
Abstract
Background ozone (O3), defined as O3 originating from transboundary transport and domestic natural precursors, has traditionally been viewed as largely unresponsive to domestic anthropogenic emissions, representing an uncontrollable baseline for a nation’s O3 pollution levels. However, this paradigm overlooks the chemical interactions between the cycled oxidants from transboundary O3 and domestic precursors. Here, we developed a novel expanded odd oxygen (Oy) tagged modeling framework to explicitly track the sources and full photochemical cycling of O3 and its radical reservoirs during a typical autumn O3 pollution episode in China. Our results demonstrated that interactions between transboundary O3 and domestic precursors accounted for 44% to 49% of surface O3 levels across Eastern China during the study period. Transboundary O3 played a dual photochemical role, simultaneously promoting O3 formation by serving as a major source of ROx radicals, while also suppressing the ozone-forming potential of domestic precursors through ROx removal and modulation of the OH turnover rate. Consequently, the interplay between background and domestic anthropogenic sources fundamentally shaped the ambient O3 formation regime. This work challenges the prevailing view of a chemically static background, redefining the “controllable” portion of O3 pollution and necessitating a reassessment of mitigation strategies from regional to intercontinental scales.
Similar content being viewed by others

Introduction
Surface ozone (O3), produced by the photochemical oxidation of volatile organic compounds (VOCs) catalyzed by nitrogen oxides (NOx ≡ NO + NO2), is a rising air pollution challenge across many parts of the globe1. This challenge is particularly evident in China, where O3 pollution has worsened despite substantial reductions of anthropogenic NOx emissions over the past decade2, highlighting the non-linear nature of O3 chemistry3,4,5 and the multiplicity of its sources6,7,8. VOC precursors are emitted from a wide range of natural and anthropogenic activities8,9,10. NOx is predominantly emitted from anthropogenic sources over industrialized regions, although natural emissions, such as soil and lightning, also contribute8,11.
In addition, O3’s multi-week tropospheric lifetime enables long-range transport across regions, countries, and continents12,13,14,15,16. This transboundary O3, combined with O3 from domestic natural precursors and other non-controllable sources (e.g., stratospheric transport and global methane oxidation), establishes a regional background O3 level that limits the air quality improvements achievable through domestic emission controls17,18. Previous studies estimated that background O3 contributes 30–90% of the lower tropospheric O3 burden in China18,19 and 50–80% in the U.S.20,21, with distinct spatio-temporal variability. Transboudary O3 accounts for 37–88% of surface O3 in Europe22, serving as a major component of the regional background O323,24.
Current conceptualizations of regional background O3, such as the policy-relevant background ozone (PRBO)25 and the emissions-influenced background ozone (EIBO)21, implicitly assume minimal chemical interaction between background O3 (particularly its transboundary component) and domestic anthropogenic emissions14,18,26,27. PRBO, defined as the O3 concentration in the absence of regional anthropogenic emissions, is quantified by “zeroing-out” domestic anthropogenic emissions in atmospheric chemistry simulations19,20,28,29,30. In contrast, EIBO represents the portion of ambient O3 from transboundary, natural, and other non-controllable sources under present-day emission conditions21. EIBO is typically quantified using “tagged” simulations that track the sources of O3 precursors31,32 or the locations of O3 formation21, thereby avoiding the chemical non-linearity issues inherent to zero-out simulations33,34,35.
While the EIBO potentially provides a more dynamic representation of background O3, existing analyses have focused mainly on interactions within domestic emission sectors, such as the interactions between anthropogenic and biogenic VOCs and NOx11,31,32,36. To date, the chemical interactions between transboundary O3 and domestic precursors remain critically under-explored, as evidenced by the lack of such analysis in major initiatives like the hemispheric transport of air pollution, despite its focus on intercontinental transport of O314,37,38,39. This same assumption of minimal interaction between transboundary O3 and domestic emissions also underlies observational studies that estimate EIBO using O3 measurements at regional upwind sites27,40,41,42.
We contend that transboundary O3 engages in complex chemical interactions with domestic anthropogenic and biogenic precursors, but that interaction has not been captured by previous source-tagging studies due to their incomplete tracking of photochemical cycling. Conventional tagged schemes only track the interconversions between O3 and reactive nitrogen species6,8,43,44, based on the traditional concept of the “odd oxygen” (Ox ≡ O3 + NO2 + NO2 reservoirs) chemical family45,46. However, O3 interconverts with a much broader range of reservoir species, as described by the “expanded odd oxygen family” Oy ( ≡ Ox + Oz)47, where Oz consists of ROx ( ≡ OH + HO2 + RO2) radicals. Within the context of the Oy cycle, O3 photolysis is not a terminal sink of O3, because its OH radical product can react with CO and VOCs to form HO2 and RO2, recycling O3 in the presence of NO47. In this way, the interconversion between Ox and Oz plays a key role in driving O3 production and cycling47. Traditional Ox-tagging would neglect this Ox-Oz interconversion and misattribute O3 sources, because the local production of O3 involving reactions between transboundary O3-derived OH and domestic precursors would be incorrectly attributed solely to domestic sources. This oversight confounds accurate quantification of transboundary O3’s contribution to local pollution and impedes effective air quality management.
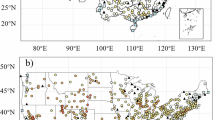
In this study, we demonstrated the complex interplay between transboundary O3 and domestic precursors and the resulting substantial impacts on surface O3, using an autumn O3 pollution period in China as a case study (Fig. 1). We developed and implemented a novel Oy-VOCs tagged framework into the WRF-Chem model48 (“Methods”). This framework uniquely tracked the full photochemical cycling of every Oy member originating from transboundary transport, from domestic anthropogenic VOCs (AVOCs), or from domestic biogenic VOCs (BVOCs). Using this advanced modeling tool, we quantified the contributions from transboundary transport, domestic sources, and their interactions to ambient O3. We explored the interaction mechanisms among these sources under different emission scenarios and evaluated the impacts of these cross–source interactions on O3 pollution mitigation.

