
Abstract
Four budding and pink-pigmented planctomycetal strains, designated SH449T, SH551T, SH467T, and SH501T, were isolated from diverse environmental sources, including Fjord Schlei (a 42 km brackish arm of the Baltic Sea), ponds and wastewater lagoons in Northern Germany, as part of an isolation campaign led by Heinz Schlesner at Kiel University between the 1980s and early 2000s. Analyses based on five phylogenetic markers indicate that these strains constitute a novel genus within the family Pirellulaceae of the phylum Planctomycetota. All four strains are aerobic, heterotrophic, mesophilic, and exhibit neutrophilic properties. Strains SH551T and SH501T possess at least one plasmid that is not present in the other two strains. Genome analyses revealed that their genomes are of comparable size to those of other members of the Pirellulaceae family, exhibiting a DNA G + C content ranging from 49 to 54%. Analysis of predicted biosynthetic gene clusters shows that all four strains possess the genetic potential to produce bioactive compounds and may therefore be regarded as putative “talented producers”. The polyphasic approach places all four novel strains in a new genus within the family Pirellulaceae, for which the name Brakhagea gen. nov. is proposed. The novel isolates represent four distinct novel species, Brakhagea baltica sp. nov. (type strain SH449T = KCTC 102025T = DSM 116586T), Brakhagea lacunae sp. nov. (type strain SH551T = KCTC 102023T = DSM 116785T = CECT 30846T), Brakhagea slesvicensis sp. nov. (type strain SH467T = KCTC 102026T = DSM 116378T), and Brakhagea aquatica sp. nov. (type strain SH501T = DSM 102088T = CECT 30906T).
Similar content being viewed by others

Introduction
Planctomycetota, a bacterial phylum within the Planctomycetota–Verrucomicrobiota–Chlamydiota (PVC) superphylum1, comprises Gram-negative bacteria with uncommon morphological and physiological characteristics that differentiate them from other bacterial taxa. These characteristics include diverse reproductive morphotypes, such as budding or binary fission, and the absence of the otherwise ubiquitous bacterial cell division protein FtsZ2,3,4,5. Some planctomycetes known as ‘bacteria of prey’ are thus far the only bacteria employing phagocytosis-like cell engulfment to eat other bacteria alive6,7. Members of the phylum Planctomycetota are characterized by a highly condensed nucleoid, the presence of cytoskeleton-like elements, extensive cytoplasmic membrane invaginations, and complex dimorphic life cycles4. The life cycle observed in many strains starts from a sessile mother cell that is attached to a surface via a holdfast structure8. Subsequently, a flagellated daughter cell originates from the mother cell, grows and eventually pinches off from the mother cell9. Morphologically, planctomycetal colonies display a wide spectrum of colors, ranging from white and beige to pink, red, or orange10, while individual cells display a variety of morphologies ranging from spherical to rod, ovoid, or pear-shaped forms11. Cells often feature crateriform structures on their cell surface with pili-like fibre structures around or associated with them. However, their fastidious nature poses significant challenges for isolation using standard microbiological media, as they are frequently outcompeted by faster-growing bacteria. The distinctive cellular biology of planctomycetes is complemented by yet untapped metabolic functions, which offer significant potential for biotechnological applications. Recent studies have demonstrated that members of the phylum display antimicrobial activity, probably by the biosynthesis of small molecules4,12. Relative to other bacterial phyla, planctomycetes can have large genomes of up to 12.4 Mbp13. Furthermore, this phylum is distinguished by containing the highest proportion of predicted genes with unknown functions4. This is partly due to the only recent development of genetic tools for the phylum Planctomycetota14,15.
Planctomycetes exhibit remarkable metabolic versatility regarding the utilization of carbon and energy sources including complex polysaccharides, enabling them to colonize a diverse array of ecological niches4. Most of the characterized strains so far are aerobic, mesophilic, and heterotrophic, thriving in environments with acidic to alkaline pH. However, certain strains, notably the members of the class “Candidatus Brocadiia”, follow a specialized metabolism involving the anaerobic oxidation of ammonium (anammox)16. Representatives of the described classes of the phylum have been identified across a broad spectrum of terrestrial habitats, including soils17,18 and peat bogs19, as well as aquatic environments such as marine waters20,21,22,23, freshwater systems24,25, sediments26,27,28, and deep-sea deposits29. Some strains are even capable of inhabiting extreme environments, including hot springs30, and desert soils31 or marine volcanic areas32,33,34,35,36,37,38, underscoring their exceptional adaptability to different ecological conditions. Additionally, in numerous instances members have been documented in (symbiotic) association with other organisms, including sponges22,39,40,41,42,43, jellyfish44, phototrophs11,45,46,47,48,49,50,51, and the gastrointestinal tract52. However, besides biotic surfaces, many members of the phylum Planctomycetota attach to abiotic surfaces as well if incubated in marine or limnic habitats46,50,53,54.
In this study, we describe a novel genus that includes four new species within the family Pirellulaceae. According to the List of Prokaryotic Names with Standing in Nomenclature (LPSN), the current family Pirellulaceae (as of March 2026) encompasses 18 genera validly published under the International Code of Nomenclature of Prokaryotes (ICNP) and includes the highest number of described species within the entire phylum. The isolates presented here belong to the strain collection of Heinz Schlesner (“SH” strains, named after his initials), a pioneering researcher of budding bacteria, including Planctomycetota, at Kiel University, Germany. Over the last two decades of the 20th century, he isolated hundreds of novel strains from various locations in Northern Germany and world-wide. Following Schlesner’s retirement in July 2002, the collection was transferred to the research group led by Michael Thomm at the Institute for Biochemistry, Genetics, and Microbiology at the University of Regensburg. In 2019, the collection of 500 strains was relocated to the Department of Microbial Interactions at Friedrich Schiller University Jena55. Advances in genome sequencing compared to the end of the last century will now enable the characterization of the novel taxa in this collection based on high-quality genome sequences in addition to the analysis of phenotypic features. The phylogenetic inference is mainly based on the analysis or single gene- and whole-genome-based phylogenetic markers that turned out to have a better resolution compared to classical chemotaxonomic analyses when applied for strains belonging to the phylum Planctomycetota39.
