
Abstract
MYSM1, a deubiquitinating enzyme, is markedly overexpressed in lung adenocarcinoma (LUAD) and correlates with advanced disease stage and poor prognosis. Functional assays demonstrated that MYSM1 promotes LUAD cell proliferation and migration in vitro and in vivo. Mechanistically, MYSM1 interacts with TRAF2 and TRAF3 via its SWIRM domain, removing K63-linked polyubiquitin chains to decrease TRAF2–TRAF3 complex stability. This process impairs NIK degradation and sustains non-canonical NF-κB activation while simultaneously promoting MAPK (p38, JNK) signal pathway. Rescue experiments indicate that inhibiting TRAF2 or TRAF3 can attenuate MYSM1-induced activation of the MAPK pathway. These findings identify MYSM1 as a novel regulator of TRAF2-TRAF3 complex stability and a potential prognostic biomarker and therapeutic target in LUAD.
Similar content being viewed by others

Introduction
According to the Global Lung Cancer Database (GLOBOCAN), lung cancer ranks first in incidence and mortality among men, and is the second most common malignancy after breast cancer—but the leading cause of cancer-related death—among women1. Globally, approximately 2 million new lung cancer cases are diagnosed each year, resulting in about 1.76 million deaths2. Lung cancer is classified into two major pathological types: non-small cell lung cancer (NSCLC), accounting for ~85% of cases, and small cell lung cancer (SCLC), representing15%3 NSCLC comprises several histological subtypes, including adenocarcinoma (~40%), squamous cell carcinoma (~25%), and large cell carcinoma (~10%), with adenocarcinoma being the most prevalent. Current treatment strategies for lung cancer—surgery, chemotherapy, radiotherapy, and their combinations—are often hindered by therapy resistance and disease progression, contributing to a dismal 5-year survival rate of only 10–20% in most countries4.
In recent decades, the emergence of precision oncology, exemplified by molecularly targeted therapies and immune checkpoint inhibitors, has improved survival for subsets of advanced lung cancer patients5. Targeted therapies, in particular, have deepened our understanding of disease biology and revealed oncogenic driver alterations that serve as therapeutic targets6,7. High-throughput sequencing has identified numerous molecular alterations in lung adenocarcinoma, some of which are actionable, marking a new era of precision medicine for this disease8. Nonetheless, additional molecular targets are urgently needed to expand personalized treatment options and improve outcomes for diverse patient populations.
Ubiquitination is a key regulatory mechanism for many signaling pathways, and its dysregulation is closely linked to cancer initiation, progression, and metastasis9. In human cells, approximately 100 deubiquitinating enzymes (DUBs) function in concert with E3 ubiquitin ligases to dynamically modulate ubiquitin signaling by removing ubiquitin from specific substrates10. The balance between ubiquitination and de-ubiquitination is critical for adapting to cellular signals and stress. DUBs are increasingly recognized as modulators of oncogenic pathways, with individual enzymes influencing multiple cancer hallmarks—sometimes in both tumor-promoting and tumor-suppressing capacities11,12. Comprehensive investigation of DUBs in relevant cancer models will facilitate the development of DUB-targeted therapies for specific patient subgroups11. In this context, identifying DUBs with functional roles in lung adenocarcinoma and elucidating their molecular mechanisms may provide novel therapeutic opportunities13,14,15.
Histone H2A was the first identified ubiquitinated protein and remains the most ubiquitinated nuclear protein16. The lysine 119 (K119) site is the predominant mono-ubiquitination site, present on ~10% of nucleosomal H2A17. De-ubiquitination at H2A K119 is catalyzed primarily by BAP1, USP16, and MYSM1 (Myb-like SWIRM and MPN domain-containing protein 1). MYSM1 enhances chromatin accessibility by specifically removing H2A K119 mono-ubiquitination, thereby promoting transcription18. Beyond chromatin remodeling, MYSM1 has been implicated in hematopoiesis, immune responses, and host defense against infection19,20,21. Emerging evidence suggests that MYSM1 also plays diverse roles in tumorigenesis: it suppresses colorectal cancer progression via H2A de-ubiquitination–mediated activation of the miR-200/CHH1 axis22, promotes prostate cancer progression by synergizing with androgen receptor signaling23, and sensitizes triple-negative breast cancer cells to cisplatin by inactivating the RSK3–phospho-BAD pathway24. However, the functional role and molecular mechanisms of MYSM1 in lung adenocarcinoma remain largely unknown. Addressing this gap is of both scientific and clinical importance, as it may reveal novel regulatory pathways and therapeutic targets for this aggressive cancer subtype.
Results
MYSM1 is upregulated in lung adenocarcinoma and predicts unfavorable patient outcomes
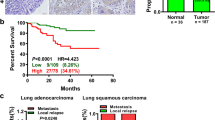
To investigate the potential role of MYSM1 in lung adenocarcinoma (LUAD), we first compared its expression in tumor versus normal tissues using publicly available transcriptomic datasets. Analysis of the GSE117049 dataset from the GEO database and TCGA LUAD data revealed significantly elevated MYSM1 mRNA levels in LUAD tissues compared with matched normal lung tissues (Fig. 1A). To assess clinical relevance, we performed survival analysis using both the GEPIA platform and the Kaplan–Meier Plotter. In both datasets, LUAD patients with high MYSM1 expression had significantly reduced overall survival compared to those with low expression (Fig. 1B). To validate these findings at the protein level, we performed immunohistochemistry (IHC) on a tissue microarray containing 87 pairs of LUAD and adjacent non-tumorous lung tissues. MYSM1 immunoreactivity was markedly higher in tumor tissues compared to adjacent non-cancerous counterparts (Fig. 1C, D). H score analysis demonstrated statistically significant upregulation of MYSM1 in LUAD. Kaplan–Meier survival analysis based on IHC scores showed that high MYSM1 protein expression correlated with poorer prognosis (Fig. 1E). Clinicopathologic correlation analysis revealed significant associations between MYSM1 expression and patient age, histopathological grade, and T stage (Supplementary Table S4). Importantly, multivariate Cox regression analysis confirmed MYSM1 as an independent prognostic factor for LUAD (Supplementary Table S5). These findings indicate that MYSM1 is both overexpressed and clinically relevant as a prognostic marker in LUAD.