Pie charts indicate the relative frequencies of O3 pollution severity in the provinces and cities of China during September 10th to October 10th, 2019.
Results
Transboundary-domestic interactions dominated surface O3 abundance over Eastern China
Using the tagged Oy-VOCs modeling framework (“Methods”), we partitioned the simulated ambient O3 over China into four major components according to individual Oy source identity (Fig. 2a): (1) domestic AVOC (DA) source: O3 produced from ROx originating from domestic AVOCs, (2) domestic BVOC (DB) source: O3 produced from ROx originating from domestic BVOCs, (3) transboundary (T) source: transboundary transport of O3 (and to a considerably smaller extent, transported foreign precursors) into China to directly becomes part of ambient O3, and (4) transboundary-domestic coupled (TDC) source: O3 generated via cross–source interaction, where some of the foreign O3 transported to China photolyzes to produce OH, interacts with domestic VOCs, OVOCs, and CO to form ROx that inherits the foreign identity, and subsequently reacts with domestic NO to form O3 (Text S1). Domestic O3 can also photolyze to become OH and subsequently cycle through the Oy family to produce O3. Therefore, DA-O3 and DB-O3 also each comprises two subcomponents, namely: O3 produced from HO2/RO2 radicals generated by photolysis of domestically sourced carbonyls, and O3 formed from reactions of domestic O3-derived OH with reactive carbon species (Fig. 2a, Text S2). In this study, we combined these two subcomponents within DA-O3 and DB-O3, respectively, because the ultimate ROx source for both subcomponents was domestic VOCs. Together, DA-O3, DB-O3, and TDC-O3 constitute “new O3 formation” within China. We focused on exploring the individual contributions of these four components and their interactions.

a Schematic of the four major components of ambient O3 in China: transboundary (T-O3), transboundary-domestic coupled O3 (TDC-O3), and O3 produced from domestic AVOCs (DA-O3) and BVOCs (DB-O3), respectively. b Source attributions for monthly mean MDA8O3 concentrations at the surface (bottom panel), in the BL (middle panel), and in the FT (top panel). Each chart displays the relative contributions of T-O3, TDC-O3, DA-O3, DB-O3, and other minor sources. Source attribution results are area-averaged for different regions in China during September 10th to October 10th, 2019.
Figure 2b quantifies the contributions of different O3 sources to regional monthly mean MDA8O3 at the surface, in the boundary layer (BL; <1 km altitude), and in the free troposphere (FT; 1 to 10 km altitude) during the study period. Across Eastern China, TDC-O3 was the single largest contributor to near-surface (surface and BL) O3 (44–49%), while T-O3 was the second-largest contributor (6–32%). Moreover, the importance of TDC-O3 relative to T-O3 was enhanced over areas of strong domestic emissions (Figs. S7, S8, and S9), indicating that domestic emissions dynamically modulated the chemical processing of foreign O3 in China. Simulations for other seasons confirmed that transboundary-domestic interactions substantially contributed to the surface O3 of China year-round, with variations driven by seasonal changes in transboundary O3 inflows, domestic emission levels, and synoptic conditions (Text S1, Fig. S10). These results demonstrated that the interactions of transboundary O3 with domestic precursors play a crucial role in shaping surface O3 levels in China.
In comparison, domestic AVOCs and BVOCs each contributed 8–33% (22–55% combined) of near-surface O3 over Eastern China, with larger biogenic contributions in the south reflecting BVOC emission patterns. We found that domestic O3 also cycled efficiently within the Oy cycle: more than 66% of DA-O3 and more than 65% of DB-O3 at the surface layer involved domestic O3-derived OH reacting with reactive carbon species (Text S2, Figs. S11 and S12). These findings align with Bates and Jacob47, demonstrating the critical role of recycling between the Ox and Oz families in O3 production on both intercontinental and regional scales. Previous studies that tagged only O3 or Ox would lose track of such recycling, leading to misattribution of O3 to its sources, whether transboundary, domestic, or involving interactions of transboundary O3 with domestic precursors.
Over western China (Fig. 2b), T-O3 dominated total ambient O3 (64–86%) throughout the troposphere, with TDC-O3 being the second largest contributor (14–34%); the contributions from DA-O3 and DB-O3 were negligible. At the national scale, T-O3 and TDC-O3 each accounted for approximately 40% of near-surface O3 during the study period, while domestic sources from AVOCs and BVOCs contributed approximately 10% each; the sum of other minor O3 sources accounted for <4% of near-surface O3 in China.
We analyzed the vertical budgets of the four major O3 components over Eastern China during the study period (Fig. S13). New O3 from TDC, DA, and DB sources were predominantly produced in the higher BL and then transported downward to the surface and upward to FT, consistent with previous studies that showed surface O3 in Eastern China was mostly produced in the BL49. In contrast, T-O3 subsided from the lower FT to the surface. In the FT over Northern (BTH and RNorth_EC) and Western China, T-O3 contributed 66% to 86% of total O3, while the contribution from TDC-O3 was considerably smaller (Fig. 2b). Conversely, over YRD, PRD, and RSouth_EC, TDC-O3 was comparable in magnitude to T-O3 in the FT. This regional contrast was consistent with the larger abundance of BVOCs in Southern China (Text S1, Fig. S14), which enhanced the photochemical processing of foreign O3 through interactions with domestic precursors.
