
Abstract
DICER1-associated sarcomas commonly exhibit cooperating mutations involving RAS signaling pathways, but the efficacy of therapies that target these mutations is unknown. Here we report two children with DICER1 tumor predisposition who presented with DICER1-associated sarcomas with cooperating, targetable mutations in HRAS or BRAF. Both had relapsed/progressed disease despite upfront multimodal therapy and were subsequently treated with molecularly targeted agents. In the first case, mutant BRAF became amplified after dual dabrafenib/trametinib therapy, presumably as a driver of acquired resistance. In the second case, a subclonal HRAS variant at diagnosis became the predominant clone at autopsy, suggesting its importance in therapy resistance. Together, these two cases provide molecular evidence of the significance of RAS/ERK signaling in DICER1-driven tumorigenesis and highlight the potential for targeting these cooperating mutations.
Similar content being viewed by others

Introduction
DICER1 tumor predisposition (OMIM 606241) is caused by pathogenic variants (PVs) in the DICER1 gene. Germline DICER1 variants were first detected in individuals affected with familial pleuropulmonary blastoma (PPB), a sarcoma of the lungs in childhood1. Subsequently, the International PPB/DICER1 Registry and others have reported additional conditions associated with DICER1 including multinodular goiter of the thyroid, differentiated thyroid carcinoma, pituitary blastoma, pineoblastoma, ciliary body medulloepithelioma, nasal chondromesenchymal hamartoma, cystic nephroma, Wilms tumor, ovarian Sertoli-Leydig cell tumor (SLCT), and cervical embryonal rhabdomyosarcoma2,3. Recently, characteristic histopathology findings and distinct methylation signatures have been described in DICER1-associated sarcomas in variable anatomical locations and lend strong evidence that they constitute a distinct sarcoma entity4,5,6. DICER1-associated sarcomas often demonstrate unique histopathologic features of variably cellular spindled cells accompanied by rhabdomyoblastic differentiation and occasional cartilaginous differentiation4,7.
The human DICER1 locus is located on chromosome 14q32.13. DICER1 is a cytoplasmic endoribonuclease within the canonical microRNA (miRNA) biogenesis pathway and is critical for the proper processing of precursor microRNA (pre-miRNA) double-stranded hairpins to their mature single-stranded forms8. The protein utilizes its ribonuclease (RNase) IIIa and IIIb domains to process a pre-miRNA into mature miRNAs. These mature miRNAs are then loaded into an Argonaute protein to form an RNA-induced silencing complex (RISC) to ultimately downregulate an mRNA target transcript through base-pair complementarity. miRNAs regulate genes involved in a variety of biological processes, including stem cell maintenance, organogenesis, and oncogenesis; therefore, failed miRNA processing is a key tumorigenic event in DICER1-mutant tumors9. In most cases, germline PVs in DICER1 are loss-of-function variants that can arise anywhere in the gene, while second-hit somatic mutations in tumors are nearly always limited to exons 24 and 25 that encode the RNase IIIb domain9. The RNase IIIa domain cleaves on the 3p side of pre-miRNA hairpins, and the RNase IIIb domain cleaves on the 5p side. For this reason, somatic mutations in the RNase IIIb domain lead to impaired production of “5p-derived” miRNAs.
It is established that the immediate molecular consequence of RNase IIIb loss-of-function mutations is an imbalance of 3p and 5p miRNAs; however, the downstream consequences that lead to tumorigenesis are unclear. Cooperative oncogenic alterations are often seen in DICER1-associated tumors. Biallelic inactivation of TP53 is the most common co-occurring event seen in DICER1-mutant sarcomas, and genetic alterations in NF1, KRAS, NRAS, FGFR4, EGFR, and PDGFRA have also been reported5,10,11,12,13,14. These findings suggest that oncogenic RAS/extracellular signal-regulated kinase (ERK) signaling may have a role in the tumorigenesis of DICER1-associated sarcomas and, if so, could represent potential therapeutic targets. There is currently no universally accepted systemic therapy approach for the treatment of patients with DICER1-associated sarcomas. Instead, standard regimens used for other cancers, such as rhabdomyosarcoma, germ cell tumor, or soft tissue sarcoma, are most often the treatment paradigms chosen for specific DICER1-associated cancers. As additional knowledge is gained regarding the molecular characterization of DICER1-associated sarcomas, an evaluation of the use of molecularly targeted therapies as precision-driven therapeutic options is necessary.