A EIF4A3 mRNA expression in LUAD and normal tissues in GSE116959 and TCGA data sets. B The relationship between MYSM1 expression and prognosis in patients with lung adenocarcinoma was analyzed online using GEPIA and the Kaplan–Meier Plotter website, and Kaplan–Meier analyses of the LUAD cohort based on univariate survival analyses were performed using the log-rank statistical test. C Representative IHC images of MYSM1 expression of high expression and low expression in LUAD tissues and normal tissues. Scale bars: 300 μm (overview), 200 μm (detail). D IHC score of MYSM1 on tissue microarrays for histochemical staining of cancer and normal tissues. E Kaplan–Meier survival curves illustrating the association between MYSM1 expression levels and overall survival in the LUAD patient cohort. Survival differences were assessed using univariate analysis with the log-rank test. F Protein expression levels of MYSM1 detected by Western blotting in the normal bronchial epithelial cell line (BEAS-2B) and four lung adenocarcinoma cell lines (H1299, PC9, A549, H1975). All data are presented as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
MYSM1 promotes proliferation of LUAD cells in vitro
To examine the functional role of MYSM1, we first profiled its expression in a panel of LUAD cell lines (H1299, PC9, H1975, A549) and the immortalized bronchial epithelial cell line BEAS-2B. MYSM1 protein levels were highest in H1299, moderately high in PC9, and lowest in A549 (Fig. 1F). Then, we designed four siRNA sequences targeting MYSM1 and validated knockdown efficiency by Western blotting in H1299 and PC9 cells, selecting the most efficient sequence for lentiviral shRNA construction (Supplementary Fig. A, B). In parallel, a MYSM1 overexpression lentiviral vector was generated for A549 cells. After puromycin selection, stable knockdown (H1299, PC9) and overexpression (A549) lines were established and validated (Fig. 2A). On this basis, we conducted a number of experiments to assess the impact of MYSM1 on the growth potential of lung adenocarcinoma cells using in vitro tests. CCK-8 assays revealed that MYSM1 knockdown significantly decreased cell proliferation in H1299 and PC9, whereas MYSM1 overexpression enhanced proliferation in A549 (Fig. 2B). The EdU assay demonstrated that knockdown of MYSM1 in H1299 and PC9 cell lines decreased the proportion of proliferating cells in lung adenocarcinoma cells (Fig. 2C). Clone formation assays, on the other hand, confirmed that knockdown of MYSM1 in H1299 and PC9 cell lines decreased the number of clonal clusters of lung adenocarcinoma cells, while overexpression of MYSM1 in A549 cell line increased the number of clonal clusters of lung adenocarcinoma cells (Fig. 2D). To minimize the likelihood of off-target effects from a single siRNA sequence, we performed a rescue experiment. MYSM1-knockdown PC9 and H1299 cells were transfected with siRNA-resistant MYSM1 overexpression plasmids. Subsequent CCK-8, EdU, and colony formation assays demonstrated that the reduced proliferative capacity observed following MYSM1 knockdown was reversed upon MYSM1 re-expression, confirming the specificity of the knockdown effect (Supplementary Fig. 1C–E). To verify the effect of MYSM1 on the proliferative capacity of lung adenocarcinoma cells in the same cell lines, we constructed MYSM1 overexpressing H1299 and PC9 cell lines. The results of CCK-8 assay showed that overexpression of MYSM1 in H1299 and PC9 cell lines was able to increased the proliferation rate of lung adenocarcinoma cells (Supplementary Fig. 1F). The EdU assay demonstrated that overexpression of MYSM1 in H1299 and PC9 cell lines increased the proportion of proliferating cells in lung adenocarcinoma cells (Supplementary Fig. 1G). And clone formation assays confirmed that overexpression of MYSM1 in H1299 and PC9 cell lines increased the number of clonal clusters of lung adenocarcinoma cells (Supplementary Fig. 1H). Similarly, we performed the same experiments in the A549 cell line overexpressing MYSM1 and obtained the same results (Fig. 2B–D). These results demonstrate that MYSM1 positively regulates LUAD cell proliferation.

A Efficiency of MYSM1 knockdown and overexpression of lentivirus in different lung adenocarcinoma cell lines (H1299, PC9, and A549). B CCK8 experiment was performed to detect the rate of cell proliferation after MYSM1 knockdown in PC9 and H1299 cells and MYSM1 overexpression in A549 cells. C EdU assay was performed to confirm the change of the proliferation ability after MYSM1 knockdown in PC9 and H1299 cells and MYSM1 overexpression in A549 cells. Scale bars: 100 μm. D Clone formation experiment was performed to detect the number of clonal clusters of forming cells after MYSM1 knockdown in PC9 and H1299 cells and MYSM1 overexpression in A549 cells. All data are presented as the mean ± SD. N = 3. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
MYSM1 enhances migration and epithelial-mesenchymal transition (EMT) in LUAD cells
Given the association of MYSM1 with aggressive clinical features, we examined its effect on cell migration. And by transwell migration assay, we found that in lung adenocarcinoma, knockdown of MYSM1 decreased the number of H1299 and PC9 cells migrating to the lower compartment (Fig. 3A, B). Subsequently, we went on to confirm that the decreased migratory ability of lung adenocarcinoma cells was due to knockdown of MYSM1 by transfection of MYSM1 overexpression plasmid in MYSM1 knockdown H1299 and PC9 cells (Supplementary Fig. 2A). Subsequently, we constructed MYSM1 overexpression cell models in the same H1299 and PC9 cells. By cell scratch assay, we found that overexpression of MYSM1 increased the migration rate of lung adenocarcinoma cells in H1299, PC9, and A549 cell lines. And by transwell migration assay, we found that in lung adenocarcinoma, overexpression of MYSM1 increased the number of H1299, PC9, and A549 cells migrating to the lower compartment (Supplementary Fig. 2C and Fig. 3C). The majority of cancer patients do not succumb to the original tumor, but rather to metastases that harm essential organs. Metastases occur when cancer cells detach from the primary tumor and spread to surrounding healthy tissues. The metastatic process involves a critical multistage mechanism known as Epithelial–Mesenchymal transition (EMT), which allows tumor cells to disseminate from their original tumor site to other parts of the body25. EMT is significantly associated with the development of tumors, their progression, metastasis, and resistance to drugs26,27,28. Western blot analysis revealed that MYSM1 knockdown upregulated the epithelial adhesion molecule E-cadherin while reducing mesenchymal markers N-cadherin, Vimentin, and EMT-inducing transcription factors Snail, Slug, and Twist. In contrast, MYSM1 overexpression reversed this pattern, downregulating E-cadherin and increasing all mesenchymal and transcriptional EMT markers (Fig. 3D and Supplementary Fig. 2B, D). qRT-PCR analyses confirmed that these protein-level changes were mirrored at the transcript level (Fig. 3E). Together, these results confirm that MYSM1 promotes the migration and EMT of lung adenocarcinoma cells.

Wound healing and transwell migration experiment without matrigel were performed to identify the change of migration ability after MYSM1 knockdown in H1299 cells (A) and PC9 cells (B) and MYSM1 overexpression in A549 cells (C). Scale bars of the Wound healing experiment: 200 μm. Scale bars of the transwell migration experiment: 100 μm. D Changes in the expression of EMT-associated indicators E-Cadherin, N-Cadherin, Vimentin, Snail, Slug, and Twist after knockdown or overexpression of MYSM1 in lung adenocarcinoma cells. E Changes in relative RNA expression levels of E-Cadherin, N-Cadherin, Vimentin, Snail, Slug, and Twist after knockdown or overexpression of MYSM1 in lung adenocarcinoma cells were detected by qRT-PCR. All data are presented as the mean ± SD. N = 3. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
MYSM1 promotes LUAD tumor growth in vivo
To validate the oncogenic role of MYSM1 under physiological conditions, we employed subcutaneous xenograft models in immunodeficient nude mice. Implantation of MYSM1-knockdown PC9 cells produced tumors with significantly reduced volumes and growth rates, and lower final weights compared to shRNA control tumors (Fig. 4A–C). Conversely, tumors derived from MYSM1-overexpressing A549 cells exhibited markedly increased volumes, accelerated growth kinetics, and greater final masses compared to vector controls (Fig. 4D–F). Next, we performed HE staining and immunohistochemical staining of tumors removed subcutaneously from nude mice. Histopathological analysis confirmed these growth differences: HE staining showed denser tumor cell populations in the MYSM1-overexpressing group, while IHC staining for the proliferation marker Ki-67 revealed significantly reduced staining intensity and positive cell percentages in MYSM1-knockdown tumors, and significantly increased Ki-67 indices in MYSM1-overexpressing tumors (Fig. 4G, H). These results confirm that MYSM1 functions as a positive regulator of LUAD tumor growth in vivo.