Materials and methods
Strains, cultivation and 16S rRNA gene sequences
All four strains, SH449T, SH501T, SH551T, and SH467T were isolated from distinct water bodies in Northern Germany by Heinz Schlesner. Strain SH449T was obtained from Fjord Schlei, a 42 km brackish arm of the Baltic Sea, strain SH501T from surface water at a lagoon in Schrevenpark pond in Kiel, strain SH551T from a village pond in Kopendorf, Fehmarn Island and strain SH467T from the wastewater aeration lagoon at a former sugar processing plant in Schleswig. Detailed methodologies and findings related to their isolation are documented in studies conducted under the supervision of Heinz Schlesner56,57. Cultivation of the strains was performed using two distinct media: M30PY for strains SH449T and SH467T, and M1aPY for strains SH501T and SH551T. The preparation of these media followed previously established protocols58. The designation “PY” denotes the supplementation of 0.25 g/L peptone and 0.25 g/L yeast extract in the media. Solid media were prepared with 15 g/L agar that was autoclaved separately in a volume of 200 mL distilled water and added to the autoclaved medium prior to pouring of the plates. The 16S rRNA genes of all four isolates were amplified via polymerase chain reaction (PCR), purified using a standardized workflow59, and subsequently sequenced at Macrogen Europe (Amsterdam, The Netherlands).
Physiological analyses
To ascertain the optimal temperature for microbial growth, 100 µL of supernatant from an exponentially growing culture, devoid of visible aggregates, was spread on agar plates. These plates were incubated in duplicates at temperatures ranging from 4 °C to 42 °C. Daily inspections were conducted, and growth was assessed by determining the time required for the formation of visible colonies or lawns. The temperature at which colonies or lawns first appeared was identified as the optimal growth temperature. The optimal pH for growth was determined in a two week 96-well plate reader experiment with continuous shaking. Media were supplemented with 100 mM of one of the following buffering agents: 2-(N-morpholino)ethanesulfonic acid (MES) for pH 5.0 and 6.0, 4-(2-hydroxyethyl)−1-piperazineethanesulfonic acid (HEPES) for pH 7.0, 7.5, and 8.0, or N-cyclohexyl-2-aminoethanesulfonic acid (CHES) for pH 9.0 and 10.0. Growth was quantified by measuring the optical density at 600 nm (OD600) using a BioTek Epoch2 microplate spectrophotometer (Agilent) at 24–28 °C depending on the determined temperature optimum of the strain. Each condition was tested in duplicates. Each measurement cycle, lasting 30 min, comprised two shaking phases of 15 min interrupted by the OD600 measurements of the entire 96-well plate (Brand Plate pureGrade™ S, transparent sterile 96-well plates). The shaking regime of the 15 min interval was changed between linear to orbital to double-orbital every 5 min. To mitigate condensation on the plate lid, a temperature gradient was established, maintaining the liquid at 24–28 °C (depending on the temperature optimum of the strain) and the lid temperature was maintained at a 2 °C higher temperature. For data analysis, the mean values of each time point were calculated, and the mean of the medium blank was subtracted. The growth rate at each pH was calculated from a selection of data points with the maximal slope of the natural logarithm of the OD600 values plotted against the cultivation time.
Light microscopy and cell size determination
Light microscopy was performed following the methodology outlined in a previous study60. In brief, cells harvested from liquid cultures at the half-maximal OD600 were mounted on a 1% (w/v) agarose cushion prepared in deionized water (dH2O). Once the culture had dried on the agarose cushion, a coverslip was applied and secured at the edges with VLAP (a 1:1:1 mixture of vaseline, lanolin, and paraffin by weight) to ensure stability. Imaging was conducted using an inverted Nikon Ti2 microscope equipped with a Nikon Plan Apo λ 100x immersion oil objective, configured with a phase ring for phase-contrast (PhC) imaging or without for differential interference contrast (DIC) imaging61. The system included a Nikon DS-Ri2 camera and NIS-Elements software (version 5.30). Three-channel RGB images were processed in FIJI62 to generate single-channel RGB images. TIFF files were subsequently analyzed in BacStalk63, with cell segmentation performed using thresholds of 25 pixels for cell size and 15 pixels for minimum cell size. A total of three biological replicates, each comprising 150 cells, were evaluated. For data visualization, results were uploaded to SuperPlotsOfData64. To enhance visualization, brightness and contrast adjustments were manually applied to PhC and DIC images.
Genomic DNA isolation, genome sequencing, annotation and analysis
Genomic DNA extraction, and quality control were conducted following established protocols60. De novo genome assembly was performed using long-read data from Oxford Nanopore, with subsequent polishing utilizing short-read data from Illumina sequencing. Details on the sequencing chemistry and bioinformatic workflow as well as tools, tool versions and optional parameters are stated in Table S1. Illumina sequencing was performed by Eurofins Genomics (Ebersberg, Germany). The genome completeness was assessed with BUSCO (version 5.8.2), while coding density and DNA G + C content were evaluated using CheckM (version 1.2.3). Following an initial annotation with Prokka (version 1.14.5), the chromosome was re-oriented to the start codon of the dnaA gene encoding the replication initiator protein, and subjected to final re-annotation using PGAP (version 2025-05−06, build 7983).
Nucleotide sequence accession numbers
The 16S rRNA gene sequences were deposited in the GenBank database under the following accession numbers: PV955651 (SH449T), PV955658 (SH551T), PV955674 (SH467T) and PV955675 (SH501T). Genome sequence information is available from NCBI under the accession numbers: SH449T: CP197409; SH551T: CP197424 (chromosome) and CP197425 (plasmid pSH551_1); SH467T: CP197412; SH501T: CP197422 (chromosome) and CP197423 (plasmid pSH501_1).
Phylogenetic and genome-based analyses
The full-length 16S rRNA gene sequences of the novel planctomycetal isolates were retrieved from the genomes annotated with Prokka and employed to identify the closest relatives via NCBI BLAST. Maximum-likelihood phylogenetic trees were constructed based on 16S rRNA gene sequences and multi-locus sequence analysis (MLSA) for the novel strains and type strains of all species within the phylum Planctomycetota, with the MLSA-based tree limited to strains belonging to the family Pirellulaceae. The 16S rRNA gene sequences of the type strains of Opitutus terrae (NCBI accession no. AJ229235), Kiritimatiella glycovorans (accession no. NR_146840), and Lentisphaera araneosa (accession no. NR_027571), representing members of the Planctomycetota-Verrucomicrobiota-Chlamydiota (PVC) superphylum outside of the phylum Planctomycetota, were used as outgroup. Sequence alignments were conducted using ClustalW65, and phylogenetic trees were reconstructed with FastTree v2.266 employing 1000 bootstrap replicates. The MLSA-based phylogeny was inferred using the autoMLST tool67 with 500 bootstrap replicates, including the genomes of Planctopirus limnophila DSM 3776T (GenBank acc. no. CP001744.1), Gimesia maris DSM 8797T (GenBank acc. no. CP042910.1) and Planctomicrobium piriforme DSM 26348T (GenBank acc. no. GCA_900113665.1), all from the family Planctomycetaceae, as outgroup. Phylogenetic trees were visualized using iTOL v668. A 16S rRNA gene sequence similarity matrix was generated using TaxonDC69 based on the ClustalW alignment used for phylogenetic tree construction. Average amino acid identities (AAI) and average nucleotide identities (ANI) were calculated using scripts from the enveomics collection70. Additional phylogenetic markers, including rpoB gene sequence similarity and percentage of conserved proteins (POCP), were determined following established methods71,72. The pangenome of selected strains was constructed using anvi’o v.8 with default parameters73. Biosynthetic gene clusters (BGCs) were predicted with antiSMASH v.8.074 in relaxed detection mode and with all extra features activated, and carbohydrate-active enzymes (CAZymes) were identified using dbCAN375.