O3 production from transboundary-domestic interactions limited by the availability of NOx, causing ROx surplus in the boundary layer of Southern China
Given that DA-O3, DB-O3 and TDC-O3 were mainly produced in the BL over Eastern China (Fig. S13), we next analyzed the spatial distributions of their production rates (P(Ox))50 and ROx dynamics in the BL, as well as the spatial correlations with surface precursor emissions (Fig. 3). Domestic AVOC emissions were spatially correlated with the emissions of anthropogenic NOx, with co-located maxima over the major urban clusters in BTH, YRD, and PRD (Fig. 3o,m). Consequently, the production rates of DA-ROx and DA-O3 both peaked over these urban clusters (Fig. 3c, i). In contrast, domestic BVOC emissions were high throughout Southern China (Fig. 3n), but DB-ROx and DB-O3 production rates were still spatially limited to locations of high NOx emissions (Fig. 3b, h). This spatial pattern reflected that the conversion of the DB-ROx to DB-O3 was inefficient in the relatively low-NOx BL over rural Southern China, resulting to accumulation of surplus DB-ROx in the BL over Southern China (Fig. 3e).

Spatial distributions of a–c tagged Ox production rates (P(Ox)), d–f tagged ROx concentrations, g–i tagged ROx production rates (P(ROx)), j–l tagged ROx loss rates (L(ROx)) attributed to transboundary-domestic interactions (top row), domestic BVOCs (middle row), and domestic AVOCs (bottom row); m–o precursor emissions of anthropogenic NOx, BVOCs, and AVOCs over Eastern China. Results are averaged in the boundary layer during the study period.
The OH produced from the photolysis of transboundary O3 reacted with domestic CO, AVOCs, and BVOCs to efficiently produce TDC-ROx in the BL over Eastern China (Fig. 3g), but the loss of TDC-ROx and the associated production of TDC-O3 were also limited by domestic NOx emissions (Fig. 3a, j). Transboundary-domestic interactions dominated the net production of ROx radicals (Fig. S15), causing a surplus of atmospheric oxidation capacity in the BL. Consequently, an extensive reservoir of TDC-ROx accumulated over Southern China. In addition, we found that the transboundary-domestic interactions produced more ROx than the DA and DB sources combined (Fig. 3d–f and S15). This impact of the interplay between transboundary O3 and domestic precursors had never been shown and has profound implications for regional air quality.
Nonlinear cross–source interactions affect OH turnover rates to modulate O3 formation
Our analyses above established that the cross-source interactions among transboundary O3 and domestic CO, VOC, and NOx precursors constitute a major source of OH, ROx, and O3 in China’s surface air. To elucidate the underlying mechanisms and quantify how perturbations of one source affect the attribution of others, we conducted sensitivity simulations comparing our BASE Oy-VOC simulation against simulations where we individually removed: (1) transboundary transport (by turning off B.C. and foreign emissions; Fig. 4a, c, e), (2) domestic AVOC emissions (Fig. 4b, d, f), and (3) domestic BVOC emissions (Fig. S16).

Comparison of the source attribution results for a-b O3 concentrations (top panel), c-d OH turnover rates (middle panel), and e-f OH concentrations (bottom panel) between the BASE simulation and the sensitivity experiments, removing a, c, e domestic AVOC emissions (SENS_NO_DAVOC, left panel) and b, d, f transboundary transport (SENS_NO_TRANSB, right panel), respectively. Results are area-averaged for different regions of China in the boundary layer during the study period.
Counter-intuitively, removing transboundary transport (predominantly of O3) led to increases of both DA-O3 and DB-O3 across China (Fig. 4a), despite transboundary-domestic interaction being a major regional ROx source. This result can be explained by weakened ROx sinks and the associated changes in the OH turnover rates, defined as the sum of reaction rates of OH with reductive species (mainly VOCs, OVOCs and CO) that convert it to HO2 and RO25,51,52. This OH turnover is a critical process in the Oy cycle, propagating conversions within the Oz reservoir and ultimately recycling O3 in the presence of NOx. Eliminating transboundary O3 weakened the scavenging of domestic ROx by transboundary-derived ROx through peroxide formation, thereby increasing DA-OH and DB-OH concentrations (Fig. 4e) and elevating their turnover rates (Fig. 4c), which enhanced domestic O3 formation. This nonlinear dynamics among ROx from different sources also caused the traditional “zero-out” method to significantly underestimate the contribution of transboundary transport to surface O3 by 18–45% over Eastern China, while overestimating domestic contributions (Fig. 4a). A similar underestimation was found when assessing the contributions of intercontinental transport to surface O3 over Western U.S. using zero-out modeling53.
In comparison, the removal of domestic AVOC emissions drove smaller but more nuanced feedback (Fig. 4b): T-O3 remained largely unchanged, but TDC-O3 decreased over BTH and YRD, and DB-O3 decreased slightly over BTH. This disparity arose because, although the removal of domestic AVOC emissions reduced OH consumption and increased transboundary-derived OH and DB-OH levels throughout China (Fig. 4f), the reduced availability of reactants also slowed the turnover rates of transboundary-derived OH over BTH and YRD and of DB-OH over BTH (Fig. 4d). Elsewhere in China, the turnover rates of TDC-OH and DB-OH were not limited by the availability of AVOC, such that TDC-O3 and DB-O3 production were not significantly affected. Zeroing out domestic AVOC emissions would therefore lead to an underestimation of background O3 over BTH and YRD, thus overestimating the improvements achievable through domestic AVOC emission control.
Removing domestic BVOC emissions led to yet different responses (Fig. S16): by greatly reducing OH consumption, ambient OH levels enhanced substantially across China during the study period (Fig. S16c). However, the turnover rates of OH from TDC and DA sources were balanced out by the lack of BVOC reactants (Fig. S16b). The net effect was that neither TDC-O3 nor DA-O3 was substantially perturbed by the removal of BVOCs (Fig. S16a), leading to a superficial agreement in DB-O3 estimates between the zero-out and tagged methods. However, this apparent consistency conceals notable discrepancies in the underlying HOx chemistry.