A Representative images of tumors in nude mice 25 days after subcutaneous injection of MYSM1 knockdown lung adenocarcinoma cells. The volume (B) and the weight (C) were lower for xenograft tumors with MYSM1 knockdown than for xenograft tumors with negative control. D Representative images of tumors in nude mice 25 days after subcutaneous injection of MYSM1 overexpression lung adenocarcinoma cells. The volume (E) and the weight (F) were higher for xenograft tumors with MYSM1 overexpression than for xenograft tumors with a negative control. G Representative images of HE staining and IHC staining of MYSM1, TRAF2, TRAF3 and Ki-67 in xenograft tumors. Scale bars: 50 μm. H Positive cell rate statistics of immunohistochemistry of tumor MYSM1, TRAF2, TRAF3, and Ki-67 in nude mice of different treatment groups. All data are presented as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
MYSM1 disrupts TRAF2–TRAF3 complex formation via K63-linked de-ubiquitination and upregulates expression of TRAF2 and TRAF3
To identify molecular interactors that could mediate MYSM1’s oncogenic activity, we performed immunoprecipitation–liquid chromatography–tandem mass spectrometry (IP–LC-MS/MS) in LUAD cells ectopically expressing MYSM1. Comparative proteomic analysis revealed 91 proteins significantly enriched in MYSM1 immunoprecipitates compared with IgG controls (Fig. 5A, Supplementary Table S6). Notably, TRAF2 and TRAF3—key adapters in NF-κB signaling and known to form functional complexes—were among the top hits, consistent with prior reports of MYSM1–TRAF3 or MYSM1–TRAF6 interactions29,30,31. Based on these findings and literature evidence, we hypothesized that MYSM1 physically interacts with TRAF2 and TRAF3 in LUAD, potentially modulating their complex stability and downstream signaling. Endogenous co-immunoprecipitation in H1299 cells (high MYSM1 expression) confirmed that MYSM1 binds both TRAF2 and TRAF3 (Fig. 5B). Exogenous co-immunoprecipitation in HEK-293T cells co-transfected with Flag–MYSM1 and HA–TRAF2 or HA–TRAF3 similarly validated these interactions (Fig. 5C). As a DUB, MYSM1 removes ubiquitin chains from target substrates. We hypothesized that MYSM1 might regulate them post-translationally. Given previous reports that MYSM1 can deubiquitinate TRAF3 and TRAF6, we hypothesized that MYSM1 similarly deubiquitinates TRAF2 and TRAF3 in LUAD. To directly test this, we co-transfected 293 T cells with Flag–MYSM1, His–ubiquitin, and HA–TRAF2 or HA–TRAF3, followed by immunoprecipitation. MYSM1 overexpression markedly reduced ubiquitin conjugation on both TRAF2 and TRAF3 (Fig. 5D). To determine the ubiquitin chain linkage targeted by MYSM1, we overexpressed either wild-type ubiquitin or single-lysine–only mutants (K6, K11, K27, K29, K33, K48, K63). Only K63-linked ubiquitin supported MYSM1-mediated de-ubiquitination of TRAF2 and TRAF3 (Fig. 5E, G). Conversely, K63R ubiquitin abrogated de-ubiquitination, whereas all other lysine-to-arginine mutants retained MYSM1 responsiveness (Fig. 5F, H), indicating that MYSM1 specifically removes K63-linked ubiquitin chains. Because de-ubiquitination of TRAF2 and TRAF3 has been implicated in regulating TRAF2–TRAF3 complex stability32, we examined whether MYSM1 affects this association. Co-immunoprecipitation revealed that MYSM1 overexpression reduced TRAF2–TRAF3 complex formation, whereas MYSM1 knockdown enhanced their interaction (Fig. 5I). Reciprocal knockdowns demonstrated that TRAF2 depletion diminished MYSM1–TRAF3 binding, and TRAF3 depletion diminished MYSM1–TRAF2 binding (Fig. 5J–L), suggesting that MYSM1 engages each protein in a non–mutually exclusive manner. Subsequently, we further explored the specific structural domains of MYSM1 that interact with TRAF2 and TRAF3 in lung adenocarcinoma, and we constructed three truncated bodies based on the three structural domains of MYSM1, SANT, SWIRM, and MPN, and transfected the full-length plasmid and the three truncated body plasmids of MYSM1, TRAF2, and TRAF3, respectively, into 293 T cells, and performed immuno co-precipitation, and the results showed that MYSM1 was interacting with TRAF2 and TRAF3 through its SWIRM structural domain(Fig. 5M). Meanwhile, we also observed MYSM1 knockdown in LUAD cells decreased TRAF2 and TRAF3 protein levels, whereas MYSM1 overexpression increased their abundance (Supplementary Fig. 3A). Consistent with this, qRT-PCR analysis showed that neither knockdown nor overexpression of MYSM1 altered TRAF2 or TRAF3 mRNA levels in LUAD cells (Supplementary Fig. 2E). Immunohistochemical analysis of xenograft tumor tissues yielded results consistent with the in vivo data, with MYSM1 expression positively correlating with TRAF2 and TRAF3 protein abundance (Fig. 4G). To assess whether MYSM1 influences TRAF2 and TRAF3 stability via the ubiquitin–proteasome pathway, MYSM1-depleted LUAD cells were treated with the proteasome inhibitor MG132 (10 μM). MG132 effectively restored TRAF2 and TRAF3 protein levels reduced by MYSM1 knockdown (Supplementary Fig. 3B). Furthermore, cycloheximide chase assays demonstrated that MYSM1 overexpression significantly prolonged the half-life of TRAF2 and TRAF3, as reflected by slower degradation rates in the presence of the protein synthesis inhibitor cycloheximide (Supplementary Fig. 3C). These findings indicate that MYSM1 disrupts TRAF2–TRAF3 complex formation and increase expression of TRAF2 and TRAF3.

A Venn diagram showed 91 proteins present in the IP group but not in the IgG group in the mass spectrometry analyses, of which MYSM1, TRAF2, and TRAF3 were all present. B Immunoprecipitation was performed in H1299 with high MYSM1 expression to detect MYSM1 interactions with TRAF2 and TRAF3. C Flag-tagged MYSM1 plasmid and HA-tagged TRAF2/TRAF3 plasmid were transfected in 293 T cells, and immunoprecipitation was performed to detect the interactions of MYSM1 with TRAF2 and TRAF3. D The interaction between MYSM1, TRAF2/3 and ubiquitin was assessed by immunoprecipitation after transfection of MYSM1, TRAF2/3, and ubiquitin granules in 293 T cells. E Western blot (WB) analysis of WCL and anti HA pull-down products derived from 293 T cells transfected with Flag-MYSM1, HA-TRAF2 and K-only ubiquitin mutants. Where indicated, 10 μM MG132 was added for 8 h before harvesting the cells. F WB analysis of WCL and anti HA pull-down products derived from 293 T cells transfected with Flag-MYSM1, HA-TRAF2 and the indicated ubiquitin KR (Lys to Arg) mutants. Where indicated, 10 μM MG132 was added for 8 h before harvesting the cells. G WB analysis of WCL and anti HA pull-down products derived from 293 T cells transfected with Flag-MYSM1, HA-TRAF3 and K-only ubiquitin mutants. Where indicated, 10 μM MG132 was added for 8 h before harvesting the cells. H WB analysis of WCL and anti HA pull-down products derived from 293 T cells transfected with Flag-MYSM1, HA-TRAF3 and the indicated ubiquitin KR (Lys to Arg) mutants. Where indicated, 10 μM MG132 was added for 8 h before harvesting the cells. I WB analysis of WCL and anti HA or flag pull-down products derived from 293 T cells transfected with HA-TRAF2, Flag-TRAF3 and Myc-MYSM1 protein constructs. 30 h post-transfection, cells were treated with 10 μM MG132 for 8 h before harvesting. J WB analysis of WCL and anti HA pull-down products derived from control and MYSM1 knockdown 293 T cells transfected with HA-TRAF2 and Flag-TRAF3 protein constructs. 30 h post-transfection, cells were treated with 10 μM MG132 for 8 h before harvesting. K WB analysis of WCL and anti HA pull-down products derived from control and TRAF2 knockdown 293 T cells transfected with HA-TRAF3 and Flag- MYSM1 protein constructs. 30 h post-transfection, cells were treated with 10 μM MG132 for 8 h before harvesting. L WB analysis of WCL and anti HA pull-down products derived from control and TRAF3 knockdown 293 T cells transfected with HA-TRAF2 and Flag-MYSM1 protein constructs. 30 h post-transfection, cells were treated with 10 μM MG132 for 8 h before harvesting. M Truncated forms of MYSM1 were constructed according to its functional domains and co-immunoprecipitation assay of truncated forms of MYSM1-Flag and TRAF2/3-HA in the whole cell lysates of 293 T cells.