Here, we report the clinical experience of two patients with confirmed recurrent and metastatic DICER1-related malignancies who received RAS pathway targeted therapies on the basis of identified cooperating mutations in their tumors. In these two patients, clinical genomic sequencing guided the use of precision-targeted therapy following standard sarcoma or solid tumor regimens. Our experience highlights the safety and potential application of RAS pathway inhibitors in patients with these tumors and suggests an opportunity to further explore the efficacy of these treatments in similar patients.
Results
Patient 1
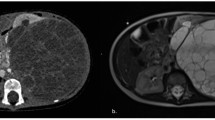
A 3-year-old girl presented with a mass on her right upper chest extending into the shoulder. The mass grew rapidly over a period of approximately 6 weeks and caused progressive loss of right hand function and significant pain in the right arm. Past medical history and family history were unremarkable. A chest CT demonstrated a large right anterior superior chest mass with remodeling of the surrounding osseous structures and encasement of the right brachial plexus, including the right C6-T1 nerve roots and the subclavian artery (Fig. 1A). Multiple pulmonary nodules were identified, ranging from 3 to 5 millimeters (mm), along with two cysts (8 mm and 5 mm) at the right lung base. There was no other radiographic evidence of distant metastases. Open biopsy of the primary lesion revealed a high-grade primitive spindle cell sarcoma with pleomorphism; sequencing of the tumor specimen via the Johns Hopkins Molecular Diagnostic Laboratory in-house next-generation sequencing (NGS) detected two DICER1 variants (p.E1813D and p.M1428fs) and a BRAF p.V600E mutation (“Primary” in Table 1). Germline testing in the patient confirmed the germline origin of the DICER1 p.M1428Cfs variant, which was subsequently also identified in paternal germline testing. Her mother and newborn sister were negative for this DICER1 variant.

A Patient 1, with DICER1-associated sarcoma and BRAF p.V600E; and B Patient 2, with pleuropulmonary blastoma and HRAS p.G13R. MRI imaging at key timepoints is shown, including available imaging during treatment with molecularly targeted agents (dabrafenib/ trametinib, Patient 1; trametinib, Patient 2) as indicated. Yellow arrows indicate tumor location. Orange arrows indicate surgical intervention.
The patient was initially treated with multi-agent neoadjuvant and adjuvant chemotherapy following a high-risk rhabdomyosarcoma protocol (ARST043115) with vincristine, irinotecan, doxorubicin, cyclophosphamide, ifosfamide, etoposide, and actinomycin-D, in addition to complete surgical resection and adjuvant radiation therapy to the primary tumor bed. Routine imaging at the completion of the planned 18-cycle treatment regimen revealed a new 1.3 by 1.2-cm lesion along the medial aspect of the left occipital horn of the lateral ventricle in the brain (Fig. 1A). A left craniotomy was performed to remove the tumor, which was confirmed again to be high-grade primitive spindle cell sarcoma. Johns Hopkins in-house NGS was performed and, again, detected the same DICER1 variants and BRAF p.V600E mutation (“First recurrence” in Table 1), suggesting its origin as a recurrence of the chest mass rather than a new primary intracranial tumor. Post-operative brain MRI showed additional areas of leptomeningeal enhancement along the right parietal lobe, concerning for residual tumor. The patient received cranial radiation to both sites. She was treated with dabrafenib and trametinib for six months, until a recurrent intracranial tumor was detected on MRI imaging. Dabrafenib and trametinib were discontinued, and a maximally safe second surgical resection was performed. NGS findings were similar to the previous recurrence, with the same DICER1 p.E1813D and BRAF p.V600E mutations, but in this specimen, amplification of the BRAF gene was also identified (Fig. 2 and “Second recurrence” in Table 1). Third-line chemotherapy was initiated with topotecan and temozolomide16 with additional proton radiation to the residual tumor. A third recurrence of intracranial disease was identified two months later. Given the 6-month progression-free interval associated with dabrafenib and trametinib, the BRAF p.V600E amplification was suspected to be a mechanism of resistance to type 1 RAF inhibition. To overcome this resistance, treatment with tovorafenib was pursued via single-patient compassionate access (prior to its Food and Drug Administration (FDA) approval). While waiting for approvals, she was treated with a regimen consisting of cyclophosphamide and vinorelbine, but she rapidly developed further clinical progression. She was enrolled in hospice care and died shortly after discontinuation of disease-directed therapy without having received additional RAF-targeted therapy.

