
Abstract
Non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) mutations initially responds to the third-generation EGFR-tyrosine kinase inhibitor (TKI) Osimertinib. However, acquired resistance inevitably develops through various mechanisms, including secondary mutations and activation of bypass signaling pathways. Nuclear translocation of EGFR has been implicated in resistance to targeted therapies, but the molecular mechanisms linking EGFR subcellular localization to Osimertinib resistance remain poorly understood. Our findings suggested that CMPK2 mediates Osimertinib resistance independently of EGFR mutations. Importantly, BRD4 inhibitor NHWD870 significantly reversed this resistance by inhibiting the nuclear translocation of EGFR and subsequent transcriptional activation of CMPK2. Moreover, upregulated APT1-mediated depalmitoylation of EGFR at C19 site was observed in Osimertinib resistant cells. BRD4 inhibitor treatment efficiently repressed viability and proliferation of Osimertinib-resistant cells, with APT1 silencing additionally enhancing these inhibitory effects. In conclusion, BRD4 inhibitor inhibited APT1-mediated depalmitoylation modification of EGFR, resulting in reduction of nuclear EGFR and subsequent downregulation of CMPK2, enhancing Osimertinib sensitivity in NSCLC. This study provides a novel therapeutic strategy for overcoming Osimertinib resistance in NSCLC treatment.
Similar content being viewed by others

Introduction
The landscape of non-small cell lung cancer (NSCLC) therapy has been profoundly transformed by the understanding of molecular mechanisms driving tumor progression and drug resistance. One such pivotal mechanism involves mutations in the epidermal growth factor receptor (EGFR), notably in the tyrosine kinase domain spanning exons 19–211,2. These mutations are implicated in about 90% of EGFR-mutant NSCLC cases, with exon 19 deletions and L858R point mutations in exon 21 being the most common1. The development and application of epidermal growth factor receptor tyrosine kinase inhibitors, including gefitinib, icotinib, and erlotinib (first-generation agents), dacomitinib, afatinib and dacomitinib (second-generation), and Osimertinib (third-generation), have significantly improved the survival and clinical outcomes of NSCLC patients harboring EGFR mutation3,4,5. While Osimertinib demonstrates superior efficacy with a median progression-free survival of ~19 months in the first-line setting6, acquired resistance inevitably develops in most patients7,8, highlighting the critical need to understand resistance mechanisms.
The role of BRD4 is increasingly recognized as pivotal in cancer biology9. For instance, BRD4 inhibitors JQ1 and ARV-771 have shown promising potential in NSCLC treatment by disrupting the BRD4-IRF1 complex formation on the PD-L1 promoter, effectively blocking cisplatin and radiation-induced PD-L1 expression and enhancing anti-tumor immune responses10. As a member of bromodomain and extra-terminal (BET) family, BRD4 is also distinguished by two tandem bromodomains, BD1 and BD2. These domains exhibit selective affinity for acetylated lysine residues on target proteins and show enhanced binding to proteins with multiple acetylation sites11,12. Importantly, this enhanced interaction facilitates the accumulation of BRD4 at transcriptionally active sites, thereby influencing both the initiation and elongation phases of gene transcription9. Moreover, publications have underscored the importance of BRD4 phosphorylation in enhancing its interaction with transcription factors, thereby promoting chromatin remodeling13. This process supports the transcriptional programs that promote tumorigenesis. Inhibitors targeting BRD4, such as NHWD870, function by inhibiting this phosphorylation, thereby obstructing the transcription of downstream target genes that are crucial in the development of tumor resistance mechanisms14. Intriguingly, the combined use of a BET degrader, which can induce the degradation of BRD4, and Osimertinib in both in vivo mouse models and in vitro lung cancer cells yields a synergistic tumor suppressive effect15. This highlights that the combination of a BRD4 inhibitor with Osimertinib may offer a potent therapeutic strategy, potentially overcoming resistance to existing therapies.
A key player in the development of Osimertinib resistance is the nuclear translocation of EGFR, a phenomenon that contributes to altered gene expression and survival pathways16. Several publications have indicated that nuclear EGFR (nEGFR) increases the expression of oncogenes, promotes protein translation, and induces resistance to radiotherapy17,18. The post-translational modifications may mediate the membrane/nuclear translocation of EGFR. Palmitoylation emerges as a significant post-translational modification in the context of NSCLC19. Palmitoylation increases the hydrophobicity of proteins and then enhances their affinity for the cell membrane, momentarily targeting cytoplasmic proteins to the membrane20. A previous study has suggested that palmitoylation of EGFR aids in its sustained membrane localization21. Notably, mutations of palmitoylated cysteine residues to alanine in the C-terminal tail are sufficient to activate EGFR signaling, facilitating cell migration and transformation22, indicating that the depalmitoylation of EGFR may reduce membrane localization and activate EGFR signaling pathways. The enzymes responsible for protein depalmitoylation, particularly acyl-protein thioesterases (APTs), have been implicated in various cancer types23,24, but their specific roles in EGFR regulation and Osimertinib resistance in NSCLC remain to be elucidated.
Cytidine monophosphate kinase 2 (CMPK2) is involved in the phosphorylation of nucleoside monophosphates and plays a crucial role in cellular nucleotide metabolism25. Alterations in CMPK2 expression or activity have been implicated in various human diseases26. Interestingly, the nuclear translocation of EGFR activated nucleotide synthesis-related genes, such as the thymidylate synthase gene promoter, leading to tumor resistance to thymidylate synthase inhibitor-based anticancer drugs27. However, it has not yet been reported whether the nEGFR in NSCLC affects Osimertinib resistance by controlling CMPK2 expression.
Herein, we found that BRD4-mediated transcriptional activation of APT1 leads to depalmitoylation of mutant EGFR, facilitating its nuclear translocation and subsequent upregulation of CMPK2. This newly identified signaling axis provides insights into the mechanisms of Osimertinib resistance and suggests potential therapeutic strategies for overcoming resistance in NSCLC.
Results
CMPK2 expression was associated with Osimertinib resistance in lung cancer cells independent of additional EGFR kinase domain mutations
To investigate the molecular mechanisms underlying Osimertinib resistance, we first generated Osimertinib-resistant cell lines (PC-9/OR and HCC827/OR) from the EGFR-mutant NSCLC cell lines PC-9 and HCC827 (both harboring EGFR 19del). Next-generation sequencing analysis revealed that these resistant cell lines do not acquire common resistance mutations in the EGFR kinase domain (amino acids 712–968), including C797S and T790M mutations, nor did they exhibit MET amplification (Fig. S1A). In addition, BRD4 inhibitors have been identified as one of the most promising drugs in NSCLC resistant cells10. To identify potential targets involved in Osimertinib resistance, we performed RNA sequencing analysis of H1975/OR cells under different treatment conditions, including Osimertinib, and the combination of Osimertinib with BRD4 inhibitor (BRDi, NHWD870). Hierarchical clustering analysis revealed distinct gene expression patterns across treatment groups (Fig. S1B). Notably, CMPK2 was significantly downregulated in cells treated with BRD4 inhibitor in combination with Osimertinib, compared with Osimertinib-treated cells (Figure S1B–D). This expression pattern raised the possibility that CMPK2 is regulated in a BRD4-dependent manner and may be involved in Osimertinib resistance. Interestingly, the sensitive cells (PC-9) were observed to exhibit a dose-dependent decrease in cell viability (Fig. 1A) and CMPK2 expression (Fig. 1B, C), while EGFR levels remained unchanged (Fig. 1C). In contrast, PC-9/OR cells showed no change in cell viability, CMPK2, or EGFR expression (Fig. 1A–C). Importantly, compared with that in PC-9 cells, enhanced nuclear localization of EGFR was presented in PC-9/OR cells (Fig. 1D), however, treatment with increased concentrations of Osimertinib did not alter the nuclear localization of EGFR in PC-9/OR cells (Fig. 1D). Furthermore, knockdown of CMPK2 in PC-9/OR cells resulted in reduced cell viability and proliferation (Fig. 1E, F), with a decrease in CMPK2 expression (Fig. 1G, H), but no change in EGFR expression (Fig. 1H). Interestingly, the addition of Osimertinib did not alter these effects (Fig. 1E–H). We also performed parallel experiments using HCC827 and HCC827/OR cells to further validate these findings. As expected, similar trends were observed in these cell lines (Fig. S2A–H). These consistent results supported a model where CMPK2 upregulation contributes to Osimertinib resistance in cells maintaining original EGFR mutations, potentially through EGFR nuclear translocation.

