
Abstract
Ovarian cancer (OC) is the deadliest gynecologic malignancy, with immune evasion contributing to its poor prognosis. This study reveals that the m6A demethylase ALKBH5 enhances OC progression and immune escape by regulating ITGB1 expression. Using bulk and single-cell RNA sequencing, deep learning, and co-expression network analysis, we identified ALKBH5 as a key regulator of m6A-modified ITGB1. Functional experiments showed that ALKBH5 promotes OC cell proliferation, metastasis, and immune evasion, while its knockdown enhances T cell-mediated cytotoxicity. Public database validation further confirmed the diagnostic value of ALKBH5 and ITGB1. Mechanistically, ALKBH5 binds to ITGB1 mRNA and removes m6A modifications, increasing its stability and expression. Targeting the ALKBH5–ITGB1 axis may offer novel diagnostic biomarkers and therapeutic strategies for OC.
Similar content being viewed by others

Introduction
Ovarian cancer (OC) is one of the deadliest gynecologic malignancies, often diagnosed in advanced stages, posing a significant challenge to women’s health1,2,3. Research has found that N6-methyladenosine (m6A) modification plays a crucial role in the initiation and progression of tumors4,5,6. m6A modification is a common epigenetic modification found on RNA, and its alterations can impact gene expression and regulation, consequently influencing cellular functions and behaviors7,8. In recent years, there has been an increasing amount of research on the mechanism of m6A modification in various cancers9,10,11. Several studies have highlighted the close association between abnormal m6A modification and processes such as growth, metastasis, and drug resistance in tumors, including OC12. However, the specific regulatory mechanisms of m6A modification in OC and its role in tumor cell behaviors and immune evasion remain not entirely understood4,6,13.
ALKBH5, an important member of m6A modification-related proteins, has attracted considerable attention for its role in OC 14. To investigate the mechanisms through which ALKBH5 regulates ITGB1 and its involvement in OC progression and immune evasion, this study employed a comprehensive set of methods and techniques. High-throughput sequencing technologies, including Bulk mRNA-seq and single-cell RNA sequencing, were used to analyze OC and normal tissue samples. Bioinformatics methods, such as deep learning, random forest models, and weighted gene co-expression network analysis, were integrated to explore the regulatory role of m6A modification. Public databases like TCGA and GEO were also used for validation to ensure the reliability and scientific rigor of the findings. In vitro cell experiments and in vivo mouse models were utilized to validate the role of ALKBH5-mediated m6A modification in promoting OC cell proliferation, metastasis, and immune escape.
The study found that the ALKBH5/m6A-ITGB1/FAK signaling axis plays a critical role in lymphangiogenesis and lymph node (LN) metastasis in OC. Targeting ITGB1 with blocking antibodies or inhibiting FAK kinase may offer promising anti-metastatic therapies. These findings provide new insights into the molecular mechanisms of LN metastasis in EOC (EOC) and may contribute to the development of novel treatments for EOC patients15.
The innovation of this study lies in its systematic exploration of how ALKBH5 regulates m6A modification levels of ITGB1, influencing OC cell proliferation, metastasis, and immune evasion, thus filling a significant gap in the field. By integrating Bulk mRNA-seq, single-cell RNA sequencing, deep learning, random forest models, and weighted gene co-expression network analysis, the study comprehensively analyzed the m6A regulatory network in OC. This research provides a scientific foundation for identifying new therapeutic targets and diagnostic biomarkers, with substantial clinical translational potential.
This study aims to further investigate the molecular mechanisms by which ALKBH5-mediated m6A modification regulates ITGB1 and its role in OC cell proliferation, metastasis, and immune evasion. Understanding the interaction between ALKBH5 and ITGB1, as well as the regulatory role of m6A modification, could identify novel therapeutic targets and diagnostic biomarkers, thereby supporting more effective OC treatments and improving patient outcomes.
Results
Discovery of 9 Cell Subpopulations in OC Tissue Using scRNA-seq
OC is a major malignant tumor significantly impacting women’s health worldwide, particularly EOC, which accounts for 90-95% of all OC cases. Its diagnosis is challenging due to subtle early symptoms, and late-stage or recurrent cases often lack effective curative treatments, resulting in a high mortality rate. Thus, there is a pressing need to explore new diagnostic methods and treatment strategies16,17.
We established an in situ OC tumor model in C57BL/6 J mice with normal immune function. By intraperitoneally injecting ID8 cells, we simulated the growth environment of OC. The results revealed that while the mice injected with ID8 cells did not show a significant difference in body weight compared to the control group of mice without cell injection (Supplementary Fig. 1A), the mice in the OC group exhibited a noticeable increase in abdominal circumference and presented symptoms of ascites (Supplementary Fig. 1B), indicating that tumor growth may lead to ascites formation. To ensure accurate assessment, we removed the skin and subcutaneous fat during abdominal circumference measurement to minimize external interference. Furthermore, the histopathological analysis demonstrated that, in comparison to the control group, the tumor tissue in the OC group exhibited typical tumor cell morphology, with tumor cells arranged haphazardly, showing significantly enlarged and irregularly shaped nuclei (Supplementary Fig. 1C). Immunohistochemistry (IHC) revealed a significantly higher proportion of Ki-67-positive cells in OC tissues compared to the sham group (Supplementary Fig. 1D). These findings suggest the successful establishment of the OC model.
In order to further investigate the cellular heterogeneity and molecular mechanisms underlying the occurrence and development of OC, we collected normal ovarian tissue (Sham group) and OC tissue from three mice for scRNA-seq analysis (Fig. 1A). The data were integrated using the Seurat package. Initially, we examined the number of genes (nFeature_RNA), mRNA molecules (nCount_RNA), and the percentage of mitochondrial genes (percent.mt) in all cells of the scRNA-seq dataset. The results revealed that the majority of cells had nFeature_RNA < 5000, nCount_RNA < 20000, and percent.mt <20% (Supplementary Fig. 2A). Using the criteria of 200 < nFeature_RNA < 5000 and percent.mt < 20%, low-quality cells were filtered out. The correlation analysis of sequencing depth indicated that after filtering, the correlation coefficient between nCount_RNA and percent.mt was -0.22, while the correlation coefficient between nCount_RNA and nFeature_RNA was 0.89 (Supplementary Fig. 2B). This suggests that the filtered cell data is of good quality and suitable for further analysis.

A Schematic diagram of single-cell sequencing process (Created with BioRender.com); B, C Overall (B) and grouped (C) visualizations of cell annotations in UMAP clustering results; D Visualization of confidence intervals for cell clustering based on UMAP, where each color represents a unique cell subset, with distinct color boundaries indicating the distribution range of each subset; E Proportion of 9 different cell subsets in each sample, represented by different colors for cell subset types; F Differential P-values in cell composition between Sham and OC group samples analyzed by t-test (Sham group: n = 3; OC group: n = 3). Quantitative data are presented as mean ± standard deviation, with between-group comparisons conducted using unpaired t-tests.
Upon further analysis of the filtered cells, we identified highly variable genes based on gene expression variance, selecting the top 2000 genes with the highest variability for downstream analysis (Supplementary Fig. 2C). The cell cycle of the samples was calculated using the CellCycleScoring function, and the data was initially normalized (Supplementary Fig. 2D). Subsequently, data was subjected to linear dimension reduction through PCA using the selected highly variable genes. Here, we present a heatmap showing the main correlated gene expression trends for PC_1 - PC_6 (Supplementary Fig. 2E) and the distribution of cells in PC_1 and PC_2 (Supplementary Fig. 2F), revealing batch effects among the samples. Batch correction using the harmony package was performed to reduce batch effects and enhance the accuracy of cell clustering (Supplementary Fig. 2G, Supplementary Fig. 3A). Additionally, an Elbow Plot was utilized to rank the standard deviations of principal components (PCs), demonstrating that PC_1-PC_15 effectively captures the information of the selected highly variable genes with significant analytical implications (Supplementary Fig. 2H). The corrected results indicate the successful mitigation of batch effects within the samples (Supplementary Fig. 2A).
Further dimensionality reduction of the top 15 principal components was performed using the UMAP algorithm. The clustering results at different resolutions were visualized with the cluster package (Supplementary Fig. 4A). Resolution 0.5 was selected for clustering based on the clustering outcomes, resulting in the identification of 16 cell clusters (Supplementary Fig. 3B-D). A heatmap was generated to display the top 10 DEGs in each cluster (Supplementary Fig. 4B). The expression patterns of marker genes for each identified cell cluster were visualized (Supplementary Fig. 3E), and cell annotation was conducted using the singleR package combined with literature review, revealing a total of 9 cell types (Supplementary Table 1): Epithelial cells, Macrophages, MSCs (Mesenchymal stem cells), CSCs (Cancer stem cells), Neutrophils, T cells, B cells, Endothelial cells, and Stromal cells (Fig. 1B-C). Consistent results were also obtained utilizing the t-SNE algorithm (Supplementary Fig. 5A-D). Additionally, the expression of marker genes for the 9 cell types in the 16 cell clusters was illustrated through dot plots and UMAP/t-SNE plots, where Cdh1 served as the marker gene for Epithelial cells, Cd72 for Macrophages, Cd90 for MSCs, Aldh1a1 for CSCs, Lcn2 for Neutrophils, Foxp3 for T cells, Cd79a for B cells, Pecam1 for Endothelial cells, and Cd18 for Stromal cells (Supplementary Fig. 3F, Supplementary Fig. 5E).
