
Abstract
Ship traffic is known as an important contributor to air pollution. Regulations aimed at reducing sulfur oxide pollution by limiting the fuel sulfur content (FSC) may also decrease primary particulate matter (PM) emitted from ships. However, there is a knowledge gap regarding how the FSC affects secondary aerosol formation. The emissions from a research ship engine operated with either low sulfur heavy fuel oil (LS-HFO) (FSC = 0.5%) or marine gas oil (MGO) (FSC = 0.01%), were photochemically processed in the oxidation flow reactor “PEAR” to achieve an equivalent photochemical age between 0 and 9 days in the atmosphere. FSC was found to have no significant impact on secondary organic aerosol formation after 3 days of aging, at 1.7 ± 0.4 g/kg for MGO and 1.5 ± 0.4 g/kg for LS-HFO. Furthermore, the composition and oxidative pathways remained similar regardless of FSC. However, because of the higher secondary SO4 formation and primary aerosol emissions, LS-HFO had significantly higher total PM than MGO.
Similar content being viewed by others

Introduction
International trade is dominated by the shipping industry, accounting for ~80% by volume1 and is expected to triple from 2019 to 20502. Currently, international shipping is responsible for about 3% of global anthropogenic greenhouse gas emissions2,3 Additionally, shipping emissions account for ~9% of global anthropogenic SOx, 17% of NOx, 4% of particulate matter <2.5 µm (PM2.5) and 4% of non-methane volatile organic compounds (NMVOCs)4,5,6.
Up to 7% of global premature mortalities are attributable to air pollution7,8,9,10. As 70% of marine traffic occurs within 400 km from the coastline, ship emissions are relevant for public health11. Air pollution from shipping prior to 2020 was estimated to cause >400 thousand premature deaths per year globally12. In 2020 the international maritime organization (IMO) introduced global regulations limiting the maximum fuel sulfur content (FSC) from 3.5 to 0.5w%. These regulations aimed to directly reduce SOx emissions from shipping by 77%13, but indirectly also lower emissions of PM by the implied ban of most heavy fuel oils (HFO)14. These new regulations are expected to reduce premature deaths due to air pollution from ships by about 30%12. Furthermore, in 2015 sulfur emission control areas (SECA) were defined, including the North Sea, Baltic Sea, and North American coastal waters, with a maximum allowed FSC of 0.1%. From July 2024, HFO defined as fuels with a kinematic viscosity >180 mm s−1 at 50 °C and a density >900 kg m−3 at 15 °C are banned in Arctic waters15.
The introduction of a lower global FSC limit has led to an increase in the use of marine gas oil (MGO), low-sulfur HFO (LS-HFO) and hybrid fuels with boiling ranges and impurities between MGO and LS-HFO. The boiling fraction of MGO, being a diesel-like fuel, has an intrinsically lower FSC than LS-HFO and is available with FSC ≤ 0.1%. Nevertheless, MGO has a significantly higher FSC than diesel used for road traffic, which has a FSC < 0.001%16.
Beyond SOx, PM, BC, NOx and NMVOCs are all emitted at high levels from marine diesel engines, which are not equipped with exhaust aftertreatment technology, such as diesel oxidation catalysts (DOC) which reduces NMHC emissions. NMVOCs participate in oxidative gas-to-particle conversion, forming secondary organic aerosol (SOA). Observational studies have found that the highest contributor to SOA formation in coastal urban areas is shipping emissions with about 38%17. The highest emission factors for hydrocarbons, black carbon (BC) and particulate mass (PM) were reported during the lowest engine loads, such as near shore maneuvering conditions16,18,19. Potential SOA precursors such as light poly aromatic hydrocarbons (PAHs) and other intermediate volatile organic compounds (IVOCs) have been reported from different fuel types with heavier fuels containing more PAHs than lighter diesel fuels. Additionally, diesel fuels were found to have less semi volatile organic compounds (SVOC) and more IVOCs than heavy fuel oils with SVOC/IVOC down to 0.085 ± 0.046 and up to 0.439 ± 0.021 respectivly20. Furthermore, PAH and oxy-PAH has been reported to contribute significantly to the organic fraction of the primary PM emitted during low engine load for HFO, diesel fuel was found to have comparatively low PAH and oxy-PAH contribution to the organic mass fraction regardless of engine load21.
