
Abstract
Biliary tract cancer (BTC) has greatly influenced patient survival for years. Nowadays, immunotherapy represents a promising breakthrough and proteomics is one of powerful technologies in biomarker research. We collected plasma and tissue samples from 37 patients with advanced BTC and 92 proteins were analyzed by proximity extension assay (PEA). Through linear mixed effect models, compared to partial response (PR) group, 8 proteins, IL7, ANGPT2, IL15, HO-1, CXCL1, CXCL5, IL33, and VEGFA, exhibited significantly higher expression in stable disease and progressive disease (SD_PD) group in response-effect analysis. It was also revealed that a subset of proteins increased over time, including PDCD1, TNFRSF4, DCN, CRTAM, VEGFR-2 and ADA in PR group and PDCD1, IL10, ADA, CD28, and PTN in SD_PD group. In interaction-effect analysis, HO-1, ANGPT2, IL15 were three significant differentially expressed proteins (DEPs). Receiver operating characteristic (ROC) analysis further demonstrated that HO-1, ANGPT2, IL15 showed high accuracy in patients with immune checkpoint blockade (ICB) treatment plus chemotherapy (AUC = 0.74). In addition, based on the obtained plasma and tissue samples, two nomogram models were constructed for predicting the prognosis of BTC by genome combined with proteomics. Collectively meaningful proteomic biomarkers are beneficial to evaluate the efficacy of immunotherapy, and these discovered biomarkers may be included in the scope of treatments’ evaluation and improvement in future study.
Similar content being viewed by others


Introduction
Biliary tract cancer (BTC) is an aggressive malignancy that includes gallbladder cancer (GBC) and cholangiocarcinoma (CCA). Previous studies have shown that the prognosis of BTC is poor. For example, as the data reported, the median overall survival (OS) of patients receiving immunotherapy was less than 1 year and readily affected by various factors1. For a long time, gemcitabine combined with cisplatin (GemCis) chemotherapy has been the main first-line standard treatment for advanced BTC; however, its clinical efficacy is not satisfactory. In recent years, the emergence of immune checkpoint inhibitors has profoundly changed the treatment of BTC2. Immunotherapy combined with chemotherapy may benefit more patients with BTC. In a randomized and double-blind phase 3 trial, statistical evidence proved that pembrolizumab plus gemcitabine and cisplatin brought promising improvements in OS3. It was also reported that gemcitabine and cisplatin plus durvalumab were evaluated as first-line treatment in patients with advanced BTC4. Our previous study evaluated the efficacy and safety of Camrelizumab plus gemcitabine and oxaliplatin (GEMOX) as a first-line treatment. The results showed that the median progression free survival rate (PFS) was 50%, and the median PFS and OS were 6.1 months and 11.8 months, respectively5.
However, some patients treated with immune checkpoint blockade (ICB) do not show durable treatment responses, highlighting the crucial need to identify predictive biomarkers for screening advantageous populations. We analyzed the relationship between genomic features and the efficacy of BTC immunotherapy. However, whole genome sequencing did not find a meaningful correlation between different response groups. No strong findings confirmed that Camrelizumab plus the GEMOX treatment was associated with circulating biomarkers, abundance of immune cell infiltration in the center or margin of tumors, and tissue or blood tumor mutation burden (TMB)6. Compared to genomics, proteomics can assist in inspecting mRNAs, proteins and metabolites under different physiological or pathological conditions7. Through differential technologies, proteomic study provides in-depth methods for exploring biomarkers, making it easier to detect diseases dynamically. To further separate differentially expressed biomarkers and improve the prediction of treatment response, we performed proteomic analysis using peripheral blood samples from patients.
Peripheral blood plasma samples were collected from 37 patients. According to the RECIST criteria, patients were divided into complete response (CR), partial response (PR), progressive disease (PD), and stable disease (SD)8. As the present study mainly aimed to find biomarkers of patients with clearly effective treatment, we divided patients into two groups according to clinical response, including 16 patients from SD_PD group(namely, 3 PD patients and 13 SD patients) and 21 patients from PR group(noteably, there were 0 CR patients). Proteome analysis was performed based on the Olink’s proximity extension assay (PEA), and the results indicated that several differentially expressed proteins (DEPs) were significant for response-effect, time-effect and interaction-effect. Additionally, by integrating proteomic results with our prior genomic analysis results, we could gain a deeper understanding of the potential molecular mechanism or signal transduction pathway between gene expression and protein function and find a more sensitive method to search for biomarkers, which may help reshape the landscape of immunotherapy response prediction in BTC patients.
Results
Patient characteristics
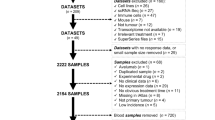
To pinpoint circulating protein biomarkers associated with ICB responses, we used PEA to analyze protein profiles in a cohort of 37 BTC patients at three distinct time points T1 (baseline), T2 (6 weeks after treatment), and T3 (6 months after treatment). ICB responders were defined as BTC patients exhibiting partial response (n = 21), while non-responders encompassed those with progressive or stable disease (n = 16) (Table 1). To identify proteins displaying significant alterations over time and distinguishing between response groups, we employed linear mixed effect models, incorporating the main effects of time and response, as well as an interaction effect between time and response. Through quality control analysis, although one sample was close to the lower detection limit, it could not be excluded from the analysis point of view; therefore, the analysis data included all samples (Fig. S1).