The analysis using scRNA-seq successfully identified 9 distinct cell types. Subsequently, employing the Seurat algorithm, we computed the confidence interval distribution and cellular composition ratios of these cell types within individual samples (Fig. 1D-E). Through T-tests, we compared the differences in cell quantities between normal and OC samples. The analysis revealed a significant reduction in Epithelial cells and Macrophages in OC samples compared to normal samples (Fig. 1F).
These findings demonstrate the successful exploration of the changes in 9 cell subtypes during the progression of OC using scRNA-seq, highlighting the pivotal role of the tumor microenvironment in the advancement and immune evasion of OC.
Integrative analysis of bulk mRNA-seq data and neural networks reveals the molecular mechanisms of OC
In order to delve deeper into the molecular mechanisms directly associated with OC, we conducted Bulk transcriptome sequencing analysis on tumor samples and normal tissues from 15 OC model mice (with ID8 cell injection) and 15 sham-operated controls (Fig. 2A). Standardization methods were applied to normalize the microarray data, and batch effects were successfully eliminated through PCA analysis to ensure the accuracy of data analysis (Fig. 2B). Differential gene analysis revealed 354 significantly DEGs, with 199 upregulated and 155 downregulated genes (Fig. 2C). The heatmap of these DEGs displayed significant differences between the OC group and the normal group (Fig. 2D).

A Schematic diagram of transcriptome sequencing process (Created with BioRender.com); B PCA of normal and OC samples (Sham = 15, OC = 15), with the x and y axes representing the two principal components after PCA reduction, the percentages of variance explained are in parentheses; C Volcano plot of 354 DEGs in normal and OC samples analysis, with red dots indicating significantly upregulated genes, blue dots indicating significantly downregulated genes, and grey dots representing genes with no differential expression; D Heat map of the top 50 significantly upregulated and downregulated genes, depicted in red for high expression and blue for low expression; E Metascape functional enrichment network diagram displaying the relationships between DEGs in normal and OC tissues and their functional classification in various biological processes, each node represents an enriched term colored by its cluster ID, and lines indicate interactions between genes, nodes with shared cluster IDs are typically closer to one another; F Circular chart of GO functional enrichment analysis of DEGs, with hierarchical levels representing functional categories, total number of genes in each category, number of significantly upregulated genes, and enrichment factors; G Enrichment analysis of DEGs in KEGG pathways, presenting pathways and the number of enriched genes within each pathway, categorized according to the top six KEGG pathway classes; H Venn diagram showing the intersection of characteristic genes from the Blue module of WGCNA analysis and feature genes selected by random forest trees; I Heat map of the expression of six core genes in the transcriptome data, with red indicating high expression and green indicating low expression; J Visualization of the neural network model, with the left input layer comprising core genes connected to an intermediate hidden layer (H1 to H5) for feature extraction and non-linear transformation, further linked to the right output layer for making final predictions on disease status; K Receiver Operating Characteristic (ROC) curve analysis based on deep learning model, evaluating predictive performance of the model on the training dataset.
Analysis of DEGs using Metascape revealed their involvement in negative regulation of cell proliferation, formation of tubular structures, cancer pathways, and cell fate determination (Fig. 2E, Supplementary Fig. 6A, Supplementary Table 2). Further, GO enrichment analysis unveiled the roles of these genes in biological processes such as myocyte differentiation and signal transduction, as well as their associations with platelet alpha granules and cell-matrix adhesion (Fig. 2F, Supplementary Table 3). KEGG analysis demonstrated that DEGs play crucial roles in pathways, including the PI3K-Akt signaling pathway and cancer-related proteoglycans (Fig. 2G).
In order to delve deeper into the key genes involved in the pathophysiology of OC, we employed Weighted Gene Co-expression Network Analysis (WGCNA) in conjunction with a random forest model for a detailed analysis of transcriptome data. Initially, clustering was conducted on a dataset consisting of 15 normal samples and 15 OC samples, whereby samples with obvious abnormalities were excluded based on a predefined threshold (Supplementary Fig. 6B). Setting the soft threshold to 4, the scale-free topology curve began to flatten out, exhibiting high average connectivity, thus enabling subsequent analysis (Supplementary Fig. 6C, D). Hierarchical clustering of genes within the OC dataset was performed based on a dissimilarity matrix, leading to the construction of a dendrogram (Supplementary Fig. 7A). Following the merger of highly correlated modules with a clustering height limit set at 0.25, six gene modules were identified for further investigation (Supplementary Fig. 7B). Subsequently, to identify modules highly associated with OC, PCA was carried out on genes within each module, with the first principal component value extracted as the module eigengene (ME). The correlation between ME values and clinical features determined modules of clinical significance. Results indicated that the Blue module exhibited a positive correlation with OC, showing the highest gene importance (Supplementary Fig. 7C, Supplementary Fig. 6E). The correlation coefficients between each module and OC are shown in Supplementary Fig. 6F. Consequently, the Brown module was selected for further analysis in this study. Additionally, utilizing random forest tree models for further refinement of DEGs, the model’s error rate was evaluated to assess stability and performance, culminating in the ranking of feature gene importance (Supplementary Fig. 7D, E). Ultimately, 30 key genes were identified and presented in Supplementary Table 4. A Venn diagram combining core module genes identified by WGCNA and feature genes from a random forest model revealed six overlapping hub genes: Alkbh5, Cxcl12, Itgb1, Lepr, Hnrnpc, and Scn7a (Fig. 2H). Expression analysis of these six core genes in the transcriptome data revealed significantly higher expression levels in the OC group compared to the normal group (Fig. 2I, Supplementary Fig. 7F). Moreover, positive correlations among these genes demonstrated strong diagnostic potential (Supplementary Fig. 7G, H).
To further elucidate the roles of these core genes in the pathological progression of OC, we employed deep learning techniques to construct a multi-layer neural network model, unveiling the profound associations between these genes and the pathological states and treatment responses of OC (Fig. 2J, K).
In conclusion, by integrating transcriptome sequencing data with deep learning technology, we have investigated the pathogenic mechanisms of OC and identified crucial core genes highly relevant to the disease. This study provides important insights and a foundation for future disease treatment and prognosis monitoring.
Validation of the diagnostic value of core genes in OC by integrating TCGA and GEO databases with neural networks
To further validate the predictive capabilities of the aforementioned six core genes, in-depth analyses were conducted in a broader public database. The heatmap displayed variations of these genes between different tumor statuses and clinical stages in the TCGA-OV cohort (Fig. 3A), with correlation analysis further strengthening the potential synergistic or regulatory relationships among these genes (Fig. 3B). To complement the lack of normal sample data in the TCGA-OV dataset, we integrated data from both TCGA-OV and GSE54388 as a test dataset. Data normalization was initially performed to ensure the accuracy of data analysis (Fig. 3C). Differential analysis revealed 774 DEGs, with 223 upregulated and 551 downregulated genes identified (Fig. 3D). Furthermore, a multi-layer neural network model was constructed to assess the learning and predictive capabilities of the core genes in the test dataset. The results indicated that four core genes (ALKBH5, CXCL12, ITGB1, SCN7A) effectively connected to multiple hidden layers in the neural network and, through feature extraction and transformation, achieved accurate prediction of disease status (Fig. 3E). The ROC curve analysis of the model further corroborated its high accuracy in OC diagnosis (Fig. 3F). In the test dataset, the expression levels of ALKBH5, CXCL12, and ITGB1 were significantly higher in the OC group compared to the normal group, whereas SCN7A expression was significantly decreased, highlighting their robust diagnostic potential (Fig. 3G, H).

A Heat map showing the expression of six core genes in different clinical stages and tumor statuses. B Circos plot depicting the expression correlations among the six core genes. C Data correction plot for the test dataset (Control, N = 6; OC, N = 392) after integrating the TCGA-OV dataset (OC, N = 376) and GSE54388 dataset (Control, N = 6; OC, N = 16), with the upper and lower graphs illustrating data distribution before and after correction. D Volcano plot of 774 DEGs between normal and OC samples, where red dots represent significantly upregulated genes, blue dots signify significantly downregulated genes, and black dots indicate genes with no expression differences. E Visualization of the neural network model, with the input layer on the left consisting of core genes connected to the middle hidden layers (H1 to H5) for feature extraction and non-linear transformations, further linked to the right output layer for making final disease status predictions. F ROC curve analysis is based on a deep learning model to evaluate the predictive performance of the test group model. G Comparison of the expression levels of four core genes in the test dataset between the normal and OC groups. H ROC curve analysis of the four core genes in the test dataset. Between-group comparisons were evaluated, where *P < 0.05 and ***P < 0.001.
In conclusion, through a comprehensive analysis of public databases, significant alterations in core genes in OC were confirmed, particularly emphasizing the highly predictive value of ALKBH5-mediated m6A modification in the diagnosis of OC.
The impact of m6A modification on OC prognosis and immune microenvironment
After a detailed analysis of the molecular mechanisms of OC and the identification of core genes highly relevant to the disease, our focus shifted to two core genes, Alkbh5 and Hnrnpc, which respectively act as “erasers” and “readers” in the process of m6A RNA methylation18. This points to the crucial role that m6A modification may play in OC development. In recent years, an increasing number of studies have revealed the connection between m6A and cancer19,20, underscoring the significance of delving into the molecular regulatory mechanisms of m6A modification networks in OC.
m6A modification involves a complex regulatory network comprising multiple “writers,” “erasers,” and “readers,” which interact with each other to collectively determine the ultimate biological effects of m6A modification21. We further gathered information from the literature on 13 major m6A RNA methylation regulatory factors, including ALKBH5 and HNRNPC, and conducted a comprehensive analysis of their roles in OC using various databases (Supplementary Table 5)22,23.