PC-9 and PC-9/OR were treated with different concentrations of Osimertinib (0, 5, 10, 20, or 40 nM), and then A cell viability was determined by CCK-8 assay; B the expression of CMPK2 was measured by qRT-PCR; C the expression of CMPK2 and EGFR was measured by western blot; D immunofluorescence staining of EGFR (green) and nuclei (DAPI, blue). Red arrows indicated representative cells with prominent nuclear EGFR accumulation. Nuclear EGFR fluorescence intensity was quantified using ImageJ (scale bar = 25 μm). E, F The viability and proliferation of PC-9/OR cells were detected by CCK-8 and colony formation after silencing CMPK2 alone or simultaneously silencing CMPK2 and treating with Osimertinib. G, H The expression of CMPK2 and EGFR was examined by qRT-PCR and western blot in PC-9/OR cells silenced CMPK2 alone or simultaneously silenced CMPK2 and treated with Osimertinib. Mean ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis was carried out by one-way ANOVA. CMPK2 cytidine monophosphate kinase 2, EGFR epidermal growth factor receptor, CCK-8 cell counting kit-8, qRT-PCR quantitative real-time polymerase chain reaction, PC-9/OR Osimertinib-resistant PC-9 cells.
BRD4 inhibition reduced CMPK2 expression and sensitized lung cancer cells to Osimertinib
Next, we began to investigate the efficacy of the BRD4 inhibitor in CMPK2-mediated Osimertinib resistance. Elevated BRD4 expression was observed in PC-9/OR cells compared to their sensitive counterparts (Fig. 2A). Then, cells were treated with different concentrations of NHWD870 (0, 0.1, 0.5, 1, 5, or 10 nM). We found no change in cell viability at concentrations low to 0.1 nM, however, a significant reduction in the viability of resistant cells at doses above 0.5 nM was observed; interestingly, NHWD870 treatment led to less impact in sensitive cells (Fig. 2B). BRD4 inhibitor (at doses above 0.5 nM) also effectively reduced BRD4 and CMPK2 expression, particularly in resistant cells (Fig. 2C). Notably, NHWD870 treatment attenuated the nuclear translocation of EGFR observed in resistant cells (Fig. 2D). In experiments combining Osimertinib with NHWD870, treatment with Osimertinib alone did not significantly affect BRD4, CMPK2, or nEGFR levels in resistant cells (Figs. 2E and S3A), nor cell viability and proliferation (Figs. S3B and 3C). However, NHWD870 treatment markedly inhibited the expression of BRD4 and CMPK2, decreased nEGFR translocation, and reduced cell viability and proliferation (Figs. S3B and 3C), indicating that BRD4 inhibitor sensitizes resistant cells to Osimertinib. Additionally, CMPK2 knockdown led to reduced CMPK2 expression without affecting nEGFR levels (Figs. S3D and 3E) in resistant cells, and also suppressed cell viability and proliferation (Figs. S3F and 3G). Importantly, NHWD870 treatment further inhibited CMPK2 and nEGFR expression and exacerbated the reduction in cell viability and proliferation in resistant cells silencing CMPK2 (Fig. S3F, G), suggesting a potential additive effect of this combination. To complement these findings, similar experiments were conducted using HCC827 and HCC827/OR cells, which showed parallel responses to BRD4 inhibition. The HCC827/OR cells also displayed elevated BRD4 expression and responded to NHWD870 treatment with reduced cell viability and decreased nEGFR localization (Fig. S4A–H). The combination of CMPK2 knockdown and BRD4 inhibition showed enhanced efficacy in these cells as well (Fig. S4I–L). These findings suggested that BRD4 inhibition reduces CMPK2 expression and nEGFR localization, and when combined with CMPK2 knockdown, may increase the sensitivity of resistant NSCLC cells to Osimertinib.

A Western blot analysis of BRD4 in PC-9 and PC-9/OR cells. Cells were treated with different concentrations of BRD4 inhibitor (NHWD870) (0, 0.1, 0.5, 1, 5, or 10 nM), and then B CCK-8 measured cell viability; C Western blot analyzed the expression of BRD4 and CMPK2; D immunofluorescence staining of EGFR (green) and nuclei (DAPI, blue). Nuclear EGFR intensity was quantified by measuring the fluorescence ratio using ImageJ (scale bar = 25 μm). PC-9/OR cells were treated with Osimertinib, NHWD870, or Osimertinib+NHWD870, then E western blot analyzed the expression of BRD4, CMPK2, and nEGFR; Mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis was carried out by a one-way ANOVA. BRD4 bromodomain-containing protein 4, CMPK2 cytidine monophosphate kinase 2, EGFR epidermal growth factor receptor, nEGFR nuclear EGFR, CCK-8 cell counting kit-8, PC-9/OR Osimertinib-resistant PC-9 cells.

A Immunofluorescence observation of EGFR entering the nucleus (scale bar = 25 μm). B The expression of EGFR in the cytoplasm and nucleus of PC-9, PC-9/OR, HCC827, and HCC827/OR cells. C Immunofluorescence showing EGFR distribution in 293T cells transfected with control or Flag-tagged nuclear EGFR (Flag-nEGFR) (scale bar = 25 μm). D ChIP assay in 293T cells transfected with Flag-nEGFR mutant fusion expression vectors was used to detect the binding of nEGFR mutants to the CMPK2 promoter region. E ChIP assay in 293T cells transfected with HA-tagged BRD4 fusion expression vectors was used to analyze the binding of BRD4 to CMPK2 promoter region. F, G Dual-luciferase reporter assay determined the interactions between nEGFR mutants and the CMPK2 promoter region or BRD4 and CMPK2 promoter region in 293T cells treated with or without BRD4 inhibitor. H Co-IP assay in 293T, PC-9/OR, or HCC827/OR cells was performed to detect the interaction between exogenous and endogenous nEGFR mutants and BRD4. I Dual-luciferase assay in PC-9/OR and HCC827/OR cells overexpressing nEGFR mutants with or without NHWD870 treatment was performed to assess the promoter activity of CMPK2. J, K qRT-PCR and western blot analysis of CMPK2 levels in PC-9/OR and HCC827/OR cells overexpressing nEGFR mutants with or without NHWD870 treatment. L, M CCK-8 and colony formation measured cell viability and proliferation of PC-9/OR and HCC827/OR cells overexpressing nEGFR mutants with or without NHWD870 treatment. Mean ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis was carried out by a one-way ANOVA. BRD4 bromodomain-containing protein 4, CMPK2 cytidine monophosphate kinase 2, EGFR epidermal growth factor receptor, nEGFR nuclear EGFR, ChIP chromatin immunoprecipitation, Co-IP co-immunoprecipitation, CCK-8 cell counting kit-8; PC-9/OR Osimertinib-resistant PC-9 cells, HCC827/OR Osimertinib-resistant HCC827 cells.