Analysis of treatment-associated function alterations
The analysis process is illustrated in Fig. 1. Subsequently, immune-related genes exhibiting differential expression were subjected to GO, KEGG, COG, PPI and subcellular localization analysis. GO analysis results showed that differentially expressed genes (DEGs) were mostly enriched in cellular process, biological regulation, immune system process, and cellular anatomical entity (Fig. 2A). The top 30 enriched functions for molecular function, biological process, and cellular component were selected individually. Among them, the most prominent projects involved nature killer cell lectin-like receptor binding, gamma-delta T cell activation, negative regulation of defense response to virus b etc. and negative regulation of immune response (Fig. 2B). KEGG analysis results showed that DEGs were mostly enriched in immune system, infectious disease (viral), and signal transduction (Fig. 2C). In addition, the KEGG results showed that the enrichment was mainly in the process of cancer, nature killer cell mediated cytotoxicity and TNF signaling pathway (Fig. 2D). COG classification showed that, in immune oncology, the majority of protein sequences were mainly concentrated in signal transduction mechanism (Fig. 2E). Meanwhile, DEPs mainly focused on inorganic ion transport and metabolism, and general function prediction only (Fig. 2F).

JPH Jiangsu Province Hospital, CHOL cholangiocarcinoma, GBC gallbladder cancer, NGS next-generation sequencing, PR partial response, SD_PD stable disease and progressive disease, PEA proximity extension assay, GO gene ontology, KEGG kyoto encyclopedia of gene and genomics, COG clusters of orthologous groups, PPI protein–protein interaction networks, ICB immune checkpoint blockade.

A Gene-ontology classification analysis. B Gene-ontology enrichment analysis showing the top 30 Gene-ontology terms. C KEGG classification analysis. D KEGG pathway enrichment analysis showing the top 30 KEGG pathways. E Immuno-oncology COG classification analysis. F COG classification analysis of SD_PD vs. PR groups. G The PPI network showing the interaction relationship of differentially expressed proteins. H Subcellular location analysis demonstrating the distribution of differentially expressed proteins. KEGG Kyoto Encyclopedia of Genes and Genomes, PPI protein–protein interaction.
To further understand the interplay among proteins with differential expression, we utilized STRING to build a PPI network by incorporating these proteins. We screened the first 10 hub proteins with the highest p-value. Among these, CXCL1, IL7, IL6, CXCL5, MICA, and MICB were upregulated (Fig. 2G). Disorders between these proteins could potentially have a strong connection to the onset and progression of BTC. Further analysis of subcellular localization revealed that the related DEPs were mainly secreted outside the cell, while the related DEPs within the cell were mainly located on the plasma membrane (Fig. 2H).
Differentially expressed proteins significant for response-effect
To investigate the association between immunotherapy response and plasma proteins, the proteins that were significant for response-effect were analyzed by using linear mixed effect models. PCA performed on all samples from the three aforementioned time points revealed observable differences between PR and SD_PD groups at each time point (Fig. 3A). Hierarchical clustering based on levels of response-effect proteins further demonstrated that SD_PD patients exhibited higher expression of a subset of proteins than PR patients (Fig. 3B). Among the 92 proteins studied in this study, 10 were found to be significant proteins in response effect (Fig. 3C). Based on our results, approximately 11% of plasma proteins were observed to exhibit significant alterations between SD_PD group and PR group. Integrating the time effect of T1, T2 and T3, there were eight DEPs with p-value less than 0.05—IL7, ANGPT2, IL15, HO-1, CXCL1, CXCL5, IL33, and VEGFA (Fig. 3D, E). At T1, the proteins HO-1 and CXCL1 showed significant differences, while at T2, IL15 and ANGPT2 exhibited differential expression. Similarly, at T3, CASP-8 displayed significant differences between the two groups (Fig. 3F–H).

A Principal component analysis of patient samples color-coded by different treatment responses. B Heatmap displaying normalized plasma expression for all proteins significant for response-effect. C The proportion graph of differentially expressed proteins significant for response-effect. D Volcano plot demonstrating differentially expressed proteins between PR group and SD_PD group. E The top eight differentially expressed proteins significant for response-effect. F Differentially expressed proteins significant for response-effect at the baseline timepoint. G Differentially expressed proteins significant for response-effect at the timepoint of 6 weeks on treatment. H Differentially expressed proteins significant for response-effect at the timepoint of 6 months on treatment.
Differentially expressed proteins significant for time-effect
Through quality control analysis, it was found that there were significant differences between the data at three different time points, which could support further analysis (Fig. 4A). Whether in SD_PD or PR groups, most of the related DEPs showed an upward trend. When examining the transition from T1 to T2, both SD_PD and PR groups showed an upward trend for most DEPs, which was subsequently confirmed by protein co-abundance analysis at each time point (Fig. 4B). Several proteins associated with immune cell activity, including PDCD1, TNFRSF4, DCN, CRTAM, VEGFR-2, and ADA, exhibited distinctive expression patterns in PR patients (Fig. 4C). Similarly, SD_PD group displayed altered expression profiles of PDCD1, ADA, CD28, IL10, and PTN. At T1 and T2, examination of plasma proteins revealed elevated levels of certain cytokines. However, at T3, the expression of these proteins in SD_PD patients displayed variable changes (Fig. 4D). Notably, most of these cytokines and cytokine receptors reached their peak expression levels at T2 in both PR and SD_PD patients, with a subsequent trend basically consistent with T2 by the T3 timepoint.

A Principal component analysis of patient samples color-coded by different time periods in PR group and SD_PD group. B Heatmap displaying normalized plasma expression for all proteins significant for time-effect in PR group and SD_PD group. C The top five differentially expressed proteins significant for time-effect in PR group. D The top five differentially expressed proteins significant for time-effect in SD_PD group.
Differentially expressed proteins significant for interaction-effect
By examining the covariance in protein levels significant for response effect in linear mixed effect models, we investigated which circulating plasma proteins might be co-regulated (Fig. 5A). Through a linear mixed effect model, 50 proteins were identified as significant for interaction effect. Among them, the values of the most significant HO-1, ANGPT2, and IL15 in SD_PD group were all higher than those in PR group at T1, T2, and T3, and the trend of alteration became more obvious with time (Fig. 5B). Receiver operating characteristic (ROC) analysis demonstrated that HO-1, ANGPT2, IL15 showed high accuracy in PR and SD_PD patients (AUC = 0.74) (Fig. 5C).