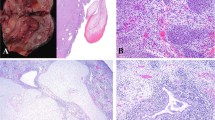
Red signals represent the BRAF gene (7q34), and green signals correspond to the chromosome 7 centromere probe (D7Z1). A, B No BRAF amplification (copy-neutral status) is observed in interphase FISH of the initial diagnosis specimen (A) or a specimen collected 14 months later (B). C Interphase FISH demonstrates BRAF amplification, with more than ten red signals corresponding to the BRAF gene (7q34) per cell, alongside two green signals indicating two copies of the chromosome 7 centromere.
Patient 2
A 4-year-old girl presented with fever, abdominal pain, weight loss, and neck pain. A chest CT demonstrated a large left posterior chest mass with significant mass effect on the mediastinum (Fig. 1B). Metastatic disease was noted in the left kidney and adjacent lymph nodes, iliac bone, and thoracic and cervical spine, the latter extending into the neural foramen and encasing the vertebral artery. An open biopsy of the pulmonary lesion revealed Type II/III PPB. The patient was initially treated with multi-agent neoadjuvant and adjuvant chemotherapy including ifosfamide, vincristine, actinomycin D, and doxorubicin (IVADo17). She underwent surgical resection with intraoperative spillage and gross residual disease. Sequencing of the tumor specimen via GEMTM Cancer Panel detected a DICER1 variant (p.G1809R) and the HRAS p.G13R mutation (“Primary” in Table 1). Further germline testing confirmed the germline origin of the DICER1 p.R676* variant.
Near the end of upfront therapy, surveillance imaging demonstrated a 2 cm structure in the medial inferior left chest along the diaphragm, along with persistent left adrenal and osseous metastases (Fig. 1B). A brain MRI revealed new focal enhancement in the right posterior parietal/occipital region. She received radiation therapy to the left chest and upper abdomen. Resection of the intracranial lesion confirmed metastatic PPB. She was then treated with vincristine, oral irinotecan, and temozolomide and radiation therapy to the right occipital lobe and subsequently developed two new metastatic lesions in the right temporo-occipital region and along the right calcarine fissure of the brain. She received additional radiation to these sites. Trametinib was added to her treatment in an attempt to target the HRAS mutation, and a response was noted at four weeks (Fig. 1B). However, further progression developed rapidly at 6 weeks following initiation of trametinib, and she died from intracranial hemorrhage and progressive thoracic disease 13 months after diagnosis of the first intracranial metastatic lesion.
Retrospective sequencing of lobectomy and postmortem tumor samples (posterior mediastinum and retroperitoneum) (“Primary subclone” and “Autopsy”, respectively, in Table 1 and Supplementary Table 1) showed HRAS p.G13R in both specimens, without any copy number changes at the HRAS locus. In the primary subclone, however, while we detected the oncogenic DICER1 variant at a high variant allele frequency (VAF), we only detected HRAS p.G13R at a low variant allele frequency, suggesting that this variant was only present at a subclonal level in the initial specimen, indicating intra-tumoral heterogeneity.