Transboundary transport of O3 suppressed the effective incremental reactivity (EIR) of domestic VOCs
A key implication of our findings is that the scavenging of domestic ROx radicals by transboundary-derived OH terminates the photochemical chain reactions responsible for propagating domestic O3 production (Fig. 4c, e). Consequently, the net O3 yield per molecule of domestically emitted VOC is suppressed in the presence of transboundary O3. We defined this crucial quantifier as the effective incremental reactivity (EIR) of a VOC species, which represents the total number of O3 molecules produced during daytime by a single VOC molecule throughout its atmospheric lifetime under actual ambient conditions, accounting for all cross-source interactions. This metric can be quantified by our novel Oy-VOC tagged modeling, which tracks the transport and complete chemical evolution of each VOC leading to O3 formation.
Figure 5 shows that, as simulated in the BASE scenario, the EIR values for individual VOC categories may vary by as much as a factor of 14 across different regions in China (Fig. 5a–e), with the highest EIRs generally associated with VOCs emitted from BTH, and the lowest EIRs associated with emissions from Western China. This spatial disparity correlated with the regional differences in NOx emissions and is consistent with the conversion of ROx to O3 being limited by the availability of NOx, shown in Figs. 3 and 5f. Within each source region, the EIR values were dependent on the reactivity and number of unsaturated bonds of VOC species, with aromatics and alkenes having larger EIR values than alkanes.

Simulated EIR for a anthropogenic alkanes, b biogenic alkanes, c anthropogenic alkenes, d biogenic alkenes, and e anthropogenic aromatics emitted from different regions of China during the study period. The EIRs were dissected for the O3 produced within the VOC source region, in downwind domestic areas, and in downwind foreign regions. Also shown are changes in EIR values in sensitivity experiments where domestic BVOC (SENS_NO_DBVOC) emissions, AVOC (SENS_NO_ABVOC) emissions, and transboundary transport (SENS_NO_TRANSB) were removed, respectively. The gray dashed lines in each panel show the MIR values for each VOC category55. f Area-averaged emissions of NOx, along with area-averaged emissions of AVOCs and BVOCs, weighted by NOx emissions.
Beyond NOx availability, EIR values were also modulated by the regional atmospheric oxidation capacity, which was in turn strongly affected by cross-source interactions. For example, removing transboundary O3 transport significantly enhanced EIR in both source and downwind regions by eliminating the nationwide ROx sink associated with transboundary-domestic coupling (Fig. 4). Zeroing out domestic AVOC or BVOC emissions would slightly increase the EIRs of VOCs from the other sources, particularly over VOC-saturated regimes like Southern China, due to reduced competition for OH radicals.
Figure 5 further compares EIRs with a widely-used metric for evaluating the ozone formation potential of VOC species54: the maximum incremental reactivity (MIR)55,56,57. The MIR is defined as the maximum difference of O3 molecular concentration for a molecule of VOC added to a baseline air mixture. MIR values are typically calculated from box model simulations under prescribed, static daytime environmental conditions, and therefore do not reflect the dynamic physical and chemical environment in which VOC photochemical react during its lifetime. More importantly, because the MIR metric does not track the full photochemical cycling of VOC’s oxidation products, its values substantially underestimated the O3 produced from AVOCs (Fig. 5). In contrast, the EIR metric realistically quantifies the O3 formation throughout a VOC’s lifetime and explicitly resolves source-receptor relationship, offering a more spatially resolved and context-sensitive assessment of VOC’s impacts on regional O3 pollution.
Non-linear responses of background ozone to domestic emission control
Finally, we quantified how the interactions between background O3 and domestic anthropogenic emissions affect the response of surface O3 to emission controls in China, providing critical insights for policy-making. Figure 6 shows simulated MDA8O3 change isopleths in response to precursor reductions (i.e., the empirical kinetic modeling approach, EKMA58) for total surface O3 and for each of its four major components. Each of the four O3 components exhibited diverse, nonlinear responses to changes in AVOC and NOx emissions. Reducing AVOC emissions decreased DA-O3 nationwide. In addition, while reducing AVOCs also slightly decreased TDC-O3 over BTH and YRD, but that benefit was counteracted by increased DB-O3. In contrast, a mild reduction of NOx emission from its current levels effectively reduced background O3 components (T-O3, TDC-O3, and DB-O3) in Southern China, but was ineffective in BTH and YRD. Table S6 quantifies the maximum achievable reduction in the monthly mean MDA8O3 concentrations attainable through complete removal of domestic NOx and AVOC emissions. We found that removing all domestic anthropogenic emissions reduced ambient MDA8O3 by 28–49% across different regions of China. Of this total reduction, 41–69% was attributed to the decrease in DA-O3, while reductions in both TDC-O3 and DB-O3 jointly accounted for 28–54%. While previous studies also identified nonlinear responses for regional upwind O3 and O3 from biogenic precursors to domestic emission controls31,32, they did not resolve the radical chemistry underlying cross–source interactions.

Results represent monthly means during September 10th to October 10th, 2019, across different regions of China (defined in Fig. 2). Relative changes are calculated with respect to the standard O3 in the base simulation without emission perturbations.
Overall, the dynamics of background O3 components shifted the total surface O3 towards a greater NOx-sensitivity regime in Southern China and a greater VOC-sensitivity in Northern China, relative to the dynamics of DA-O3 alone. For instance, in the NOx-limited PRD and RSouth_EC areas, a 50% AVOC emission reduction would increase DB-O3, partially offsetting the reduction in DA-O3. Thus, concurrent reduction of NOx emissions is necessary to suppress DB-O3. Conversely, in the VOC-limited BTH, NOx reduction alone would initially increase T-O3 and TDC-O3, such that concurrent AVOC controls would also be required for near-term O3 mitigation. These results demonstrated that cross-source interactions substantially alter optimal emission control pathways, underscoring the need for integrated, region-specific strategies that account for the complex behavior of background O3.