A Heatmap displaying the correlation between plasma protein expression for all interaction-effect proteins. B The top three differentially expressed proteins significant for interaction-effect. C ROC curse analysis of the combined protein biomarker for predicting treatment responses. ROC receiver operating characteristic.
Construction of prognostic model of BTC by genome combined with proteomics
To further study the influence of genes and proteins on the prognosis of BTC, we constructed a prognosis model of BTC by genomics combined with proteomics. First, we performed enrichment analysis on the mutant gene set. For the analysis of plasma samples, in SD_PD group, GO analysis results showed that Ras protein signal transduction and ERBB2 signaling pathway were crucial and the KEGG results showed that the enrichment was prominent in the process of PI3K-Akt signaling pathway (Fig. 6A). The other side, in PR group, positive regulation of MAPK cascade was vital in GO analysis and PI3K-Akt signaling pathway was significant as well in KEGG analysis (Fig. 6B). For the analysis of tissue samples, we also performed GO and KEGG analysis on SD_PD group and PR group respectively. There was a high degree of duplication between the research results and those of plasma samples (Fig. 6C, D).

A Gene-ontology enrichment analysis and KEGG pathway enrichment analysis showing the top 20 terms from plasma in SD_PD group. B Gene-ontology enrichment analysis and KEGG pathway enrichment analysis showing the top 20 terms from plasma in PR group. C Gene-ontology enrichment analysis and KEGG pathway enrichment analysis showing the top 20 terms from tissue in SD_PD group. D Gene-ontology enrichment analysis and KEGG pathway enrichment analysis showing the top 20 terms from tissue in PR group.
In addition, we integrated the data of mutant genes, proteins and clinical features (sex, age and PDL1) to identify the features significantly related to PFS and OS. In view of plasma samples, the NTRK1 gene, and the CD27 and CRTAM proteins were protective factors while the Akt1, CDKN1A, CTCF and BAI3 genes were high-risk factors in PFS (Fig. 7A). In OS, IL1 and VEGFR2 were protective factors, while the BRIP1, GRM3, TET2 genes were relatively high-risk factors (Fig. 7B). We further used features significantly related to PFS and OS obtained in the previous step to construct nomograms. PFS was used to predict the survival rate of 6, 9 and 12 months and OS was used to predict the survival rate at 1, 1.5, and 2 years. Based on the plasma samples obtained, we constructed two nomogram models for predicting BTC for related genes and proteins. In PFS, we found that the gene NTRK1 was the most important influencing factor, and normal NTRK1 meant a higher risk of death (Fig. 7C). In OS, the protein IL1 as a protective factor had the greatest influence on the long-term survival rate (Fig. 7D). Similarly, we also conducted GO and KEGG analyses on tissue samples (Fig. 7E, F). Subsequently, we created nomogram models. In PFS, the gene CTNNB1 and the protein CRTAM were the two essential influencing factors (Fig. 7G). In OS, mutations in the genes TNFAIP3 and NRAS greatly increased the risk of death (Fig. 7H). In summary, the short-term survival rate was relatively high, and the long-term survival rate was not very optimistic.

A Proportional hazards model analysis of plasma samples in PFS. B Proportional hazards model analysis of plasma samples in OS. C Nomogram of plasma samples in PFS. D Nomogram of plasma samples in OS. E Proportional hazards model analysis of tissue samples in PFS. F Proportional hazards model analysis of tissue samples in OS. G Nomogram of tissue samples in PFS. H Nomogram of tissue samples in OS.
Discussion
BTC, including intrahepatic, perihilar, and distal CCA as well as GBC, is one of the lethal of malignancies9. Since the rise of immunotherapy in the treatment of BTC, a variety of immune monotherapy/combination therapies have been explored and achieved relevant results. In 2020, we carried out a single-arm, open-label phase II study, and the evaluation showed that Camrelizumab plus the GEMOX treatment had good anti-tumor activity and acceptable safety. According to this result, the regimen of Camrelizumab plus GEMOX was recommended by the Chinese Society of Clinical Oncology (CSCO) Guidelines for Diagnosis and Treatment of Biliary Malignancies in 2023 as the first-line treatment of BTC (level II recommendation). At present, many biomarkers, including PD-L1, tumor mutation burden (TMB), mismatch repair-deficiency (dMMR) and microsatellite instability (MSI), have been proved to be related to the curative effects of immunotherapy for solid tumors, and there are also many reports on biomarkers related to evaluation of immunotherapy in BTC10. In previous studies, we preliminarily analyzed the findings between immunotherapy efficacy and common biomarkers in patients with BTC, and found that there was no association between response and TMB, blood TMB, immune promotion scores or immune cells (P > 0.05), with the exception of PFS, which was associated with blood TMB. Compared with genomics, the composition of proteomics is larger and more complex, thus helping study life activities from more angles and depths. As an emerging method, proteomics plays an important role in biological science research, disease diagnosis and drug development. So in this study, we mainly analyzed the predictive effect of plasma proteome on immunotherapy for BTC and verified the value of proteomics.
Currently, it has been found that the genome diversity and heterogeneity of BTCs are closely related to tumor origin regions and epidemiological risk factors, which also has a certain impact on their prognosis11. However, whether the differences in molecular pathology may lead to the differences in the efficacy of immunotherapy is still inconclusive. For instance, TOPAZ-1 and KEYNOTE-966 studies have shown that the pathological type of intrahepatic cholangiocarcinoma had a better benefit from OS, but no correlation between pathological type and curative effect of immunotherapy has been found3,12. Therefore, in this study, intrahepatic, perihilar, distal CCA, and GBC were enrolled at the same time.