BRD4 inhibition attenuated nuclear EGFR-associated CMPK2 transcriptional activation
Next, we began to explore how BRD4 inhibitors restore Osimertinib sensitivity in EGFR-mutant NSCLC cells. In PC-9/OR and HCC827/OR cells, an increase in nEGFR and a corresponding decrease in cytoplasmic EGFR were observed, contrasting with the sensitive cells where EGFR remained predominantly cytoplasmic (Fig. 3A, B). To further investigate nEGFR’s role, we transfected 293T cells with Flag-nEGFR. Immunofluorescence analysis revealed that while control cells show EGFR distribution in both cytoplasm and cell membrane, Flag-nEGFR-transfected cells exhibit increased nEGFR localization without altering membrane EGFR expression (Fig. 3C). Using these transfected cells, ChIP analysis detected binding of nEGFR and BRD4 to the CMPK2 promoter, with enhanced occupancy upon nEGFR or BRD4 overexpression (Fig. 3D, E). Moreover, the binding of nEGFR to the CMPK2 WT promoter increased luciferase activity upon nEGFR overexpression, an effect reduced by the addition of NHWD870. This suggested that BRD4 inhibitor may reduce CMPK2 promoter activity enhanced by nEGFR (Fig. 3F). Similarly, BRD4 binding to the CMPK2 WT promoter enhanced luciferase activity upon BRD4 overexpression, which was repressed upon adding NHWD870, indicating that the BRD4 inhibitor attenuates BRD4-mediated transcriptional activation of CMPK2 (Fig. 3G). Moreover, PPA-Pred2 prediction (https://www.iitm.ac.in/bioinfo/PPA_Pred/prediction.html#) suggested that the free energy of binding between BRD4 and nEGFR mutants is −9.83 kcal/mol, implying a potential stable interaction. Supporting this, Co-IP analysis detected a physical association between BRD4 and nEGFR in 293T and Osimertinib-resistant cells (Fig. 3H). We then observed that nEGFR overexpression increases CMPK2 promoter activity (Fig. 3I) and CMPK2 mRNA and protein levels (Fig. 3J, K) in Osimertinib-resistant cells, as well as promoted cell viability (Fig. 3L) and proliferation (Fig. 3M). However, these effects were reversed by treating of NHWD870 (Fig. 3I–M). Taken together, BRD4 inhibition suppressed both nEGFR accumulation and its transcriptional activation of CMPK2, contributing to restored drug sensitivity.
Depalmitoylation of EGFR at C19 site was elevated in Osimertinib-resistant lung cancer cells
A previous study has indicated that the depalmitoylation of EGFR may reduce its membrane localization22, we therefore further investigated its palmitoylation levels in Osimertinib-resistant cells. The results revealed that EGFR palmitoylation levels are higher in Osimertinib sensitive cells than that in the resistant cells (Fig. 4A). The palmitoylation inhibitor 2-BP decreased EGFR palmitoylation in sensitive cells (Fig. 4B), the palmitoyltransferase activator Palm B restored palmitoylation in resistant cells (Fig. 4C). Subsequently, a conservation analysis of cysteine residues in EGFR across different species (human, mouse and rat) highlighted the high conservation of the C19 palmitoylation site (Fig. 4D). When 293T cells were transfected with HA-tagged EGFR mutants at the C19 site, there was a loss of palmitoylation, indicating the critical role of this site in the process (Fig. 4E). Similarly, transfection with the C19 site mutant EGFR did not alter palmitoylation levels in PC-9/OR and HCC827/OR cells, whereas EGFR WT transfection increased its palmitoylation (Fig. 4F). Furthermore, EGFR WT enhanced CMPK2 promoter activity, while the mutant EGFR had no significant effect (Fig. 4G). EGFR WT increased CMPK2 expression, whereas the mutant EGFR did not influence CMPK2 expression (Fig. 4H). Thus, the palmitoylation of EGFR regulated CMPK2 levels in Osimertinib-resistant cells.

A Acyl-RAC assay of EGFR palmitoylation in PC-9, PC-9/OR, HCC827, and HCC827/OR cells. B Examination of EGFR palmitoylation in PC-9 and HCC827 cells treated with 2-BP (100 µM) for 0, 2, or 4 h using Acyl-RAC assay. C Acyl-RAC assay of EGFR palmitoylation in PC-9/OR and HCC827/OR cells treated with Palmitate B (Palm B, 50 μM) for 0, 2, 4, and 8 h. D Conservation analysis of cysteine residues in EGFR protein sequences across different species (human, mouse, and rat). E Acyl-RAC assay of palmitoylation levels in 293T cells transfected with HA-tagged EGFR mutants at the C19 palmitoylation site. F Acyl-RAC assay of palmitoylation levels in PC-9/OR, and HCC827/OR cells transfected with HA-tagged wild-type or C19 palmitoylation site mutant EGFR. G Dual-luciferase assay assessed the impact of wild-type or mutant EGFR transfection on CMPK2 promoter activity in PC-9/OR, and HCC827/OR cells. H qRT-PCR and western blot analysis of CMPK2 levels in PC-9/OR and HCC827/OR cells transfected with wild-type or mutant EGFR. Mean ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis was carried out by a student’s t-test or a one-way ANOVA. EGFR epidermal growth factor receptor, Acyl-RAC acyl-resin-assisted capture, 2-BP 2-bromopalmitate, Palm B Palmitate B, CMPK2 cytidine monophosphate kinase 2, qRT-PCR quantitative real-time polymerase chain reaction, PC-9/OR Osimertinib-resistant PC-9 cells, HCC827/OR Osimertinib-resistant HCC827 cells, HAM hydroxylamine hydrochloride.
APT1 promoted EGFR depalmitoylation and contributes to Osimertinib resistance in NSCLC cells
Palmitoylation modification involves attaching a palmitoyl group via a thioester bond to a cysteine residue in the proteins, while depalmitoylation is mainly achieved by the APTs, which cleaves the thioester bond28. Herein, APT1 (acyl-protein thioesterase 1) was observed to be up-expressed in NSCLC. We therefore explored the role of APT1 in mediating the depalmitoylation of EGFR in Osimertinib-resistant NSCLC cells. The results indicated an increased expression of APT1 in PC-9/OR cells compared to PC-9 cells (Fig. 5A). Subsequently, APT1 expression was inhibited by transfecting with shAPT1 (Fig. 5B). Immunofluorescence studies revealed that this knockdown leads to a reduction in the colocalization of APT1 and EGFR at the cell membrane (Fig. 5C). Moreover, EGFR palmitoylation levels remained unchanged in PC-9 cells after APT1 knockdown, while the palmitoylation of EGFR in PC-9/OR cells increased (Fig. 5D). Immunofluorescence observations showed that knockdown of APT1 leads to a downregulation of EGFR nuclear translocation in PC-9/OR cells, an effect not observed in sensitive cells (Fig. 5E). To further investigate this phenomenon, we examined the effect of APT1 knockdown on EGFR phosphorylation status and subcellular localization. Western blot analysis revealed that PC-9 sensitive cells lacked nEGFR expression, and APT1 knockdown has no effect on nEGFR or p-EGFR levels in these cells. In contrast, PC-9/OR resistant cells exhibited substantial n-EGFR expression, and APT1 knockdown significantly reduces nEGFR levels while increasing p-EGFR expression (Fig. 5F). This result was further confirmed by analyzing EGFR cytoplasm and nucleus distribution in resistant and sensitive cells (Fig. S5A). Additionally, knockdown of APT1 in PC-9/OR cells caused a reduction of CMPK2 mRNA and protein levels, while no significant change was observed in PC-9 cells (Fig. S5B). Functionally, knockdown of APT1 did not affect the viability and proliferation of sensitive cells, while resulting in an inhibition in cell viability and proliferation in PC-9/OR cells (Fig. S5C, D). APT1 knockdown suppressed viability and proliferation in resistant cells, but had no effect in sensitive cells (Fig. S5C–F). Interestingly, APT1 knockdown partially restored Osimertinib sensitivity in PC-9/OR cells, as evidenced by reduced viability and proliferation (Fig. S5E, F). To validate these findings in an additional cell model, we performed parallel experiments with HCC827 and HCC827/OR cells. Similar results were observed, with increased APT1 expression in resistant cells and APT1 knockdown effects on EGFR palmitoylation, nuclear translocation, and cell viability showing consistent patterns (Fig. S6A–L). Taken together, by influencing the palmitoylation status of EGFR, APT1 directly impacted the downstream signaling pathways, including the nuclear translocation of EGFR and the expression of CMPK2, thereby affecting Osimertinib sensitivity in NSCLC cells.