SOA from IVOCs, such as naphthalene, representative of combustion-derived IVOCs, have been shown to have adverse effect on human health22. Even with a change from HFO to LS-HFO or MGO it has been predicted that shipping will remain a significant source of anthropogenic aerosol23. A recent study explored the impact of aging on HFO emissions at 65% engine load. The ship was equipped with a sulfur scrubber and a DOC as exhaust aftertreatment, and showed no significant SOA formation24.
Therefore, in order to explore the SOA formation potential for other engine conditions and better understand the impacts of new IMO regulations on SOA formation, our study investigates the impacts of aging on low sulfur fuels such as LS-HFO and MGO fuel at low engine load.
Results
SOA formation from LS-HFO and MGO photooxidation
Figure 1 illustrates the chemical composition of aerosols for both LS-HFO and MGO. At 0 days of equivalent photochemical aging the fresh aerosol has an organic mass to particle mass (OM/PM) of 0.47 for LS-HFO and 0.34 for MGO, respectively. The MGO thus contains relatively less organic and more soot, however the total aerosol emitted is much lower for MGO than for LS-HFO. The LS-HFO emissions of POA were >5 times higher than those of the MGO, while BC emissions were ~3 times higher in LS-HFO than in MGO. Other non-refractory PM components including SO4 and NO3 were negligible for the unaged total PM.

The left axis indicates the emission factor in grams of PM measured per kg fuel consumed and the organic enhancement factor (EROA) The left set of the graph represents the LS-HFO results and the right the MGO aerosol emissions, both with a photochemical age displayed in atmospheric equivalent days at the bottom.
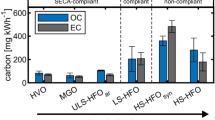
The fresh particles had a geometric mean diameter of 75 ± 6 nm for MGO and 94 ± 3 nm for LS-HFO (Supplementary Table S13), which is comparable to fresh particles emitted from a ship engine running on HFO published by Karjalainen et al.24. The most notable change from previous studies is that there is no SO4 contribution to the fresh aerosol emissions due to the fuel used in this study LS-HFO(FSC = 0.5%) in comparison to Karjalainen et al.24 with an FSC of 1.9%. HFO with a FSC of 0.12% has previously been found to emit 2.57 ± 0.41 g/kg total primary PM at 25% engine load19, this is comparable to the emission factor of LS-HFO of this study 2.05 ± 0.51 g/kg, both significantly higher than the primary PM measured for MGO at 0.78 ± 0.24 g/kg. Furthermore, HFO 350 (FSC = 2.7%) representative of pre-2020 global fuels have been reported the in literature to emit 1.9 g/kWh at 25% engine load21 compared to the 0.6 ± 0.15 g/kWh and 0.20 ± 0.06 g/kWh emitted by LS-HFO and MGO respectively. All emission factors can be found as g/kWh and g/kg in the supplementary table S15.
Ozonolysis had a negligible impact on the aerosol in the PEAR in terms of chemical composition, particle size and PM. As the emissions were photochemically processed in the PEAR, the aerosol composition changed. At 1.5 days the aerosol composition is dominated by organics with an OM/PM of 0.60 for LS-HFO and 0.69 for MGO. BC is comparatively not affected by photochemical aging and the total BC mass remains constant at 1.1 ± 0.2 g/kg for LS-HFO and 0.4 ± 0.1 g/kg for MGO for all oxidation experiments. The increase in total mass can be attributed to SO4 and SOA formation (Fig. 1). The SOA emission factor for both MGO and LS-HFO is similar and reaches a maximum of 1.5 ± 0.4 g/kg LS-HFO and 1.7 ± 0.4 g/kg MGO after 3.3 and 4.7 days of equivalent photochemical aging respectively, though smaller aging steps would be required to determine the specific maximum. However, the majority of SOA is formed within the first 1.5 days of equivalent photochemical aging. Some results from recent studies have suggested the majority of secondary aerosol (SA) from shipping is even formed within the first 3 h after emission25. While photochemical aging increases SOA formation equally for the two fuels, SO4 aerosol formation is greatly enhanced in the LS-HFO emissions with an emission factor after 8.7 days of photochemical aging 6.20 ± 0.83 g/kg, more than 4 times higher than that of MGO at 1.44 ± 0.19 g/kg after 8.9 days of photochemical aging. This increase in SO4 aerosol formation is linked to the higher FSC of LS-HFO and thus emission of SO2 compared to MGO as seen in Table S3. It is however clear that whilst FSC is 50 times lower for MGO compared to LS-HFO, the decrease in emitted SO2 is only 5 times lower than the emissions of LS-HFO.