Discussion
We demonstrated that transboundary O3 engages in complex, dualistic interactions with domestic emissions of anthropogenic and biogenic precursors, acting both as a major source and a major sink of ROx throughout China and substantially affecting regional photochemical chemistry and surface O3 levels. Furthermore, the presence of transboundary O3 substantially suppressed the O3 formation potential of domestic AVOC emissions. This realization overthrew the prevailing view that background O3 is mostly inert to changes in domestic anthropogenic emissions, fundamentally changing our perspective on “controllable” O3 levels and demands a reassessment of mitigation strategies. Our new perspective of background O3 is especially important under future climate scenarios that project enhanced biogenic emissions and intensified transboundary transport in a future warmer climate59,60. Our study focused on attributing O3 to transboundary O3 and domestic VOC sources, therefore the current protocol was not designed to quantify the contribution of NO emissions to O3 formation. However, our Oy-based source attribution framework is inherently expandable. Future applications can incorporate other critical O3 drivers, such as NOx, HONO, and reactive halogens, to build a comprehensive, multi-faceted understanding of O3 production and its controls on regional, continental, and intercontinental scales.
Methods
Standard WRF-Chem simulation of an O3 pollution episode over China in Fall 2019
We used the WRF-Chem model (v3.8)48 to simulate surface O3 over China during September 10th to October 10th, 2019, preceded by a 10-day model initialization. This period was characterized by extensive O3 pollution across Eastern China, with the observed maximum daily 8-h average surface O3 levels (MDA8O3) exceeding the World Health Organization’s Air Quality Guideline (100 μg m−3)61 on >50% of days across most provinces and major cities (Fig. 1). Our simulations (domain in Fig. 1) were configured with 20-km horizontal resolution and 30 vertical layers up to 50 hPa, with ten layers resolving the lowest 1 km. Meteorology was nudged toward observations every 3 h62. Chemical initial and boundary conditions were from a CAM-chem global simulation. Monthly anthropogenic emissions were from the Multi-resolution Emission Inventory for China (MEIC) for 201963,64. The molar fraction of NO2 in NOx combustion emissions varies considerably with factors such as fuel type, combustion temperature, air–fuel ratio, and the use of catalytic end-of-pipe treatments (Text S3). In this study, a ratio of 90% NO to 10% NO2 was adopted65. Hourly biomass burning emissions were from the Fire Inventory from NCAR (FINN)66 for 2019. Meteorology-dependent emissions (BVOCs and soil NOx) were calculated online67. Full model configurations for the standard simulation are detailed in Table S1.
Development and implementation of an Oy-VOCs tagged source attribution framework
We developed an Oy-VOCs tagged mechanism for the regional atmospheric chemistry modeling (RACM) mechanism68 in WRF-Chem, adopting the definition of Oy species from Bates and Jacob47. Each standard reaction involving VOCs and Oy species in the RACM mechanism was modified to one or more tagged reactions, facilitating explicit tracking of the respective chemical evolutions of the reactive carbon content (i.e., the tagged carbon thread) and the Oy family (i.e., the tagged Oy thread) originating from transboundary transport and domestic VOCs. The basic assumptions of the Oy-VOCs tagging protocol are as follows:
(1) NO is not a member of the Oy family, and NO emissions (which are predominantly anthropogenic in China) do not contribute directly to Oy. In the reactions between HO2/RO2 and NO, the resulting NO2 product, and that NO2’s subsequent O3 product, inherit the source identity of the HO2/RO2 reactant. This approach allowed for a clear attribution of VOC emissions on O3 formation.
(2) For reactions with no net change in the total Oy molar amount (i.e., reactions where a certain number of moles of Oy reactants produced the same number of moles of Oy products), the Oy products retained the source identity of the Oy reactant. For example, the reactions VOC+Oy and CO + OH merely transform existing Oy species to another (e.g., transforms OH to HO2 or RO2). In these reactions, the Oy products inherit the identity of the Oy reactant (tagged Oy thread), while non-Oy organic products inherit the VOC reactant’s identity (tagged carbon thread).
(3) For reactions with net production in Oy, the newly formed Oy products inherit the source identity of the non-Oy reactant. For example, in the photolysis of an OVOC from a specific VOC precursor, the ROx products would inherit the identity of that VOC precursor. Thereby, VOC sources imprinted their identity on Oy only when their OVOC products photolyzed to produce new HO2 or RO2 radicals.
O3 source attribution results derived from our protocol depend on the exact definition of Oy family, particularly the member species included and the stoichiometric coefficients applied in Oy molar amount calculations (e.g., O3 = 1 unit of Oy, each ROx species = 0.5 unit of Oy). The Oy-VOCs tagging protocol builds upon the Ox-VOCs framework previously established by Butler et al.8. A key advancement of our method is the explicit tracking of Oy, which preserves the source identity of O3 throughout its photochemical cycling within the Oy family. In total, the tagged species include 16 primary VOCs (including 3 primary oxygenated volatile organic compounds, OVOCs), 6 secondary VOCs, and 42 Oy species (Table S2). The 237 standard reactions in RACM were expanded to 593 tagged reactions (Table S3). All other atmospheric processes, including transport, gas-particle partitioning (only considering gaseous HNO3), and dry/wet deposition, were identical to the standard simulation. A process analysis (PA) module69,70 was implemented to quantify contributions of individual physical and chemical processes to simulated concentrations of tagged species.