Considering that SD group is a kind of patients with high heterogeneity, including enlargement or reduction of lesions, it is still doubtful whether it can reflect the effective treatment. In order to explore the biomarkers of patients who had achieved objective remission, patients were divided into response (CR or PR) and non-response (SD or PD) groups.
For the purpose of predicting prognosis biomarkers, we sought DEPs based on time, response and interaction effects. Compared with genomics research, our findings provide a relatively affordable, multi-dimensional, and profound method that helps clinicians predict the therapeutic response in BTC. In this study, enrichment analysis was used to express the potential interactions among these DEPs. GO analysis revealed that genes related to DEPs were mainly involved in cellular process, biological regulation, and immune system process. A large number of studies have proved that the immune system is closely related to cancer metabolism. For instance, both the release of cancer metabolites and the expression of immune molecules have wide effects in the regulation of antitumor immune response13. KEGG analysis revealed that the related genes were mainly involved in diverse cancer-related pathways such as nature killer cell mediated cytotoxicity and cytokine-cytokine receptor interaction. Subsequently, to find the co-existence pattern among DEPs, we built PPI and 10 hub proteins with high value were found from the network. Among these, VEGFA plays an important role in vascular development and angiogenesis, and several non-endothelial functions of VEGF have also been established, which may have an impact on the use and design of cancer treatment14. Another significant protein is MIC, including MICA and MICB, and previous research has found evidence of an association between MICA/MICB and human cancer. Antibodies targeting the MICA’s proteolytic shedding domain proves to greatly inhibit tumor growth in multiple fully immunocompetent mouse models15. Besides, various studies have shown that the remaining hub proteins are closely associated with cancer.
Further, we noted the presence of proteins associated with immune response. Traditional proteomic research mainly focuses on the static expression levels of proteins, that is, the composition of proteins at a given time point. However, the protein group in an organism is dynamic, not only in time, but is also influenced by physiological state, drug action and other factors. Dynamic proteomics focus on the temporal and spatial changes of protein group, revealing the dynamic behavior, interaction and regulatory network of proteins. This analysis method can obtain more comprehensive information by tracking the change of proteins’ expression level or monitoring the dynamic process of proteins’ post-translation modification and help us understand the timing regulation and complexity of biological systems. So, we set three time points: baseline, 6 weeks after treatment, 6 months after treatment.
In response-effect, based on the comprehensive analysis of all three time points, differentially expressed proteins between SD_PD and PR groups were IL7, ANGPT2, IL15, HO-1, CXCL1, CXCL5, IL33, and VEGFA. Respectively, HO-1 and CXCL1 showed significant difference at baseline. They mainly reflected the differences between tumor features and subject situations, and may be used as biomarkers to predict the initial curative effect. MUC-16, IL-15, ANGPT2, FGF2, and LAP TGF-β-1 showed difference after 6 weeks of treatment. They were closely related to the growth of tumor cells. Among them, the differential expression of MUC-16(CA125), recognized as a mature tumor marker and having certain value in evaluating curative effect and prognosis16, was the most distinct. Moreover, IL-15, ANGPT2 were consistent with the DEPs obtained by synthesizing three time points. They mainly reflected the changes of patients’ internal environment during treatment, and they may also have predictive effects on curative effect. After 6 months of treatment, CASP-8 was the only DEP detected. CASP-8 could induce or inhibit cell death both. This was closely related to the survival rate of patients in the later stage of treatment. In addition, DEPs changed with the development of the disease course. It might be related to the change of related cytokines and signaling pathways caused by immunotherapy. With the progress of the disease course and curative effect, different cytokines showed their more significant impacts at different time points. In overall consideration, we thought the dynamic monitoring of these DEPs could also play a certain role in reflecting the curative effect of immunotherapy.
In addition, in time-effect, SD_PD group showed changes in the expression profiles of PDCD1, IL10, ADA, CD28, and PTN and PR group showed changes of PDCD1, TNFRSF4, DCN, CRTAM, and ADA. Among them, IL10, CD28, PTN, TNFRSF4, DCN and CRTAM may be valuable DEPs to distinguish SD_PD and PR groups. They affect the development of tumors through different functions. IL10, CD28, TNFRSF4(OX40) can enhance anti-tumor immunity by influencing immune response, such as activating T cells, macrophages17,18,19. Studies have shown that the PTN gene may be a proto-oncogene, which is highly expressed in many tumors and closely related to tumor angiogenesis and metastasis20. DCN is a biological factor with anti-lymphangiogenic activity, which mediates its role in tumor inhibition by binding with various cell surface receptors21. CRTAM plays a key role in promoting NK cells to kill tumor cells in the immune process, presenting a potential tumor targeting site22. The dynamic changes of these DEPs in time may be due to the dynamic progression of the disease, so they may indicate the severity of the disease.
As the dynamic changes of tumors are affected by many factors, we set up an interaction-effect model to identify more comprehensive and suitable biomarkers. In this model, HO-1, ANGPT2 and IL15 showed significant AUC values for BTC treatment (AUC = 0.74) and all the three proteins kept a stable differential expression trend at three time points. Meanwhile, these three proteins were coincident with the eight DEPs found in the response-effect model, and they also expressed as DEPs at individual T1, T2 time points. This provided evidence and explanations for the reliability of interaction-effect analysis results. In summary, we suggested that these three collective proteomic biomarkers may be beneficial to dynamically monitoring the efficacy of immunotherapy.