Results and discussion
Phylogenetic inference
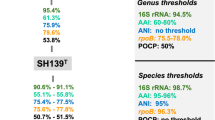
Initial nucleotide BLAST analyses of the 16S rRNA gene sequences of the four strains SH449T, SH551T, SH467T, and SH501T demonstrated the highest sequence similarity (< 90%) to Aureliella helgolandensis Q31aT in the family Pirellulaceae (Fig. 1). This level of similarity is substantially below the established 94.5% threshold for 16S rRNA gene sequence similarity used to delineate a new genus72, indicating a genus-level relationship within the Pirellulaceae family. Furthermore, comparisons of POCP and AAI values between the four isolates and A. helgolandensis Q31aT revealed maximum similarities of 42.4% and 50.4%, respectively. These values fall well below the recognized thresholds of 50% for POCP and 60% for AAI used for genus delineation72, thereby providing robust support for the proposal of a novel genus (Fig. 1).

Comparison of phylogenetic markers for genus and species delineation. Markers used: 16S rRNA gene sequence identity (16S rRNA), average amino acid identity (AAI), average nucleotide identity (ANI), sequence similarity of a partial sequence of the rpoB gene (rpoB), percentage of conserved proteins (POCP).
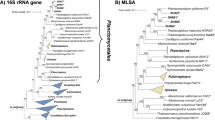
The 16S rRNA gene sequence similarity between strains SH449T and SH551T is 99.4%, while that between strain SH467T and SH501T is 99.6%, both exceeding the 98.7% threshold typically used to delineate bacterial species on the basis of 16S rRNA gene sequence similarity72(Fig. 1). Both phylogenetic trees show clustering patterns that reflect these similarities (Fig. 2). However, the MLSA-based phylogenetic tree suggests more distant clustering; with SH449T and SH551T forming a closer-related clade separate from the clade of SH467T and SH501T (Fig. 2). In a comparative analysis of phylogenetic markers between strains SH449T and SH551T, ANI, AAI, and rpoB gene sequence similarity values were determined to be 79.2%, 76.6%, and 83.0%, respectively. Likewise, for strains SH467T and SH501T, the corresponding ANI, AAI, and rpoB similarity values were 79.6%, 82.1%, and 85.3%, respectively. These values fall substantially below the established thresholds for species delineation, which are 95% for ANI, 95–96% for AAI, and 96.3% for rpoB71,72(Fig. 1). Consequently, these findings provide robust evidence that strains SH449T, SH551T, SH467T, and SH501T represent four distinct novel species. Comparative analysis of the clades encompassing SH449T/SH551T and SH467T/SH501T indicates notably high POCP (> 50.0%) and AAI (> 60.0%) values, clearly confirming that all four novel isolates belong to the same novel genus. Similar observations that strains belong to separate species despite 16S rRNA gene sequence similarities exceeding the threshold occur occasionally in the phylum Planctomycetota. The 16S rRNA gene sequence identity of Planctopirus limnophila Mü290T and Planctopirus ephydatiae spb1T is 99.99% and chemotaxonomic measures fell short in distinguishing the two species. Only the analysis using whole genome-based phylogenetic markers revealed that they indeed represent two separate species39.

Phylogenetic placement. (A) Maximum likelihood phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic relationship of the novel isolates and other members of family Pirellulaceae. Bar, 0.1 substitutions per nucleotide position. (B) Multi-locus sequence analysis (MLSA)-based phylogenetic tree constructed with the genomes of characterized members in the family Pirellulaceae. The tree was computed based on a set of at least 30 single-copy gene-encoding proteins in a maximum likelihood approach with 500 bootstrap replications. Bar, 0.1 substitution per amino acid position. Bootstrap values for both trees are given at the nodes (in %). Phylogenetic trees were visualized with iTOL v6. In both trees, the collapsed clade Rhodopirellula includes the genera Allorhodopirellula, Aporhodopirellula and Neorhodopirellula (that we do not regard as distinct genera), while the collapsed clade Stieleria includes the genus Roseiconus (the Roseiconus species were recently re-named Stieleria). NCBI RefSeq accession numbers of the genome sequences used for tree reconstruction are provided in Table S2.
Genomic characteristics
In comparison to the genome of A. helgolandensis Q31aT, which has a size of 8.44 Mbp, the genomes of the four novel isolates are notably smaller, with strain SH467T exhibiting a genome size of only 5.87 Mbp (Table 1). Strains SH551T and SH501T harbor a plasmid, whereas strains SH449T and SH467T lack any extrachromosomal elements. The DNA G + C content of the novel strains ranges from 49.4 to 54.2%, which is slightly below the value obtained for the genome of A. helgolandensis Q31aT (55.3%). In terms of numbers of protein-coding genes per Mbp, relative numbers of hypothetical protein-encoding genes, coding density, and numbers of tRNA genes, the genomes of strains SH449T and SH551T exhibit only minor differences when compared to the more pronounced differences observed between strains SH467T and SH501T (Table 1). Across all four strains, no variation in the copy numbers of 5S, 16S, and 23S rRNA genes can be observed, which are consistently present in single copy per genome. In contrast, in A. helgolandensis Q31aT, these genes are present in two copies each. Among the novel strains, strain SH501T exhibits the highest tRNA gene count, with 81 tRNA genes, while strain SH467T harbours only 61 tRNA genes.