Discussion
Germline DICER1 PVs increase an individual’s risk for benign and malignant tumors. Many of these are sarcomas, including PPB, embryonal rhabdomyosarcoma (ERMS), anaplastic sarcoma of the kidney, and primary intracranial sarcomas (PIS), among others2,3. There has not been a prospective clinical study to determine the optimal management of non-PPB DICER1-associated sarcomas, given the rarity and heterogeneity of these cancers; however, regimens used for PPB, such as VAIA (vincristine, actinomycin-D, ifosfamide, doxorubicin) and IVADo, are often used for DICER1-associated sarcomas. Here, we report on the clinical outcomes of two patients with metastatic DICER1-associated sarcomas who had transient responses to molecularly targeted therapies used in the relapsed setting. Our clinical experiences highlight the potential of targeted therapeutics in the management of DICER1 sarcomas with known targetable mutations.
Retrospective studies have attempted to assess clinical outcomes associated with chemotherapy regimens in the treatment of DICER1-associated malignancies. The European Cooperative Study Group for Pediatric Rare Tumors (EXPeRT) analyzed a cohort of 52 pediatric patients with Type II or III PPB and reported that all patients received chemotherapy, including mostly standard rhabdomyosarcoma regimens18. Moreover, patients who received doxorubicin-containing regimens (IVADo or VAIA) had significantly better 5-year progression-free survival (PFS) compared to all other regimens (70% versus 31.3%, p = 0.01). In a retrospective analysis from the International PPB/DICER1 Registry, outcomes associated with various chemotherapy regimens, specifically the IVADo regimen, were evaluated in 314 children diagnosed with Type II or III PPB who received upfront chemotherapy17. Compared to historical cohorts, treatment with IVADo was associated with an estimated 23% and 35% decrease in progressive disease and in death, respectively, although these differences were somewhat mitigated by trends in time. Despite intensive treatment, however, children with PPB type II and III with distant metastasis face 3-year overall survival (OS) of 40% and 0%, respectively17. Given the limited knowledge, many patients with DICER1-associated sarcomas are treated on similar general soft tissue sarcoma regimens10,11. A new prospective study for PPB is evaluating the impact of the addition of camptothecins to an IVADo-based regimen for newly diagnosed children with Types II and III PPB (Children’s Oncology Group (COG) ARAR2331).
A growing number of studies have unveiled recurrent genetic alterations in DICER1-associated sarcomas, which may suggest putative targets for therapeutic interventions. Mutations or deletions in TP53 are the most common co-occurring genetic alterations found in DICER1-associated PPB and intracranial sarcomas5,10,11,13, but less commonly in SLCT or sarcomas within the genitourinary tract11,12. Genomic alterations in RAS signaling pathways are frequently found in DICER1-associated sarcomas. For example, KRAS and NRAS mutations have been identified in DICER1-mutated PIS, ERMS, and PPB5,6,10,11,13. Additional mutations identified in DICER1-associated sarcomas that lead to dysregulated RAS signaling pathways include inactivating alterations in NF15,10,11 and BRAF (other than p.V600E)11. A recent comprehensive molecular study on DICER1-associated sarcomas identified recurrent alterations in TP53 (32/80, 40%), KRAS (17/80, 21%), NRAS (6/80, 8%), and NF1 (8/80, 10%)6.
Both patients in this series had TP53 alterations in at least one tumor specimen as well as an oncogenic mutation in the RAS/ERK signaling pathway. Patient 1 had the BRAF p.V600E mutation, and Patient 2 had the HRAS p.G13R mutation. BRAF p.V600E represents the most frequent oncogenic mutation in BRAF and is found in about half of melanomas, 40% of papillary thyroid cancers, and about 10% of colorectal cancers19. While the majority of oncogenic RAS mutations are in KRAS (commonly found in pancreas, lung, and colon carcinomas), bladder urothelial carcinomas, head and neck squamous cell carcinomas, and rhabdomyosarcomas more frequently harbor HRAS mutations20. Oncogenic mutations in HRAS, NRAS and KRAS occur at similar hotspot codons, including G12, G13, and Q61, and less frequently A14621. Neither of the mutations identified in our patients (BRAF p.V600E or HRAS p.G13R) has been previously published in DICER1-associated malignancies, despite being bona fide driver mutations in other cancers.