Firstly, we evaluated the relative expression of these factors in OC patients using the GEPIA database. The results revealed that the expression of HNRNPC, ALKBH5, and WTAP was relatively high, while that of RBM15 was relatively low (Supplementary Fig. 8A). Upon further assessment of the prognostic value of these regulatory factors in OC patients, we observed a significant association between low expression of WTAP and longer overall survival (OS) (Supplementary Fig. 8B). Analysis of progression-free survival (PFS) indicated that with the exception of ZC3H13 and HNRNPC, the remaining factors were significantly associated with disease progression. Compared to the low-expression group of ALKBH5, the high-expression group had a 56% increased risk of disease progression or death (HR = 1.56) (Supplementary Fig. 8C). Additionally, analysis using the Timer database revealed a correlation between immune cell infiltration and the expression profile of m6A modification regulatory factors, highlighting the crucial role of these regulatory factors in the tumor microenvironment (Supplementary Fig. 9).
In conclusion, m6A RNA methylation and its regulatory factors play a crucial role in the progression and prognosis assessment of OC, as well as in regulating the tumor microenvironment. Particularly, the role of ALKBH5 is emphasized, as its expression levels not only exhibit significant heterogeneity but are also closely associated with disease progression and survival of patients. These findings not only deepen our understanding of the pathogenesis of OC but also provide new insights for future diagnosis and treatment strategies.
ALKBH5 regulates ITGB1 through m6A demethylation to influence OC progression
Analyzing the single-cell expression data mentioned earlier, we found that Alkbh5 is widely expressed across all identified cell types (Fig. 4A). To further elucidate its role in OC progression, we utilized the “monocle” software package to construct a pseudo-temporal trajectory, simulating the dynamic changes in cell states. By mapping information from the originating tissue onto the trajectory plot, we observed that cells undergo continuous state changes during their differentiation or development processes (Fig. 4B). The pseudo-temporal trajectory analysis revealed that at the initial stage of the trajectory, both CSCs and MSCs cluster together, and as the trajectory progresses, more immune cells such as Macrophages and T cells were observed moving towards the trajectory’s endpoint, while Epithelial cells increased (Fig. 4C, D). The expression of Alkbh5 in the trajectory analysis exhibited dynamic changes closely associated with cell state transitions, highlighting its regulatory role as an “eraser” of m6A modification in determining cell fate (Fig. 4E, F).

A Violin plot showing the expression of Alkbh5 in various cell types. B The trajectory skeleton plot is colored by pseudo-time, where each point represents a cell. C, D The trajectory skeleton plot is colored by cell type, along with its split trajectory skeleton plot, with different cell types represented by different colors. E Expression trends of Alkbh5 in different pseudo-temporal states. F Expression trends of Alkbh5 relative to the pseudo-time trajectory. G Comparison of the expression cell numbers of six core genes in scRNA-seq data pseudo-temporal trajectory analysis. H Predicted target genes of ALKBH5 potential m6A modification by the RM2Target database. I Predicted m6A methylation sites of ITGB1 by the SRAMP database and local secondary structure information around the mutation site with the highest confidence score (Score = 0.744); J The potential m6A methylation site of ITGB1 and its surrounding secondary structure diagram.
Further examination of the dynamic expression of six core genes during disease progression revealed that Itgb1 was expressed in most cells (N = 2045) (Fig. 4G). The RM2Target database further indicated that ALKBH5 may influence the function of ITGB1 by regulating m6A modifications on it (Fig. 4H). Additionally, predictions from the SRAMP database suggested that there are 24 m6A modification sites on ITGB1 (Supplementary Table 6), indicating that ALKBH5 may regulate the activity of ITGB1 through demethylation (Fig. 4I, J).
These results collectively demonstrate the critical role of ALKBH5 in regulating ITGB1 through m6A demethylation in OC progression, emphasizing the widespread expression of ALKBH5 as an “eraser” of m6A modification within the OC cell population and its potential regulatory impact on ITGB1 function.
ALKBH5 promotes proliferation and metastasis of OC both in vivo and in vitro
To investigate the role of ALKBH5 in OC, we initially assessed the expression levels of ALKBH5 in normal ovarian cells (IOSE-80) and OC cells (A2780 and SK-OV-3). The results revealed a significant increase in ALKBH5 mRNA levels in cancer cells (Fig. 5A). Subsequently, ALKBH5 was stably knocked down in OC cells A2780 and SK-OV-3 using two independent interference sequences (Supplementary Fig. 10A, B). CCK-8 assays demonstrated that ALKBH5 knockdown inhibited cell growth (Fig. 5B). EdU staining indicated a significant decrease in cell proliferation upon ALKBH5 knockdown (Fig. 5C). Cell cycle analysis revealed cell arrest at the G2/M phase (Fig. 5D). Similarly, colony formation assays showed suppressed clonogenic ability after ALKBH5 knockdown (Fig. 5E). Moreover, Transwell experiments demonstrated significant impairment in cell migration and invasion abilities following ALKBH5 knockdown (Fig. 5F). These findings indicate the crucial role of ALKBH5 in regulating the proliferation, migration, and invasion of OC cells.

A mRNA expression levels of ALKBH5 in normal ovarian cells and OC cells detected by RT-qPCR; B Cell growth assays using CCK8 to assess the growth of A2780 and SK-OV-3 cells after knocking down ALKBH5; (C) EdU incorporation assay to determine the proportion of positively labeled cells in each group, shown at a scale of 25 µm; D Flow cytometry analysis of cell cycle changes and the ratio of G1/S and G2/M phases in each group; E Colony formation assay and quantification of colony numbers for two groups of cells; F Transwell assay measuring changes in migration and invasion abilities of cells in each group, quantified at a scale of 50 µm; G−I Quantitative analysis of tumor size and weight for subcutaneous xenograft tumors in each group (n = 5); J Evaluation of Ki-67 and Caspase-3 positive staining in tumor tissues using H&E and immunohistochemistry staining, with quantification provided at a scale of 20 µm; K Representative images and quantitative information of intraperitoneal metastatic tumors in each group. Data are presented as mean ± standard deviation and intergroup differences were assessed using unpaired t-tests. *P < 0.05 compared to the control group, **P < 0.01, ***P < 0.001, and “ns” indicates no statistical difference between the groups. Cell experiments were performed in triplicate, and each animal experiment involved five mice per group.
To assess the oncogenic role of ALKBH5 in vivo in OC, ALKBH5-knocked down A2780 OC cells and control cells were subcutaneously injected into nude mice. After 4 weeks, the mice were euthanized, and the tumors were excised. It was observed that tumors in the ALKBH5 knockdown group were significantly smaller than those in the control group (Fig. 5G). Additionally, the average tumor volume and euthanized weight were significantly reduced in ALKBH5 knockdown mice compared to the control group (Fig. 5H, I). The proliferation and apoptosis indices of these solid tumors were further evaluated. HE staining revealed that tumor cell structures were sparser, with smaller and evenly distributed nuclei in the ALKBH5 knockdown group compared to the control sh-NC group. Furthermore, immunohistochemical staining demonstrated a decrease in Ki-67 signal and an increase in caspase-3 activity in tumor cells of the ALKBH5 knockdown group compared to control cells (Fig. 5J). Moreover, the injection of ALKBH5 knockdown cells significantly abolished the ability of OC cells to form secondary tumors in the peritoneal cavity (Fig. 5K).
In conclusion, ALKBH5 plays a crucial regulatory role in the proliferation, migration, and invasion of OC cells. It promotes tumor growth and metastasis in vivo, emphasizing its significant oncogenic role in OC development.
ALKBH5 promotes immune evasion in OC
In clinical studies, the progression and metastasis of cancer often rely on evading immune surveillance, particularly evading T cell-mediated immune responses against tumors24,25. Given the observed deceleration in tumor growth and improved survival rates after knocking down ALKBH5, we hypothesize that ALKBH5 may promote immune evasion mechanisms by regulating the activity and function of T cells, thereby impacting the development and spread of OC.
To assess the impact of ALKBH5 on T cells, we conducted in vitro cytotoxicity assays mediated by T cells using a co-culture system, where splenocytes extracted from C57BL/6 J mice were co-cultured with mouse OC cells (ID8). Prior to co-culturing with tumor cells, T cells were activated and expanded using anti-CD3/CD28 antibodies and IL-2. The percentage of CD3+ T cells in the lymphocytes used for the experiments varied, and T cell #17, reaching 95.3%, was selected for further experiments (Supplementary Fig. 11A). The results indicated that ALKBH5 knockdown OC cells were more susceptible to T cell-mediated killing (Supplementary Fig. 11B-C), accompanied by an increase in apoptotic tumor cell numbers (Supplementary Fig. 11D). Furthermore, flow cytometry analysis identified the proportions of CD3+ T cells (representing all T cells), CD4+ T cells (helper T cells), and CD8+ T cells (cytotoxic T cells). The findings revealed a significant increase in the proportions of CD3+ T cells and CD8+ T cells after ALKBH5 knockdown, while the proportion of CD4+ T cells decreased significantly (Supplementary Fig. 11E). Additionally, levels of IFNγ and IL-2 were elevated after knocking down ALKBH5 compared to the respective control cells (Supplementary Fig. 11F). Immune evasion refers to the ability of tumor cells to escape recognition and attack by the host immune system, thereby promoting their survival and expansion. In our study, ALKBH5 knockdown OC cells exhibited increased susceptibility to T cell-mediated cytotoxicity, along with elevated secretion of cytolytic cytokines, indicating that ALKBH5 depletion suppresses immune evasion in OC.
