AP-2α suppression weakens the transcriptional boosting effect of ELK1 by causing mTOR phosphorylation. Phosphorylated mTOR also stops ferroptosis and lowers TFEB while activating the YAP pathway. ELK1 is a key player in the process of hippocampal cell ferroptosis because it encourages the activation of the mTOR/CREB/YAP signaling pathway and stops TFEB production. This slows down the progression of vascular dementia.
Although this study preliminarily revealed through in vivo and in vitro experiments the mechanism by which ELK1 suppresses ferroptosis in hippocampal cells by regulating the mTOR/CREB/YAP/TFEB signaling pathway, we also acknowledge its limitations. The intricate cellular microenvironment, neurovascular unit interactions, and systemic immune regulation present in the in vivo hippocampus cannot be entirely replicated by cultured hippocampal neurons in vitro, despite their ability to emulate significant pathological characteristics of vascular dementia under hypoxic, low-serum, and iron-treated conditions. To enhance the clinical translational relevance of our findings, subsequent research may employ in vivo conditional knockout/overexpression models, co-culture systems, or organoid-like hippocampal slice cultures to further validate the functionality of this pathway in conditions that closely resemble physiological or pathological states. Future research may further study the principal molecules of these signaling pathways and their distinct activities in many clinical contexts, providing more comprehensive scientific rationales for targeted therapy strategies. Additionally, research using animal models and clinical trials may validate the safety and efficacy of these therapeutic strategies, offering novel concepts and methodologies for the treatment of vascular dementia.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Chang, W. E. & Chang, C. H. Vascular cognitive impairment and dementia. Continuum (Minneap Minn). 28 (3), 750–780 (2022).
O'brien, J. T. & Thomas, A. Vascular dementia. Lancet 386 (10004), 1698–1706 (2015).
Sachdev, P. et al. Diagnostic criteria for vascular cognitive disorders: a VASCOG statement. Alzheimer Dis. Assoc. Disord. 28 (3), 206–218 (2014).
Iadecola, C. et al. Vascular cognitive impairment and dementia: JACC scientific expert panel. J. Am. Coll. Cardiol. 73 (25), 3326–3344 (2019).
Livingston, G. et al. Dementia prevention, intervention, and care: 2020 report of the lancet commission. Lancet 396 (10248), 413–446 (2020).
Slotnick, S. D. The hippocampus and long-term memory. Cogn. Neurosci. 13 (3–4), 113–114 (2022).
Duering, M. et al. Neuroimaging standards for research into small vessel disease-advances since 2013. Lancet Neurol. 22 (7), 602–618 (2023).
Snyder, J. S. et al. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 476 (7361), 458–461 (2011).
Belkhelfa, M. et al. The involvement of neuroinflammation and necroptosis in the hippocampus during vascular dementia. J. Neuroimmunol. 320, 48–57 (2018).
Tiedt, S. et al. The neurovascular unit and systemic biology in stroke - implications for translation and treatment. Nat. Rev. Neurol. 18 (10), 597–612 (2022).
Heneka, M. T. et al. Neuroinflammation in alzheimer disease. Nat. Rev. Immunol. 25 (5), 321–352 (2025).
Zhang, X. & Wu, M. Fast-spiking parvalbumin-positive interneurons: new perspectives of treatment and future challenges in dementia. Mol. Psychiatry. 30 (2), 693–704 (2025).
Chae, S. et al. The dopamine analogue CA140 alleviates AD pathology, neuroinflammation, and rescues synaptic/cognitive functions by modulating DRD1 signaling or directly binding to Abeta. J. Neuroinflamm. 21 (1), 200 (2024).
Wheeler, M. A. et al. Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell 176 (3), 581–96e18 (2019).
Gallegos, D. A. et al. Chromatin regulation of neuronal maturation and plasticity. Trends Neurosci. 41 (5), 311–324 (2018).
Lee, H. et al. Cell Type-Specific transcriptomics reveals that mutant Huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron 107 (5), 891–908e8 (2020).