It is not yet known how mutations in DICER1 and in components of RAS pathways interact and perhaps amplify signals to drive tumorigenesis. Preclinical studies show that the addition of Dicer1 mutations to Ras-mutated mouse models of malignancies confers accelerated tumorigenesis or decreased survival22,23,24. Furthermore, it has been reported that Dicer1 haploinsufficiency promotes the development of distant metastases in a preclinical mouse model of undifferentiated pleomorphic sarcoma (UPS) expressing Kras-G12D but not those expressing Braf-V600E23, suggesting additional complexity in the way distinctive alterations within the RAS/ERK pathways cooperate with DICER1 mutations. Alternatively, DICER1 mutations in the RNase IIIb domain trigger increases in RAS/ERK signaling output genes in DICER1-mutated differentiated thyroid cancers, suggesting that dysfunctional DICER1 alone drives RAS signaling pathways25. It is therefore reasonable to postulate that additional mutations within the RAS pathways potentiate dysregulated signaling and could lead to more aggressive behavior. Additional preclinical work suggests that RAS/ERK pathways also lead to phosphorylation, and thus activation, of DICER1, contributing to tumor development and invasion26. Additional studies are needed to gain deeper insight into the relationship between DICER1 and specific RAS/ERK pathway mutations in malignancies.
The finding of genomic alterations in RAS/ERK pathways raises the potential for therapeutic intervention with targeted inhibition of its dysregulated signaling. A number of RAF inhibitors (RAFi) and MEK inhibitors (MEKi) are currently FDA approved for use in genomically selected cancers as well as Neurofibromatosis type 1 (NF1)-associated plexiform neurofibroma19,27,28,29. Patient 1 received the combination of dabrafenib (RAFi) and trametinib (MEKi), and Patient 2 received trametinib (MEKi), treatment recommendations that were guided by tumor genetic profiling (BRAF p.V600E and HRAS p.G13R, respectively). Both patients experienced relatively short periods of stabilized disease following targeted agent administration prior to the rapid progression of their disease burden. Previously published cases on the use of targeted agents in DICER1-associated malignancies include a young adult patient with an anaplastic sarcoma of the kidney with biallelic somatic DICER1 mutations and concurrent activating PDGFRA p.D842V mutation, treated with avapritinib, resulting in significant response but intolerable toxicities14. An additional report highlighted a 2-year-old child with a histologic diagnosis of initially localized PPB (no germline or somatic DICER1 variant was identified) who then had a second disease recurrence as brain metastases that demonstrated ETV6::NTRK3 fusion and was treated with larotrectinib for four cycles prior to progression of the disease and death30. In our cases, both patients had already received multimodal therapies and had aggressive clinical courses before targeted agents were used as treatment regimens. Therefore, we are unable to draw broadly applicable conclusions on the optimal timing and clinical scenario in which targeted therapeutics may be beneficial in these cancers. Additionally, it is not yet known if there is benefit to the use of molecularly targeted agents in combination with specific conventional chemotherapy or other agents such as immunotherapy, which has shown success in other cancer types31,32,33. Further studies are needed to understand the potential role of targeted therapies in this patient population, as there is not currently a uniformly accepted standard of care systemic treatment for patients with DICER1-associated sarcomas.
In these two cases, we further evaluated subsequent tumor specimens following treatment with targeted therapy, which allowed us to characterize molecular responses to the targeted therapy. In the post-treatment tumor specimen of Patient 1, there was emergence of BRAF copy number amplification, which is a known mechanism of resistance to type 1 RAF inhibition in cancer cells harboring the BRAF p.V600E mutation34. Based on these findings, we gained compassionate use access to tovorafenib, a pan-RAF inhibitor shown to overcome resistance to type 1 RAF inhibitors (e.g., vemurafenib, dabrafenib)35, but unfortunately, rapid intracranial disease progression precluded administration of this agent prior to her abrupt death. Conversely, in the post-treatment tumor specimen from Patient 2, the HRAS mutation remained detectable with a VAF of 65.2%. While MEK inhibition was used in both cases based on tumor genomic status, it is possible that a MEKi alone was insufficient to overcome the aggressive biology and advanced disease burden.