Pangenome reconstruction and evaluation of genome-encoded functions
To visualize the genome-based similarity among the compared strains, a pangenome was constructed and analyzed. This pangenome, encompassing the genomes of all five species including A. helgolandensis Q31aT, comprises 14,479 genes, with 1,057 genes being conserved across all five strains (core genome) and 2,038 genes being conserved among the four novel isolates (Fig. 3). The remaining genes are either unique to individual strains (singletons) or not conserved across all strains. The singleton gene count for strains SH449T, SH551T, SH467T, and SH501T is 1588, 1440, 869, and 1308, respectively. Beyond the core genome (represented in the 3–4 o’clock segment in the pangenome visualization), the isolates SH467T and SH501T share additional genes (5 o’clock in the pangenome visualization). Likewise, the other two isolates SH449T, and SH551T, share separate additional genes (6 o’clock in the pangenome visualization). These findings align with the close species-level relationship among the novel isolates and their more distant relationship to A. helgolandensis Q31aT. Smaller genomes, like that of strain SH467T (5.8 Mbp), which likely encode fewer accessory functions compared to many other members in the class Planctomycetia (genome sizes of up to 12.4 Mbp), are vital for pinpointing conserved, yet uncharacterized genes in future studies.

Pangenome reconstruction. Each open circle represents the pangenome of all strains but is colored darker when the gene is present in the respective genome. The heatmap in the upper right corner indicates the degree of relationship based on ANI values (ANI ≤ 70%, pale orange to ANI = 100%, bright orange).
Genome mining for secondary metabolite-associated biosynthetic gene clusters (BGCs) using antiSMASH identified 7–12 BGCs each in the genomes of the four novel isolates (Table 1). Of these, four to five BGCs are likely associated with terpenoid or terpenoid precursor biosynthesis. Other predicted BGCs include genes encoding putative type I or type III polyketide synthases or are linked to the synthesis of short peptides formed by non-ribosomal peptide synthetase (NRPS)-like enzymes. The putative synthesis of resorcinols is unique to strain SH551T, while synthesis of indoles is specific to strain SH501T. Across all four novel isolates, the identification of CAZyme-encoding genes resulted in 120–200 hits per genome, with comparable numbers of hits in the classes of glycoside hydrolases and glycosyltransferases, though these numbers were lower than in A. helgolandensis Q31aT (Table 1). This is not surprising since the genome of A. helgolandensis Q31aT is at least 1 Mbp larger than that of the newly identified isolates. The counts of genes encoding polysaccharide lyases or proteins with auxiliary activities fall below ten per genome for all five genomes analyzed. This indicates that all five strains can in principle degrade complex carbohydrates into simpler sugars, however, the exact substrate spectrum should be determined in future cultivation experiments and cannot be predicted from the genome-based analysis. Strains SH449T and SH467T possess a single CRISPR array each with 34 and 45 spacers, respectively. Genes encoding phage-derived components (e.g., phage tail tape measure, minor head, and tail tube proteins) suggest prophage regions in the genomes. Conversely, the novel isolates SH551T and SH501T also contain genes encoding putative phage proteins but lack CRISPR arrays. The here described set of closely related strains could be valuable for investigating defense mechanisms or phage-host co-evolution in the phylum Planctomycetota. Noteworthy, the presence of plasmids in the two strains SH551T and SH501T that lack CRISPR array might suggest a potential functional relationship worth addressing further.
Physiological characterization
In both, agar plate and liquid cultures, all four strains exhibit light to dark pink pigmentation (Fig. 4), contrasting the lucid white appearance of their closest relative, A. helgolandensis Q31aT. Strains SH551T, SH467T, and SH501T produce circular, convex colonies characterized by entire margins. In contrast, colonies of strain SH449T exhibit either circular or irregular morphologies and a more mucoid consistency (Fig. 4). Strain SH449T exhibits growth within a temperature range of 18–32 °C, with an optimum at 28 °C, and demonstrates tolerance to alkaline conditions up to a pH of 10.0, with an optimal pH of 8.0. In contrast, strain SH551T displays growth between 21 and 28 °C, with an optimum at 28 °C, and prefers a pH of 7.5 for optimal growth (Table 2). Strains SH467T and SH501T demonstrate growth at temperatures up to a maximum of 37 °C, with an optimal growth temperature of 28 °C. Both, strains SH467T and SH501T, exhibit optimal growth at a pH of 7.5. Additionally, strain SH467T is capable of biomass formation at an alkaline pH of 10.0 (Table 2). The aerobic heterotrophic lifestyle of A. helgolandensis Q31aT is also followed by the four novel strains.

Appearance of colonies on plates. Colonies of the four strains have the same color, but colonies differ in size, shape and consistency. Strains SH467T and SH501T form smaller circular colonies, whereas colonies of strains SH449T and SH551T are much bigger, irregular and have a slimier consistency.
Phenotypic characterization
Microscopically, cells of the analyzed strains were observed to be pear-shaped (Fig. 5A, C, E, G). Out of all four, only strains SH449T and SH501T were able to form rosette-like aggregates (Fig. 5A and E). Cells have a cell length and width in a range of 1.4 to 1.7 μm and 1.1 μm, respectively. Their pear-shaped morphology is a shared feature with their current closest relative A. helgolandensis Q31aT; however, Q31aT cells were longer, but similar in width44. In accordance with strain Q31aT and most members of the order Pirellulales44, cells of all four strains divide through an asymmetric cell division type (polar budding) in which a small daughter cell emerges on a larger mother cell’s pole (Fig. 5A, C, E, G), grows over time and eventually pinches off from the mother cell.

Cell morphology and cell sizes. Phase contrast (PhC) and differential interference contrast (DIC) images of dividing cells of strains SH449T (A), SH467T (C), SH501T (E), and SH551T (G). Smaller daughter cells emerging from larger mother cells are depicted in the early division stage. Cell size analysis of strains SH449T (B), SH467T (D), SH501T (F), and SH551T (H) were determined from three biological replicates. Larger circles indicate mean values of each replicate. The scale bars represent 2 μm.
Conclusion
In this study, we introduce four novel species belonging to a novel genus in the family Pirellulaceae, expanding the known diversity of this understudied bacterial group through high-quality genome sequencing and robust phylogenomic analyses. While the determined genome sequences enabled precise species delineation and genus-level novelty based on five analysed phylogenetic markers, the work relies primarily on these molecular data for taxonomic placement, with physiological and biochemical characterizations limited to standard growth conditions and basic phenotypic traits. Chemotaxonomic profiling including fatty acid, polar lipid, and quinone analyses was not performed, representing a key limitation that precludes deeper insights into cell-wall architecture and membrane adaptations typical of planctomycetes. This approach aligns with the ongoing transition in prokaryotic taxonomy, where maturing genome sequencing technologies increasingly shift species identification toward data accumulation rather than purely phenotypic novelty. Nevertheless, the absence of functional genomic profiling highlights a broader challenge in the field that is predominantly related to the high number of proteins of unknown function encoded in planctomycetotal genomes. Future investigations could leverage these genomes for the analysis of secondary metabolite biosynthesis capabilities, catabolic activities or essential cell biological processes such as cell division to bridge the gap between gene content and phenotypic expression. By explicitly documenting these methodological boundaries, the present work maintains scientific rigor while providing foundational genomic resources that will facilitate subsequent multi-omics and experimental studies. Overall, the description of this novel genus contributes valuable biodiversity data to the broader microbial ecology community, paving the way for integrated taxonomic and functional explorations in the phylum Planctomycetota. For the novel taxa, we propose the names Brakhagea baltica gen. nov., sp. nov., represented by the type strain SH449T, Brakhagea lacunae sp. nov., represented by the type strain SH551T, Brakhagea slesvicensis sp. nov., represented by the type strain SH467T, and Brakhagea aquatica sp. nov., represented by the type strain SH501T.