Figure 1 shows that photochemical aging beyond 4.7 days does not yield additional SOA formation, and a further increase in total aerosol results from SO4 formation. After 9 days of equivalent photochemical aging, the LS-HFO aerosol is in a SO4 dominated state where OM/PM decreased to 0.23. For MGO, however, the aerosol remains organic-dominated with OM/PM > 0.5. This is further highlighted by the organic aerosol enhancement ratio, which indicates the 5-fold increase in organics for the MGO, whereas LS-HFO only increases 2-fold compared to POA. However, this is largely due to the higher initial POA observed with LS-HFO as the maximal total organic aerosol formation after photochemical aging is 2.46 ± 0.33 g/kg, and 2.33 ± 0.31 g/kg for LS-HFO and MGO respectively. Thus, the use of MGO significantly decreases the total aerosol mass concentration as black carbon and secondary SO4 aerosol mass is reduced compared to LS-HFO. However, a change from LS-HFO to MGO does not reduce total organic aerosol formation, resulting in an organic dominated aerosol.
Elemental composition and oxidation pathway
While more than 4.7 days of equivalent photochemical aging does not increase organic aerosol (OA) mass for either MGO or LS-HFO, the composition of the OA changes significantly. Initial photochemical aging from 0 to 4.7 days increases both f43 (fraction of 43 m/z of total OA mass spectrum, mostly \({C}_{2}{H}_{3}{O}^{+}\) and \({C}_{3}{H}_{7}^{+}\) representing less oxidized, hydrocarbon like aerosols) and f44 (fraction of 44 m/z to total OA mass, mostly \(C{O}_{2}^{+}\), associated with highly oxidized, aged aerosols) fraction, consistent with the increase in OA mass (Fig. 2A, B)26,27.

A, B show a triangle plot as first published by Ng et al.35 illustrating the f43 and f44 of the organic aerosol (OA). A Shows the impact of aging on these two fractions for LS-HFO and B shows the same for MGO. The photochemical age of the aerosol is illustrated by color as seen on the right. C, D illustrate a Van Krevelen plot36,38 with hydrogen to carbon ratio on the y axis and oxygen to carbon ratio on the x axis, showing the chemical composition of the OA: the OA regression line is shown with dotted lines, and the dashed lines highlight the area where ambient aerosol typically fall within.
However, an equivalent age >4.7 days results in a decrease of f43 and an increase of f44, similar to observations from EURO2 diesel vehicles28. This coincides with the plateau in SOA formation that was observed. This observation is valid for both MGO and LS-HFO. All aged aerosols fall within the triangular space, representing the occurrence of ambient OA, as atmospheric aerosols follow a characteristic trajectory within this region, because oxidation processes predominantly increase oxygenated functional groups (increasing F44) while fragmenting less oxidized organic compounds(decreasing f43), leading to constrained aging pathways in the diagram26.
The similarity between the OA from MGO and LS-HFO is further supported by the elemental ratios depicted as a Van Krevelen diagram (Fig. 2C, D)27,29. The main difference in the elemental composition was found in the fresh aerosol, with the H/C ratio of (\(1.79\pm 0.05\)) for MGO and (\(1.67\pm 0.03\)) for LS-HFO aerosol, resulting in a slope from fresh aerosol to 3.3 days of equivalent photochemical aging of -0.97 ± 0.08 (r2 = 0.97) and -0.89 ± 0.04 (r2 = 0.98) for MGO and LS-HFO, respectively. This result suggests that the initial photochemical aging from fresh to maximum SOA formation is equivalent to carboxylic acid formation with no fragmentation or carbonyl and hydroxyl addition on different carbons27. Further after 3–4 days of photochemical aging there is no significant difference in the elemental ratios between two fuels. The slope of both MGO and LS-HFO after 3 days of age is -0.54 ± 0.02 (r2 = 0.99). Photochemical aging beyond the maximum SOA formation point thus suggests carboxylic acid formation with fragmentation. This coincides with the decrease in f43 but continued increase in f44 observed in the triangle plot suggesting acid formation27. The fragmentation effectively results in a lower SOA yield, which leads to a net zero mass change in SOA at higher photochemical age. Based on KinSim30 simulation the difference in oxidation is not a result of the difference in RO2 fate as HO2-RO2 reactions accounted for the >50% of lost RO2 and HO accounting for >25% at all ages for both fuels (Supplementary Table S9–10).