MYSM1 activates non-classical NF-κB/P38/JNK signaling pathway in lung adenocarcinoma
In order to investigate through which signaling pathway MYSM1 affects tumor development in lung adenocarcinoma, we performed transcriptomics in lung adenocarcinoma after knockdown of MYSM1, and then after comparing the genes that underwent up-regulation and down-regulation after knockdown of MYSM1 and pathway enrichment, we found that among the tumor-related signaling pathways, the correlation of AGE-RAGE ranked the most highly (Fig. 6A). On the one hand, the KEGG data site and related Literature show that the NF-κB signaling pathway and the JNK/P38 signaling pathway in the MAPK signaling pathway are located downstream of the AGE-RAGE signaling pathway33,34, and on the other hand, the KEGG data site shows that the MYSM1 downstream genes TRAF2 and TRAF3 are located upstream of the non-classical NF-κB/P38/JNK signaling pathways and have been reported to be closely related to these pathways35 (Fig. 6B). On this basis, we hypothesized that MYSM1 in lung adenocarcinoma may promote lung adenocarcinoma development by affecting the non-classical NF-κB/P38/JNK signaling pathway. By WB validation, we found that knockdown of MYSM1 in lung adenocarcinoma cells could result in down-regulation of the expression of key molecules in the non-classical NF-κB pathway, NIK, phosphorylated IKKα, phosphorylated NF-κB P100, and NF-κB P52, and also of ASK1, phosphorylated P38 in the P38 signaling pathway, as well as MEKK1 and phosphorylated JNK in the JNK signaling pathway (Fig. 6C and Supplementary Fig. 3D). Together, these experimental results also confirm that MYSM1 can activate the non-classical NF-κB/P38/JNK signaling pathway in lung adenocarcinoma.

A Lung adenocarcinoma cells after MYSM1 knockdown were subjected to transcriptomic sequencing, and KEGG pathway enrichment was performed on the differentially expressed genes obtained by sequencing. Statistical significance is indicated by different colors. The size of the circle represents the number of genes. B KEGG pathway enrichment sites showed that the AGE-RAGE signaling pathway contains TRAF2/3 and non-classical NF-κB signaling pathways, the P38 signaling pathway and the JNK signaling pathway. C Altered key components in the non-canonical NF-κB, p38, and JNK signaling pathways following MYSM1 knockdown or overexpression in lung adenocarcinoma cells (H1299, PC9 and A549). D Evaluation of the knock-down efficiency of TRAF2 and TRAF3 four small interfering sequences by Western blotting in A549 cell line. E. The effect of TRAF2/TRAF3 knockdown on the action of MYSM1 on P38/JNK signaling pathway was tested by rescue assay. F. The effect of TRAF2/TRAF3 knockdown on the action of MYSM1 on the EMT-related molecules was tested by rescue assay.
MYSM1 promotes lung adenocarcinoma proliferation and migration through TRAF2 and TRAF3
We first designed four small interfering RNA (siRNA) sequences targeting TRAF2 and TRAF3, respectively. Western blot (WB) analysis was used to assess knockdown efficiency, and the most effective sequences for each target were selected for subsequent functional assays (Fig. 6D). To evaluate the impact on EMT, WB analysis of EMT-related markers was performed. Silencing TRAF2 or TRAF3 reversed the MYSM1 overexpression–induced activation of P38/JNK pathway, upregulation of mesenchymal markers (N-cadherin, Vimentin, Snail, Slug, Twist) and downregulation of the epithelial marker E-cadherin (Fig. 6E, F). We next examined whether TRAF2 and TRAF3 are required for MYSM1-driven proliferation of LUAD cells. CCK-8 assays demonstrated that knockdown of either TRAF2 or TRAF3 abrogated the MYSM1-induced increase in proliferation rate (Fig. 7A). Consistently, EdU incorporation assays showed that depletion of TRAF2 or TRAF3 reduced the proportion of cells in the proliferative phase elevated by MYSM1 overexpression (Fig. 7B). Colony formation assays further confirmed that silencing TRAF2 or TRAF3 significantly diminished the increase in clonogenic capacity conferred by MYSM1 overexpression (Fig. 7C). We then assessed cell motility. Scratch wound healing assays revealed that TRAF2 or TRAF3 knockdown attenuated the MYSM1-induced acceleration of wound closure. Similarly, Transwell migration assays confirmed that depletion of TRAF2 or TRAF3 reduced the number of migrating cells increased by MYSM1 overexpression (Fig. 7D). Finally, in vivo xenograft experiments were performed in nude mice. Knockdown of TRAF2 or TRAF3 in MYSM1-overexpressing LUAD cells significantly reduced tumor growth rate, final tumor volume, and tumor weight compared to MYSM1-overexpressing controls (Fig. 7E–G). Histological analysis by hematoxylin–eosin (HE) staining further supported the conclusion that TRAF2 and TRAF3 are required for the proliferative advantage conferred by MYSM1 in vivo (Supplementary Fig. 3E). Collectively, these findings demonstrate that MYSM1 promotes LUAD cell proliferation, migration, and EMT in vitro and tumor growth in vivo through TRAF2- and TRAF3-dependent mechanisms.

A CCK8 was performed to confirm the effect of knockdown of TRAF2 and TRAF3 on the upregulation of MYSM1 for the proliferative capacity of lung adenocarcinoma in A549 cells. B EdU assay was performed to confirm the effect of knockdown of TRAF2 and TRAF3 on the upregulation of MYSM1 for the proliferative capacity of lung adenocarcinoma in A549 cells. Scar bars: 100 μm. C Clone formation experiments was performed to confirm the effect of knockdown of TRAF2 and TRAF3 on the upregulation of MYSM1 for the proliferative capacity of lung adenocarcinoma in A549 cells. D Wound healing and transwell migration assay without matrigel of the rescue experiment were performed to identify the effect of knockdown of TRAF2 and TRAF3 on the upregulation of MYSM1 for the migration capacity of lung adenocarcinoma in A549 cell. Scale bars of Wound healing experiment: 200 μm. Scale bars of transwell migration experiment: 100 μm. E Representative images of tumors in nude mice 25 days after subcutaneous injection of MYSM1 overexpression and TRAF2 and TRAF3 knockdown lung adenocarcinoma cells. TRAF2/3 knockdown will reduce the increase in size (F) and weight (G) of xenograft tumors due to MYSM1 overexpression. All data are presented as the mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Discussion
Our study identifies the DUB MYSM1 as a novel regulator of TRAF2 and TRAF3 complex in lung adenocarcinoma (LUAD). We demonstrate that MYSM1 physically interacts with both TRAF2 and TRAF3, decrease the complex stability via de-ubiquitination, and thereby sustaining their protein levels. This promotes activation of the non-canonical NF-κB, p38, and JNK signaling pathways, collectively driving LUAD initiation and progression (Supplementary Fig. 3F). Clinically, MYSM1 expression was markedly elevated in LUAD tissues and correlated with unfavorable patient prognosis. High MYSM1 expression was significantly associated with age, histopathological grade, and T stage in LUAD patients, suggesting its potential as a biomarker of tumor aggressiveness. Functional in vitro and in vivo assays further confirmed that MYSM1 promotes both the proliferative and migratory capacities of LUAD cells, underscoring its critical role in tumor progression. Although we did not directly assess drug resistance or tumor initiation in this study, prior reports indicate that NF-κB signaling enhances anti-apoptotic programs, while JNK and p38 pathways mediate stress-adaptive responses36,37. These findings raise the possibility that MYSM1-driven destabilization of TRAF2/3 complex could elevate apoptotic thresholds and promote survival under cytotoxic or targeted therapy conditions. The potential contribution of MYSM1 to therapeutic resistance warrants further investigation. Because our current models utilized established LUAD cell lines and xenografts, we cannot infer a role for MYSM1 in tumor initiation. This limitation is acknowledged, and future work will employ early-lesion models and lung organoid systems to explore MYSM1’s function in the earliest stages of tumorigenesis.