Description of Brakhagea gen. nov.
Brakhagea (Brak.ha’ge.a. N.L. fem. n. Brakhagea, named to honor Prof. Axel A. Brakhage for his groundbreaking contributions in microbiology).
Members of the genus are heterotrophic, thrive in aerobic conditions, are mesophilic, and range from neutrophilic to slightly alkaliphilic. Cells are pink-pigmented, pear-shaped (drop-like shape) and divide by polar budding. Genomes have a DNA G + C content in the range of 49–54%. The genus is part of the family Pirellulaceae, order Pirellulales, class Planctomycetia, phylum Planctomycetota. The type species of the genus is Brakhagea baltica.
Description of Brakhagea baltica sp. nov.
Brakhagea baltica (bal’ti.ca. M.L. fem. adj. baltica, baltic, referring to its isolation from Fjord Schlei, a part of Baltic Sea).
Cells are ellipsoidal to pear-shaped with an average length and width of 1.4 × 1.1 μm. Cells are aerobic. The type strain is SH449T (= KCTC 102025T = DSM 116586T). It was isolated from Fjord Schlei, a 42 km brackish arm of the Baltic Sea. The type strain grows optimally at a pH of 8.0 (range 7.0–10.0) and a temperature of 28 °C (range 18–32 °C). The genome of the type strain is 7.05 Mb in size and has a DNA G + C content of 51.6%. The type strain lacks plasmids.
Description of Brakhagea lacunae sp. nov.
Brakhagea lacunae (la.cu’nae. L. gen. n. lacunae, of a pond, referring to the isolation from a village pond).
Cells are ellipsoidal to pear-shaped, with an average length and width of 1.6 × 1.1 μm. Cells are aerobic. The type strain is SH551T (= KCTC 102023T = DSM 116785T = CECT 30846T). It was isolated from a village pond in Kopendorf (Fehmarn Island), Northern Germany. It grows optimally at a pH of 7.5 (range 6.0–8.0) and a temperature of 28 °C (range 21–28 °C). The genome of the type strain is 7.25 Mb in size with a DNA G + C content of 49.4%. A single plasmid is present.
Description of Brakhagea slesvicensis sp. nov.
Brakhagea slesvicensis (sles.vi.cen’sis. M.L. fem. adj. slesvicensis, of Slesvicum, the latin name of Schleswig, a city in Northern Germany, from which the type strain was isolated).
Cells are ellipsoidal to pear-shaped, with an average length and width of 1.6 × 1.1 μm. Cells are aerobic. The type strain is SH467T (= KCTC 102026T = DSM 116378T). It was isolated from a wastewater aeration lagoon of a sugar processing plant in Schleswig, Northern Germany. It grows optimally at a pH of 7.5 (range 6.0–10.0) and a temperature of 28 °C (range 18–37 °C). The genome of the type strain is 5.87 Mb in size and has a DNA G + C content of 54.2%. The type strain lacks plasmids.
Description of Brakhagea aquatica sp. nov.
Brakhagea aquatica (a.qua’ti.ca. L. fem. adj. aquatica; pertaining to water, reflecting the isolation from surface water in a lagoon within a pond).
Cells are ellipsoidal to pear-shaped, with an average length and width of 1.7 × 1.1 μm. Cells are aerobic. The type strain is SH501T (= KCTC 102088T = CECT 30906T). It was isolated from Schrevenpark pond (Kiel) in Northern Germany. The type strain grows optimally at a pH of 7.5 (range 6.0–9.0) and a temperature of 28 °C (range 21–37 °C). The genome of the type strain is 6.70 Mb in size, has a DNA G + C content of 53.1% and includes a plasmid.
Data availability
The 16 S rRNA gene sequences were deposited in the GenBank database under the following accession numbers: PV955651 (SH449T), PV955658 (SH551T), PV955674 (SH467T) and PV955675 (SH501T). Genome sequence information is available from NCBI under the accession numbers: SH449T: CP197409; SH551T: CP197424 (chromosome) and CP197425 (plasmid pSH551_1); SH467T: CP197412; SH501T: CP197422 (chromosome) and CP197423 (plasmid pSH501_1).
References
Wagner, M. & Horn, M. The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr. Opin. Biotechnol. 17, 241–249. https://doi.org/10.1016/j.copbio.2006.05.005 (2006).
Wiegand, S., Jogler, M. & Jogler, C. On the maverick Planctomycetes. FEMS Microbiol. Rev. 42, 739–760. https://doi.org/10.1093/femsre/fuy029 (2018).
Mahajan, M., Seeger, C., Yee, B. & Andersson, S. G. E. Evolutionary Remodeling of the Cell Envelope in Bacteria of the Planctomycetes Phylum. Genome Biol. Evol. 12, 1528–1548. https://doi.org/10.1093/gbe/evaa159 (2020).
Wiegand, S. et al. Cultivation and functional characterization of 79 planctomycetes uncovers their unique biology. Nat. Microbiol. 5, 126–140. https://doi.org/10.1038/s41564-019-0588-1 (2020).
Jogler, C. et al. Identification of proteins likely to be involved in morphogenesis, cell division, and signal transduction in Planctomycetes by comparative genomics. J. Bacteriol. 194, 6419–6430. https://doi.org/10.1128/jb.01325-12 (2012).
Wurzbacher, C. E. et al. Candidatus Uabimicrobium helgolandensis-a planctomycetal bacterium with phagocytosis-like prey cell engulfment, surface-dependent motility, and cell division. mBio 15, e0204424. https://doi.org/10.1128/mbio.02044-24 (2024).
Shiratori, T., Suzuki, S., Kakizawa, Y. & Ishida, K. -i. Phagocytosis-like cell engulfment by a planctomycete bacterium. Nat. Commun. 10, 5529 (2019).
Jogler, C., Glöckner Frank, O. & Kolter, R. Characterization of Planctomyces limnophilus and Development of Genetic Tools for Its Manipulation Establish It as a Model Species for the Phylum Planctomycetes. Appl. Environ. Microbiol. 77, 5826–5829. https://doi.org/10.1128/AEM.05132-11 (2011).