Many studies have revealed that these cytokines are valuable in tumor growth and immunotherapy. For instance, HO-1 is inducible and widely distributed in various tissues and cells of mammals23. In normal tissues and cells, HO-1 has anti-inflammatory, antioxidant, anti-apoptotic and anti-proliferative effects on a variety of cells. Meanwhile, HO-1 can regulate the acquired immune response by inhibiting the activation, proliferation and function of T cells. It has been found that the signaling pathways regulating HO-1 are also closely related to tumors, such as the Keap1/Nrf2 and PI3K/Akt signaling pathways24,25. HO-1 promotes tumor cell metastasis by stimulating angiogenesis. At the same time, it has been proven that vascular endothelial growth factor (VEGF) is one of the main factors affecting tumor angiogenesis, and its regulation is influenced by HO-126. Another important protein, ANGPT2, belongs to the angiopoietin (Ang) family and is a growth factor that regulates the growth and maturation of blood vessels during angiogenesis27. Vascular endothelial growth factor (VEGF) and angiopoietin (Ang) are both involved in tumor angiogenesis. ANGPT2 first destroys the stability of resting blood vessels, so that VEGF can drive the proliferation and chemotaxis of vascular buds28. ANGPT2 can also promote angiogenesis, tumor formation, and metastasis through the ANGPT2/Tie2 signaling pathway29. Furthermore, IL15 is a cytokine with a wide range of immunomodulatory activities. It activates CD8+ T cells, natural killer (NK) cells and NKT cells, and can promote the formation of anti-tumor antibodies. In various experimental animal tumor models, IL15 therapy has shown enhanced anti-tumor effects30,31. However, in some cases, IL15 may also have side effects, including inducing autoimmunity and promoting the proliferation, survival and spread of some tumor cells32. Therefore, it suggests that we should be cautious about its application in tumor immunotherapy. In addition, it was previously found that CXCL1 promotes the proliferation and migration of prostate, gastric and oral squamous carcinoma33,34,35, suggesting the feasibility of further investigation of function in BTC. According to published data in previous journals, blockade of CXCL1 contributes to the protection against pancreatic ductal adenocarcinoma (PDA) as the tumor microenvironment associated with RIP1/RIP3 signaling partly depends on the necroptosis-induced expression of CXCL136. Hence, we speculate that highly-expressed CXCL1 in BTC non-responders has corresponding signaling pathway or immunoreaction mechanisms.
Linear analysis of interaction effect revealed that HO-1, ANGPT2 and IL15 were the most significant differentially expressed proteins. At T1, T2, and T3, HO-1 levels in SD_PD group were all evidently higher than those in PR group. With time, HO-1 in both groups showed a constant increasing trend. This suggests that ICB treatment may affect the curative effect by affecting HO-1 or related pathways, and the longer the time, the more obvious the effect. According to some studies, in glioblastoma and prostate cancer, the expression level of HO-1 is positively correlated with the proliferation of tumor cells37,38. In addition, HO-1 can inhibit the proliferation of tumor cells to some extent. For instance, in breast cancer, HO-1 expression plays a negative regulatory role in tumor cell growth39. Therefore, the different expression of HO-1 may be associated with different tissue sources. In our study, HO-1 expression was negatively correlated with tumor. Anyway, it is certain that HO-1 is closely related to tumor and may have some influence on tumor proliferation, metastasis and apoptosis. Another important protein, ANGPT2, showed a weak change trend over time in PR group, but increased continuously in SD_PD group. However, the value of ANGPT2 in SD_PD group was still higher than that in PR group at all three time points. Recent studies have found that ANGPT2 expression in gastric cancer (GC) and hepatocellular carcinoma (HCC) tissues is significantly higher than that in normal tissues40,41. These findings are consistent with the results of our study, indicating that ANGPT2 may play a key role as a prognostic marker in tumors and can be used as a breakthrough point for poor curative effect. The last one, IL15, consistent with the first two groups, was higher in SD_PD group than in PR group. However, in both groups, the increasing trend was more evident from T1 to T2, and it was basically unchanged from T2 to T3. These results may be related to the effect of our treatment and suggest that IL15 is hoped to be regarded as a vital biomarker for early curative effect.
Our present research delivers breakthroughs owing to the use of Olink PEA, a highly sensitive detection method based on nucleic acid. Currently, the advantages of this technology have been proven in cancer treatment; it can screen for effective proteins more efficiently and precisely. In our previous related research, no significant differential genes were identified through genomic analysis6, and Cox model and nomogram analysis did not strongly find any differential genes as important influencing factors. However, through proteomic analysis, we found differential proteins from different angles. This indicated that proteomics may be more sensitive than genomics. In this study, plasma proteomics across serial timepoints shed light on immunotherapy non-responses in BTC patients treated with immunotherapy. Instead of focusing on individual protein biomarkers or small cohorts without paired timepoints, our results demonstrated that proteomic has a better prediction effect than the variation of an individual protein and some significantly expressed proteins provided potential ideas for the ICB treatment of BTC.
This study provides a novel perspective for targeted therapy of BTC and helps to identify more effective drugs to improve the sensitivity of immunotherapy. However, this study still has some limitations. With limited cases from a single hospital, some inevitable bias and confounding factors may exist during the course. In addition, although there is no definite impact in our research, hematological parameters may be affected by some concomitant medications and pathophysiologic status. At the same time, many questions surrounding key cytokine interactions in the TME remain unresolved. As signal pathways are complex reactions involving a series of cytokines such as receptors, ligands and transcription factors, it is more suitable to discuss the influencing factors of a single signal pathway additionally. The purpose of this study is to discuss the DEPs obtained under various interactions, so the part of pathways is not discussed in depth. There is no transparent difference in genomics between SD_PD group and PR group, but there are differences in proteomics, suggesting that there may be changes in the modification level after translation. In the future, we will explore underlying mechanisms and signaling pathways associated with those differential proteins that play a regulatory role in tumor immunity.