There is a growing repertoire of molecularly targeted agents directed at oncogenic RAS/ERK signaling, many in early phase clinical trials or approved in other RAS-driven cancer types. The clinical experiences described above are the first attempts to molecularly characterize clinical responses in patients with DICER1-associated sarcomas harboring RAS/ERK signaling pathway mutations. More preclinical and clinical studies are needed to determine the most effective, precision-driven therapeutic strategies for these rare cancers.
Methods
Case reports
Two pediatric patients treated for DICER1-associated sarcomas were identified for this case series. Retrospective chart review was conducted to extract clinical data relevant to each case. The research has been conducted in compliance with all relevant regulations including the Declaration of Helsinki. Written informed consent was obtained from the patients’ parents prior to publication of this case series.
Next-generation sequencing (NGS)
NGS for Patient 1 was conducted by the Clinical Laboratory Improvement Amendments (CLIA)-certified Johns Hopkins Molecular Diagnostic Laboratory at the Johns Hopkins Hospital (Baltimore, MD), as described previously36. Targeted NGS was performed on formalin-fixed, paraffin-embedded (FFPE) malignant tissue sections to analyze the coding regions of cancer-related genes with the Johns Hopkins Solid Tumor Panel (https://pathology.jhu.edu/test-directory/ngs-solid-tumor-panel).
The primary tumor for Patient 2 had NGS conducted by the CLIA-certified clinical laboratory Ashion Analytics, LLC. Genomic DNA was extracted from the FFPE tumor sample and prepared using the KAPA Hyper Prep Kit (KAPA Biosystems) to create genomic libraries. Once the genomic libraries are created, a custom SureSelect XT Target Enrichment System (Agilent Technologies) is used to select specific genomic regions that are proprietary to the Ashion GEM Cancer Panel. Captured libraries are then clustered on a flow cell and sequenced using the Illumina HiSeq 2500 instrument. Sequence data were then analyzed using validated bioinformatic tools (Ashion pipeline version 2.0).
Whole-exome sequencing
For the primary subclone and autopsy specimens from Patient 2, DNA was extracted from banked formalin-fixed, paraffin-embedded (FFPE) scrolls using the QIAamp DNA FFPE Tissue Kit (Qiagen, cat. 56404). Whole-exome sequencing and low-passage whole-genome sequencing were performed at Azenta Life Science (South Plainfield, NJ). Reads were aligned to the GRCh38 reference genome with BWA-MEM. Variants were called from whole-exome sequencing using Genome Analysis ToolKit (GATK) v3.7 and annotated using SnpSift. Copy number changes were called from low-passage whole-genome sequencing using CNVkit.
Fluorescence in situ hybridization (FISH) on FFPE tumor specimens
FISH analysis was performed on interphase nuclei using a disease-specific probe panel targeting the BRAF gene, along with a centromere probe for chromosome 7 (D7Z1), following the FFPE protocol provided by the manufacturer (Empire Genomics Inc., Depew, NY, USA). A total of 100 nuclei were evaluated within the tumor areas marked by H&E staining. The assessment was performed by two independent technologists, who were blinded to each other’s results, using a Zeiss Axioscope fluorescence microscope (Zeiss Inc., White Plains, NY, USA). Data analysis was carried out using Cytovision software (Leica Inc., Buffalo Grove, IL, USA). The specimen was deemed abnormal if the observed results exceeded the laboratory-established threshold for the probe set.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. Raw data files are available upon request from the corresponding author. NGS and WES data were generated will be deposited in dbGaP for public use.