A Western blot analysis of APT1 expression in PC-9 and PC-9/OR cells. B The knockdown efficiency of shAPT1 in PC-9 and PC-9/OR cells was measured by western blot. C Immunofluorescence assessment of the colocalization between APT1 and EGFR in cells transfected with sh-APT1 (scale bar = 25 μm). D Acyl-RAC assay analyzed the palmitoylation status of EGFR at C19 site in cells transfected with shAPT1. E Immunofluorescence staining of EGFR (green) and nuclei (DAPI, blue) in cells transfected with shAPT1. Red arrows indicated cells with enhanced nuclear EGFR accumulation in shAPT1 groups. Nuclear EGFR intensity was quantified as the ratio of EGFR fluorescence within DAPI-defined regions to total cellular EGFR intensity using ImageJ (scale bar = 25 μm). F Western blot analysis of nEGFR, p-EGFR, and t-EGFR expression in cells transfected with shAPT1. Mean ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis was carried out by a student’s t-test or a one-way ANOVA. APT1 acyl protein thioesterase 1, EGFR epidermal growth factor receptor, nEGFR nuclear EGFR, p-EGFR phosphorylated EGFR, t-EGFR total EGFR, Acyl-RAC acyl-resin-assisted capture, CMPK2 cytidine monophosphate kinase 2, PC-9/OR Osimertinib-resistant PC-9 cells.
BRD4 inhibitor restored Osimertinib sensitivity by suppressing CMPK2 transcription via modulation of nEGFR and APT1
Next, we examined whether the BRD4 inhibitor NHWD870 restores Osimertinib sensitivity by regulating nEGFR and APT1 in vitro. To assess the role of EGFR palmitoylation at the C19 site in nuclear translocation and CMPK2 regulation, PC-9/OR cells were transfected with either WT EGFR or a C19-site mutant, followed by NHWD870 treatment. Transfection with WT EGFR increased CMPK2 levels and promoter activity, but these effects were not observed after transfecting with mutant EGFR. Importantly, NHWD870 treatment reduced CMPK2 expression and promoter activity in both scenarios (Fig. 6A–C). Furthermore, the nuclear translocation of EGFR was enhanced with WT EGFR transfection, an effect not seen after transfecting with mutant EGFR. NHWD870 treatment effectively decreased this nuclear translocation (Fig. 6D). As expected, EGFR WT transfection promoted the viability and proliferation of Osimertinib-resistant cells, while NHWD870 treatment blocked these effects (Fig. 6E, F). Additional experiments involving APT1 knockdown followed by NHWD870 treatment revealed a decrease in both APT1 and CMPK2 expression, with the combination of APT1 knockdown and NHWD870 showing the greatest reduction (Fig. 6G, H). Similarly, APT1 knockdown alone reduced CMPK2 promoter activity, and this effect was further amplified when combined with NHWD870 (Fig. S7A). This combination also led to an increase in EGFR palmitoylation (Fig. S7B) and a decrease in its nuclear translocation (Fig. S7C), along with a significant reduction in cell viability and proliferation (Fig. S7D, E). To confirm the consistency of these findings, we conducted similar experiments using HCC827/OR cells, which demonstrated comparable results regarding the effects of EGFR WT/mutant transfection and NHWD870 treatment on CMPK2 expression, EGFR nuclear translocation, and cell viability. Likewise, the combination of APT1 knockdown with NHWD870 showed enhanced effects in HCC827/OR cells (Fig. S8A–M). These findings position as a potential strategy to counteract resistance via the APT1/nEGFR/CMPK2 axis.

PC-9/OR cells were transfected with HA-tagged C19 palmitoylation site mutant EGFR, followed by treating with BRD4 inhibitor. The groups include: NC, WT-HA-EGFR, Mut-HA-EGFR, NC + NHWD870, WT-HA-EGFR + NHWD870, and Mut-HA-EGFR + NHWD870, and then A, B CMPK2 levels were measured by qRT-PCR and western blot. C Dual-luciferase reporter assay analyzed CMPK2 promoter activity. D Immunofluorescence observed the nuclear translocation of EGFR (scale bar = 25 μm). E, F CCK-8 and colony formation determined cell viability and proliferation. PC-9/OR cells were transfected with shAPT1, followed by treating with BRD4 inhibitor, the groups include: shNC, shAPT1, NHWD870+shNC, and NHWD870+shAPT1, and then G CMPK2 levels were measured by qRT-PCR, H APT1 and CMPK2 levels were measured by western blot. Mean ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis was carried out by a one-way ANOVA. BRD4 bromodomain-containing protein 4, CMPK2 cytidine monophosphate kinase 2, nEGFR nuclear EGFR, APT1 acyl protein thioesterase 1, HA hemagglutinin, NC negative control, WT wild-type, Mut mutant, NHWD870 BRD4 inhibitor, qRT-PCR quantitative real-time polymerase chain reaction, Acyl-RAC acyl-resin-assisted capture, CCK-8 cell counting kit-8, PC-9/OR Osimertinib-resistant PC-9 cells.
BRD4 inhibitor enhanced the Osimertinib sensitivity in vivo
To investigate the efficacy of BRD4 inhibitor NHWD870 in enhancing Osimertinib sensitivity in vivo, mice were injected with PC-9/OR or HCC827/OR cells, followed by treatment with Osimertinib and NHWD870. The results revealed that while Osimertinib alone has minimal impact on tumor size, NHWD870 treatment significantly reduces tumor size, with the most pronounced effect seen in the combination treatment (Fig. 7A). Ki67 expression showed no significant change with Osimertinib alone. In contrast, NHWD870 treatment led to a notable decrease in Ki67 expression, indicating reduced tumor proliferation. The combination therapy yielded the most significant reduction in Ki67 expression (Fig. 7B). Furthermore, treatment with Osimertinib alone did not markedly alter the expression of BRD4, APT1, and CMPK2. However, Combination therapy further reduced CMPK2 expression compared to NHWD870 alone (Fig. 7C), consistent with in vitro data. Osimertinib treatment did not significantly affect the nuclear co-localization of BRD4 and EGFR. In contrast, NHWD870 treatment reduced their co-localization in the nucleus (Fig. 7D). Additionally, Osimertinib treatment alone did not change the palmitoylation status of EGFR. However, NHWD870 treatment resulted in an increase in EGFR palmitoylation (Fig. 7E). These in vivo findings align with cellular phenotypes, supporting further exploration of BRD4 inhibition as a combinatorial strategy with Osimertinib.

Mice were divided into four groups by injecting with PC-9/OR or HCC827/OR cells and treating with BRD4 inhibitor (n = 6): control, Osimertinib, NHWD870, Osimertinib + NHWD870, and then the following analyses were conducted: A Live imaging observed tumor size. B Ki67 staining assessed tumor proliferation (scale bar = 50 μm). C IHC staining detected the expression of BRD4, APT1, and CMPK2 in tumors (scale bar = 50 μm). D Fluorescence dual-staining determined the localization of BRD4 and EGFR in cell nucleus (scale bar = 50 μm). E Acyl-RAC assay analyzed the palmitoylation modification of EGFR. Mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis was carried out by a one-way ANOVA. BRD4 bromodomain-containing protein 4, APT1 acyl protein thioesterase 1, CMPK2 cytidine monophosphate kinase 2, EGFR epidermal growth factor receptor, IHC immunohistochemistry, NHWD870 BRD4 inhibitor, Acyl-RAC acyl-resin-assisted capture, PC-9/OR Osimertinib-resistant PC-9 cells, HCC827/OR Osimertinib-resistant HCC827 cells, HAM hydroxylamine hydrochloride.