In conclusion, ALKBH5 promotes immune evasion in OC by modulating the activity and function of T cells, subsequently accelerating tumor growth.
ALKBH5 eliminates m6A modification on ITGB1 mRNA through a transcriptional mechanism
ALKBH5 demethylates mRNA to promote or suppress proliferation in various cancers26,27. Through preceding bioinformatics analysis, we hypothesized that ITGB1 may be a crucial downstream target regulated by ALKBH5. ITGB1, a member of the integrin family, contributes to embryonic development, tumor metastasis, and angiogenesis28,29.
To investigate whether the ALKBH5 protein regulates the expression of ITGB1 mRNA, we initially examined the interaction between these two molecules. The ability of ALKBH5 to bind ITGB1 mRNA was confirmed by RIP-qPCR (Fig. 6A). Our study results demonstrate that overexpression of ALKBH5 enhances ITGB1 mRNA expression, while conversely, knockdown of ALKBH5 significantly suppresses the levels of ITGB1 mRNA (Fig. 6B, C, Supplementary Fig. 10E). m6A modification can influence RNA metabolism, including RNA stability and translation efficiency30. RNA stability assays indicated that in OC cells overexpressing ALKBH5, the expression of ITGB1 mRNA remained stable (Fig. 6D). The regulatory relationship between ALKBH5 and ITGB1 protein expression was validated through Western Blot (Fig. 6E-F). Protein stability experiments revealed that the overexpression of ALKBH5 does not affect the stability of ITGB1 protein (Fig. 6G). These results indicate that ALKBH5 regulates the expression of ITGB1 protein and mRNA through a post-transcriptional mechanism.

A RIP-qPCR experiments confirming the binding of ALKBH5 to ITGB1 mRNA; B, C PCR analysis to assess the expression changes of ITGB1 in OC cells with overexpression and knockdown; D RNA stability assay at specified time points with Actinomycin D treatment in ALKBH5 overexpressing and NC cells, along with qPCR analysis of ITGB1 mRNA expression; E, F Western blot analysis to examine the expression levels and quantitative information of ALKBH5 and ITGB1 in OC cells with overexpression and knockdown; G Protein stability assay at specified time points in ALKBH5 overexpressing and cycloheximide (CHX) treated NC cells; Western blot analysis (left) and quantification (right) of ITGB1 protein expression; H, I Dot blot experiments analyzing the impact of ALKBH5 knockdown and overexpression on m6A modification levels in cells; J Estimation of the impact of ALKBH5 overexpression on ITGB1 m6A levels using MeRIP-qPCR analysis. Cell experiments were repeated thrice. Error bars represent the standard deviation of the mean. Statistical significance was assessed using a two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
Given that ALKBH5 can modulate RNA metabolism by demethylating m6A in ITGB1 mRNA, we focused on investigating ALKBH5’s regulation of m6A modification. m6A dot-blot analysis revealed that downregulation of ALKBH5 in OC cells A2780 significantly increased the m6A modification levels on mRNA, whereas ALKBH5 overexpression had the opposite effect (Fig. 6H, I). MeRIP-qPCR experiments confirmed that the knockdown of ALKBH5 elevated the m6A modification levels on ITGB1 mRNA, while ALKBH5 overexpression reduced these levels (Fig. 6J).
These findings suggest that ALKBH5 can remove the m6A modification on ITGB1 mRNA through its demethylase activity, thereby promoting its post-transcriptional modification.
Knockdown of ITGB1 reverses ALKBH5 overexpression-induced proliferation, metastasis, and immune escape of OC in vitro and in vivo
The role of ITGB1 in regulating tumor cell migration has been acknowledged31, yet its specific function in OC cell migration remains unclear. Therefore, we stably knocked down ITGB1 in OC cells A2780 and SK-OV-3 (Supplementary Fig. 10C, D). CCK-8 assays revealed the inhibitory effect of ITGB1 knockdown on OC cell growth, which was further confirmed by EdU staining showing reduced cell proliferation (Supplementary Fig. 12A, B). Cell cycle analysis demonstrated cell arrest at the G2/M phase (Supplementary Fig. 12C). Colony formation assays further confirmed the negative impact of ITGB1 knockdown on cell clonogenicity (Supplementary Fig. 12D). Additionally, Transwell experiments indicated that ITGB1 knockdown significantly weakened the migration and invasion capabilities of OC cells (Supplementary Fig. 12E). These results underscore the crucial role of ITGB1 in sustaining the proliferation, migration, and invasion abilities of OC cells, highlighting the potential value of ITGB1 as a therapeutic target for OC.
To validate the impact of ALKBH5 regulation of ITGB1 on OC cell functions, we knocked down ITGB1 (sh-IT#2, hereinafter referred to as sh-IT) and overexpressed ALKBH5 (oe-ALK) in OC cells A2780, and examined their expression levels (Supplementary Fig. 10E, F). In vitro functional experiments revealed that ALKBH5 overexpression significantly enhanced the viability (Fig. 7A), proliferation (Fig. 7B), and clonogenic capacity (Fig. 7C) of OC cells, promoted cell cycle progression (Fig. 7D), and significantly increased cell migration and invasion capabilities (Fig. 7E). However, the knockdown of ITGB1 reversed these effects promoted by ALKBH5 overexpression (Fig. 7A–E).

A Cell growth of each group was measured by CCK8 assay; B EdU assay results and quantitative analysis of positive cells in each group (scale bar: 25 μm); C Colony formation assay and quantitative data of each group; D Flow cytometry analysis of cell cycle distribution in each group and the ratio of G1/S and G2/M phases; E Changes in migration and invasion capabilities of each group of cells detected by Transwell assay and their quantitative data (scale bar: 50 μm); F−H Comparative analysis of subcutaneous tumor growth and quantitative analysis of tumor size and weight (n = 5); I Immunohistochemical staining to evaluate the presence of Ki-67, Caspase-3, CD3, and CD8 positive staining in tumor tissues, along with quantitative data (scale bar: 20 μm); J Representative images of intraperitoneal metastatic tumors in each group with corresponding quantitative information. Data are expressed as mean ± standard deviation and intergroup comparisons were made using unpaired t-test. *P < 0.05, **P < 0.01, and ***P < 0.001 for comparison between two groups. Five mice were included in each group for animal experiments.
Furthermore, in in vivo experiments, we monitored the tumor growth of OC cells in nude mice among different treatment groups. It was observed that knocking down ITGB1 significantly inhibited the tumor growth caused by ALKBH5 overexpression, leading to reduced tumor volume and weight (Fig. 7F–H). Immunohistochemistry showed an increase in Ki-67 positive cells, a decrease in caspase-3 activity, and reduced infiltration of CD3+ and CD8 + T lymphocytes in the ALKBH5 overexpression group, whereas ITGB1 knockdown reversed these effects (Fig. 7I). Additionally, ALKBH5 overexpression markedly enhanced the formation of secondary tumors in the peritoneal cavity, while ITGB1 knockdown effectively suppressed this process (Fig. 7J). This result indicates that immune infiltration of tumor cells is influenced and promoted by ALKBH5 overexpression, confirming the association between ALKBH5 and immune evasion.
In conclusion, the knockdown of ITGB1 reverses the promoting effects of ALKBH5 overexpression on OC cell proliferation, migration, invasion, and immune escape, revealing potential regulatory mechanisms of ALKBH5 and ITGB1 in OC development.
Discussion
The role of m6A modification in tumor development has long been a subject of interest. m6A modification plays a crucial role in various cancers, and our study further reveals its impact on the biological behavior of OC. In contrast to previous studies, we have identified, for the first time, the significant role of ALKBH5-mediated m6A modification in regulating ITGB1 in the development of OC, offering a new direction for in-depth research in this field14. Through single-cell transcriptomic analysis, we further characterized the distribution and regulation of m6A-related genes across different cell populations, providing new insights into tumor heterogeneity and intercellular signaling32. Our findings align with other studies and collectively reveal critical mechanisms involving m6A in OC development and immune evasion33,34. We found that ALKBH5-mediated m6A modification not only affects the proliferation and metastasis of OC cells but also plays a role in regulating immune evasion mechanisms. These findings are consistent with previous research results, further deepening our understanding of m6A modification in tumor immune evasion33,34. Additionally, through cellular and in vivo experiments, this study confirms the significant influence of m6A modification on the development of OC, providing a theoretical basis for future treatment strategies based on m6A modification. Our study further elucidates the significance and specific regulatory mechanism of the m6A demethylase ALKBH5 in modulating ITGB1 in OC. Previous research has shown that ALKBH5 can regulate target gene expression through m6A demethylation, thereby influencing tumor cell proliferation, migration, and invasion by enhancing or suppressing cancer cell self-renewal or by modulating autophagy to alter the tumor microenvironment15. In contrast to prior research, we delve more profoundly into the interaction between ALKBH5 and ITGB1 in OC, offering a fresh perspective on the association between m6A modification and oncological biology15. By focusing on the ALKBH5–ITGB1 axis and immune-related pathways, our findings offer novel mechanistic understandings that may inform new therapeutic approaches. Ultimately, our findings contribute novel insights into the role of m6A modification in OC development, revealing the significance of ALKBH5-mediated m6A modification in regulating ITGB1 in OC. This not only has instructive implications for OC treatment but also holds promise for the development of new therapeutic strategies. Nonetheless, the limitations of this study encompass flaws in experimental models and constraints in data analysis. Future research should refine the experimental design, delve deeper into the relationship between m6A modification and immune escape, strengthen functional studies on ALKBH5, and provide more precise targets and methods for treating OC.