By mass spectrometry analysis and combining with related literature, we hypothesized that MYSM1 may interact with TRAF2 and TRAF3 in lung adenocarcinoma. We further confirmed that MYSM1 can interact with TRAF2 and TRAF3 in lung adenocarcinoma by endogenous and exogenous immunoprecipitation experiments. Subsequently, we found that MYSM1 interacts with TRAF2 and TRAF3 proteins, and then destabilizes TRAF2/3 complex via k63-linked de-ubiquitination. We also found that the expression of TRAF2 and TRAF3 increased with the upregulation of MYSM1 expression. We hypothesized that the increase in free TRAF2 and TRAF3 may be due to the decreased stability of the TRAF2/3 complex (Supplementary Fig. 3F). In addition to this, we found that MYSM1 promotes the protein stability of TRAF2 and TRAF3 and prevents degradation by the proteasome. Emerging evidence indicates that K63-linked chains can act as “seeds” for forming branched ubiquitin chains, thus facilitating substrate recognition by the proteasome38. Considering the E3 ubiquitin ligase HUWE1 generates K48 branches on K63 chains formed by TRAF6, yielding K48-K63 branched chains39, thereby, removal of K63-linked polyubiquitin chains from TRAF2 and TRAF3 by MYSM1 increases these stability. This de-ubiquitination preserves TRAF2 and TRAF3 within the cell, prolonging its availability for signaling and shifting the balance toward sustained activation of downstream pathways. Consistent with our biochemical mapping, truncation analysis revealed that MYSM1 engages TRAF2 and TRAF3 via its SWIRM domain. This observation aligns with prior reports showing that the SWIRM domain mediates MYSM1’s interaction with TRAF family proteins, including the TRAF3/6 complex29. The SWIRM domain is known to facilitate protein–protein interactions and may serve as a structural scaffold that positions MYSM1 in proximity to ubiquitinated TRAF substrates. In the context of our findings, this domain-specific binding likely underlies MYSM1’s ability to selectively deubiquitinate TRAF2 and TRAF3. Together, these results suggest that the SWIRM domain is a critical determinant of MYSM1’s substrate specificity and functional impact in LUAD.
The TRAF (tumor necrosis factor receptor-associated factor) family, comprising TRAF 1 through 7, consists of cytoplasmic adapter proteins that modulate signaling pathways associated with various receptors, including those within the TNF-R superfamily, Toll-like receptors, NOD-like receptors, RIG-I-like receptors, and cytokine receptors. TRAF proteins serve dual roles as adapter proteins and E3 ubiquitin ligases, thereby influencing receptor signaling. This modulation results in the activation of both classical and non-classical nuclear factor-κB (NF-κB) pathways, as well as mitogen-activated protein kinase (MAPK) pathways, which encompass ERK1/2, JNK1/2, and p38, in addition to interferon regulatory factor (IRF) pathways, including IRF3, IRF5, and IRF7. Signaling pathways that utilize TRAF (TNF receptor-associated factor) play a crucial role in numerous essential cellular functions, encompassing survival, growth, differentiation, activation, and responses to stress across diverse cell types. Numerous signaling pathways that rely on TRAF have been linked to the development of cancer40,41. Combined with the transcriptome sequencing and pathway enrichment results after knockdown of MYSM1, we further hypothesized that MYSM1 activates non-classical NF-κB in the AGE-RAGE signaling pathway and JNK and P38 signaling pathways in the MAPK signaling pathway in lung adenocarcinoma. We acknowledge that there is substantial overlap and crosstalk between the classical and non-classical NF-κB pathways. Our transcriptome sequencing data indicate that MYSM1 influences the AGE–RAGE signaling pathway, within which both the classical and non-classical NF-κB cascades are embedded, suggesting that MYSM1 may also modulate the classical pathway. However, based on our mass spectrometry results and immunoprecipitation validation, we focused our mechanistic investigations on TRAF2 and TRAF3. These proteins are well-established upstream regulators of the non-classical NF-κB pathway, primarily through activation of its key signaling mediator, NIK. Therefore, in this study, we concentrated on elucidating MYSM1’s role in regulating the non-classical NF-κB pathway, while acknowledging the potential involvement of the classical pathway for future exploration. By detecting key molecular proteins in the pathway through WB experiments, we further confirmed that MYSM1 was able to activate JNK and P38 signaling pathways in non-classical NF-κB and MAPK signaling pathways in lung adenocarcinoma.
To test whether TRAF2/3 influence MYSM1’s chromatin-associated de-ubiquitinase activity, we performed rescue assays with TRAF2 or TRAF3 knockdown. WB for histone H2A Lys119 ubiquitination (H2AK119ub1) showed no detectable change upon TRAF2 or TRAF3 depletion, indicating that MYSM1’s H2A de-ubiquitinase activity is not measurably affected by TRAF2/3 in LUAD (Fig. 6F). Based on our experimental data and supporting literature, TRAF2 and TRAF3 can form a complex that regulates the non-canonical NF-κB pathway40. In contrast, within the MAPK pathways (p38 and JNK), TRAF2 and TRAF3 function independently, as knockdown of either protein impairs pathway activation42,43,44. In summary, when MYSM1 expression is low, the TRAF2/3 complex remains stable, preventing activation of NIK and keeping the non-canonical NF-κB pathway inactive. The levels of free TRAF2 and TRAF3 are also low, resulting in nonactive of the p38 and JNK pathways. In contrast, when MYSM1 expression is high, MYSM1 destabilizes TRAF2/3 complex via k63-linked de-ubiquitination, thereby activating NIK and the non-canonical NF-κB pathway. Concurrently, the abundance of free TRAF2 and TRAF3 increase, leading to activation of the p38 and JNK pathways (Supplementary Fig. 3F).
Finally, to elucidate the role of TRAF2 and TRAF3 in MYSM1-mediated signaling and LUAD progression, we conducted a series of functional and mechanistic assays. Our findings demonstrate that MYSM1 facilitates LUAD advancement by activating JNK and p38 signaling via TRAF2/3 through the non-canonical NF-κB and MAPK pathways. Nonetheless, this study has certain limitations that warrant consideration. First, as E3 ubiquitin ligases, the TRAF family shares a common RING structural domain, which also suggests that TRAF family proteins are involved in the process of ubiquitination44,45. In tumors, the coordination between ubiquitination and de-ubiquitination regulates ubiquitin substrates or signaling, which in turn has an impact on tumor development. According to the literature, the levels and signaling of IL-33R are mutually regulated by the K27-linked polyubiquitination and de-ubiquitination processes involving TRAF6 and USP38, which become critical for triggered inflammatory responses in the lungs and pulmonary fibrosis46. On the premise that MYSM1 can interact with TRAF2/TRAF3 in lung adenocarcinoma, as demonstrated in the previous stage, whether the two can act as DUBs and ubiquitinating enzymes, respectively, on the same ubiquitin substrates or signals, which in turn can coordinate their actions on signaling and regulation in tumors, also requires our further subsequent investigation. Second, to further delineate the structural determinants of MYSM1 function, future work should employ point mutations targeting key residues within the SWIRM domain to assess their impact on MYSM1’s interaction with TRAF2/3, its deubiquitinating activity, and downstream signaling. Such mutations could help distinguish whether the observed effects on MAPK and non-canonical NF-κB activation are strictly SWIRM-dependent or whether other regions contribute to complex formation and pathway modulation. This approach would also clarify whether MYSM1’s structural integrity is essential for its role in promoting LUAD progression. Third, knockout of MYSM1 by CRISPR/Cas9-related techniques should also be used to scientifically validate the effect of MYSM1 on lung adenocarcinoma development. Partial depletion by RNA interference leads to a moderate decrease in TRAF2/3 stability and a corresponding reduction in MAPK and non-canonical NF-κB activity, resulting in attenuated proliferation and migration, but with residual signaling that permits partial recovery over time. In contrast, genetic ablation of MYSM1 abolishes its deubiquitinating activity. The presence of residual MYSM1 in knockdown cells may allow for compensatory mechanisms—such as upregulation of alternative de-ubiquitinases—whereas knockout cells have limited capacity for compensation, leading to more profound and sustained biological effects.