Discussion
EGFR mutation occurs in a considerable number of NSCLC cases, especially Asian cases29, and therefore has been established as one of driver oncogenes for NSCLC30. The third-generation EGFR-TKI Osimertinib has granted approval for clinical application in NSCLC patients with EGFR T790M mutation31, and has made a great breakthrough in clinical treatment. However, resistance to Osimertinib poses a substantial obstacle. Intriguingly, recent evidence suggested that the nuclear translocation of EGFR may play a critical role in the development of Osimertinib resistance16. In this study, BRD4 was found to increase APT1 expression, inducing the depalmitoylation of EGFR and subsequently upregulation of nEGFR, upregulated nEGFR activated CMPK2 expression and enhanced Osimertinib resistance in NSCLC.
CMPK2 is a mitochondrial enzyme that plays a crucial role in the synthesis of deoxyribonucleotides, which are essential for DNA replication and repair32. A previous study has indicated an interesting conclusion, nEGFR induces the activation of thymidylate synthase gene promoter, a nucleotide synthesis-related gene, leading to drug resistance in tumor27. Herein, our data supported a role for CMPK2 in Osimertinib resistance independent of secondary EGFR mutations, adding to the complexity of resistance mechanisms. More importantly, we introduced the BRD4 inhibitor NHWD870, which significantly reduced CMPK2 expression and enhanced sensitivity to Osimertinib in resistant cells. Targeting BRD4-related modification sites or enzymes may be an effective strategy for cancer prevention and treatment33. The inhibitor targeting BRD4 has displayed sensitivity in Osimertinib-tolerant persisters34. Indeed, inhibition of BRD4 has been suggested to enhance Osimertinib sensitivity in both in vivo mouse models and in vitro lung cancer cells15. Our study highlighted that BRD4 induces Osimertinib resistance by regulating CMPK2, providing a new target for an in-depth understanding of the complexity of drug resistance mechanisms.
EGFR localization can change between the cell membrane, cytoplasm, and nucleus35. Notably, increasing publications confirmed the key roles of nEGFR in cancer development36,37, including in drug-resistant cancer cells. For instance, nuclear expression of ErbB-3 (a receptor tyrosine kinase of EGFR) can be a prognostic marker or a therapeutic target for advanced prostate cancer with resistance to androgen ablation38. Rong et al. suggested that nEGFR can be recognized as a key factor in the process of Osimertinib resistance16. Here, we observed that the upregulated BRD4 in Osimertinib-resistant cells promotes nEGFR levels, thereby activating CMPK2 activity. Therefore, the BRD4 inhibitor NHWD870 impeded the transcription of CMPK2 mediated by the nuclear translocation of EGFR, thereby restoring Osimertinib sensitivity. These findings added to the evidence that nEGFR blockade may attenuate certain resistance pathways in EGFR-mutant NSCLC.
The dynamic and reversible process of palmitoylation influences protein properties such as accumulation, distribution, function, secretion, and stability by modifying membrane affinity39. Previous research has uncovered lots of cancer-related proteins that undergo palmitoylation40,41, suggesting that targeting this post-translational modification or the enzymes catalyzing it could be an efficacious approach for cancer prevention and treatment strategies.
The palmitoylation of EGFR in tumorigenesis has received increasing attention. It has been suggested that the palmitoyltransferase DHHC20 palmitoylates EGFR on the C-terminal domain and played important roles in signal regulation during oncogenesis42. Hence, we explored the palmitoylation of EGFR in Osimertinib-resistant cells and observed the upregulation of EGFR depalmitoylation at C19 site. Moreover, palmitoylation could be reversed by APTs, including APT1 and APT243. Here, we thoroughly analyzed the enzymes responsible for depalmitoylation in NSCLC and confirmed the upregulation of APT1 in NSCLC. Intriguingly, we observed and subsequently verified the interaction between BRD4 and APT1, suggesting that BRD4 transcriptionally regulates APT1 expression. Functionally, the combination of APT1 knockdown and BRD4 inhibitor treatment further decreased nEGFR levels and repressed the viability and proliferation of Osimertinib-resistant cells. These findings shed light on the novel role of the BRD4 axis in modulating EGFR activity through APT1-mediated depalmitoylation, providing a new perspective on how post-translational modifications of EGFR can influence drug sensitivity in NSCLC.
While our findings elucidate a potential BRD4/APT1/nEGFR/CMPK2 axis in Osimertinib resistance, several limitations should be acknowledged. First, the reliance on cell line models may not fully recapitulate the tumor microenvironment or intratumoral heterogeneity observed in patients. Second, although we demonstrated correlative relationships between BRD4 inhibition, nEGFR reduction, and CMPK2 downregulation, direct mechanistic links (e.g., whether BRD4 transcriptionally regulates APT1 or whether CMPK2 is the sole critical downstream effector) remain to be experimentally validated. Third, the in vivo efficacy was evaluated only in xenograft models; patient-derived xenografts or genetically engineered mouse models might provide more clinically relevant validation. Finally, the therapeutic window and potential toxicity of combining BRD4 inhibitors with Osimertinib in humans require further investigation, as systemic BRD4 inhibition could affect normal tissues. Addressing these limitations in future studies will strengthen the translational potential of targeting this pathway.
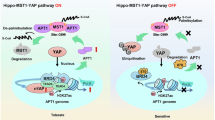
In conclusion, BRD4 enhanced the nuclear translocation of mutant EGFR by APT1-mediated depalmitoylation, thereby increasing CMPK2 levels and subsequent mitochondrial nucleotide metabolism, ultimately resulting in Osimertinib resistance in NSCLC. Based on this mechanism, the BRD4 inhibitor was demonstrated to restore Osimertinib sensitivity through regulating APT1/nEGFR/CMPK2 axis (Fig. 8). This discovery not only enhances our understanding of the mechanisms underlying Osimertinib resistance but also points to potential therapeutic interventions targeting EGFR palmitoylation.

In the resistant state (left panel), APT1 mediates the depalmitoylation of EGFR, facilitating its nuclear translocation. Within the nucleus, nEGFR interacts with BRD4 to form a transcriptional complex at the CMPK2 promoter region. This interaction leads to transcriptional activation of CMPK2, which catalyzes the conversion of ATP + CMP to ADP + CDP. Elevated CMPK2 expression ultimately confers resistance to Osimertinib. In contrast, the sensitive state (right panel) shows palmitoylated EGFR anchored to the cell membrane, where it remains accessible to Osimertinib inhibition. The BRD4 inhibitor prevents BRD4 from interacting with acetylated histones, thereby disrupting the transcriptional activation of CMPK2. Consequently, CMPK2 expression is reduced, and cells maintain sensitivity to Osimertinib. BRD4 bromodomain-containing protein 4, NSCLC non-small cell lung cancer, APT1 acyl protein thioesterase 1, nEGFR nuclear EGFR, CMPK2 cytidine monophosphate kinase 2, EGFR epidermal growth factor receptor, ATP adenosine triphosphate, CMP cytidine monophosphate, ADP adenosine diphosphate, CDP cytidine diphosphate, S-CoA stearoyl-CoA, Pol II RNA polymerase II.
Methods
RNA extraction and next-generation sequencing
Total RNA was extracted from cell lines using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), with RNA integrity verified by Agilent 2100 Bioanalyzer (#5067-1511, Agilent Technologies, Santa Clara, CA, USA). Polyadenylated mRNA was enriched using NEBNext Poly(A) mRNA Magnetic Isolation Module (#E7490, New England Biolabs, Ipswich, MA, USA), and strand-specific libraries were prepared with NEBNext Ultra II Directional RNA Library Prep Kit (#E7760, New England Biolabs). RNA fragmentation (94 °C, 8 min) generated 200–300 bp fragments for cDNA synthesis using ProtoScript II Reverse Transcriptase (#M0368, New England Biolabs) and dUTP-based strand marking. Adapter-ligated libraries were size-selected (200–500 bp, BluePippin, Sage Science, Beverly, MA, USA) and quantified by Quantitative PCR (qPCR). Paired-end sequencing (2 × 150 bp) was performed on Illumina NovaSeq 6000 (Illumina, San Diego, CA, USA) using sequencing-by-synthesis technology to a depth of 50 million reads per sample (Q30 > 85%), with demultiplexing using bcl2fastq2 v2.20 (Illumina).