In this study, we implemented the Oy-VOCs tagged framework to resolve Oy contributed by six sectoral sources: AVOCs, BVOCs, wildfire VOCs (FVOCs), Oy from initial conditions (I.C.), Oy from boundary conditions (B.C.), and an “Extra” source to account for the direct emissions of NO2 from anthropogenic and wildfire sources. Furthermore, AVOCs and BVOCs were tagged for seven geographical origins (Fig. 1), including the Beijing-Tianjin-Hebei area (BTH), the Yangtze River Delta area (YRD), the Pearl River Delta area (PRD), the remaining parts of Northeastern China (RNorth_EC) and Southeastern China (RSouth_EC), Western China (WestCN), and foreign regions. Foreign and domestic emissions of FVOCs and NO2, as well as B.C. for O3 and other species, were also individually tagged.
Evaluation of the WRF-Chem simulations and the tagged framework
We evaluated the standard and tagged O3 simulations against hourly surface measurements of O3 and NO2 across China (Fig. S1). The model captured the observed pollutant concentrations and their spatiotemporal variability (Fig. S2, Table S4). Simulated O3 showed normalized mean biases (NMB) of 9–24% with hourly correlations (R) of 0.8–0.9 against observations. For simulated NO2, the NMB ranged from −40 to 7% with R values of 0.6–0.8. The summed concentrations of tagged VOCs, O3, ROx, and other Oy species from different sources in the tagged simulation were near identical to those in the standard simulation across the troposphere (R = 1.0; NMB < 3%, with minor upper-tropospheric imbalances; Figs. S3–S6), validating our tagged source methodology. The impacts of chemical I.C. were negligible after the 10-day spin-up. In addition, we found that foreign sources primarily affect China through transboundary transport of O3; transboundary transport of CO and NMVOC precursors has relatively minor impacts on surface O3 levels in China.
Data availability
All the datasets used in this study are publicly accessible. The Final Operational Global Analysis (FNL) data (https://gdex.ucar.edu/datasets/d083002/) used for meteorological initial and boundary conditions, and National Centers for Environmental Prediction (NCEP) ADP Global Surface Observational Weather Data (https://gdex.ucar.edu/datasets/d461000/) used for observational nudging of surface meteorology were obtained from National Center for Atmospheric Research (NCAR) Geoscience Data Exchange (GDEX) Archive. The Community Atmosphere Model with Chemistry (CAM-chem) output data (https://www.acom.ucar.edu/cesm/subset.shtml) used for chemical initial and boundary conditions, and the Fire Inventory from NCAR (FINN) data (https://www.acom.ucar.edu/Data/fire/) used for wildfire emissions were provided by NCAR Research Data Archive. The Multi-resolution Emission Inventory (MEIC) data used for anthropogenic emissions were downloaded from MEIC official website (http://meicmodel.org.cn/?page_id=45&lang=en). Model of Emissions of Gases and Aerosols from Nature (MEGAN) data used for biogenic emissions were retrieved from NCAR Weather Research and Forecasting model coupled to Chemistry (WRF-Chem) Pre-processors Archive (https://www.acom.ucar.edu/wrf-chem/download.shtml).
References
Health Effects Institute. State of Global Air 2025: A Report on Air Pollution and Its Role in the World’s Leading Causes of Death. (Boston, 2025).
Wang, W. et al. Long-term trend of ozone pollution in China during 2014–2020: distinct seasonal and spatial characteristics and ozone sensitivity. Atmos. Chem. Phys. 22, 8935–8949 (2022).
Zheng, B. et al. Trends in China’s anthropogenic emissions since 2010 as the consequence of clean air actions. Atmos. Chem. Phys. 18, 14095–14111 (2018).
Li, K. et al. Anthropogenic drivers of 2013–2017 trends in summer surface ozone in China. Proc. Natl. Acad. Sci. USA 116, 422 (2019).
Wang, W. et al. Ozone pollution mitigation strategy informed by long-term trends of atmospheric oxidation capacity. Nat. Geosci. 17, 20–25 (2024).
Li, P. et al. Source attribution of near-surface ozone trends in the United States during 1995–2019. Atmos. Chem. Phys. 23, 5403–5417 (2023).
Wang, P., Chen, Y., Hu, J., Zhang, H. & Ying, Q. Source apportionment of summertime ozone in China using a source-oriented chemical transport model. Atmos. Environ. 211, 79–90 (2019).
Butler, T., Lupascu, A., Coates, J. & Zhu, S. TOAST 1.0: tropospheric ozone attribution of sources with tagging for CESM 1.2. 2. Geosci. Model Dev. 11, 2825–2840 (2018).
Li, L. et al. Source apportionment of surface ozone in the Yangtze River Delta, China in the summer of 2013. Atmos. Environ. 144, 194–207 (2016).
Zhao, Y. et al. A comprehensive provincial-level VOCs emission inventory and scenario analysis for China: Enhanced sectoral resolution through GAINS-China model. Atmos. Environ. X 25, 100316 (2025).
Lu, X. et al. The underappreciated role of agricultural soil nitrogen oxide emissions in ozone pollution regulation in North China. Nat. Commun. 12, 5021 (2021).
Wang, T. et al. Ground-level ozone pollution in China: a synthesis of recent findings on influencing factors and impacts. Environ. Res. Lett. 17, 063003 (2022).
Qu, K. et al. The effect of cross-regional transport on ozone and particulate matter pollution in China: a review of methodology and current knowledge. Sci. Total Environ. 947, 174196 (2024).
Fiore, A. M. et al. Multimodel estimates of intercontinental source-receptor relationships for ozone pollution. J. Geophys. Res. Atmos. 114, D04301 (2009).
Zhang, Y. et al. Contributions of World Regions to the Global Tropospheric Ozone Burden Change From 1980 to 2010. Geophys. Res. Lett. 48, e2020GL089184 (2021).
Shen, L. et al. Atmospheric transport drives regional interactions of ozone pollution in China. Sci. Total Environ. 830, 154634 (2022).
Jaffe, D. A. et al. Scientific assessment of background ozone over the US: implications for air quality management. Elem. Sci. Anthr. 6: 56 (2018).