Rivas-Marin, E. et al. Non-essentiality of canonical cell division genes in the planctomycete Planctopirus limnophila. Sci. Rep. 10, 66. https://doi.org/10.1038/s41598-019-56978-8 (2020).
Kallscheuer, N. et al. Pink- and orange-pigmented planctomycetes produce saproxanthin-type carotenoids including a rare C(45) carotenoid. Environ. Microbiol. Rep. 11, 741–748. https://doi.org/10.1111/1758-2229.12796 (2019).
Lage, O. M. & Bondoso, J. Planctomycetes and macroalgae, a striking association. Front. Microbiol. 5, 267–267. https://doi.org/10.3389/fmicb.2014.00267 (2014).
Kumar, G. et al. Gemmata algarum, a Novel Planctomycete Isolated from an Algal Mat, Displays Antimicrobial Activity. Mar. Drugs. 22, 10. https://doi.org/10.3390/md22010010 (2023).
Kulichevskaya, I. S. et al. Fimbriiglobus ruber gen. nov., sp. nov., a Gemmata-like planctomycete from Sphagnum peat bog and the proposal of Gemmataceae fam. nov. Int. J. Syst. Evol. Microbiol. 67, 218–224. https://doi.org/10.1099/ijsem.0.001598 (2017).
Overmann, J., Abt, B. & Sikorski, J. Present and Future of Culturing Bacteria. Annu. Rev. Microbiol. 71, 711–730. https://doi.org/10.1146/annurev-micro-090816-093449 (2017).
Rivas-Marín, E., Canosa, I., Santero, E. & Devos, D. P. Development of Genetic Tools for the Manipulation of the Planctomycetes. Front. Microbiol. 7, 914. https://doi.org/10.3389/fmicb.2016.00914 (2016).
Kuenen, J. G. Anammox bacteria: from discovery to application. Nat. Rev. Microbiol. 6, 320–326. https://doi.org/10.1038/nrmicro1857 (2008).
Buckley, D. H., Huangyutitham, V., Nelson, T. A., Rumberger, A. & Thies, J. E. Diversity of Planctomycetes in soil in relation to soil history and environmental heterogeneity. Appl. Environ. Microbiol. 72, 4522–4531. https://doi.org/10.1128/aem.00149-06 (2006).
Kaushik, R. et al. Paludisphaera soli sp. nov., a new member of the family Isosphaeraceae isolated from high altitude soil in the Western Himalaya. Antonie van Leeuwenhoek. 113, 1663–1674 (2020).
Kulichevskaia, I. S., Pankratov, T. A. & Dedysh, S. N. Detection of representatives of the Planctomycetes in Sphagnum peat bogs by molecular and cultivation methods. Mikrobiologiia 75, 389–396 (2006).
Kumar, D. et al. Gimesia chilikensis sp. nov., a haloalkali-tolerant planctomycete isolated from Chilika lagoon and emended description of the genus Gimesia. Int. J. Syst. Evol. Microbiol. 70, 3647–3655 (2020).
Gaurav, K. et al. Phylo-taxogenomics of the genus Tautonia with descriptions of Tautonia marina sp. nov., Tautonia rosea sp. nov., and emended description of the genus. Syst. Appl. Microbiol. 44, 126229 (2021).
Kumar, G. et al. A genomic overview including polyphasic taxonomy of Thalassoroseus pseudoceratinae gen. nov., sp. nov. isolated from a marine sponge, Pseudoceratina sp. Antonie Van Leeuwenhoek 115, 843–856 (2022).
Rivas-Marin, E. et al. Thalassoglobus polymorphus sp. nov., a novel Planctomycete isolated close to a public beach of Mallorca Island. Antonie Van Leeuwenhoek 113, 1915–1926. https://doi.org/10.1007/s10482-020-01437-y (2020).
Kumar, G. et al. Aquisphaera insulae sp. nov., a new member in the family Isosphaeraceae, isolated from the floating island of Loktak lake and emended description of the genus Aquisphaera. Antonie Van Leeuwenhoek 114, 1465–1477 (2021).
Lhingjakim, K. L. et al. Paludisphaera rhizosphaereae sp. nov., a new member of the family Isosphaeraceae, isolated from the rhizosphere soil of Erianthus ravennae. Antonie van Leeuwenhoek 115, 1073–1084 (2022).
Lage, O. M. & Bondoso, J. Bringing Planctomycetes into pure culture. Front. Microbiol. 3, 405. https://doi.org/10.3389/fmicb.2012.00405 (2012).
Sreya, P., Gaurav, K., Ahmed, S., Sasikala, C. & Ramana, C. V. Blastopirellula sediminis sp. nov. a new member of Pirellulaceae isolated from the Andaman and Nicobar Islands. Antonie Van Leeuwenhoek 116, 463–475 (2023).
Rivas-Marin, E. et al. Maioricimonas rarisocia gen. nov., sp. nov., a novel planctomycete isolated from marine sediments close to Mallorca Island. Antonie Van Leeuwenhoek 113, 1901–1913. https://doi.org/10.1007/s10482-020-01436-z (2020).
Storesund, J. E. & Øvreås, L. Diversity of Planctomycetes in iron-hydroxide deposits from the Arctic Mid Ocean Ridge (AMOR) and description of Bythopirellula goksoyri gen. nov., sp. nov., a novel Planctomycete from deep sea iron-hydroxide deposits. Antonie Van Leeuwenhoek 104, 569–584. https://doi.org/10.1007/s10482-013-0019-x (2013).
Elcheninov, A. G. et al. Thermogemmata fonticola gen. nov., sp. nov., the first thermophilic planctomycete of the order Gemmatales from a Kamchatka hot spring. Syst. Appl. Microbiol. 44, 126157. https://doi.org/10.1016/j.syapm.2020.126157 (2021).
Andrew, D. R. et al. Abiotic factors shape microbial diversity in Sonoran Desert soils. Appl. Environ. Microbiol. 78, 7527–7537. https://doi.org/10.1128/aem.01459-12 (2012).
Kumar, G. et al. Stratiformator vulcanicus gen. nov., sp. nov., a marine member of the family Planctomycetaceae isolated from a red biofilm in the Tyrrhenian Sea close to the volcanic island Panarea. Antonie Van Leeuwenhoek 116, 995–1007. https://doi.org/10.1007/s10482-023-01860-x (2023).
Kallscheuer, N. et al. Mucisphaera calidilacus gen. nov., sp. nov., a novel planctomycete of the class Phycisphaerae isolated in the shallow sea hydrothermal system of the Lipari Islands. Antonie Van Leeuwenhoek 115, 407–420. https://doi.org/10.1007/s10482-021-01707-3 (2022).