Transcriptomic data analysis
Raw reads were trimmed with Trimmomatic v0.39 (Usadel Lab, RWTH Aachen University, Germany) (ILLUMINACLIP:2:30:10) and aligned to GRCh38 using HISAT2 v2.2.1 (Center for Computational Biology, Johns Hopkins University, Baltimore, MD, USA) (parameters: dta rna-strandness RF). Transcript abundance was quantified as TPM values with StringTie v2.2.1 (Center for Computational Biology, Johns Hopkins University), and differential expression analysis was performed using DESeq2 v1.34.0 (Bioconductor) (thresholds: |log2FC| > 1, FDR < 0.05). Hierarchical clustering of differentially expressed genes (DEGs) was visualized by heatmaps (ComplexHeatmap v2.10.0, Bioconductor, z-score normalized TPM), and volcano plots (ggplot2 v3.3.5, R Foundation, Vienna, Austria) highlighted significant DEGs (FDR < 0.05).
Cell culture and treatment
PC-9 (RIKEN Cell Bank, Ibaraki, Japan) and HCC827 (ATCC, VA, USA), harboring a deletion in exon-19, were kept in RPMI1640 medium containing 10% (v/v) fetal bovine serum, 50 g/mL streptomycin, and 100 U/mL penicillin (all from Gibco, Carlsbad, USA) at 37 °C supplied with 5% CO2. Osimertinib-resistant cells (PC-9/OR and HCC827/OR) were established through continuous exposure to gradually increasing concentrations of Osimertinib (0, 5, 10, 20, or 40 nM, Selleck Chemicals, Houston, TX, USA) over a period of ~6 months. Initially, multiple resistant clones (n = 5) were generated for each parental cell line, and the most stable ones were selected for further experiments. Cancer cells and Osimertinib resistance cells were treated with different concentrations BRD4 inhibitor (NHWD870, 0, 0.1, 0.5, 1, 5, or 10 nM), Palmitate B (Palm B, 50 μM) for indicated time, or 2-bromopalmitate (2-BP, 100 µm) for indicated time as needed.
Cell transfection
Short hairpin RNA (shRNA) targeted CMPK2, APT1 (shCMPK2, shAPT1) or Synthetic scrambled oligonucleotide sequences, procured from GenePharma (Shanghai, China), were inserted into the pGLVH1 vector backbone for cloning purposes. Plasmid transfection into PC-9/OR and HCC827/OR cells was achieved through Lipofectamine 3000 (Invitrogen).
CCK-8 assay
Following treatment, the cells were dispensed into 96-well plates, with each well accommodating a seeding density of 5000 cells. After an overnight incubation period, a colorimetric cell viability assay was performed by introducing CCK-8 reagent (10 μL, Beyotime, China) into each well. Subsequent incubation for 2 h at 37 °C allowed for the formation of a formazan dye, the absorbance of which was quantified at 450 nm using a microplate spectrophotometer (BioRad, Hercules, CA, USA).
Colony formation assay
After trypsinizing and counting, treated cells were placed into in six-well plates (600 cells/well) and maintained for 2 weeks, allowing colony formation. During this period, the medium was replaced every 3 days to maintain optimal growth conditions. Next, cells underwent fixation in a 4% paraformaldehyde solution for 15 min. Following fixation, they were subjected to staining with a 0.1% crystal violet reagent. The stained cells were visualized and enumerated under a microscopic instrument. Colonies were defined as groups of more than 50 cells.
Immunofluorescence
The immunofluorescence assay was conducted to evaluate EGFR expression, its nuclear translocation, and its colocalization with APT1. Initially, cells were seeded on coverslips in a 24-well plate and allowed to adhere overnight. After reaching appropriate confluency, cellular fixation was executed utilizing a 4% paraformaldehyde reagent (15 min). Subsequently, permeabilization was facilitated through the employment of a 0.1% Triton X-100 solution (10 min). To mitigate nonspecific binding, cells underwent blocking with a 5% bovine serum albumin solution (1 h), after which they were subjected to incubation with primary antibodies (4 °C, overnight) against EGFR (ab52894, Abcam, Cambridge, UK), Flag (F3165, Sigma-Aldrich, St. Louis, MO, USA) and APT1 (A07249, Boster, Pleasanton, CA, USA). For Flag-nEGFR expression visualization in 293T cells, anti-EGFR or anti-Flag antibodies were used to confirm nuclear localization after transfection. Subsequently, cells were rinsed again for three times followed by incubation with indicated secondary antibodies (room temperature, dark, 1 h). DAPI was applied for nuclei staining. After final washes, coverslips were mounted on slides with an anti-fade mounting medium. Fluorescence microscopy was utilized to visualize and capture images of the stained cells. Quantitative analysis of EGFR nuclear localization was performed using ImageJ software (NIH, Bethesda, MD, USA) by measuring the nuclear-to-total EGFR fluorescence intensity ratio from at least 50 cells per condition across three independent experiments.
Subcellular fractionation
Treated cells were lysed in a lysis buffer and homogenized by 30 strokes in a tightly fitting Dounce homogenizer. Next, Nuclei were pelleted at 1500 × g for 5 min, and the supernatant was taken as the cytoplasmic fraction. of EGFR distribution in cytoplasm and nucleus was determined vis Western blot.
Chromatin immunoprecipitation (ChIP)
ChIP was performed by constructing with Flag-tagged nEGFR mutants and HA-tagged BRD4 in 293T cells. For Flag-nEGFR expression, 293T cells were transfected with the Flag-nEGFR plasmid, which resulted in increased nEGFR localization. Specifically, cells were cross-linked with 1% formaldehyde and quenched with glycine. After washing, nuclei were isolated and sonicated to shear DNA into fragments of ~200–500 base pairs. The fragmented chromatin was then subjected to immunoprecipitation using specific antibodies: anti-Flag nEGFR (F3165, Sigma-Aldrich), anti-HA BRD4 (ab9110, Abcam), anti-IgG (as a negative control, ab218427, Abcam). Following overnight incubation at 4 °C, protein G magnetic beads were added to bind the antibody-chromatin complexes. The beads were then washed to remove nonspecifically bound DNA. The bound DNA-protein complexes were eluted from the beads and treated with RNase and proteinase K to purify the DNA. Finally, qRT-PCR was performed on the purified DNA using primers specific to the CMPK2 promoter region.
Dual-luciferase reporter assay
Wild-type (WT) or mutated (MUT) CMPK2 promoter sequences containing the binding regions for BRD4 and nEGFR were constructed in Genechem Co., Ltd (Shanghai, China) and then inserted into a pGL3 vector (Promega Corporation) as luciferase reporter gene vectors. CMPK2-WT contains the intact binding sites for BRD4 and nEGFR in the CMPK2 promoter region, while CMPK2-MUT contains specific mutations in these binding sites to disrupt transcription factor binding. It is important to note that as nEGFR lacks a direct DNA binding domain, it requires interaction with other transcription factors like BRD4 to exert its transcriptional regulatory functions, as described in a previous study44. CMPK2 WT/MUT were transfected into 293T, PC-9/OR, or HCC827/OR cells overexpressing nEGFR or BRD4. Post-transfection, cells were treated with NHWD870. Similarly, EGFR WT/MUT transfected into PC-9/OR, or HCC827/OR cells transfected C19 palmitoylation site mutant EGFR vectors. After 48 h, cells were lysed and subjected to the Dual-Luciferase Reporter Assay according to the manufacturer’s protocol. Firefly luciferase activity was measured as a reporter of promoter activity, normalized to Renilla luciferase activity, serving as an internal control.