Chen, C. et al. A comprehensive review of tropospheric background ozone: definitions, estimation methods, and meta-analysis of its spatiotemporal distribution in China. Atmos. Chem. Phys. 25, 15145–15169 (2025).
Ye, X. et al. Spatial and temporal variations of surface background ozone in China analyzed with the grid-stretching capability of GEOS-Chem High Performance. Sci. Total Environ. 914, 16909 (2024).
Dolwick, P. et al. Comparison of background ozone estimates over the western United States based on two separate model methodologies. Atmos. Environ. 109, 282–296 (2015).
Lefohn, A. S. et al. Estimates of background surface ozone concentrations in the United States based on model-derived source apportionment. Atmos. Environ. 84, 275–288 (2014).
Garatachea, R. et al. National and transboundary contributions to surface ozone concentration across European countries. Commun. Earth Environ. 5, 588 (2024).
Derwent, R. G., Witham, C. S., Utembe, S. R., Jenkin, M. E. & Passant, N. R. Ozone in Central England: the impact of 20 years of precursor emission controls in Europe. Environ. Sci. Policy 13, 195–204 (2010).
Ni, R., Lin, J., Yan, Y. & Lin, W. Foreign and domestic contributions to springtime ozone over China. Atmos. Chem. Phys. 18, 11447–11469 (2018).
U.S. EPA: Air quality criteria for ozone and related photochemical oxidants (final report, 2006), U.S. Environmental Protection Agency, Washington, DC, EPA/600/R-05/004aF-cF, https://cfpub.epa.gov/ncea/risk/recordisplay.cfm?deid=149923 (last access: 4 November 2025).
Vingarzan, R. A review of surface ozone background levels and trends. Atmos. Environ. 38, 3431–3442 (2004).
Reid, N., Yap, D. & Bloxam, R. The potential role of background ozone on current and emerging air issues: an overview. Air Qual. Atmos. Health 1, 19–29 (2008).
Fiore, A. et al. Variability in surface ozone background over the United States: implications for air quality policy. J. Geophys. Res.-Atmos. 108, 4787 (2003).
Fiore, A. M. et al. Estimating North American background ozone in U.S. surface air with two independent global models: variability, uncertainties, and recommendations. Atmos. Environ. 96, 284–300 (2014).
Lu, X. et al. Exploring 2016-2017 surface ozone pollution over China: source contributions and meteorological influences. Atmos. Chem. Phys. 19, 8339–8361 (2019).
Kang, M., Zhang, H. & Ying, Q. Enhanced summertime background ozone by anthropogenic emissions—implications on ozone control policy and health risk assessment. Atmos. Environ. 314, 120116 (2023).
Wang, R. et al. The reward and penalty for ozone pollution control caused by natural sources and regional transport: a case study in Guangdong province. Sci. Total Environ. 949, 174984 (2024).
Clappier, A., Belis, C. A., Pernigotti, D. & Thunis, P. Source apportionment and sensitivity analysis: two methodologies with two different purposes. Geosci. Model Dev. 10, 4245–4256 (2017).
Cohan, D. S., Hakami, A., Hu, Y. & Russell, A. G. Nonlinear response of ozone to emissions: source apportionment and sensitivity analysis. Environ. Sci. Technol. 39, 6739–6748 (2005).
Dunker, A. M., Yarwood, G., Ortmann, J. P. & Wilson, G. M. Comparison of source apportionment and source sensitivity of ozone in a three-dimensional air quality model. Environ. Sci. Technol. 36, 2953–2964 (2002).
Wang, Y. et al. Soil emissions of reactive oxidized nitrogen reduce the effectiveness of anthropogenic source control in China. Environ. Sci. Technol. 58, 21015–21024 (2024).
Galmarini, S. et al. Technical note: coordination and harmonization of the multi-scale, multi-model activities HTAP2, AQMEII3, and MICS-Asia3: simulations, emission inventories, boundary conditions, and model output formats. Atmos. Chem. Phys. 17, 1543–1555 (2017).
Jonson, J. E. et al. The effects of intercontinental emission sources on European air pollution levels. Atmos. Chem. Phys. 18, 13655–13672 (2018).
Huang, M. et al. Impact of intercontinental pollution transport on North American ozone air pollution: an HTAP phase 2 multi-model study. Atmos. Chem. Phys. 17, 5721–5750 (2017).
Zhang, X. et al. Long-term variations in surface ozone at the Longfengshan regional atmosphere background station in Northeast China and related influencing factors. Environ. Pollut. 348, 123748 (2024).
Xiao, J., Wang, T., Wang, Y., Yang, Q. & Shi, Y. Analysis of ozone time series variation in atmospheric background area in China. Res. Environ. Sci. 35, 2128–2135 (2022).
Chakraborty, T., Beig, G., Dentener, F. J. & Wild, O. Atmospheric transport of ozone between Southern and Eastern Asia. Sci. Total Environ. 523, 28–39 (2015).
Butler, T., Lupascu, A. & Nalam, A. Attribution of ground-level ozone to anthropogenic and natural sources of nitrogen oxides and reactive carbon in a global chemical transport model. Atmos. Chem. Phys. 20, 10707–10731 (2020).
Lupaşcu, A., Otero, N., Minkos, A. & Butler, T. Attribution of surface ozone to NOx and volatile organic compound sources during two different high ozone events. Atmos. Chem. Phys. 22, 11675–11699 (2022).
Wang, Y., Logan, J. A. & Jacob, D. J. Global simulation of tropospheric O3-NOx-hydrocarbon chemistry: 2. Model evaluation and global ozone budget. J. Geophys. Res. Atmos. 103, 10727–10755 (1998).
Crutzen, P. J., Lawrence, M. G. & Pöschl, U. On the background photochemistry of tropospheric ozone. Tellus B Chem. Phys. Meteorol. 51, 123–146 (1999).