Methods
Ethics approval
The study was approved by relevant regulatory and independent ethics committee of the First Affiliated Hospital with Nanjing Medical University (Approval number: 2017-SR-291) and done in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines. All patients provided written, informed consent before study entry.
Patients
Thirty-seven patients who were diagnosed with advanced BTC between March 2015 and August 2019 and received Camrelizumab plus the GEMOX treatment from NCT03486678 trial were retrospectively screened from our previous study5. The inclusion criteria were as follows: pathological diagnosis of CCA or GBC; stage IV; sufficient tumor tissue to allow next-generation sequencing (NGS); treatment-naive when sampling. All 37 patients were sequenced for histological genomics, ctDNA and plasma proteomics. This study was performed in accordance with the Declaration of Helsinki and ethical standards of the institutional research committees. All participating patients have provided written informed consent for the collection of blood samples.
Samples collection and preparation
Tumor tissues were collected at baseline and whole blood was collected prior to ICB treatment, 6 weeks after treatment, and 6 months after treatment, and 3 mL of plasma was isolated after centrifuging the tubes for 30 min at room temperature. Plasma was stored at −80 °C for further use and thawed at the time of proteomic analysis. Tissue slides were first analyzed to evaluate the tumor content and percentage. Only samples with tumor purity >20% on histopathological assessment were eligible for genomic profiling. Proteomics and genomics laboratories in China that are certified by the Clinical Laboratory Improvement Amendments sequenced tumor tissues and matched normal specimens.
Plasma proteomic immunoprofiling
After plasma proteins were extracted and qualified, we used PEA to evaluate the abundance of proteins in the plasma. In total, 92 proteins were analyzed. According to Olink, PEA is a highly sensitive and specific targeted proteomic detection technology that is based on antibody immunoassay and qPCR and NGS. A pair of oligonucleotide-labeled antibody probes was bound to the target protein for each one to be detected. When antibodies are combined with targeted proteins, oligonucleotides in close proximity hybridize in a pair-wise manner. The addition of DNA polymerase leads to a proximity-dependent DNA polymerization event, generating a unique PCR target sequence, which is proportional to the initial concentration of the target protein. Using PEA technology, we skillfully converted protein quantification into DNA quantification, which could be detected and quantified using microfluidic real-time PCR instrument (Biomark HD, Fluidigm). Patients’ plasma samples were evenly distributed across four 96-well plates, and the resulting Ct-data was then quality controlled and normalized using a set of internal and external controls according to Olink’s recommendations. The final assay read-out was presented in Normalized Protein eXpression (NPX) values, which was an arbitrary unit on a log2-scale where a high value corresponded to a higher protein expression.
Pre-processing of plasma proteomic data
Unless otherwise stated, the analysis of plasma proteomic data was conducted in R (version 4.0.3) programming environment. Sample 18AB00006BP-1 was close to the lower detection limit, but from an analytical point of view, it might not be excluded. In conclusion, the analysis data contained all samples.
Dimensionality reduction and visualization of proteomic data
The Olink principal component analysis (PCA) plot function in the OlinkAnalyze package in R was used to perform principal component analysis on the plasma proteomic data for dimensionality reduction and visualize principal components.
Linear mixed effect models and mean different estimates
We used linear mixed-effect models to identify proteins that significantly changed over time and between responses. The models included a main effect of time, a main effect of responder status, and a main effect of interaction between time and responder status. The statistical significance of the three model terms was assessed through an F-test, employing Satterthwaite degrees of freedom and executed with the olink_lmer function in the OlinkAnalyze package in the R programming language. To mitigate the risk of false discoveries, all p-values were adjusted using the Benjamini–Hochberg method to maintain a false discovery rate (FDR) of 5%.
Correlation scatterplots
Pearson’s correlations were computed using the cor function in R to determine the estimated mean differences between groups. Additionally, scatterplots were generated using the ggplot package in R to visually represent the estimates for each cohort. To identify statistically significant assays, the main effects of response, time and interaction needed to surpass the FDR threshold for the F-test, and the Tukey adjusted p-value for estimated mean difference had to be less than 0.05.
Gene ontology (GO) and kyoto encyclopedia of gene and genomics (KEGG) analysis
GO and KEGG are commonly used gene sets in gene function enrichment analysis. GO refers to the mining of gene function categories significantly related to biological problems. The results of GO enrichment analysis by ggplot2 (Lianchuan Biological Company) are displayed in the form of scatter plots (bubble plots), which consist of rich factors (rich factor = S protein number/B protein number) on the abscissa and GO Term on the ordinate. KEGG analysis is mainly aimed at analyzing metabolic pathways in organisms. Pathway enrichment analysis takes KEGG pathway as the unit, and uses a hypergeometric test to determine the pathway that is significantly enriched in the significant differentially expressed proteins or genes compared with the whole proteome or genome background. The results of KEGG enrichment analysis by ggplot2 (Lianchuan Biological Company) are displayed in the form of scatter plot (bubble plot), consisting of rich factor on the abscissa and KEGG Term on the ordinate.
Clusters of orthologous groups (COG) analysis
COG database is a database for homologous classification of gene products, which can identify orthologous genes earlier and come from a large number of comparisons of protein sequences of various organisms. In the COG enrichment analysis diagram, the abscissa shows the classification contents of COG and the ordinate shows the number of proteins.
Protein–protein interaction networks (PPI) analysis
In this data, Search Tool for the Retrieval of Interacting Genes (STRING) database was used to analyze the interaction of identified proteins. Corresponding or near-source species in the database were extracted and then the sequence of the differential protein was compared with the extracted sequence by blast and the corresponding interaction information was obtained.