As TRAF2 and TRAF3 also function as E3 ubiquitin ligases, MYSM1’s stabilization of TRAF2 and TRAF3 might affect the balance between substrate ubiquitination and signaling activation. We found that MYSM1 promoted the activation of both MAPK (JNK, P38) and non-canonical NF-κB pathways. Because TRAF2 and TRAF3 also act as E3 ubiquitin ligases, our data indicate that MYSM1 overexpression in LUAD shifts the equilibrium between substrate ubiquitination and signal output by increasing TRAF2 and TRAF3 while reducing TRAF2–TRAF3–cIAP complex assembly. Mechanistically, increased TRAF2 and TRAF3 maintains a substantial pool of TRAF2 and TRAF3 available for signaling even when complex formation is diminished. Reduced complex assembly, in turn, compromises NIK ubiquitination and proteasomal degradation, thereby sustaining non-canonical NF-κB (RelB/p52) activation. In parallel, the accumulation of free TRAF3 and TRAF2 supports independent activation of MAPK p38 and JNK pathways. Functionally, our rescue data suggest that although both cascades are engaged, MAPK signaling (p38/JNK) is the primary terminal determinant of MYSM1-dependent LUAD phenotypes. These MAPK branches exert strong effects on both proliferation and migration/EMT, with proliferation showing the larger effect size.
Our study identifies MYSM1 as a key oncogenic regulator in lung adenocarcinoma, functioning through direct interaction with TRAF2 and TRAF3. By catalyzing K63-linked de-ubiquitination, MYSM1 reduces TRAF2–TRAF3 complex formation, enhances TRAF2 and TRAF3 expression, and sustains activation of the non-canonical NF-κB and MAPK (p38, JNK) pathways. Clinically, MYSM1 overexpression correlates with aggressive disease features and poor prognosis in LUAD patients. While these findings establish MYSM1 as a promising therapeutic target, future work should be focused on the drug resistance, tumor initiation and point mutants establishment to define its signaling and oncogenic activities.
Methods
Samples collected from clinical sources
The lung adenocarcinoma (LUAD) tissue microarray (HLugA180Su11) was sourced from Shanghai Outdo Biotech Company. The tissue microarray contains 87 pairs of cancers and corresponding normal tissues, along with clinical and prognostic details of the associated patients, and was approved and cleared by the relevant ethical committee (SHYJS-CP-2206001). This part of the experimental manipulation follows the guidelines of the World Medical Association’s Declaration of Helsinki and the associated ethical standards.
Cultivation and treatment of cells
The cell lines BEAS-2B, H1299, A549, PC9, H1975, and 293 T utilized in this research were obtained from the cell repository of the Chinese Academy of Sciences and were subjected to STR identification. The cell lines were maintained in RPMI-1640 medium (Gibco) or Dulbecco’s Modified Eagle Medium (Gibco) within an incubator maintained at 37 °C and supplemented with 5% CO2. All media utilized contained 10% fetal bovine serum (FBS) by volume. PC9 stable cells and H1299 stable cells were transfected with Sh-NC and Sh-MYSM1 lentiviruses from Jikai Co., while A549 stable cells were transfected with overexpression-MYSM1 lentiviruses and vector lentiviruses from Jikai Co. Puromycin was employed to select the transfected cells over a period of 7 days. GenePharma Co., Ltd. provided the siRNA-MYSM1, siRNA-TRAF2, siRNA-TRAF3 and their respective negative controls. We used jetPRIME transfection reagent (Polyplus) to transiently transfect cells with siRNAs following the provided instructions, and conducted further experiments 48 h post-transfection. Supplementary Table S1 contains the siRNA sequences. Similarly, the plasmids involved in this experiment were all transfected with the jetPRIME co-transfection reagent to the appropriate mass into the corresponding cells and the relevant experiments were performed 48 h after transfection. A scrambled or non-targeting siRNA control was used in knockdown experiments, and an empty vector was used as a control in overexpression experiments.
Western blotting
Cellular lysis was performed using RIPA buffer (Beyotime), which was supplemented with 1% protease and phosphatase inhibitors, to facilitate the extraction of proteins. Proteins were isolated using an SDS-PAGE gel and then moved to a polyvinylidene difluoride membrane via an electrical transfer method. In order to mitigate nonspecific binding of antibodies, the membrane was subjected to a blocking procedure for 1 h using a 5% nonfat milk solution, prior to being incubated with primary antibodies overnight at a temperature of 4 °C. Following incubation with the appropriate secondary antibodies, the membranes were subsequently analyzed utilizing an enhanced chemiluminescence system.
Cell counting kit-8 (CCK-8) assay
Lentivirus-transfected cells were maintained in a 96-well plate and subjected to incubation periods of 1, 2, 3, and 4 days. Subsequently, CCK-8 (APExBIO, #K1018) was introduced to the cells. The absorbance was quantified at 450 nm utilizing a microplate reader following a 2 h incubation period in the absence of light. The experiment was carried out independently on three separate occasions.
5-Ethynyl-2’-deoxyuridine (EdU) assay
Cells transfected with an appropriate amount of lentivirus were seeded in a 96-well plate. After an overnight culture, Edu staining was performed utilizing the Edu in vitro kit (Beyotime, #C0075S), adhering to the specified protocols. Subsequently, the labeled cells were imaged using a fluorescence microscope. The experiment was carried out independently on three separate occasions.
Colony formation assay
In a six-well plate, an appropriate amount of H1299, A549, and PC9 cells transfected with lentivirus was inoculated. The cells were subjected to fixation using 4% paraformaldehyde for a duration of 30 min, after which they were stained with crystal violet for 20 min, following a culture period of 14 days. Subsequently, we washed the cells three times and counted the colonies formed. The experiment was carried out independently on three separate occasions.
Wound-healing assay
Cells that were transfected at an appropriate amount were plated in a six-well plate until they were fully grown in the wells. Subsequently, a linear incision was made in the cell monolayer utilizing the tip of a 10 mL pipette. Following three washes with phosphate-buffered saline, the wells were substituted with serum-free medium. The width of the wound was assessed and quantified using a microscope at the initial time point (0 h) and again after a duration of 24 h. The experiment was carried out independently on three separate occasions.
Transwell assay
Cells that had been transfected with the requisite amount were introduced into the upper chamber of the transwell apparatus, which contained a serum-free medium. Conversely, the lower chamber was supplemented with 700 mL of medium enriched with 20% FBS. The cells located in the lower chamber underwent fixation for a duration of 30 min, followed by a staining process lasting 20 min, after which they were visualized using a microscope after 2 days. The experiment was carried out independently on three separate occasions.
In vivo experiments
The female mice provided by VitoLiva Animal Company were randomly assigned to either test or comparison groups to create a xenograft tumor model. Each experimental group contained five mice (n = 5). The number was determined based on a priori power calculation (α = 0.05, 80% power) to detect a ≥ 30% difference in tumor size between groups, while also considering ethical guidelines to minimize animal use. Then, the transfected PC9 cells or A549 cells with the corresponding lentivirus were subcutaneously implanted in the right armpit area. Tumor sizes were assessed every 5 days, and their volumes were determined using the formula: V = (length × width²)/2. After 28 days, the volume and weight of each tumor were recorded and measured. Tumor volume and weight were measured by investigators blinded to group allocation to reduce assessment bias. The mice were randomly allocated and maintained under specific pathogen-free conditions throughout the study. All experiments were performed in accordance with the guidelines established by the Qilu Hospital of Shandong University Committee on Animal Care and Use. At the end of each in vivo experiment, the mice were euthanized by carbon dioxide (CO₂) inhalation in accordance with in animal care guidelines. The Medical Ethics Committee at Qilu Hospital of Shandong University granted approval and oversight for all conducted experiments (DWLL-2024-029).