Through this study, we look forward to further investigating the role of m6A modification in OC, exploring the roles of different cell types in tumor development, and discussing additional potential targets regulated by ALKBH5-mediated m6A modification, offering robust support for personalized therapy and precision medicine practices. The findings of this study will serve as essential references for the continued progress in the field of oncological biology and clinical treatment. The ALKBH5-mediated ITGB1 m6A modification pathway plays a critical role in the initiation, progression, and immune evasion of OC. Targeting this axis not only enhances our understanding of OC molecular pathogenesis but also provides valuable opportunities for developing novel diagnostic biomarkers and therapeutic strategies. The translational potential of this pathway is reflected in the following aspects: (1) Diagnostic value: It serves as a prognostic biomarker and a potential target for non-invasive detection of OC. (2) Therapeutic value: It holds promise as a novel target for small-molecule drug development and immunotherapy. (3) Combination therapy: Its modulation in conjunction with existing chemotherapeutic and immunotherapeutic approaches may enhance treatment efficacy and overcome drug resistance. Thus, the ALKBH5–ITGB1 signaling axis represents a highly promising target for translational cancer research and is expected to facilitate more precise and effective treatment options for patients with OC in the future.
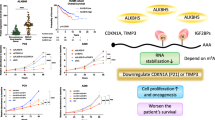
Based on the aforementioned results, we can preliminarily conclude that ALKBH5 mediates the elimination of m6A modification on ITGB1 mRNA through a transcriptional mechanism, thereby regulating the expression and stability of ITGB1, promoting proliferation, migration, and invasion of OC cells, influencing T cell function to facilitate immune escape, and consequently accelerating tumor growth and metastasis (Fig. 8). This study combines deep transfer learning and single-cell multi-omics technology for the first time, delving into the role of ALKBH5-mediated ITGB1 m6A modification in OC and revealing its crucial involvement in disease progression. Moreover, this research validates the diagnostic value of core genes and m6A regulatory factors through public databases and multi-layer neural network models while experimentally confirming the roles of ALKBH5 and ITGB1 in the biological functions of OC cells, thus providing new molecular targets for the diagnosis and treatment of OC. Nonetheless, there are some limitations. Firstly, despite employing various techniques and methods to elucidate the roles of ALKBH5 and ITGB1 in OC, further research is needed to explore their detailed molecular mechanisms. Secondly, this study primarily focuses on a single m6A regulatory factor and its target gene, suggesting that future investigations should consider other potential regulatory factors and target genes in OC, along with their interactions. Additionally, this study heavily relies on in vitro and in vivo laboratory experiments, lacking clinical trial validation. To better assess their clinical potential in OC patients, prospective clinical studies involving larger patient cohorts are necessary. Future research should address these issues to obtain a more comprehensive understanding of OC pathogenesis, thereby facilitating the development of optimal treatment strategies for OC.

(Created with BioRender.com).
Methods
Building and grouping of OC mouse models
The experimental procedures and animal use protocols were approved by the Institutional Animal Ethics Committee of Shengjing Hospital of China Medical University. SPF-grade 8 week-old female C57BL/6 J mice (n = 219, Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China) weighing 20−24 g, 6 week-old BALB/c mice (n = 211, Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China) weighing 17−21 g, and 6 week-old BALB/c Nude mice (n = 401, Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China) weighing 17−21 g were purchased. The mice were housed under standard environmental conditions: temperature of 23 ± 2°C, humidity of 55 ± 5%, and a 12 h light/dark cycle. After a 1-week acclimatization period, the experiments commenced.
Single-cell and whole-transcriptome sequencing: The procedure involved shaving the unilateral back hair of C57BL/6 J female mice, extending from the upper to lower limbs and from the side to the spine, with further disinfection and anesthesia. The midpoint between the spine and the axillary midline, along with the point of intersection between the spine and the perpendicular line to the axillary midline, was identified. A portion of the skin was lifted with forceps, the skin and muscles were incised, hemostasis was achieved, and the ovaries and fallopian tubes were located. Mouse ovarian surface epithelial cancer cells ID8 (SCC145, Merck, USA) in the logarithmic growth phase were prepared as a cell suspension at a concentration of 2 × 10⁷/mL and injected into both ovarian bursae of mice to establish an orthotopic OC mouse model. Abdominal circumference was measured after removing the skin and subcutaneous fat to avoid external interference and accurately reflect ascites accumulation. Equal volumes of PBS were injected into both ovarian bursae of control mice as the sham operation group. On day 32, the mice were euthanized by orbital exsanguination, and tumor tissues as well as normal ovarian tissues were aseptically harvested, placed in cryotubes, and stored at –80°C35,36. Each group consisted of 5 mice (n = 5). The experimental groups were: Sham group (normal control) and OC group (tumor tissue).
In vivo tumor occurrence assessment: Human OC cells A2780 (SCC257, Merck, USA) were infected with shRNA or empty vectors to produce a cell suspension of 5 × 106/ml, which was subcutaneously implanted into the subcutaneous tissues of the flank of BALB/c Nude mice (0.2 ml per mouse) to establish an OC subcutaneous xenograft mouse model. The tumor dimensions were measured and recorded every 3 days using a caliper; tumor volume (V) was calculated as length × width2/2. After 4 weeks, the mice were euthanized, and tumors were dissected, photographed, and weighed37,38. Each experimental group consisted of n = 5 mice. The experimental groups were sh-NC (A2780 cells infected with sh-NC), sh-ALK#1 (A2780 cells infected with sh-ALKBH5#1), sh-ALK#2 (A2780 cells infected with sh-ALKBH5#2), oe-NC (A2780 cells infected with oe-NC), oe-ALK (A2780 cells infected with oe-ALKBH5), oe-ALK + sh-NC (A2780 cells co-infected with oe-ALKBH5 and sh-NC), and oe-ALK + sh-IT (A2780 cells co-infected with oe-ALKBH5 and sh-ITGB1).
In vivo tumor metastasis evaluation: A2780 cells were infected with shRNA, overexpression plasmids, or empty vectors to create a cell suspension at a concentration of 1 × 106/ml, which was injected into the abdominal cavity of BALB/c mice to establish an OC intraperitoneal metastasis model. After 4 weeks, the mice were euthanized, and the number of metastatic nodules was counted37. Each experimental group consisted of n = 5 mice. The experimental groups were sh-NC (A2780 cells infected with sh-NC), sh-ALK#1 (A2780 cells infected with sh-ALKBH5#1), sh-ALK#2 (A2780 cells infected with sh-ALKBH5#2), oe-NC (A2780 cells infected with oe-NC), oe-ALK (A2780 cells infected with oe-ALKBH5), oe-ALK + sh-NC (A2780 cells co-infected with oe-ALKBH5 and sh-NC), and oe-ALK + sh-IT (A2780 cells co-infected with oe-ALKBH5 and sh-ITGB1).
scRNA-seq
Normal ovarian tissue and tumor tissue were collected in mice, with three samples in each group. The tissues were dissociated into single-cell suspensions using trypsin (9002-07-7, Sigma Aldrich, USA) and captured as individual cells using the C1 Single-Cell Auto Prep System (Fluidigm, Inc., South San Francisco, CA, USA). Subsequently, the captured cells were lysed within the chip, releasing mRNA, which was then reverse-transcribed to generate cDNA. The lysed and reverse-transcribed cDNA underwent pre-amplification on a microfluidic chip for subsequent sequencing. The amplified cDNA was used for library construction and subjected to single-cell sequencing on the HiSeq 4000 Illumina platform (parameters: paired-end reads, read length 2 × 75 bp, ~20,000 reads per cell)39.
Data analysis was performed using the R software package “Seurat.” Cells were filtered based on the criteria of 200 < nFeature_RNA < 5000 & percent.mt < 20, and the top 2000 highly variable genes were selected for further analysis40.
To reduce the dimensionality of the scRNA-Seq dataset, principal component analysis (PCA) was conducted based on the top 2000 highly variable genes. The top 20 principal components were selected for downstream analysis using the Elbowplot function in the Seurat package. Cell subgroups were identified with default resolution (res = 1) using the FindClusters function in Seurat. Subsequently, the t-distributed Stochastic Neighbor Embedding (t-SNE) algorithm was applied to reduce the nonlinear dimensionality of the scRNA-seq sequencing data. Marker genes for various cell subgroups were identified using the Seurat package and annotated using the “SingleR” package41. Cell communication analysis was performed using the “CellChat” package in R.
Differentially expressed genes (DEGs) in the scRNA-Seq dataset were identified using the “Limma” package in R. DEGs between normal and OC samples were filtered with criteria of |log2FC | > 1 & P.adjust < 0.0542.