Kallscheuer, N. et al. Blastopirellula retiformator sp. nov. isolated from the shallow-sea hydrothermal vent system close to Panarea Island. Antonie Van Leeuwenhoek 113, 1811–1822. https://doi.org/10.1007/s10482-019-01377-2 (2020).
Wiegand, S. et al. Updates to the recently introduced family Lacipirellulaceae in the phylum Planctomycetes: isolation of strains belonging to the novel genera Aeoliella, Botrimarina, Pirellulimonas and Pseudobythopirellula and the novel species Bythopirellula polymerisocia and Posidoniimonas corsicana. Antonie Van Leeuwenhoek 113, 1979–1997. https://doi.org/10.1007/s10482-020-01486-3 (2020).
Peeters, S. H. et al. Three marine strains constitute the novel genus and species Crateriforma conspicua in the phylum Planctomycetes. Antonie Van Leeuwenhoek 113, 1797–1809. https://doi.org/10.1007/s10482-019-01375-4 (2020).
Rensink, S. et al. Description of the novel planctomycetal genus Bremerella, containing Bremerella volcania sp. nov., isolated from an active volcanic site, and reclassification of Blastopirellula cremea as Bremerella cremea comb. nov. Antonie Van Leeuwenhoek 113, 1823–1837. https://doi.org/10.1007/s10482-019-01378-1 (2020).
Kallscheuer, N. et al. Rubinisphaera italica sp. nov. isolated from a hydrothermal area in the Tyrrhenian Sea close to the volcanic island Panarea. Antonie Van Leeuwenhoek 113, 1727–1736. https://doi.org/10.1007/s10482-019-01329-w (2020).
Kohn, T. et al. Planctopirus ephydatiae, a novel Planctomycete isolated from a freshwater sponge. Syst. Appl. Microbiol. 43, 126022. https://doi.org/10.1016/j.syapm.2019.126022 (2020).
Kallscheuer, N. et al. Cultivation-Independent Analysis of the Bacterial Community Associated With the Calcareous Sponge Clathrina clathrus and Isolation of Poriferisphaera corsica gen. nov., sp. nov., Belonging to the Barely Studied Class Phycisphaerae in the Phylum Planctomycetes. Front. Microbiol. 11, 602250. https://doi.org/10.3389/fmicb.2020.602250 (2020).
Kumar, G. et al. Crateriforma spongiae sp. nov., isolated from a marine sponge and emended description of the genus Crateriforma. Antonie Van Leeuwenhoek 114, 341–353. https://doi.org/10.1007/s10482-020-01515-1 (2021).
Kallscheuer, N. et al. Description of Stieleria mannarensis sp. nov., isolated from a marine sponge, and proposal to include members of the genus Roseiconus in the genus Stieleria. Antonie van Leeuwenhoek 118, 111 (2025).
Kumar, G., Radha, V., Jagadeeshwari, U. & Sasikala, C. & Venkata Ramana, C. Bacterial communities of sponges from the wetland ecosystem of Little Rann of Kutch, India with particular reference to Planctomycetes. 3 Biotech 10, 478 (2020).
Kallscheuer, N. et al. Aureliella helgolandensis gen. nov., sp. nov., a novel Planctomycete isolated from a jellyfish at the shore of the island Helgoland. Antonie Van Leeuwenhoek 113, 1839–1849. https://doi.org/10.1007/s10482-020-01403-8 (2020).
Kohn, T. et al. The Microbiome of Posidonia oceanica Seagrass Leaves Can Be Dominated by Planctomycetes. Front. Microbiol. 11, 1458. https://doi.org/10.3389/fmicb.2020.01458 (2020).
Godinho, O. et al. Bremerella alba sp. nov., a novel planctomycete isolated from the surface of the macroalga Fucus spiralis. Syst. Appl. Microbiol. 44, 126189. https://doi.org/10.1016/j.syapm.2021.126189 (2021).
Vitorino, I. et al. Alienimonas chondri sp. nov., a novel planctomycete isolated from the biofilm of the red alga Chondrus crispus. Syst. Appl. Microbiol. 43, 126083. https://doi.org/10.1016/j.syapm.2020.126083 (2020).
Wiegand, S. et al. Additions to the genus Gimesia: description of Gimesia alba sp. nov., Gimesia algae sp. nov., Gimesia aquarii sp. nov., Gimesia aquatilis sp. nov., Gimesia fumaroli sp. nov. and Gimesia panareensis sp. nov., isolated from aquatic habitats of the Northern Hemisphere. Antonie Van Leeuwenhoek 113, 1999–2018. https://doi.org/10.1007/s10482-020-01489-0 (2020).
Jogler, C. et al. Tautonia plasticadhaerens sp. nov., a novel species in the family Isosphaeraceae isolated from an alga in a hydrothermal area of the Eolian Archipelago. Antonie Van Leeuwenhoek 113, 1889–1900. https://doi.org/10.1007/s10482-020-01424-3 (2020).
Wiegand, S. et al. Analysis of Bacterial Communities on North Sea Macroalgae and Characterization of the Isolated Planctomycetes Adhaeretor mobilis gen. nov., sp. nov., Roseimaritima multifibrata sp. nov., Rosistilla ulvae sp. nov. and Rubripirellula lacrimiformis sp. nov. Microorganisms 9, 1494. https://doi.org/10.3390/microorganisms9071494 (2021).
Sandargo, B. et al. Stieleriacines, N-Acyl Dehydrotyrosines From the Marine Planctomycete Stieleria neptunia sp. nov. Front. Microbiol. 11, 1408. https://doi.org/10.3389/fmicb.2020.01408 (2020).
Cayrou, C., Sambe, B., Armougom, F., Raoult, D. & Drancourt, M. Molecular diversity of the Planctomycetes in the human gut microbiota in France and Senegal. Apmis 121, 1082–1090. https://doi.org/10.1111/apm.12087 (2013).
Salbreiter, M. et al. Three Planctomycetes isolated from biotic surfaces in the Mediterranean Sea and the Pacific Ocean constitute the novel species Symmachiella dynata gen. nov., sp. nov. and Symmachiella macrocystis sp. nov. Antonie Van Leeuwenhoek 113, 1965–1977. https://doi.org/10.1007/s10482-020-01464-9 (2020).
Kallscheuer, N. et al. Rhodopirellula heiligendammensis sp. nov., Rhodopirellula pilleata sp. nov., and Rhodopirellula solitaria sp. nov. isolated from natural or artificial marine surfaces in Northern Germany and California, USA, and emended description of the genus Rhodopirellula. Antonie Van Leeuwenhoek. 113, 1737–1750. https://doi.org/10.1007/s10482-019-01366-5 (2020).