Co-immunoprecipitation (Co-IP)
Co-IP was performed in extracts of 293T, PC-9/OR, or HCC827/OR cells. Briefly, cells were lysed in lysis buffer, placed on ice for 20 min and centrifugated to obtain supernatant. The supernatant was transferred to a new tube, with a fraction pipetted out as Input. Following a 1-h incubation with below antibodies: anti-Flag nEGFR (F3165, Sigma-Aldrich) and anti-IgG (ab218427, Abcam) at 4 °C, immune complexes were captured overnight on protein G magnetic beads. Coimmunoprecipitates were resolved by SDS-PAGE and measured by Western blotting as described below using anti-BRD4 (ab128874, Abcam).
Acyl resin-assisted capture (Acyl-RAC)
Acyl-RAC assay was used to isolated and measured palmitoylated EGFR in treated cells and mouse tissues. Briefly, membrane fractions prepared from treated cells dissolved in the buffer containing 100 mM HEPES, pH 7.5, 1 mM EDTA, 2.5% SDS at a concentration of 1 mg protein/mL. To block free thiols, samples were incubated with 0.3% S-methyl methanethiosulfonate for 20 min at 42 °C. The proteins were then precipitated using pre-chilled acetone at −20 °C for 1 h, followed by two washes with 70% cold acetone. The precipitated proteins were resuspended in the buffer containing 100 mM HEPES, pH 7.5, 1 mM EDTA, and 1% SDS. Afterwards, samples were diluted to equal concentrations. For affinity purification of palmitoylated proteins 450 μL lysate was combined with 200 μL of 100 mM HEPES pH 7.4 with 1 mM EDTA, 300 μL of 1 M NH2OH pH 7.4 or 150 mM Tris·HCl pH 7.4 as a negative control, and 30 μL thiopropyl sepharose and incubated at room temperature for 3 h. Finally, the fractions eluted in the buffer containing 100 mM HEPES, pH 7.5, 1 mM EDTA, 1% SDS were treated with SDS loading buffer at 37 °C for 1 h and subjected to SDS-PAGE for western blot analysis.
In vivo mouse model
All murine experiments were performed in accordance with protocols sanctioned by the Animal Ethics Committee of Xiangya Hospital, Central South University (No.:2023030370). BABL/c nude mice (aged 6 weeks and weighing between 16 and 20 g) purchased from human SJA laboratory animal Co., Ltd (Hunan, China) were subcutaneously injected into the flanks with a total of 5 × 106 PC-9/OR or HCC827/OR cells suspended in 200 μL of Matrigel/PBS, and then fed with food and water ad libitum and at specific pathogen-free conditions (20 °C; 60% humidity and alternating 12-h light/dark cycles). Afterwards, the cohort of tumor-bearing mice was stratified into four experimental groups when tumor volume reached an average size of 200 mm3 (n = 6): control, Osimertinib, NHWD870, and Osimertinib + NHWD870. Osimertinib (1.25 mg/kg) was given once per day by oral gavage. NHWD870 (1.5 mg/kg) was given intraperitoneally every 3 days. Combined treatment (Osimertinib+NHWD870) was given half the dose of each. The tumor volumes were observed and recorded every 2 days. After 3 weeks of treatment, mice were euthanized by CO2 asphyxiation followed by cervical dislocation to ensure death, and tumor tissues were collected for subsequent experiments.
Immunohistochemistry (IHC)
Antigen unmasking was achieved by subjecting the tissue sections to heat-mediated retrieval in a 10 mM citrate buffer solution, using a microwave oven for 3 min. The sections were then incubated overnight at 4 °C with primary antibodies targeting Ki-67 (ab16667, Abcam), BRD4 (ab128874, Abcam), anti-APT1 (A07249, Boster), and anti-CMPK2 (PA5-34461, ThermoFisher). Subsequent to primary antibody labeling, the sections were exposed to poly-peroxidase-conjugated anti-mouse/rabbit IgG secondary antibodies. The immunogenic reaction was developed by employing 3,3′-diaminobenzidine (DAB; Bioss, Beijing, China) as the chromogenic substrate, followed by counterstaining with hematoxylin. The stained sections were then examined under a light microscope for visualization and analysis.
qRT-PCR
Subsequent to the treatment regimen, total RNA was isolated from samples using Trizol reagent (Invitrogen), adhering to the manufacturer’s protocol. The extracted RNA underwent reverse transcription to cDNA using the Prime-Script RT-PCR master mix (Takara, Tokyo, Japan). qRT-PCR was employed to evaluate mRNA expression levels of CMPK2, utilizing SYBR Green qPCR (Applied Biosystems, Carlsbad, CA, USA; 4309155) as the detection method. GAPDH was regarded as the internal control. The gene expression levels were presented as fold changes relative to the expression levels of appropriate controls using the 2–ΔΔCt method. Please see Table 1 for primer sequences.
Western blot analysis
After extracted from indicated cells and tissues, proteins were subjected for concentration determination, and then equal amounts of protein samples were isolated in 10% SDS-polyacrylamide gels. Isolated proteins were then transferred to a polyvinylidene difluoride membrane, which was subjected to an incubation with 5% skim milk over 1 h. Afterwards, after three times washes with PBS, the membrane was subjected for an incubation with primary antibodies overnight at 4 °C, followed by another incubation with horseradish peroxidase-conjugated secondary antibody (1 h, sc-2004, Santa Cruz Biotechnology). The antibody-reactive bands were detected with ECL reagent (Millipore, Billerica, MA, USA). Band intensities were quantified using ImageJ. For nuclear protein extraction, the Nuclear and Cytoplasmic Extraction Reagents kit (NE-PER, #78833, Thermo Scientific) was used according to the manufacturer’s instructions. Histone H3 (ab1791, Abcam) was used as a nuclear fraction loading control, while β-actin (ab8245, Abcam) was used as a cytoplasmic or total protein loading control. The following primary antibodies were used: anti-EGFR (total EGFR, t-EGFR, ab52894, Abcam), anti-phospho-EGFR (p-EGFR, Tyr1068, #3777, Cell Signaling Technology, Danvers, MA, USA), anti-BRD4 (ab128874, Abcam), anti-APT1 (A07249, Boster), and anti-CMPK2 (PA5-34461, ThermoFisher). nEGFR was detected using the anti-EGFR antibody (ab52894, Abcam) in nuclear fractions.
Statistical analysis
The collected data were summarized as mean ± standard deviation. GraphPad Prism 8.0 software was employed for statistical computations. Survival distributions were estimated using the Kaplan-Meier method, and differences in overall survival (OS) between groups were assessed through log-rank tests. Comparisons between two groups were conducted using Student’s t-test, while multiple group comparisons were performed via one-way analysis of variance (ANOVA). A p value threshold of 0.05 was considered statistically significant for all analyses.
Data availability
We have instead uploaded it to the OSF database. Please visit the website https://osf.io/cmb7s/files/osfstorage for details.
References
Sequist, L. V. et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 41, 2869–2876 (2023).
Wu, Y. L. et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 15, 213–222 (2014).
Park, K. et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol. 17, 577–589 (2016).
Wu, Y. L. et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 18, 1454–1466 (2017).
Cross, D. A. et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 4, 1046–1061 (2014).
Watanabe, K. et al. The whole picture of first-line Osimertinib for EGFR mutation-positive advanced NSCLC: real-world efficacy, safety, progression pattern, and posttreatment therapy (Reiwa Study). JTO Clin. Res. Rep. 5, 100720 (2024).
Westover, D. et al. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann. Oncol. 29, i10–i19 (2018).
Nishiyama, A. et al. MET amplification results in heterogeneous responses to osimertinib in EGFR-mutant lung cancer treated with erlotinib. Cancer Sci. 111, 3813–3823 (2020).