Bates, K. & Jacob, D. J. An expanded definition of the odd oxygen family for tropospheric ozone budgets: implications for ozone lifetime and stratospheric influence. Geophys. Res. Lett. 47, e2019GL084486 (2020).
Grell, G. A. et al. Fully coupled “online” chemistry within the WRF model. Atmos. Environ. 39, 6957–6975 (2005).
Ye, F. et al. Integrated process analysis retrieval of changes in ground-level ozone and fine particulate matter during the COVID-19 outbreak in the coastal city of Kannur, India. Environ. Pollut. 307, 119468 (2022).
Young, P. J. et al. Tropospheric ozone assessment report: assessment of global-scale model performance for global and regional ozone distributions, variability, and trends. Elem. Sci. Anthr. 6, 10 (2018).
Kleinman, L. I. et al. Ozone production efficiency in an urban area. J. Geophys. Res. Atmos. 107, ACH 23–12 (2002).
Cho, C. et al. Experimental chemical budgets of OH, HO2, and RO2 radicals in rural air in western Germany during the JULIAC campaign 2019. Atmos. Chem. Phys. 23, 2003–2033 (2023).
Guo, Y. et al. Long-lived species enhance summertime attribution of North American ozone to upwind sources. Environ. Sci. Technol. 51, 5017–5025 (2017).
Wu, R. & Xie, S. Spatial distribution of ozone formation in China derived from emissions of speciated volatile organic compounds. Environ. Sci. Technol. 51, 2574–2583 (2017).
Zhang, Y. et al. Developing the maximum incremental reactivity for volatile organic compounds in major cities of Central-Eastern China. J. Geophys. Res. Atmos. 127, e2022JD037296 (2022).
Carter, W. P. Development of an improved chemical speciation database for processing emissions of volatile organic compounds for air quality models. Center for Environmental Research and Technology (CE-CERT), University of California, Riverside. http://www.engr.ucr.edu/~carter/emitdb (2008).
Wang, M. et al. Maximum incremental reactivity for volatile organic compounds in three city clusters of China: quantification, variability, and implications for ozone control. Atmos. Environ. 361, 121459 (2025).
Sillman, S., Logan, J. A. & Wofsy, S. C. The sensitivity of ozone to nitrogen oxides and hydrocarbons in regional ozone episodes. J. Geophys. Res. Atmos. 95, 1837–1851 (1990).
Chen, W. et al. Assessment of background ozone concentrations in China and implications for using region-specific volatile organic compounds emission abatement to mitigate air pollution. Environ. Pollut. 305, 119254 (2022).
Yang, Y. et al. Impact of temperature on the biogenic volatile organic compound (BVOC) emissions in China: a review. J. Environ. Sci. 159, 649–660 (2025).
Kan, H. World Health Organization air quality guidelines 2021: implication for air pollution control and climate goal in China. Chinese Med. J. 135, 513–515 (2022).
Liu, Y. et al. An implementation of obs-nudging-based FDDA into WRF for supporting ATEC test operations. 2005 WRF user workshop. Paper 10.7. https://www2.mmm.ucar.edu/wrf/users/docs/How_to_run_obs_fdda.html (2005).
Zhang, Q. et al. Asian emissions in 2006 for the NASA INTEX-B mission. Atmos. Chem. Phys. 9, 5131–5153 (2009).
Li, M. et al. MIX: a mosaic Asian anthropogenic emission inventory under the international collaboration framework of the MICS-Asia and HTAP. Atmos. Chem. Phys. 17, 935–963 (2017).
Garin, F. Mechanism of NOx decomposition. Appl. Catal. A Gen. 222, 183–219 (2001).
Wiedinmyer, C. et al. The Fire INventory from NCAR (FINN)—a high resolution global model to estimate the emissions from open burning. Geosci. Model Dev. Discuss 3, 2439–2476 (2010).
Guenther, A. et al. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature). Atmos. Chem. Phys. 6, 3181–3210 (2006).
Stockwell, W. R., Kirchner, F., Kuhn, M. & Seefeld, S. A new mechanism for regional atmospheric chemistry modeling. J. Geophys. Res. Atmos. 102, 25847–25879 (1997).
Tao, W. et al. Effects of urban land expansion on the regional meteorology and air quality of eastern China. Atmos. Chem. Phys. 15, 8597–8614 (2015).
Wang, H. et al. Seasonality and reduced nitric oxide titration dominated ozone increase during COVID-19 lockdown in eastern China. npj Clim. Atmos. Sci. 5, 24 (2022).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2023YFC3706205), the National Natural Science Foundation of China (42325504, 42305188), the Shenzhen Science and Technology Program (KQTD20210811090048025, JCYJ20220818100611024), and the High-level Special Funds (G03034K006). Computational resources were supported by the Center for Computational Science and Engineering at the Southern University of Science and Technology.
Author information
Authors and Affiliations
Contributions
Conceptualization: W.T. and T.M.F.; Funding acquisition: T.M.F. and M.S.; Supervision: T.M.F.; Methodology: W.T.; Investigation: W.T., A.Z., T.J., J.M., X.W., H.S., J.L., H.S., Y.C., R.N., and Y.G.; Formal analysis: W.T.; Software: W.T.; Visualization: W.T.; Writing-original draft: W.T. and T.M.F.; Writing—review and editing: W.T., T.M.F., J.L., H.S., Y.C., R.N., A.Z., Y.G., T.J., J.M., X.W., H.S., and M.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tao, W., Fu, TM., Liu, J. et al. Complex interplay between transboundary ozone and domestic emissions shapes surface ozone pollution in China. npj Clim Atmos Sci 9, 107 (2026). https://doi.org/10.1038/s41612-026-01379-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41612-026-01379-8