Kallscheuer, N., Wurzbacher, C. E., Schmitz, R. A. & Jogler, C. In the footsteps of Heinz Schlesner and Peter Hirsch: Exploring the untapped diversity of the phylum Planctomycetota in isolates from the 1980s to the early 2000s. Syst. Appl. Microbiol. 47, 126486. https://doi.org/10.1016/j.syapm.2023.126486 (2024).
Griepenburg, U. et al. Phylogenetic diversity, polyamine pattern and DNA base composition of members of the order Planctomycetales. Int. J. Syst. Bacteriol. 49 Pt 2, 689–696. https://doi.org/10.1099/00207713-49-2-689 (1999).
Ward, N., Rainey, F. A., Stackebrandt, E. & Schlesner, H. Unraveling the extent of diversity within the order Planctomycetales. Appl. Environ. Microbiol. 61, 2270–2275. https://doi.org/10.1128/aem.61.6.2270-2275.1995 (1995).
Schlesner, H. The Development of Media Suitable for the Microorganisms Morphologically Resembling Planctomyces spp., Pirellula spp., and other Planctomycetales from Various Aquatic Habitats Using Dilute Media. Syst. Appl. Microbiol. 17, 135–145. https://doi.org/10.1016/S0723-2020(11)80042-1 (1994).
Rast, P. et al. Three Novel Species with Peptidoglycan Cell Walls form the New Genus Lacunisphaera gen. nov. in the Family Opitutaceae of the Verrucomicrobial Subdivision 4. Front. Microbiol. 8, 202. https://doi.org/10.3389/fmicb.2017.00202 (2017).
Wurzbacher, C. E. et al. Planctoellipticum variicoloris gen. nov., sp. nov., a novel member of the family Planctomycetaceae isolated from wastewater of the aeration lagoon of a sugar processing plant in Northern Germany. Sci. Rep. 14, 5741. https://doi.org/10.1038/s41598-024-56373-y (2024).
Haufschild, T. et al. An untargeted cultivation approach revealed Pseudogemmatithrix spongiicola gen. nov., sp. nov., and sheds light on the gemmatimonadotal mode of cell division: binary fission. Sci. Rep. 14, 16764. https://doi.org/10.1038/s41598-024-67408-9 (2024).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 9, 676–682. https://doi.org/10.1038/nmeth.2019 (2012).
Hartmann, R., van Teeseling, M. C. F., Thanbichler, M., Drescher, K. & BacStalk A comprehensive and interactive image analysis software tool for bacterial cell biology. Mol. Microbiol. 114, 140–150. https://doi.org/10.1111/mmi.14501 (2020).
Goedhart, J. SuperPlotsOfData-a web app for the transparent display and quantitative comparison of continuous data from different conditions. Mol. Biol. Cell. 32, 470–474. https://doi.org/10.1091/mbc.E20-09-0583 (2021).
Thompson, J. D., Gibson, T. J. & Higgins, D. G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinf. (Chap. 2), Unit23. https://doi.org/10.1002/0471250953.bi0203s00 (2002).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. https://doi.org/10.1093/molbev/msp077 (2009).
Alanjary, M., Steinke, K. & Ziemert, N. AutoMLST: an automated web server for generating multi-locus species trees highlighting natural product potential. Nucleic Acids Res. 47, W276–w282. https://doi.org/10.1093/nar/gkz282 (2019).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–w296. https://doi.org/10.1093/nar/gkab301 (2021).
Tarlachkov, S. & Starodumova, I. TaxonDC: calculating the similarity value of the 16S rRNA gene sequences of prokaryotes or ITS regions of fungi. J Bioinf. Genomics 3, 1. https://doi.org/10.18454/jbg.2017.3.5.1 (2017).
Rodriguez-R, L. M. & Konstantinidis, K. T. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes, PeerJ Preprints 4, e1900v1. https://doi.org/10.7287/peerj.preprints.1900v1 (2016).
Bondoso, J., Harder, J. & Lage, O. M. rpoB gene as a novel molecular marker to infer phylogeny in Planctomycetales. Antonie Van Leeuwenhoek 104, 477–488 (2013).
Qin, Q. L. et al. A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 196, 2210–2215. https://doi.org/10.1128/JB.01688-14 (2014).
Delmont, T. O. & Eren, A. M. Linking pangenomes and metagenomes: the Prochlorococcus metapangenome. PeerJ 6, e4320. https://doi.org/10.7717/peerj.4320 (2018).
Blin, K. et al. antiSMASH 8.0: extended gene cluster detection capabilities and analyses of chemistry, enzymology, and regulation. Nucleic Acids Res. 53, W2-W38. https://doi.org/10.1093/nar/gkaf334 (2025).
Zheng, J. et al. dbCAN3: automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 51, W115–W121. https://doi.org/10.1093/nar/gkad328 (2023).
Acknowledgements
The authors thank Vera Thiel (DSMZ, Braunschweig, Germany), MariCarmen Macián (CECT, Valencia, Spain) and the staff from the Korean Collection for Type Cultures (KCTC) for help during deposition of the strains and the Jena School for Microbial Communication (JSMC) for continuous support. Help from Emanuel Petre and Hanan Alderzy during genomic DNA extraction, DNA quality control and library preparation is greatly acknowledged. The authors also thank Aharon Oren for checking the etymology for the names of the novel taxa.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was funded by the German Research Foundation (DFG, Deutsche Forschungsgemeinschaft) under Project-ID 239748522 (ChemBioSys) and Germany´s Excellence Strategy under Project-ID 390713860 - EXC 2051. Funded by the Landesgraduiertenstipendium of the Free State of Thuringia awarded by the Friedrich Schiller University Jena. Funding from the Carl Zeiss Stiftung is greatly acknowledged.
Author information
Authors and Affiliations
Contributions
GK revived, identified and cultivated the strains of the collection, took care of the deposition and performed the determination of the temperature and pH range and optimum for growth and wrote the manuscript; NK performed genome annotation, analyses of phylogeny and genome-encoded features and contributed to writing of the manuscript; JH performed genome sequencing and assembly; TH performed light microscopy and cell size determination. CJ supervised the study and contributed to data analysis. All authors have worked on the final version of the manuscript and agreed to the submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kumar, G., Kallscheuer, N., Hammer, J. et al. The novel genus Brakhagea gen. nov. is constituted by four planctomycetal species isolated from aquatic environments in Northern Germany. Sci Rep 16, 12750 (2026). https://doi.org/10.1038/s41598-026-47393-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-47393-x