Bulk RNA extraction and sequencing
Normal ovarian tissues and tumor tissues from mice were collected (15 samples per group), and total RNA was isolated using Trizol reagent (15596026, Invitrogen, Carlsbad, CA, USA). The RNA sample concentration and purity were assessed using an Agilent 2100 Bioanalyzer (G2939B, Agilent, USA). Total RNA samples meeting the following criteria were selected for downstream experiments: RNA Integrity Number (RIN) ≥ 7.0, 28S:18S ratio ≥ 1.5. The sequencing libraries were generated and sequenced by CapitalBio Technology (Beijing, China). In brief, 5 μg of RNA was used for each sample. The Ribo-Zero™ Magnetic Kit (MRZB12424, Epicentre Technologies Corp, USA) was utilized to deplete ribosomal RNA (rRNA) from the total RNA. The NEB Next Ultra RNA Library Prep Kit (#E7775S, NEB, USA) was employed for library construction for sequencing on the Illumina platform. Subsequently, RNA fragments were enzymatically fragmented into ~300 base pair (bp) fragments in NEB Next First Strand Synthesis Reaction Buffer (5×). The first-strand cDNA was synthesized using reverse transcriptase primers and random primers, and the second-strand cDNA was synthesized in NEB Next Second Strand Synthesis Reaction Buffer supplemented with dUTP Mix (10×). End repair of cDNA fragments, including polyadenylation and adapter ligation, was performed. After ligating the Illumina sequencing adapters, the second strand of cDNA was digested using USER Enzyme (#M5508, NEB, USA) to generate strand-specific libraries. Library DNA was then amplified, purified, and enriched by PCR. Subsequently, the libraries were assessed using the Agilent 2100 Bioanalyzer and quantified using the KAPA Library Quantification Kit (KK4844, KAPA Biosystems). Finally, paired-end sequencing was carried out on the NextSeq CN500 (Illumina), achieving an average sequencing depth of 50 × 43,44.
Quality control of sequencing data and differential gene analysis
The quality of paired-end reads of raw sequencing data was assessed using FastQC software version 0.11.8. Preprocessing of the raw data was performed using Cutadapt software version 1.18 to remove Illumina sequencing adapters and poly(A) tail sequences. Reads with an N content exceeding 5% were filtered out using a perl script. The FASTX Toolkit software version 0.0.13 was employed to extract reads with a base quality of at least 20 and accounting for 70% of the bases. BBMap software was utilized to fix any issues with the paired-end sequences. Subsequently, the filtered high-quality read fragments were aligned to the mm10 reference genome provided by Gencode using hisat2 software version 0.7.12. Differential gene expression analysis between normal and OC samples was conducted with the “Limma” package in R software under the criteria of |log2FC | >1.5 and a P-value of <0.05 to identify DEGs. Differential gene expression heatmaps were generated using the “heatmap” package, volcano plots were created by the “ggplot2” package, and GO and KEGG enrichment analyses were carried out using the “clusterProfiler” package45,46. The potential m6A modification target genes of ALKBH5 and m6A methylation sites of ITGB1 were explored through the RM2Target (http://rm2target.canceromics.org/#/home) and SRAMP (http://www.cuilab.cn/sramp) databases47.
Cell culture and transfection
Mouse ovarian epithelial cancer cells ID8 (SCC145, Merck, USA), human normal ovarian epithelial cells IOSE-80 (IM-H366, Yimo Biotech, Xiamen, China), human OC cells A2780 (SCC257, Merck, USA), and human OC cells SK-OV-3 (CBP60291, Nanjing Kebai Biotechnology Co., Ltd., Jiangsu, China) were cultured in DMEM medium (11965092, Gibco, USA) containing 10% FBS (TMS-016, Sigma Aldrich, USA), 10 μg/mL streptomycin (85886, Sigma Aldrich, USA), and 100 U/mL penicillin (9073-60-3, Sigma Aldrich, USA).
Spleen cells were extracted from C57BL/6 J mice, and T cells and CD8 + T cells were separated from the spleen using Mouse T Cell Isolation Kit (8804-6820-74, ThermoFisher, USA) and Mouse CD8 + T Cell Isolation Kit (8804-6822-74, ThermoFisher, USA), respectively. The cells were cultured in RPMI-1640 medium (R4130, Sigma, USA) and activated by adding CD3 antibody (2 μg/mL; 11-0032-82, 1:50, ThermoFisher, USA), CD28 antibody (1 μg/mL, 12-0281-82, ThermoFisher, USA), and interleukin-2 (IL-2, 5 ng/mL; 402-ML-100/CF, R&D Systems, USA). Activated T cells, CD8 + T cells, and mouse OC cells ID8 were co-cultured at a ratio of 4:1.
The cells were maintained in a humidified incubator (Heracell™ Vios 160i CO2 Incubator, Thermo Scientific™, Germany) at 37 °C with 5% CO2. Passaging was performed when the cells reached 80%−90% confluency for continuation of the culture48,49,50,51.
Groups: sh-NC (transfected with sh-NC), sh-ALK#1 (transfected with sh-ALKBH5#1), sh-ALK#2 (transfected with sh-ALKBH5#2), sh-IT#1 (transfected with sh-ITGB1#1), sh-IT#2 (transfected with sh-ITGB1#2), NC (transfected with oe-NC), ALKBH5 (transfected with oe-ALKBH5).
Lentivirus
The method of lentivirus infection is based on the construction of corresponding cell lines. Recombinant lentivirus particles and virus packaging reagents, including sh-NC, sh-ALKBH5, sh-ITGB1, oe-NC, and oe-ALKBH5, were purchased from Shanghai JimaGen (Shanghai, China). These were then co-transfected into HEK-293T cells (CRL-3216, ATCC, USA) using lipofectamine 2000 (11668500, Invitrogen, USA). After 48 h of cell culture, the virus titer was determined using a p24 ELISA kit (VPK-108-H, Cell Biolabs, Inc., USA) and adjusted to 109 TU/mL. Subsequently, 5 × 105 cells were seeded in 6-well plates, and when the cell density reached 50-70%, the medium containing an appropriate amount of packaged lentivirus (MOI = 10, working titer ~5 × 106 TU/mL) and 5 μg/mL polybrene (TR-1003, Merck, USA) was added for transfection. Following a 4 h transfection period, an equal amount of medium was added to dilute the polybrene. After 24 h, fresh medium was replaced, and 48 h post-transfection, the transfection efficiency was observed through a luciferase reporter gene, followed by selection with 10 μg/mL puromycin (A1113803, Gibco, Grand Island, NY, USA) to obtain stably transduced cell lines. Cells were collected when no further deaths occurred in the puromycin-containing medium, and the overexpression and knockdown efficiency were confirmed by RT-qPCR. The silencing sequences are presented in Supplementary Table 752.
Flow cytometry
Cells were isolated from mouse tumor tissues and fixed and permeabilized using the Cytofix/Cytoperm Plus kit from BD (555028, USA). Cell staining was performed using rabbit anti-CD3 (11-0032-82, 1:50, ThermoFisher, USA), rabbit anti-CD8 (47-0081-82, 1:50, ThermoFisher, USA), rabbit anti-CD4 (47-0042-82, 1:50, ThermoFisher, USA), rabbit anti-IFN-γ (48-7311-82, 1:50, ThermoFisher, USA), and rabbit anti-IL2 (16-7021-85, 1:50, ThermoFisher, USA) antibodies. Flow cytometric data of the stained cells were acquired using FlowJo CE software and a flow cytometer. Subsequent data analysis was conducted using FlowJo software51,53.
Flow cytometry was employed to detect cell cycle changes. Cells were co-cultured at a concentration of 1 × 106/ml and seeded into 6-well plates with 4 mL per well. Cells were collected by centrifugation, washed twice with PBS, fixed in 70% ethanol, stored overnight at 4°C, and washed with PBS before staining. The cells were then stained with 500 μL of Propidium Iodide (PI) (P8080, Cell Signaling Technology, Beijing, China), mixed thoroughly, and incubated at 4°C in the dark for 30 min. Flow cytometry was used to detect fluorescence at an excitation wavelength of 488 nm, distinguishing different stages of the cell cycle (G0/G1 phase, S phase, G2/M phase)54,55.
Flow cytometry was utilized to detect cell apoptosis. In brief, co-cultured cells (1 × 105/well) were collected and washed in chilled PBS, followed by staining in the dark for 15 min with a detection kit (APOAF-20TST, Sigma-Aldrich, USA). Subsequently, the pellet was resuspended in 400 μL of binding buffer and stained with 5 μL of Annexin-V as provided in the kit. Cell analysis was performed using a flow cytometer56. Cells in the upper right quadrant with the phenotype Annexin V + PI+ represented late apoptotic cells; cells in the lower right quadrant with the phenotype Annexin V + PI- represented early apoptotic cells; cells in the upper left quadrant with the phenotype Annexin V-PI+ represented necrotic cells; and cells in the lower left quadrant with the phenotype Annexin V-PI- denoted viable cells57.
CCK-8 assay for cell proliferation and colony formation experiment
The CCK-8 assay kit (C0037, Beyotime, Shanghai, China) was utilized to assess cell proliferation. Cells from different logarithmic growth phases were taken, adjusted to a concentration of 5 × 104 cells/mL, seeded into 96-well culture plates with 100 μL of cell suspension per well, and then incubated for 24, 48, 72, and 96 h in a CO2 incubator. At specific time points, 10 μL of CCK-8 solution was added to each well, followed by a 2 h incubation at 37°C, and the absorbance value at 450 nm (A) was measured using a Multiskan FC microplate reader (Thermo Fisher, 51119080, USA). Each group was run in triplicates, and the average was calculated57,58.
For the colony formation experiment, cells from the exponential growth phase were taken and passaged using standard digestion methods to obtain a cell suspension with over 95% single cells. The cells were counted and diluted to an appropriate concentration with a medium, and 5 mL of cell suspension containing 100 cells per dish (60 mm in diameter) was seeded. The dish was gently rocked in a cross pattern to ensure even cell distribution. The dishes were then incubated at 37°C with 5% CO2 for 2–3 weeks. When visible colonies appeared in the dishes, the culture was terminated, the medium was removed, and the dishes were washed twice with PBS and air-dried. The colonies were fixed with methanol for 15 min, air-dried after methanol removal, stained with Giemsa stain for 10 min, rinsed gradually with running water to remove excess stain, and air-dried. Colonies containing >10 cells were counted either by the naked eye or under a microscope (low magnification). The colony formation rate was calculated using the formula: Colony formation rate = (number of colonies/number of seeded cells) × 100%58,59.