Hematoxylin–eosin (HE) staining
Tumor specimens were fixed in 4% paraformaldehyde at room temperature for 24–48 h, dehydrated through graded ethanol series, cleared in xylene, and embedded in paraffin. Paraffin blocks were sectioned at a thickness of 4 μm using a microtome. Sections were deparaffinized in xylene, rehydrated through graded ethanol to distilled water, and stained with hematoxylin for 5 min. After rinsing in running tap water, sections were differentiated in 1% acid alcohol, blued in alkaline solution, and counterstained with eosin for 2 min. Finally, sections were dehydrated through graded ethanol, cleared in xylene, and mounted with neutral resin. Images were captured using a light microscope.
Immunohistochemical staining and result analysis
Utilize a 4% paraformaldehyde solution for the preservation of lung adenocarcinoma (LUAD) tissue and murine tumor tissue. Incubate at 60 °C for an hour, then deparaffinize and rehydrate the samples before extracting antigens. Incubate the samples with a peroxidase blocking solution at ambient temperature for a duration of 10 min. Following this, perform a 2 h incubation at 37 °C utilizing 5% normal goat serum in PBST. Subsequently, proceed with an overnight incubation at 4 °C with the primary antibodies MYSM1, TRAF2, TRAF3, and Ki67. Prior to re-staining with hematoxylin, apply an HRP-conjugated secondary antibody to each slide, wash three times with TBST, and develop with DAB solution. Then, capture images of the slides using an inverted microscope and perform statistical analysis of the positive cell rate for the staining results. For analysis, IHC scoring was manually performed by three individuals: one board-certified pathologist, who identified tumor regions, and two additional scorers, who evaluated staining. The scorers assessed staining intensity and the percentage of positive cells to calculate the H-score for each tissue core. All slides were evaluated in a blinded manner with respect to clinical outcomes to prevent bias. Inter-observer variability was minimized through the use of a predefined scoring protocol, with all scoring conducted independently and blinded to clinical information.
Immunoprecipitation (IP) assay
Lung adenocarcinoma cells and 293 T cells were lysed in IP buffer, and after centrifugation, IP-grade antibodies against MYSM1, TRAF2, TRAF3, or IgG antibodies, as well as tag antibodies such as Flag, HA, or His of the same quality were subjected to the supernatant and placed on a rotating device at 4 °C for a duration of 1 h. Next, an appropriate amount of Protein A/G magnetic beads was added to the mixture and allowed to incubate overnight at 4 °C on a rotating device. The following day, we used a magnetic rack to remove the supernatant, washed the IP lysate mixture on the rotating device for 5 min, repeating this process five times. After removing the supernatant with the magnetic rack, the precipitated mixture was combined with the loading buffer for Western blot analysis. Finally, we detected the expression of relevant indicators using Western blotting, choosing a primary antibody from a different species than the previously added antibodies during the primary antibody incubation to avoid the influence of light and heavy chains. Supplementary Table S2 contains details about the primary and secondary antibodies used in this assay.
Quantitative polymerase chain reaction (qPCR)
Cellular mRNA was obtained using the Cellular RNA Rapid Extraction Kit from SparkJade (AC0205). Then, reverse transcribed into cDNA and subjected to qPCR for MSYM1, TRAF2, TRAF3, and EMT-related molecules. The upstream and downstream primer sequences used in the experiment are shown in Supplementary Table S3.
Protein stability assay
A549 cells were treated with the protein synthesis inhibitor cycloheximide (CHX, 50 μg/mL, MedChemExpress, MCE, # HY-12320), with DMSO used as the vehicle control, to assess the effect of MYSM1 on TRAF2 and TRAF3 protein stability. Cells were harvested at 0, 2, 4, and 8 h post-CHX treatment, and total proteins were extracted. Western blot analysis was then performed to quantify MYSM1, TRAF2, and TRAF3 expression levels.
Ubiquitination level detection assay
Proteins were isolated from H1299 and PC9 cells following an 8-h exposure to the proteasome inhibitor MG-132, and the levels of MYSM1, TRAF2, and TRAF3 proteins were detected using Western blotting.
Analysis through transcriptomic sequencing, mass spectrometry for protein characterization, and bioinformatics
The transcriptome sequencing was conducted by LC Biotechnology Co., Ltd. Through transcriptome sequencing, mRNA transcripts that were expressed differently in MYSM1 knockout H1299 cells compared to control H1299 cells were detected. KEGG pathway enrichment maps for these transcripts were generated using the OmicStudio platform (https://www.omicstudio.cn/tool).
Jikai Company conducted the mass spectrometry analysis. We first conducted immunoprecipitation on A549 cells stably overexpressing MYSM1, and then collected the precipitated magnetic beads following centrifugation for analysis via mass spectrometry. Mass spectrometry analysis was employed to examine the protein constituents that interact with MYSM1.
For the TCGA dataset, MYSM1 RNA-Seq expression data (HTSeq-FPKM) for TCGA-LUAD were obtained directly from the UCSC Xena platform (https://xena.ucsc.edu/), which hosts uniformly processed TCGA datasets with standardized normalization. For the GEO dataset GSE117049, we focused our analysis on the sub-series GSE116959, which contains the relevant expression profiles of lung adenocarcinoma and normal tissues. Raw data were downloaded from the GEO repository, and probe-level intensities were preprocessed and normalized using the limma R package with normalization. The MYSM1 survival analyses were conducted using the KMplotter (https://kmplot.com/analysis/) online platform with probe 238644_at, applying the “auto-select best cutoff” option to determine the optimal expression threshold for patient stratification.
Statistical analysis
All statistical analyses were performed utilizing GraphPad Prism software. A range of statistical methods were applied for comparative analysis, including the log-rank test, Kaplan–Meier survival analysis, Pearson chi-square test, two-tailed Student’s t-test, Fisher’s exact test, and Pearson correlation analysis, as specified.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Code availability
This study did not involve the development of any custom code. All analyses were performed using standard, publicly available software tools, as described in the Methods section.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Thai, A. A., Solomon, B. J., Sequist, L. V., Gainor, J. F. & Heist, R. S. Lung cancer. Lancet 398, 535–554 (2021).
Travis, W. D. et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 10, 1243–1260 (2015).
Konoshenko, M., Lansukhay, Y., Krasilnikov, S. & Laktionov, P. MicroRNAs as predictors of lung-cancer resistance and sensitivity to cisplatin. Int. J. Mol. Sci. 23, https://doi.org/10.3390/ijms23147594 (2022).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 70, https://doi.org/10.3322/caac.21590 (2020).
Sadee, W., Wang, D., Hartmann, K. & Toland, A. E. Pharmacogenomics: driving personalized medicine. Pharm. Rev. 75, 789–814 (2023).
Lheureux, S., Denoyelle, C., Ohashi, P. S., De Bono, J. S. & Mottaghy, F. M. Molecularly targeted therapies in cancer: a guide for the nuclear medicine physician. Eur. J. Nucl. Med Mol. Imaging 44, 41–54 (2017).
Comprehensive molecular profiling of lung adenocarcinoma. Nature. 511, 543–550. https://doi.org/10.1038/nature13385 (2014).
Deng, L., Meng, T., Chen, L., Wei, W. & Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 5, 11 (2020).
Sun, T., Liu, Z. & Yang, Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol. Cancer 19, 146 (2020).
Dewson, G., Eichhorn, P. J. A. & Komander, D. Deubiquitinases in cancer. Nat. Rev. Cancer 23, 842–862 (2023).
Ren, J. et al. Deubiquitylating enzymes in cancer and immunity. Adv. Sci.10, e2303807 (2023).