References
Hill, D. A. et al. DICER1 mutations in familial pleuropulmonary blastoma. Science 325, 965 (2009).
de Kock, L., Wu, M. K. & Foulkes, W. D. Ten years of DICER1 mutations: Provenance, distribution, and associated phenotypes. Hum. Mutat. 40, 1939–1953 (2019).
Schultz, K. A. P. et al. DICER1 and Associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin. Cancer Res. 24, 2251–2261 (2018).
McCluggage, W. G. & Foulkes, W. D. DICER1-sarcoma: an emerging entity. Mod. Pathol. 34, 2096–2097 (2021).
Koelsche, C. et al. Primary intracranial spindle cell sarcoma with rhabdomyosarcoma-like features share a highly distinct methylation profile and DICER1 mutations. Acta Neuropathol. 136, 327–337 (2018).
Kommoss, F. K. F. et al. Genomic characterization of DICER1-associated neoplasms uncovers molecular classes. Nat. Commun. 14, 1677 (2023).
Warren, M. et al. Expanding the spectrum of dicer1-associated sarcomas. Mod. Pathol. 33, 164–174 (2020).
Macrae, I. J. et al. Structural basis for double-stranded RNA processing by Dicer. Science 311, 195–198 (2006).
Foulkes, W. D., Priest, J. R. & Duchaine, T. F. DICER1: mutations, microRNAs and mechanisms. Nat. Rev. Cancer 14, 662–672 (2014).
Cardona, A. F. et al. DICER1-associated central nervous system sarcoma: A comprehensive clinical and genomic characterization of case series of young adult patients. Neurooncol. Pract. 10, 381–390 (2023).
Kamihara, J. et al. DICER1-associated central nervous system sarcoma in children: comprehensive clinicopathologic and genetic analysis of a newly described rare tumor. Mod. Pathol. 33, 1910–1921 (2020).
Kommoss, F. K. F. et al. Clinicopathologic and molecular analysis of embryonal rhabdomyosarcoma of the genitourinary tract: evidence for a distinct DICER1-associated subgroup. Mod. Pathol. 34, 1558–1569 (2021).
Pugh, T. J. et al. Exome sequencing of pleuropulmonary blastoma reveals frequent biallelic loss of TP53 and two hits in DICER1 resulting in retention of 5p-derived miRNA hairpin loop sequences. Oncogene 33, 5295–5302 (2014).
Antonescu, C. R. et al. DICER1-associated anaplastic sarcoma of the kidney with coexisting activating PDGFRA D842V mutations and response to targeted kinase inhibitors in one patient. JCO Precis. Oncol. 6, e2100554 (2022).
Weigel, B. J. et al. Intensive multiagent therapy, including dose-compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation, in patients with high-risk rhabdomyosarcoma: a report from the Children’s Oncology Group. J. Clin. Oncol. 34, 117–122 (2016).
Le Teuff, G. et al. Phase II study of temozolomide and topotecan (TOTEM) in children with relapsed or refractory extracranial and central nervous system tumors including medulloblastoma with post hoc Bayesian analysis: a European ITCC study. Pediatr. Blood Cancer 67, e28032 (2020).
Schultz, K. A. P. et al. Outcomes for children with type II and type III pleuropulmonary blastoma following chemotherapy: a report from the international PPB/DICER1 Registry. J. Clin. Oncol. 41, 778–789 (2023).
Bisogno, G. et al. Treatment and prognostic factors in pleuropulmonary blastoma: an EXPeRT report. Eur. J. Cancer 50, 178–184 (2014).
Yaeger, R. & Corcoran, R. B. Targeting alterations in the RAF-MEK pathway. Cancer Discov. 9, 329–341 (2019).
Moore, A. R. et al. RAS-targeted therapies: is the undruggable drugged?. Nat. Rev. Drug Discov. 19, 533–552 (2020).
Dunnett-Kane, V. et al. Germline and sporadic cancers driven by the RAS pathway: parallels and contrasts. Ann. Oncol. 31, 873–883 (2020).