EdU staining
EdU staining is employed to examine cell proliferation. Cells from different groups in the logarithmic growth phase were adjusted to a concentration of 1 × 104 cells/mL, seeded into a 96-well plate, and triplicated. After transfection for 24 h, cells were harvested. Subsequently, cells were stained with EdU (C0071S, Beyotime, Shanghai, China) for 2 h at a final concentration of 50 μmol/L, washed thrice with PBS, and then stained with DAPI staining solution (C1002, Beyotime Biotechnology, Shanghai) for 15 min at room temperature. Slides were mounted, and 6−10 random fields were observed under a fluorescence microscope (BX63, Olympus, Japan). The number of positive cells in each field was recorded. The EdU labeling rate (%) was calculated as follows: % EdU labeling = (number of positive cells / (number of positive cells + number of negative cells)) × 100%60,61.
Evaluation of cell migration and invasion using transwell assays
In the experiments, migration and invasion assays were conducted using 8 μm Transwell chambers (3422, Corning, USA) in a 24-well plate. For invasion assays, prior to the experiment, 100 μL of ECM gel (E1270, Sigma-Aldrich, Germany) was coated in each chamber and then incubated at 37°C for 2 h. Cells were trypsinized, washed twice with PBS, resuspended in serum-free medium, and adjusted to a cell density of 3 × 105 cells/mL. Three chambers were used for each group, with 200 μL of cell suspension added to each chamber, and 700 μL of complete culture medium was added to the bottom chamber. The chambers were then incubated at 37°C with 5% CO2 for 48 h. After fixation with methanol for 30 min, the chambers were stained with 0.05% crystal violet (G1062, Solarbio, Beijing, China) for 5 min. The cells inside the chambers were wiped off with a cotton swab, and images were captured under an inverted microscope (IX73, OLYMPUS, Japan) for observation. ImageJ software was utilized for image processing and quantification. Gel coating in the chambers was not required for migration assays, while all other experimental procedures were identical to those for invasion assays. At least four random microscopic fields were counted for cells in each experiment62,63.
Quantification of total m6A in RNA
The total m6A levels in RNA were measured utilizing the EpiQuik m6A RNA Methylation Kit (Colorimetric) (P-9005-48, Epigentek, USA). In brief, total RNA was extracted from the cells of interest and quantified using a Nanodrop 2000 (Thermo Fisher Scientific, USA). Subsequently, 200 ng of total RNA and the binding solution were added to each well and incubated at 37°C for 1.5 h. Following this, capture antibody, detection antibody, enhancer solution, color development solution, and stop solution were sequentially added to each well, and the reactions were allowed to proceed for a specified time. Finally, the absorbance values at 450 nm were measured using a microplate reader (Molecular Devices, USA) to compare the relative m6A levels in each group64.
Analysis of methylated RNA immunoprecipitation (MeRIP)-PCR
The MeRIP detection of m6A modification was performed using the Magna MeRIP™ m6A Kit (17-10499) from MERCK, following the manufacturer’s instructions. In essence, total RNA (300 μg) was fragmented into ~100-nucleotide-long pieces, and m6A antibody (10 μg, ab286164, Abcam, UK) was pre-coated onto protein A/G magnetic beads (88802, Thermo Fisher Scientific, USA). Subsequently, the RNA fragments were thoroughly mixed with the m6A antibody-coated beads and incubated under rotation conditions for 2 h. The methylated RNA fragments obtained from immunoprecipitation were eluted for PCR analysis65.
RNA-binding protein immunoprecipitation (RIP)-PCR analysis
The RNA immunoprecipitation (RIP) experiment was conducted using the Magna RIP® RNA-Binding Protein Immunoprecipitation Kit from MERCK (17-700) following the manufacturer’s instructions. The cell lysates were treated overnight at 4°C with beads coated with ALKBH5 antibody (5 μg, 16837-1-AP, Proteintech, USA). Post-immunoprecipitation, the beads were washed five times with high or low salt buffer. The RNA-protein-magnetic bead complexes were washed and eluted using proteinase K digestion buffer (AM2546, Thermo Fisher Scientific, USA). Immunoprecipitated RNA was extracted using the Trizol (15596026, Thermo Fisher Scientific, USA)-chloroform (J67241-AP, Thermo Fisher Scientific, USA) method. Subsequently, the enriched RNA was quantitatively analyzed using PCR and normalized to the input. Details of the antibodies used in the RIP assay are provided in Supplementary Table 815.
Determination of spot imprints
RNA was incubated with an equal amount of 20× sodium citrate buffer (P4922, Merck, USA) at 95°C for 5 min. Subsequently, 100, 200, or 400 ng of Poly(A) RNA was applied to a Hybond N membrane (RPN1520B, GE Healthcare). After UV crosslinking for 30 min and blocking with 5% milk, the membrane was incubated overnight at 4°C with anti-m6A antibody (ab286164, Abcam, UK) and detected using a secondary antibody conjugated with horseradish peroxidase HRP (P0202, Biyuntian, Shanghai, China). Imaging was carried out using an ECL working solution (Omt-01, Beijing Omegajade Pharmaceutical Technology Co., Ltd., Beijing, China)65.
Analysis of RNA stability
To measure the RNA stability of various cell groups, we treated the cells with Actinomycin D (SBR00013, Merck, USA) at a concentration of 5 μg/ml. After incubating for a specified period, cell samples were collected at 2, 4, 6, and 8 h, followed by RNA extraction from each sample for real-time quantitative polymerase chain reaction using GAPDH as an internal reference66.
Analysis of protein stability
To assess the protein stability of different cell groups, we supplemented cells with cycloheximide (CHX) (CAS: 66-81-9, Merck, USA) at a concentration of 100 μg/ml. After a designated incubation period, total protein was extracted at specific time points, and the expression of ITGB1 protein was detected using Western blot analysis15.
Hematoxylin and eosin staining
Tumor tissues from mice were embedded in paraffin and sectioned. The sections were sequentially immersed in xylene I for 10 min, xylene II for 10 min, absolute ethanol I for 5 min, absolute ethanol II for 5 min, 95% ethanol for 5 min, 90% ethanol for 5 min, 80% ethanol for 5 min, 70% ethanol for 5 min, followed by distilled water rinsing. After deparaffinization and rehydration, the sections were stained with Harris hematoxylin for 10–30 min, rinsed in tap water, differentiated in 1% hydrochloric acid alcohol briefly, rinsed again in tap water, counterstained with 0.6% ammonia water, and rinsed in running water. The sections were then stained with eosin for 1–3 min. Subsequently, they were dehydrated and cleared in 95% ethanol I for 5 min, 95% ethanol II for 5 min, absolute ethanol I for 5 min, absolute ethanol II for 5 min, xylene I for 5 min, and xylene II for 5 min, before being air-dried after removal from xylene and mounted with neutral resin. The tissue morphology was observed under a microscope (BX63, Olympus, Japan), and the experiment was replicated three times. Five random fields were chosen for cellular morphology and structural analysis. All chemicals used in the experiment were purchased from Sangon Biotech (Guangzhou, China)67.
Immunohistochemical staining (IHC)
Mouse tumor tissues were fixed, decalcified, dehydrated in graded alcohols, embedded in paraffin, and cut into 5 µm thick sections. The sections were baked at 60°C for 20 min, followed by successive immersion in xylene, with two changes of xylene for 15 min each, dehydration in absolute alcohol for 5 min, another change of absolute alcohol for 5 min, hydration in 70% and 95% alcohols for 10 min each. Each section was then treated with 3% H2O2 for 10 min at room temperature to block endogenous peroxidase activity. Subsequently, antigen retrieval was performed by treating the sections with citrate buffer in a microwave for 3 min, followed by incubation with antigen retrieval solution at room temperature for 10 min, and washed with PBS thrice. Normal goat serum blocking solution (C-0005, Shanghai Haoran Biotechnology Co., Ltd., Shanghai, China) was applied to the tissue sections at room temperature for 20 min, followed by incubation with primary antibodies: rabbit anti-Ki-67 (ab15580, 5 µg/ml, Abcam), rabbit anti-Caspase-3 (ab184787, 1:1000, Abcam), rabbit anti-CD3 (ab16669, 1:150, Abcam), rabbit anti-CD8 (ab217344, 1:2000, Abcam) in a mixture overnight at 4°C. The next day, the sections were incubated with goat anti-rabbit IgG (ab6721, 1:1000, Abcam) secondary antibody for 30 min, followed by incubation with streptavidin-biotin complex (SABC) (SA0041, Solarbio, Beijing, China) for 30 min in a 37°C incubator. DAB chromogen solution from the DAB staining kit (DA1010, Solarbio, Beijing, China) was applied to the sections for 6 min to visualize the immunoreactivity, followed by counterstaining with hematoxylin (C0107, Beyotime, Shanghai, China) for 30 s. Subsequently, the sections were dehydrated in 70%, 80%, 90%, 95% ethanol, and absolute ethanol for 2 min each, immersed twice in xylene for 5 min each for transparency, before mounting with neutral resin. The sections were examined under a light microscope (BX63, Olympus, Japan), and the experiment was repeated three times. PBS was used as a negative control instead of the primary antibody. Five different fields of view from the IHC images were selected for quantitative analysis, counting the number of cells with brown staining signal representing positive staining cells, calculating the percentage of positive cells as the ratio of brown-stained cells to total cells multiplied by 100%68.