Discussion
Implementing a global FSC limit to the current SECA level could incentivize the use of cleaner shipping fuels instead of residual fuel oils, potentially lowering emissions of total PM, BC, POA and sulfate. However, this may not impact the SOA mass formed after atmospheric aging.
It was observed that both POA and BC were reduced when changing from LS-HFO to MGO, the fresh PM at emission point would therefore be significantly reduced by changing from current global FSC regulations to SECA compliant FSC fuels. The use of MGO over LS-HFO in harbor areas could decrease primary PM by >50%. A change to MGO would comparatively to pre-2020 fuel types such as HFO (FSC = 1.9%) be a 3-fold reduction in primary PM19.
The majority of SOA was formed after only 1.5 days of equivalent photochemical aging while maximum SOA formation was reached at 3.3 days of equivalent photochemical age. Thus, SOA will presumably form close to the emission point, i.e. harbors and nearby populated areas, considering the emission scenario of low engine load. The impact of these emissions will thus primarily be in near-coastal regions, having adverse health effects on 2.85 billion people globally31. Whilst total PM would be expected to decrease with a change from LS-HFO to MGO as BC, POA and SO4 containing aerosols decrease.
Notably the SOA levels would not be expected to decrease in these areas with a change from LS-HFO to MGO. Rather, such a fuel change could lead to an increase in SOA mass formation but not total organic aerosol emissions.
Secondary sulfate formation proceeded much slower than SOA formation but continued to increase in mass during photochemical aging to the maximum of almost 9 days of equivalent photochemical age. The reduction in sulfate aerosol associated with changing from LS-HFO to MGO would thus have an environmental and air quality impact further from the emission point than the OA.
Regarding the composition of SOA, a high similarity was found for H/C, O/C, f43 and f44 for the LS-HFO and MGO. This suggests that the precursor for the SOA is similar between the two fuels, which are likely alkylated aromatic mono- to tricyclic structures32. Reducing aromatic VOC emissions would improve air quality and lessen the health burden associated with ship emissions during harbor maneuvers .
Methods
A four-stroke single-cylinder research diesel engine (80 kW nominal power) was operated on either MGO (FSC = 0.01 S w%) or LS-HFO (FSC = 0.5 S w%) at low engine load (20 kW, 25% of nominal power), representing docking maneuvers. Raw exhaust emissions were connected to diluters via heated tubing (constant at 350 °C), see Fig. 3. A series configuration of a porous tube diluter and an ejector diluter was employed to achieve a dilution ratio of 1:250. The dilution ratio was determined by measuring the CO2 concentrations in the raw exhaust, the diluted exhaust, and the dilution air. The dilution ratio was regulated by automatically adjusting the flow rate of the porous tube diluter, while maintaining a constant flow rate for the ejector diluter.

The dotted line signifies heated tubing(350°C). Only Either HFO or MGO was added to the engine.
The diluted (1:250) exhaust sample passed through the oxidation flow reactor “Photochemical Emission Aging flow tube Reactor” (PEAR), which was operated at a flow rate of 100 lpm33. At these flow conditions, the mean residence time in the PEAR is 70 s. At the entrance to the PEAR 10ppm of ozone was added in addition to water vapor keeping the relative humidity (RH) at ~50%. The photochemical aging was controlled through UV-lamp(254 nm) intensity, water vapor supply for 50% relative humidity and constant ozone (10ppm) addition in order to obtain atmospheric aging equivalent to 0–9 days (supplementary table S8). The photochemical age was determined with d9-butanol decay measured by a Proton-transfer-reaction mass spectrometry (PTR-MS) based on the principle of the “photochemical clock”34. The Kinsim30,35,36,37,38 model was used to verify the atmospheric relevancy by simulating the chemistry within the PEAR, by determining the RO2 fate to remove speculations on non-tropospheric chemistry occurring in the OFR. For the KinSim modeling SO2 was used as a surrogate for THC as suggested in literature38 (supplementary table S8-10).
An additional ejector diluter was added after the PEAR for a further 10-fold dilution before online instruments, resulting in a total dilution ratio of 1:2500 for aerosol instruments presented here. Aerosol phase measurement were done with High-Resolution Time of Flight Aerosol Mass Spectrometer (HR-ToF-AMS)39 for obtaining the chemical composition of the non-refractory aerosol, aethalometer for BC and a scanning mobility particle sizer (SMPS) for particle number concentration and size distribution. SMPS data were treated with the AIM V. 10.3.1.0 software.