Park, H. B., Hwang, S. & Baek, K. H. USP7 regulates the ERK1/2 signaling pathway through deubiquitinating Raf-1 in lung adenocarcinoma. Cell Death Dis. 13, 698 (2022).
Yang, M., Wei, Y., He, X. & Xia, C. The deubiquitinating protein OTUD6B promotes lung adenocarcinoma progression by stabilizing RIPK1. Biol. Direct 19, 46 (2024).
Wang, X. et al. The deubiquitinase USP10 regulates KLF4 stability and suppresses lung tumorigenesis. Cell Death Differ. 27, 1747–1764 (2020).
Goldknopf, I. L. et al. Isolation and characterization of protein A24, a “histone-like” non-histone chromosomal protein. J. Biol. Chem. 250, 7182–7187 (1975).
Gu, Y. et al. The histone H2A deubiquitinase Usp16 regulates hematopoiesis and hematopoietic stem cell function. Proc. Natl. Acad. Sci. USA 113, E51–E60 (2016).
Jiang, X. X. et al. Control of B cell development by the histone H2A deubiquitinase MYSM1. Immunity 35, 883–896 (2011).
Belle, J. I. et al. MYSM1 maintains ribosomal protein gene expression in hematopoietic stem cells to prevent hematopoietic dysfunction. JCI Insight. 5, https://doi.org/10.1172/jci.insight.125690 (2020).
Tian, M. et al. MYSM1 represses innate immunity and autoimmunity through suppressing the cGAS-STING pathway. Cell Rep. 33, 108297 (2020).
Panda, S. & Gekara, N. O. The deubiquitinase MYSM1 dampens NOD2-mediated inflammation and tissue damage by inactivating the RIP2 complex. Nat. Commun. 9, 4654 (2018).
Chen, X. et al. MYSM1 inhibits human colorectal cancer tumorigenesis by activating miR-200 family members/CDH1 and blocking PI3K/AKT signaling. J. Exp. Clin. Cancer Res. 40, 341 (2021).
Sun, J. et al. MYSM1-AR complex-mediated repression of Akt/c-Raf/GSK-3β signaling impedes castration-resistant prostate cancer growth. Aging 11, 10644–10663 (2019).
Guan, X. et al. MYSM1 induces apoptosis and sensitizes TNBC cells to cisplatin via RSK3-phospho-BAD pathway. Cell Death Discov. 8, 84 https://doi.org/10.1038/s41420-022-00881-1 (2022).
Geiger, T. R. & Peeper, D. S. Metastasis mechanisms. Biochim. Biophys. Acta. 1796, 293–308 (2009).
Kalluri, R. & Weinberg, R. A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 119, 1420–1428 (2009).
Brabletz, T., Kalluri, R., Nieto, M. A. & Weinberg, R. A. EMT in cancer. Nat. Rev. Cancer 18, 128–134 (2018).
Williams, E. D., Gao, D., Redfern, A. & Thompson, E. W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 19, 716–732 (2019).
Panda, S., Nilsson, J. A. & Gekara, N. O. Deubiquitinase MYSM1 regulates innate immunity through inactivation of TRAF3 and TRAF6 complexes. Immunity 43, 647–659 (2015).
Gardam, S., Sierro, F., Basten, A., Mackay, F. & Brink, R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity 28, 391–401 (2008).
Vashisht, M. et al. TRAF2/3 deficient B cells resist DNA damage-induced apoptosis via NF-κB2/XIAP/cIAP2 axis and IAP antagonist sensitizes mutant lymphomas to chemotherapeutic drugs. Cell Death Dis. 14, 599 (2023).
Lu, C. H. et al. USP17 mediates macrophage-promoted inflammation and stemness in lung cancer cells by regulating TRAF2/TRAF3 complex formation. Oncogene 37, 6327–6340 (2018).
Subedi, L., Lee, J. H., Gaire, B. P. & Kim, S. Y. Sulforaphane inhibits MGO-AGE-mediated neuroinflammation by suppressing NF-κB, MAPK, and AGE-RAGE signaling pathways in microglial cells. Antioxidants 9, https://doi.org/10.3390/antiox9090792 (2020).
Nonaka, K. et al. Advanced glycation end-products increase IL-6 and ICAM-1 expression via RAGE, MAPK and NF-κB pathways in human gingival fibroblasts. J. Periodontal Res. 53, 334–344 (2018).
Dawuti, A. et al. Salvianolic acid A alleviates heart failure with preserved ejection fraction via regulating TLR/Myd88/TRAF/NF-κB and p38MAPK/CREB signaling pathways. Biomed. Pharmacother. 168, 115837 (2023).
Pua, L. J. W. et al. Functional roles of JNK and p38 MAPK signaling in nasopharyngeal carcinoma. Int. J. Mol. Sci. 23, https://doi.org/10.3390/ijms23031108 (2022).
Li, L. et al. BAY11-7082 targets RNF25 to reverse TRIP4 ubiquitination-dependent NF-κB activation and apoptosis resistance in renal cell carcinoma. Int J. Biol. Sci. 21, 4410–4427 (2025).
Ohtake, F., Tsuchiya, H., Saeki, Y. & Tanaka, K. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc. Natl. Acad. Sci. USA 115, E1401–E1408 (2018).
Ohtake, F., Saeki, Y., Ishido, S., Kanno, J. & Tanaka, K. The K48-K63 branched ubiquitin chain regulates NF-κB signaling. Mol. Cell 64, 251–266 (2016).
Yang, X. D. & Sun, S. C. Targeting signaling factors for degradation, an emerging mechanism for TRAF functions. Immunol. Rev. 266, 56–71 (2015).
Lalani, A. I., Zhu, S., Gokhale, S., Jin, J. & Xie, P. TRAF molecules in inflammation and inflammatory diseases. Curr. Pharm. Rep. 4, 64–90 (2018).
Qu, L., Ma, X. & Wang, F. The roles of gut microbiome and metabolites associated with skin photoaging in mice by intestinal flora sequencing and metabolomics. Life Sci. 341, 122487 (2024).
Xie, L. et al. Cardiomyocyte-specific knockout of ADAM17 alleviates doxorubicin-induced cardiomyopathy via inhibiting TNFα-TRAF3-TAK1-MAPK axis. Signal Transduct. Target Ther. 9, 273 (2024).
Lee, S. et al. Glutathione peroxidase-1 regulates ASK1-dependent apoptosis via interaction with TRAF2 in RIPK3-negative cancer cells. Exp. Mol. Med. 53, 1080–1091 (2021).
Das, A. et al. The structure and ubiquitin binding properties of TRAF RING heterodimers. J. Mol. Biol. 433, 166844 (2021).
Yi, X. M., Li, M., Chen, Y. D., Shu, H. B. & Li, S. Reciprocal regulation of IL-33 receptor-mediated inflammatory response and pulmonary fibrosis by TRAF6 and USP38. Proc. Natl. Acad. Sci. USA 119, e2116279119 (2022).
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (No. 82472814) and the Taishan Scholar Program of Shandong Province (No. ts201712087).
Author information
Authors and Affiliations
Contributions
Wenhao Yu: Conceptualization, writing original draft, visualization, methodology, formal analysis, Data Curation. Luyuan Ma: Writing, Review and Editing. Rongyang Li: Software. Dingqiang Zhu: Resources. Chenghao Qu: Data Curation. Jingyi Han: Visualization. Yu Tian, Hui Tian and Jinghui Liang: Conceptualization, Supervision, Funding acquisition, Project administration. Correspondence to Yu Tian, Hui Tian or Jinghui Liang.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, W., Ma, L., Li, R. et al. MYSM1 promotes lung adenocarcinoma progression via TRAF2/3-mediated activation of MAPK and non-canonical NF-κB pathways. npj Precis. Onc. 9, 337 (2025). https://doi.org/10.1038/s41698-025-01116-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01116-z