Donati, B., Lorenzini, E. & Ciarrocchi, A. BRD4 and cancer: going beyond transcriptional regulation. Mol. Cancer 17, 164 (2018).
Wang, J. et al. BRD4-IRF1 axis regulates chemoradiotherapy-induced PD-L1 expression and immune evasion in non-small cell lung cancer. Clin. Transl. Med. 12, e718 (2022).
Wu, S. Y. & Chiang, C. M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 282, 13141–13145 (2007).
Shi, J. & Vakoc, C. R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 54, 728–736 (2014).
Devaiah, B. N. et al. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 23, 540–548 (2016).
Yin, M. et al. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat. Commun. 11, 1833 (2020).
Liu, C. et al. The novel BET degrader, QCA570, is highly active against the growth of human NSCLC cells and synergizes with osimertinib in suppressing osimertinib-resistant EGFR-mutant NSCLC cells. Am. J. Cancer Res. 12, 779–792 (2022).
Rong, X. et al. Molecular mechanisms of tyrosine kinase inhibitor resistance induced by membranous/cytoplasmic/nuclear translocation of epidermal growth factor receptor. J. Thorac. Oncol. 14, 1766–1783 (2019).
Porcelli, L. et al. The EGFR pathway regulates BCRP expression in NSCLC cells: role of erlotinib. Curr. Drug Targets 15, 1322–1330 (2014).
Dittmann, K. et al. New roles for nuclear EGFR in regulating the stability and translation of mRNAs associated with VEGF signaling. PLoS ONE 12, e0189087 (2017).
Zhang, Y. et al. Potential role of S-palmitoylation in cancer stem cells of lung adenocarcinoma. Front Cell Dev. Biol. 9, 734897 (2021).
Aicart-Ramos, C., Valero, R. A. & Rodriguez-Crespo, I. Protein palmitoylation and subcellular trafficking. Biochim. Biophys. Acta. 1808, 2981–2994 (2011).
Ali, A. et al. Fatty acid synthase mediates EGFR palmitoylation in EGFR mutated non-small cell lung cancer. EMBO Mol. Med. 10, e8313 (2018).
Runkle, K. B. et al. Inhibition of DHHC20-mediated EGFR palmitoylation creates a dependence on EGFR signaling. Mol. Cell 62, 385–396 (2016).
Kwon, H. et al. Flotillin-1 palmitoylation turnover by APT-1 and ZDHHC-19 promotes cervical cancer progression by suppressing IGF-1 receptor desensitization and proteostasis. Cancer Gene Ther. 30, 302–312 (2023).
Bian, J. et al. Identification and prognostic biomarkers among ZDHHC4/12/18/24, and APT2 in lung adenocarcinoma. Sci. Rep. 14, 522 (2024).
Xu, Y., Johansson, M. & Karlsson, A. Human UMP-CMP kinase 2, a novel nucleoside monophosphate kinase localized in mitochondria. J. Biol. Chem. 283, 1563–1571 (2008).
Wu, W. et al. Endothelial Gata6 deletion reduces monocyte recruitment and proinflammatory macrophage formation and attenuates atherosclerosis through Cmpk2-Nlrp3 pathways. Redox Biol. 64, 102775 (2023).
Kim, H. P. et al. Lapatinib, a dual EGFR and HER2 tyrosine kinase inhibitor, downregulates thymidylate synthase by inhibiting the nuclear translocation of EGFR and HER2. PLoS ONE 4, e5933 (2009).
Zhang, M. et al. A STAT3 palmitoylation cycle promotes T(H)17 differentiation and colitis. Nature 586, 434–439 (2020).
Midha, A., Dearden, S. & McCormack, R. EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: a systematic review and global map by ethnicity (mutMapII). Am. J. Cancer Res. 5, 2892–2911 (2015).
Le, X. et al. Dual EGFR-VEGF pathway inhibition: a promising strategy for patients with EGFR-mutant NSCLC. J. Thorac. Oncol. 16, 205–215 (2021).
Greig, S. L. Osimertinib: first global approval. Drugs 76, 263–273 (2016).
Zhong, Z. et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018).
Liu, N. et al. Post-translational modifications of BRD4: therapeutic targets for tumor. Front Oncol. 12, 847701 (2022).
Criscione, S. W. et al. The landscape of therapeutic vulnerabilities in EGFR inhibitor osimertinib drug tolerant persister cells. NPJ Precis. Oncol. 6, 95 (2022).
Bazzani, L. et al. PGE2/EP3/SRC signaling induces EGFR nuclear translocation and growth through EGFR ligands release in lung adenocarcinoma cells. Oncotarget 8, 31270–31287 (2017).
Wang, S. C. & Hung, M. C. Cytoplasmic/nuclear shuttling and tumor progression. Ann. N. Y. Acad. Sci. 1059, 11–15 (2005).
Wei, H., Zhu, Z. & Lu, L. Inhibition of EGFR nuclear shuttling decreases irradiation resistance in HeLa cells. Folia Histochem. Cytobiol. 55, 43–51 (2017).
Wang, S. C. & Hung, M. C. Nuclear translocation of the epidermal growth factor receptor family membrane tyrosine kinase receptors. Clin. Cancer Res. 15, 6484–6489 (2009).
Fhu, C. W. & Ali, A. Protein lipidation by palmitoylation and myristoylation in cancer. Front Cell Dev. Biol. 9, 673647 (2021).
Ivaldi, C. et al. Proteomic analysis of S-acylated proteins in human B cells reveals palmitoylation of the immune regulators CD20 and CD23. PLoS ONE 7, e37187 (2012).
Chen, B. et al. Protein lipidation in cell signaling and diseases: function, regulation, and therapeutic opportunities. Cell Chem. Biol. 25, 817–831 (2018).
Kadry, Y. A., Lee, J. Y. & Witze, E. S. Regulation of EGFR signalling by palmitoylation and its role in tumorigenesis. Open Biol. 11, 210033 (2021).
Tian, L. et al. Distinct acyl protein transferases and thioesterases control surface expression of calcium-activated potassium channels. J. Biol. Chem. 287, 14718–14725 (2012).
Zhu, J. et al. Nuclear epidermal growth factor receptor (nEGFR) in clinical treatment. Heliyon 10, e40150 (2024).
Acknowledgements
We sincerely appreciate Prof. Yin Mingzhu for their invaluable guidance during the project initiation phase, whose expert advice significantly contributed to the feasibility assessment of this research. This work was supported by Hunan Provincial Natural Science Foundation of China (No.: 2023JJ30970), Changsha Municipal Natural Science Foundation (No.: kq2208396), National Natural Science Foundation of China (No.: 82203694) and Beijing Xisike Clinical Oncology Research Foundation (No.: Y-2024AZ(EGFR)MS-0141).
Author information
Authors and Affiliations
Contributions
Guarantor of integrity of the entire study: Heng Zhang. Study concepts: Wolong Zhou, Shaoqiang Wang. Study design: Heng Zhang, Shaoqiang Wang. Definition of intellectual content: Heng Zhang. Literature research: Zhenyu Zhang, Linfeng Li, Hang Lin. Experimental studies: Zhenyu Zhang, Linfeng Li, Jiebo Zhu, Hang Lin. Data acquisition: Zhenyu Zhang, Linfeng Li, Jiebo Zhu and Hang Lin. Data analysis: Wolong Zhou, Heng Zhang. Statistical analysis: Wolong Zhou, Shaoqiang Wang. Manuscript preparation: Wolong Zhou, Shaoqiang Wang, Heng Zhang. Manuscript editing: Wolong Zhou, Shaoqiang Wang, Heng Zhang. Manuscript review: Heng Zhang.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, W., Wang, S., Zhang, Z. et al. Restoration of Osimertinib sensitivity in lung cancer through BRD4 inhibitor-mediated depalmitoylation of mutant EGFR via APT1. npj Precis. Onc. 9, 305 (2025). https://doi.org/10.1038/s41698-025-01048-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01048-8