Optically defined black carbon (BC), “equivalent black carbon” (eBC), was quantified by the Aethalometer (AE33-7, Aerosol Magee Scientific), which measures light attenuation through a filter tape at 7 wavelengths (UV–IR, 370–950 nm). Using different corrections, such as multiple-scattering correction and loading compensation, attenuation coefficients were converted to absorption coefficients, and further to black carbon concentrations. A mass absorption coefficient (MAC) was used to quantify light absorption as a mass concentration. Absorption at 880 nm is generally assumed to be representative of BC as NOx inference is low. Thus, the equivalent black carbon mass concentrations (\({eBC}\)) was calculated (Eq.1)
where \(A\) is the surface area of the filter spot, \(\Delta {ATN}\) the change of attenuation at 880 nm during a time interval \(\Delta t\), \({F}_{{meas}}\) the measured flow rate, \(\zeta\) the leakage factor (default value 0.01), \(k\) the real-time loading compensation parameter40 \({AT}{N}_{t}\) the attenuation at time \(t\) for 880 nm, \(C\) the multiple-scattering coefficient (here \(C\) = 2.80 based on reference absorption measurement by PAX-870 during the campaign), and \({MAC}\) the mass absorption coefficient (here \({MAC}(880{nm})\) = 5.47 m2/g, based on reference refractory black carbon (rBC) measurement by LII 300 and elemental carbon (EC) measurement by DRI 2015 during the campaign).
AMS data was treated in Tof-AMS Analysis Toolkit 1.65 (squirrel), and HR analysis was performed with ToF-AMS HR Analysis 1.25 (PIKA). Elemental analysis was performed by the Aiken method41. The AMS was equipped to handle particles with an aerodynamic diameter between 42 nm and 645 nm. With small aerosol diameters, (supplementary table S13), falling outside the size range of the AMS. The total mass lost in the AMS compared to the SMPS is available in supplementary table S14 as mass per kg fuel consumed. The AMS consistently measures <88 ± 10% of the SMPS for LS-HFO and below 70 ± 8% of the SMPS for MGO. Additionally, the AMS aerosol mass is compared to the SMPS with eBC subtracted as eBC is not detectable in the AMS. When subtracting the eBC it is found that the AMS mass would account for up to 108 ± 24% of the total mass observed in the SMPS, as eBC particles falling outside the SMPS measuring range (7.5 nm–300 nm) would be overcounted compared to the SMPS. Resulting in a potential overrepresentation of eBC in the size range 7.5 nm–300 nm for the LS- HFO, this is considered within the uncertainties.
Moreover, to the best of our knowledge no information is available on the density of aged aerosol from ships emissions. However, aged aerosol from solid fuel combustion demonstrate a particle size dependent increase in effective density with differences between fuels42. Therefor, the mass of the aerosol is determined by the SMPS, with an assumed density of 1.5 + -0.2 g/cm3. Composition of the aerosol is then calculated based on AMS and aethalometer as in Eq. 2, assuming consistent composition across the measured monomodal size distributions.
Where \({{EF}}_{X}\) is the emission factor of an aerosol group, \({Con}{c}_{x}\) is the concentration of the aerosol group measured by the AMS, \({{Conc}}_{{AMS}}\) is the total concentration of aerosols measured by the AMS. \(E{F}_{{SMPS}}\) is the emission factor determined by the SMPS and \(E{F}_{{eBC}}\) is the emission factor of eBC determined by aethalometer. Measured concentrations (Cx) in g per m3 were converted to emission factors based on the measured fuel consumption (Fc) in kg per hour and exhaust volume (Vex) in m3 per hour, see Eq.3
Primary organic aerosol (POA) was determined as the organic fraction with no photochemical aging. The SOA fraction was calculated by subtracting the POA from the total OA mass at a given simulated age Eq. 4. Organic enhancement factor was calculated as the fractional increase of organics, see Eq. 5.
Where POA is the total organic mass at 0 days of atmospheric aging and OMx is the total organic mass at X days of atmospheric aging.
Data availability
The datasets associated with the current study are available from the corresponding authors [an.paul@fz-juelich.de or T.Hohaus@fz-juelich.de] on reasonable request.