Kumar, M. S. et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 23, 2700–2704 (2009).
Mito, J. K. et al. Oncogene-dependent control of miRNA biogenesis and metastatic progression in a model of undifferentiated pleomorphic sarcoma. J. Pathol. 229, 132–140 (2013).
Hanna, J. A. et al. Genetic context of oncogenic drivers dictates vascular sarcoma development in aP2-Cre mice. J. Pathol. 257, 109–124 (2022).
Ricarte-Filho, J. C. et al. DICER1 RNase IIIb domain mutations trigger widespread miRNA dysregulation and MAPK activation in pediatric thyroid cancer. Front. Endocrinol.14, 1083382 (2023).
Aryal, N. K. et al. Dicer1 phosphomimetic promotes tumor progression and dissemination. Cancer Res 79, 2662–2668 (2019).
Zhong, L. et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduct. Target Ther. 6, 201 (2021).
Gross, A. M. et al. Selumetinib in children with inoperable plexiform neurofibromas. N. Engl. J. Med. 382, 1430–1442 (2020).
Moertel, C. L. et al. ReNeu: a pivotal, phase IIb trial of mirdametinib in adults and children with symptomatic neurofibromatosis type 1-associated plexiform neurofibroma. J. Clin. Oncol. 43, 716–729 (2025).
Ferguson, M. J. et al. Previously unreported somatic variants in two patients with pleuropulmonary blastoma with metastatic brain recurrence. Pediatr. Blood Cancer 68, e28825 (2021).
Ye, F. et al. Advancements in clinical aspects of targeted therapy and immunotherapy in breast cancer. Mol. Cancer 22, 105 (2023).
Singh, M. et al. Advancements in combining targeted therapy and immunotherapy for colorectal cancer. Trends Cancer 10, 598–609 (2024).
Gotwals, P. et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 17, 286–301 (2017).
Corcoran, R. B. et al. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci. Signal 3, ra84 (2010).
Nakamura, A. et al. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 73, 7043–7055 (2013).
Resch, E. E. et al. Relapse-free survival in a pediatric patient with recurrent EZH2-mutant melanoma treated with adjuvant tazemetostat. NPJ Precis. Oncol. 9, 48 (2025).
Acknowledgements
The patients’ parents in each case have given consent for publication of their cases. We thank the patients and their families for allowing us to participate in their care, providing patient-reported outcomes, and giving us permission to share their journeys. Our patients and their families are our greatest teachers. The International PPB/DICER1 Registry is supported by the Pine Tree Apple Classic Fund, Children’s Minnesota Foundation and Rein in Sarcoma. This analysis was supported by a grant from the Children’s Minnesota Internal Research Grant Program. This research used computational resources provided by the BioHPC cluster computing facility at UT Southwestern, supported by a grant from the Cancer Prevention and Research Institute of Texas (RP150596). This research was also supported by a gift from Kids Helping Kids to the pediatric sarcoma research program at Johns Hopkins University.
Author information
Authors and Affiliations
Contributions
Idea conception: L.Z., K.S.C., K.A.S., and C.A.P. Manuscript writing, data collection: L.Z., P.M., S.Z., Y.Z., K.S.C., K.A.S., and C.A.P. Figure creation: L.Z., P.M., Y.Z., K.A.S., and C.A.P. Significant contribution to clinical patient care: L.Z., D.R.O., S.R.M., J.R., Y.M., K.A.S., and C.A.P. Paper review and revisions: L.Z., P.M., S.Z., S.C.M., J.M.G., C.G.L., Y.Z., W.M., D.R.O., S.R.M., J.R., Y.M., K.S.C., K.A.S., and C.A.P.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, L., Mallinger, P.H.R., Zhou, S. et al. RAS pathway targeted therapy in patients with DICER1-associated sarcomas. npj Precis. Onc. 9, 232 (2025). https://doi.org/10.1038/s41698-025-01026-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01026-0
This article is cited by
-
Multiple drugs
Reactions Weekly (2025)