Cox proportional-hazards models and visualization of prognosis prediction
The genes and proteins pertaining to the enrichment of pathways and molecular functions in KEGG and GO were analyzed using the least absolute shrinkage and selection operator (LASSO) method. Univariate Cox analysis was used to analyze the selected genes or proteins. The coefficients of each prognostic molecule in the risk-scoring model were derived from the corresponding univariate analysis of these identified biomarkers. The data of mutated genes, proteomics, and clinical features (including sex, age, and PDL1) were integrated to identify the features significantly associated with PFS and OS (p < 0.05). Using the features obtained in the previous step, a nomogram was constructed to assess each patient’s survival, with PFS in 6, 9, and 12 months, and OS in 1, 1.5, and 2 years. The nomogram offers convenience in predicting the prognosis of immunotherapy according to the molecular characteristics in plasma and tumor tissues.
Statistical analysis
All statistical analyses of plasma proteomic were performed with R package (version 4.0.3). We considered p-values of less than 0.05 to be statistically significant. F-test, Tukey test and Satterthwaite test were used for intergroup comparison as needed. We applied hypergeometric distribution to test the significance of a functional class in a group of co-expression or differentially expressed genes. LASSO was used in KEGG and GO analyses to identify genes and proteins pertaining to enrichment of pathway. Univariate Cox analysis was performed to analyze the selected genes or proteins. Clinical outcomes were previously defined5.
Data availability
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study that are not in the article and Supplementary Material are available from the corresponding author (Xiaofeng Chen) on reasonable request.
Code availability
Some or all data, models, or code generated or used during the study are available from the corresponding author by request.
Abbreviations
- Ang:
-
angiopoietin
- BTC:
-
biliary tract cancer
- CCA:
-
cholangiocarcinoma
- COG:
-
clusters of orthologous group
- CTLs:
-
cytotoxic T lymphocytes
- DEGs:
-
differentially expressed genes
- DEPs:
-
differentially expressed proteins
- EBaLCC:
-
Epstein–Barr virus-associated intrahepatic cholangiocarcinoma
- FDR:
-
false discovery rate
- GBC:
-
gallbladder cancer
- GC:
-
gastric cancer
- GemCis:
-
gemcitabine combined with cisplatin
- GEMOX:
-
gemcitabine and oxaliplatin
- GO:
-
Gene Oncology
- HCC:
-
hepatocellular carcinoma
- ICB:
-
immune checkpoint blockade
- ILC2s:
-
group 2 innate lymphoid cells
- KEGG:
-
Kyoto Encyclopedia of Gene and Genomics
- LAK:
-
lymphokine-activated killer
- LASSO:
-
least absolute shrinkage and selection operator
- LUAD:
-
lung adenocarcinoma
- NGS:
-
next-generation sequencing
- NK:
-
natural killer
- NPX:
-
normalized protein eXpression
- OS:
-
overall survival
- PCA:
-
principal component analysis
- PDA:
-
pancreatic ductal adenocarcinoma
- PEA:
-
proximity extension assay
- PEA:
-
proximity extension assay
- PFS:
-
progression free survival rate
- PPI:
-
protein–protein interaction networks
- PR:
-
partial response
- ROC:
-
receiver operating characteristic
- SD_PD:
-
stable disease and progressive disease
- STRING:
-
Search Tool for the Retrieval of Interacting Genes
- TMB:
-
tumor mutation burden
- TME:
-
tumor microenvironment
- VEGF:
-
vascular endothelial growth factor
References
Danese, M. D. et al. Treatment patterns and survival in locally advanced or metastatic biliary tract cancer using SEER medicare data. Gastro Hep Adv. 2, 580–587 (2023).
Carlino, M. S., Larkin, J. & Long, G. V. Immune checkpoint inhibitors in melanoma. Lancet 398, 1002–1014 (2021).
Kelley, R. K. et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 401, 1853–1865 (2023).
Oh, D. Y. et al. Gemcitabine and cisplatin plus durvalumab with or without tremelimumab in chemotherapy-naive patients with advanced biliary tract cancer: an open-label, single-centre, phase 2 study. Lancet Gastroenterol. Hepatol. 7, 522–532 (2022).
Chen, X. et al. Camrelizumab plus gemcitabine and oxaliplatin (GEMOX) in patients with advanced biliary tract cancer: a single-arm, open-label, phase II trial. J. Immunother. Cancer 8, e001240 (2020).
Chen, X. et al. Genomic alterations in biliary tract cancer predict prognosis and immunotherapy outcomes. J. Immunother. Cancer 9, e003214 (2021).
Patterson, S. D. Proteomics: evolution of the technology. Biotechniques 35, 440–444 (2003).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Valle, J. W. et al. Biliary tract cancer. Lancet 397, 428–444 (2021).
Luchini, C. et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: a systematic review-based approach. Ann. Oncol. 30, 1232–1243 (2019).
McNamara, M. G. et al. Landmark survival analysis and impact of anatomic site of origin in prospective clinical trials of biliary tract cancer. J. Hepatol. 73, 1109–1117 (2020).
Oh, D. Y. et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evid. 1, EVIDoa2200015 (2022).
Xia, L. et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 20, 28 (2021).
Wiszniak, S. & Schwarz, Q. Exploring the Intracrine Functions of VEGF-A. Biomolecules https://doi.org/10.3390/biom11010128 (2021).
Ferrari de Andrade, L. et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 359, 1537–1542 (2018).
Chen, X. et al. MUC1 and MUC16: critical for immune modulation in cancer therapeutics. Front. Immunol. 15, 1356913 (2024).
Jensen, S. M. et al. Signaling through OX40 enhances antitumor immunity. Semin. Oncol. 37, 524–532 (2010).
Guo, Y. et al. Metabolic reprogramming of terminally exhausted CD8(+) T cells by IL-10 enhances anti-tumor immunity. Nat. Immunol. 22, 746–756 (2021).