References
Gray, N., McDonagh, S., O’Shea, R., Smyth, B. & Murphy, J. D. Decarbonising ships, planes and trucks: an analysis of suitable low-carbon fuels for the maritime, aviation and haulage sectors. Adv. Appl. Energy 1, 100008 (2021).
OECD. Ocean Shipping and Shipbuilding. https://www.oecd.org/ocean/topics/ocean-shipping/ (2018).
IMO. Fourth IMO Greenhouse Gas Study. https://www.imo.org/en/ourwork/Environment/Pages/Fourth-IMO-Greenhouse-Gas-Study-2020.aspx (2018).
Shi, Z. et al. Perspectives on shipping emissions and their impacts on the surface ocean and lower atmosphere: an environmental-social-economic dimension. Elementa. Sci. Anthropocene 11, 00052 (2023).
McDuffie, E. E. et al. A global anthropogenic emission inventory of atmospheric pollutants from sector- and fuel-specific sources (1970–2017): an application of the community emissions data system (CEDS). Earth Syst. Sci. Data 12, 3413–3442 (2020).
European, C. et al. GHG Emissions of all World – 2021 report. https://edgar.jrc.ec.europa.eu/report_2021 (2021).
Murray, C. J. L. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet 396, 1223–1249 (2020).
Corbett, J. J. et al. Mortality from ship emissions: a global assessment. Environ. Sci. Technol. 41, 8512–8518 (2007).
Mueller, N., Westerby, M. & Nieuwenhuijsen, M. Health impact assessments of shipping and port-sourced air pollution on a global scale: a scoping literature review. Environ. Res. 216, 114460 (2023).
Organization, W. H. Burden of Disesase From Teh Joint Effects of Household and Ambient Air Pollution for 2016’. https://cdn.who.int/media/docs/default-source/air-quality-database/aqd-2018/ap_joint_effect_bod_results_may2018.pdf?sfvrsn=4dc44c26_3 (2018).
Contini, D. & Merico, E. Recent advances in studying air quality and health effects of shipping emissions Atmosphere 12, 92 (2021).
Sofiev, M. et al. Cleaner fuels for ships provide public health benefits with climate tradeoffs. Nat. Commun. 9, 406 (2018).
IMO. IMO 2020 – Cutting Sulphur Oxide Emissions. https://www.imo.org/en/MediaCentre/HotTopics/Pages/Sulphur-2020.aspx (2025).
Yuan, T. et al. Abrupt reduction in shipping emission as an inadvertent geoengineering termination shock produces substantial radiative warming. Commun. Earth Environ. 5, 281 (2024).
IMO. Marine Environment Protection Committee (MEPC) 75, 16-20 November (virtual session). https://www.imo.org/en/MediaCentre/MeetingSummaries/Pages/MEPC-75th-session.aspx (2020).
Aakko-Saksa, P. T. et al. Reduction in greenhouse gas and other emissions from ship engines: current trends and future options. Prog. Energy Combust. Sci. 94, 101055 (2023).
Jang, E., Choi, S., Yoo, E., Hyun, S. & An, J. Impact of shipping emissions regulation on urban aerosol composition changes revealed by receptor and numerical modelling. npj Clim. Atmos. Sci. 6, 52 (2023).
Chu-Van, T. et al. A comparison of particulate matter and gaseous emission factors from two large cargo vessels during manoeuvring conditions. Energy Rep. 5, 1390–1398 (2019).
Anderson, M., Salo, K., Hallquist, Å. M. & Fridell, E. Characterization of particles from a marine engine operating at low loads. Atmos. Environ.101, 65–71 (2015).
Tang, R. et al. Detailed speciation of semi-volatile and intermediate-volatility organic compounds (S/IVOCs) in marine fuel oils using GC × GC-MS Int. J. Environ. Res. Public Health 20, 2508 (2023).
Sippula, O. et al. Particle emissions from a marine engine: chemical composition and aromatic emission profiles under various operating conditions. Environ. Sci. Technol. 48, 11721–11729 (2014).
Offer, S. et al. Effect of atmospheric aging on soot particle toxicity in lung cell models at the air–liquid interface: differential toxicological impacts of biogenic and anthropogenic secondary organic aerosols (SOAs). Environ. Health Perspect. 130, 027003 (2022).
Kuittinen, N. et al. Shipping remains a globally significant source of anthropogenic PN emissions even after 2020 sulfur regulation. Environ. Sci. Technol. 55, 129–138 (2021).