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1), D672–d7 (2025).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. Publ. Protein Soc. 28 (11), 1947–1951 (2019).
Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30 (2000).
Khoshnam, S. E. et al. Memory deficits and hippocampal inflammation in cerebral hypoperfusion and reperfusion in male rats: neuroprotective role of vanillic acid. Life Sci. 211, 126–132 (2018).
Kalaria, R. N., Akinyemi, R. & Ihara, M. Stroke injury, cognitive impairment and vascular dementia. Biochim. Biophys. Acta. 1862 (5), 915–925 (2016).
Fu, P. et al. Effect of ferroptosis on chronic cerebral hypoperfusion in vascular dementia. Exp. Neurol. 370, 114538 (2023).
Hase, Y. et al. Hippocampal capillary pericytes in post-stroke and vascular dementias and alzheimer’s disease and experimental chronic cerebral hypoperfusion. Acta Neuropathol. Commun. 12 (1), 29 (2024).
Xie, Z. et al. Activated AMPK mitigates diabetes-related cognitive dysfunction by inhibiting hippocampal ferroptosis. Biochem. Pharmacol. 207, 115374 (2023).
Lee, K. K. et al. Ferritin-nanocaged copper arsenite minerals with oxidative stress-amplifying activity for targeted cancer therapy. J. Control Release. 361, 350–360 (2023).
Ohshima, T. et al. NCOA4 drives ferritin phase separation to facilitate macroferritinophagy and microferritinophagy. J. Cell. Biol. 221, 10 (2022).
Park, E. & Chung, S. W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell. Death Dis. 10 (11), 822 (2019).
Zeng, Y. et al. Nickel induces blood-testis barrier damage through ROS-mediated p38 MAPK pathways in mice. Redox Biol. 67, 102886 (2023).
Huang, P. et al. DNMT1 and Sp1 competitively regulate the expression of BACE1 in A2E-mediated photo-oxidative damage in RPE cells. Neurochem Int. 121, 59–68 (2018).
Liu, G. Y & Sabatini, D. M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell. Biol. 21 (4), 183–203 (2020).
Kumari, S., Dhapola, R. & Reddy, D. H. Apoptosis in alzheimer’s disease: insight into the signaling pathways and therapeutic avenues. Apoptosis 28 (7–8), 943–957 (2023).
Jiang, X. et al. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell. Biol. 22 (4), 266–282 (2021).
Zhang, Y. et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 12 (1), 1589 (2021).
Liu, L. et al. Autophagy-Dependent ferroptosis as a therapeutic target in cancer. ChemMedChem 16 (19), 2942–2950 (2021).
Dikic, I. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell. Biol. 19 (6), 349–364 (2018).
Shariq, M. et al. PRKAA2, MTOR, and TFEB in the regulation of lysosomal damage response and autophagy. J. Mol. Med. (Berl.) 102 (3), 287–311 (2024).
Funding
We thank the grants from 2022 Hebei Province Government-funded Excellent Talents Project in Clinical Medicine(GrantNo.2022-180-5) and Scientific and Technological Innovation 2030-Major Project Subject of “Brain Science and Brain-inspiredResearch”(Grant No.2021ZD0201807).
Author information
Authors and Affiliations
Contributions
J.X, M.M.L and Q.Q.Q conceived the design concept and wrote the first draft; J.A, YNX, T.Y.G and Z.J.T performed the cell cultures and completed the experiments; W.H.C, R.L, Y.H.D, Y.Z.C and P.Y.L managed and analysed the data. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was approved by the Medical Ethics Committee of Hebei General Hospital, Approval No. 2025-DW-018.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, J., Liu, M., Qi, Q. et al. ELK1 suppressed the progression of vascular dementia via modulating mTOR/CREB/YAP/TFEB signaling induced ferroptosis in hippocampal cells. Sci Rep 16, 11088 (2026). https://doi.org/10.1038/s41598-026-40339-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-40339-3