Khan, M., Arooj, S. & Wang, H. Soluble B7-CD28 family inhibitory immune checkpoint proteins and anti-cancer immunotherapy. Front Immunol. 12, 651634 (2021).
Papadimitriou, E. et al. Roles of pleiotrophin in tumor growth and angiogenesis. Eur. Cytokine Netw. 20, 180–190 (2009).
Mondal, D. K. et al. Decorin suppresses tumor lymphangiogenesis: a mechanism to curtail cancer progression. Proc. Natl Acad. Sci. USA 121, e2317760121 (2024).
Boles, K. S. et al. The tumor suppressor TSLC1/NECL-2 triggers NK-cell and CD8+ T-cell responses through the cell-surface receptor CRTAM. Blood 106, 779–786 (2005).
Keyse, S. M. & Tyrrell, R. M. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl Acad. Sci. USA 86, 99–103 (1989).
Huang, C. Y. et al. Attenuation of lipopolysaccharide-induced acute lung injury by hispolon in mice, through regulating the TLR4/PI3K/Akt/mTOR and Keap1/Nrf2/HO-1 pathways, and suppressing oxidative stress-mediated ER stress-induced apoptosis and autophagy. Nutrients https://doi.org/10.3390/nu12061742 (2020).
Hamdulay, S. S. et al. Celecoxib activates PI-3K/Akt and mitochondrial redox signaling to enhance heme oxygenase-1-mediated anti-inflammatory activity in vascular endothelium. Free Radic. Biol. Med. 48, 1013–1023 (2010).
Cisowski, J. et al. Role of heme oxygenase-1 in hydrogen peroxide-induced VEGF synthesis: effect of HO-1 knockout. Biochem. Biophys. Res. Commun. 326, 670–676 (2005).
Jary, M. et al. Molecular description of ANGPT2 associated colorectal carcinoma. Int. J. Cancer 147, 2007–2018 (2020).
Yang, S. et al. Immunoregulation and clinical significance of neutrophils/NETs-ANGPT2 in tumor microenvironment of gastric cancer. Front. Immunol. 13, 1010434 (2022).
Xie, J. Y. et al. Angiopoietin-2 induces angiogenesis via exosomes in human hepatocellular carcinoma. Cell Commun. Signal. 18, 46 (2020).
Cheng, L. et al. Hyper-IL-15 suppresses metastatic and autochthonous liver cancer by promoting tumour-specific CD8+ T cell responses. J. Hepatol. 61, 1297–1303 (2014).
Evans, R. et al. IL-15 mediates anti-tumor effects after cyclophosphamide injection of tumor-bearing mice and enhances adoptive immunotherapy: the potential role of NK cell subpopulations. Cell Immunol. 179, 66–73 (1997).
Jakobisiak, M., Golab, J. & Lasek, W. Interleukin 15 as a promising candidate for tumor immunotherapy. Cytokine Growth Factor Rev. 22, 99–108 (2011).
Wei, L. Y. et al. Reciprocal activation of cancer-associated fibroblasts and oral squamous carcinoma cells through CXCL1. Oral. Oncol. 88, 115–123 (2019).
Chen, X. et al. Complementary action of CXCL1 and CXCL8 in pathogenesis of gastric carcinoma. Int. J. Clin. Exp. Pathol. 11, 1036–1045 (2018).
Kuo, P. L. et al. CXCL1/GROα increases cell migration and invasion of prostate cancer by decreasing fibulin-1 expression through NF-κB/HDAC1 epigenetic regulation. Carcinogenesis 33, 2477–2487 (2012).
Seifert, L. et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 532, 245–249 (2016).
Castruccio Castracani, C. et al. Heme oxygenase-1 and carbon monoxide regulate growth and progression in glioblastoma cells. Mol. Neurobiol. 57, 2436–2446 (2020).
Yu, F. & Bai, T. Sevoflurane activates the IL-6/HO-1 pathway to promote macrophage M2 polarization and prostate cancer lung metastasis. Int. Immunopharmacol. 113, 109380 (2022).
Lin, C. W. et al. Heme oxygenase-1 inhibits breast cancer invasion via suppressing the expression of matrix metalloproteinase-9. Mol. Cancer Ther. 7, 1195–1206 (2008).
Yang, S. et al. Neutrophil extracellular traps promote angiogenesis in gastric cancer. Cell Commun. Signal 21, 176 (2023).
Huang, Y. H. et al. MiR-29a curbs hepatocellular carcinoma incidence via targeting of HIF-1α and ANGPT2. Int. J. Mol. Sci. 23, 1636 (2022).
Acknowledgements
The authors thank all the patients, their families and the institutions for supporting this study. Thanks to all the peer reviewers for their opinions and suggestions.
Author information
Authors and Affiliations
Contributions
Xiaofeng Chen, Xiangcheng Li, and Rong Wang designed the study and participated in coordination and project control. Qiuyu Wu, Weici Liu, Lei Fan, Yitong Tian, Hao Qian, Yuting Ding, Xinyi Zhang, Jiaguang Zhang, and Xiaofeng Wu collected the data and conducted the analysis. Shiyun Cui, Hejian Zheng, Yiyang Xu, Yucheng Cai, and Qiuyu Wu wrote the original draft. Xiaofeng Chen, Shiyun Cui, Yiyang Xu, Qiuyu Wu, and Weici Liu, Yucheng Cai revised the paper. Xiaofeng Chen and Shiyun Cui and got financial support. All authors reviewed and approved the final edition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cui, S., Zheng, H., Xu, Y. et al. Plasma proteomic biomarkers predict therapeutic responses in advanced biliary tract cancer patients receiving Camrelizumab plus the GEMOX treatment. npj Precis. Onc. 9, 102 (2025). https://doi.org/10.1038/s41698-025-00879-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-00879-9