Karjalainen, P. et al. Real-world particle emissions and secondary aerosol formation from a diesel oxidation catalyst and scrubber equipped ship operating with two fuels in a SECA area. Environ. Pollution 292, 118278 (2022).
Gryspeerdt, E., Goren, T. & Smith, T. W. P. Observing the timescales of aerosol–cloud interactions in snapshot satellite images. Atmos. Chem. Phys. 21, 6093–6109 (2021).
Ng, N. L. et al. Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry. Atmos. Chem. Phys. 10, 4625–4641 (2010).
Ng, N. L. et al. Changes in organic aerosol composition with aging inferred fromaerosol mass spectra. Atmos. Chem. Phys. 11, 6465–6474 (2011).
Heringa, M. F. et al. A new method to discriminate secondary organic aerosols from different sources using high-resolution aerosol mass spectra. Atmos. Chem. Phys. 12, 2189–2203 (2012).
Krevelen, D. W. V. Graphical-statistical method for the study of structure and reaction processes of coal. Fuel 29, 269–284 (1950).
Peng, Z. & Jimenez, J. L. KinSim: A research-grade, user-friendly, visual kinetics simulator for chemical-kinetics and environmental-chemistry teaching. J. Chem. Edu. 96, 806–811 (2019).
Reimann, L., Vafeidis, A. T. & Honsel, L. E. Population development as a driver of coastal risk: Current trends and future pathways. Camb. Prism. Coast. Futures 1, e14 (2023).
Czech, H. et al. A chemometric investigation of aromatic emission profiles from a marine engine in comparison with residential wood combustion and road traffic: Implications for source apportionment inside and outside sulphur emission control areas. Atmos. Environ. 167, 212–222 (2017).
Ihalainen, M. et al. A novel high-volume photochemical emission aging flow tube reactor (PEAR). Aerosrol. Sci. Technol. 53, 276–294 (2019).
Barmet, P. et al. OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber. Atmos. Meas. Tech. 5, 647–656 (2012).
Peng, Z. et al. Model evaluation of new techniques for maintaining high-NO conditions in oxidation flow reactors for the study of OH-initiated atmospheric chemistry. ACS Earth Space Chem. 2, 72–86 (2018).
Peng, Z. & Jimenez, J. L. Modeling of the chemistry in oxidation flow reactors with high initial NO. Atmos. Chem. Phys. 17, 11991–12010 (2017).
Peng, Z. et al. Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling. Atmos. Chem. Phys. 16, 4283–4305 (2016).
Peng, Z. et al. HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling. Atmos. Meas. Tech. 8, 4863–4890 (2015).
Decarlo, P. F. et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 78, 8281–8289 (2006).
Drinovec, L. et al. The “dual-spot” aethalometer: an improved measurement of aerosol black carbon with real-time loading compensation. Atmos. Meas. Tech. 8, 1965–1979 (2015).
Aiken, A. C., Decarlo, P. F. & Jimenez, J. L. Elemental Analysis of Organic Species with Electron Ionization High-Resolution MassSpectrometry. Anal. Chem. 79, 8350–8358 (2007).
Leskinen, J. et al. Photochemical aging induces changes in the effective densities, morphologies, and optical properties of combustion aerosol particles. Environ. Sci. Technol. 57, 5137–5148 (2023).
Acknowledgements
We Thank the Helmholtz international Laboratory aeroHEALTH (InterLabs-0005; https://aerohealth.eu) and the ULTRHAS Horizon Europe project (agreement 955390) for supporting this work.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.P. wrote the main manuscript and prepared all figures. T.K., Z.F. and M.I. wrote parts of the methods segments in the main manuscript.U.E. prepared Supplementary Table 1–2. Data acquisition was conducted by A.P., T.K., Z.F., M.I., U.E. and H.C.All authors contributed to data analysis and interpretation.Supervision and funding by T.H.,H.C., O.S., Y.R., B.B., A.K. and R.Z.All authors revised the manuscript.Critical revision by T.H., H.C. T.K., Z.F., Y.R. and O.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Synopsis: Atmospheric oxidation of ship engine exhaust, reveals significant impact of new regulations but highlights secondary organic aerosols as key pollutant.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Paul, A., Kokkola, T., Fang, Z. et al. The impact of photochemical aging on secondary aerosol formation from a marine engine. npj Clim Atmos Sci 8, 106 (2025). https://doi.org/10.1038/s41612-025-00985-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41612-025-00985-2