Detection of target gene expression by RT-qPCR
Total RNA was extracted using a Trizol reagent (15596026, Invitrogen, USA) as per the manufacturer’s instructions. The RNA was reverse transcribed into cDNA using the PrimeScript RT reagent Kit (RR047A, Takara, Japan). The synthesized cDNA was then subjected to RT-qPCR analysis using the Fast SYBR Green PCR kit (11736059, Thermo Fisher Scientific (China) Co., Ltd., Shanghai, China) with three replicates per well. GAPDH was used as an internal control, and relative expression levels were calculated using the 2-ΔΔCt method. The experiment was repeated thrice. The primer sequences utilized for RT-qPCR in our study are detailed in Supplementary Table 9, with the primers synthesized by Takara69.
Western blot analysis
Total protein was extracted from cells and tissues. Cells and tissues were digested using trypsin (T4799-5G, Sigma-Aldrich, USA) and collected. Enhanced RIPA lysis buffer containing protease inhibitors (AR0108, Wuhan Boshide Company, Wuhan, China) was used for cell lysis, and the protein concentration was measured using a BCA protein quantification kit (AR1189, Wuhan Boshide Company, Wuhan, China). The proteins were separated by SDS-PAGE and transferred to a PVDF membrane. The membrane was blocked with 5% BSA (9048-46-8, Sigma-Aldrich, USA) at room temperature for 1 h, followed by overnight incubation with diluted primary antibodies (see Supplementary Table 8 for details) at 4°C. The membrane was washed three times with PBST (3 × 5 min) and then incubated at room temperature for 1 h with Anti-Mouse-HRP secondary antibody (7076, 1/5000; CST, USA) or Anti-Rabbit-HRP secondary antibody (7074, 1/5000; CST, USA). The membrane was washed again three times with PBST (3 × 5 min). PBST was removed, and an appropriate amount of ECL working solution (Omt-01, Beijing Omijia Medical Technology Co., Ltd., Beijing, China) was added. The membrane was incubated at room temperature for 1 min for membrane development; excess ECL reagent was removed, the membrane was sealed with plastic wrap, placed in a dark box, and exposed to X-ray film for 5−10 min for visualization and imaging. The Western blot image bands were quantified for grayscale using Image J analysis software, with GAPDH (ab7291, 1:10000, Abcam, UK) used as an internal control for total cellular proteins70,71,72. All blots derive from the same experiment and were processed in parallel and the full un-cropped WB images are available at Supplementary Figs. 13-33.
Statistical analysis
All data were analyzed using SPSS 22.0 statistical software (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 7.0. Descriptive statistics for continuous variables were expressed as means ± standard deviation (Mean ± SD). Group comparisons were performed using independent t-tests, while comparisons among multiple groups were conducted using one-way analysis of variance (ANOVA). Homogeneity of variances was assessed using Levene’s test, and when the variances were determined to be homogeneous, pairwise comparisons were made using Dunnett’s t-test and the LSD-t test. In cases of inhomogeneous variances, Dunnett’s T3 test was utilized. A statistical significance level was set at P < 0.05 for determining differences between groups.
Code availability
Custom R scripts were used in this study for data generation, preprocessing, and analysis, including differential expression analysis, clustering, and visualization. These scripts are not publicly available but can be obtained from the corresponding author upon reasonable request. The analyses were conducted using R software version 4.2.1, primarily employing packages such as Seurat, limma, clusterProfiler, and ggplot2, with default or recommended parameters unless otherwise specified in the Methods section.
Data availability
The raw sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA1255838 and submission ID SUB15284108. Bulk transcriptomic RNA-seq data: Control group: SRR33323495, SRR33323494, SRR33323483, SRR33323472, SRR33323465, SRR33323464, SRR33323463, SRR33323462, SRR33323461, SRR33323460, SRR33323493, SRR33323492, SRR33323491, SRR33323490, SRR33323489. Treatment group: SRR33323488, SRR33323487, SRR33323486, SRR33323485, SRR33323484, SRR33323482, SRR33323481, SRR33323480, SRR33323479, SRR33323478, SRR33323477, SRR33323476, SRR33323475, SRR33323474, SRR33323473. Single-cell transcriptomic RNA-seq data. Control group: SRR33323471, SRR33323470, SRR33323469. Treatment group: SRR33323468, SRR33323467, SRR33323466. These datasets are publicly available at the NCBI SRA and can be accessed using the above accession numbers.
References
Kumari, A. et al. TGFβ signaling networks in ovarian cancer progression and plasticity. Clin. Exp. Metastasis 38, 139–161 (2021).
Punzón-Jiménez, P., Lago, V., Domingo, S., Simón, C. & Mas, A. Molecular management of high-grade serous ovarian carcinoma. IJMS 23, 13777 (2022).
Asangba, A. E. et al. Diagnostic and prognostic potential of the microbiome in ovarian cancer treatment response. Sci. Rep. 13, 730 (2023).
Chen, J., Fang, Y., Xu, Y. & Sun, H. Role of m6A modification in female infertility and reproductive system diseases. Int. J. Biol. Sci. 18, 3592–3604 (2022).
Xu, F. et al. FBW7 suppresses ovarian cancer development by targeting the N6-methyladenosine binding protein YTHDF2. Mol. Cancer. 20, 45 (2021).
Nie, S. et al. ALKBH5-HOXA10 loop-mediated JAK2 m6A demethylation and cisplatin resistance in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 40, 284 (2021).
Ye, L. et al. RNA epigenetic modifications in ovarian cancer: the changes, chances, and challenges. WIREs RNA. 14, e1784 (2023).
Zhao, S. et al. The potential regulatory role of RNA methylation in ovarian cancer. RNA Biol. 20, 207–218 (2023).
Jiang, X. et al. The role of m6A modification in the biological functions and diseases. Sig. Transduct. Target Ther. 6, 74 (2021).
Deng, L. J. et al. m6A modification: recent advances, anticancer targeted drug discovery and beyond. Mol. Cancer. 21, 53 (2022).
Oerum, S., Meynier, V., Catala, M. & Tisné, C. A comprehensive review of m6A/m6Am RNA methyltransferase structures. Nucleic Acids Res. 49, 7239–7255 (2021).
Jin, Q., Qu, H. & Quan, C. New insights into the regulation of METTL3 and its role in tumors. Cell Commun. Signal 21, 334 (2023).
Li, J., Zhang, H. & Wang, H. N1-methyladenosine modification in cancer biology: Current status and future perspectives. Comput. Struct. Biotechnol. J. 20, 6578–6585 (2022).
Wu, X. et al. Captive ERVWE1 triggers impairment of 5-HT neuronal plasticity in the first-episode schizophrenia by post-transcriptional activation of HTR1B in ALKBH5-m6A dependent epigenetic mechanisms. Cell Biosci 13, 213 (2023).
Sun, R. et al. ALKBH5 activates FAK signaling through m6A demethylation in ITGB1 mRNA and enhances tumor-associated lymphangiogenesis and lymph node metastasis in ovarian cancer. Theranostics 13, 833–848 (2023).
Torre, L. A. et al. Ovarian cancer statistics, 2018. CA A Cancer J. Clin. 68, 284–296 (2018).
Karlan, B. Y., Alvarez, R. D. & Strauss, J. F. Evolving poaradigms in research and care in ovarian cancers. Obstet. Gynecol. 128, 771–774 (2016).
Zhao, W. et al. Epigenetic regulation of m6a modifications in human cancer. Mol. Ther. Nucleic Acids 19, 405–412 (2020).
Liu, Z. X., Li, L. M., Sun, H. L. & Liu, S. M. Link between m6A modification and cancers. Front. Bioeng. Biotechnol. 6, 89 (2018).
He, L. et al. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer. 18, 176 (2019).
Wang, Y. et al. Four m6A RNA Methylation Gene Signatures and Their Prognostic Values in Lung Adenocarcinoma. Technol. Cancer Res. Treat. 21, 15330338221085373 (2022).
Garcias Morales, D. & Reyes, J. L. A. birds'-eye view of the activity and specificity of the mRNA m6A methyltransferase complex. WIREs RNA 12, e1618 (2020).
Hu, B. bin. et al. N6-methyladenosine (m6A) RNA modification in gastrointestinal tract cancers: roles, mechanisms, and applications. Mol. Cancer 18, 178 (2019).
Gajewski, T. F., Schreiber, H. & Fu, Y. X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013).
Ma, L. et al. Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity. Cell 186, 3148–3165.e20 (2023).
Guo, X. et al. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A-YTHDF2-dependent manner. Mol. Cancer 19, 91 (2020).
Jiang, Y. et al. RNA demethylase ALKBH5 promotes ovarian carcinogenesis in a simulated tumour microenvironment through stimulating NF-κB pathway. J. Cell. Mol. Med. 24, 6137–6148 (2020).
Brakebusch, C., Hirsch, E., Potocnik, A. & Fässler, R. Genetic analysis of beta1 integrin function: confirmed, new and revised roles for a crucial family of cell adhesion molecules. J. Cell Sci. 110, 2895–2904 (1997).
Mettouchi, A. & Meneguzzi, G. Distinct roles of β1 integrins during angiogenesis. Eur. J. Cell Biol. 85, 243–247 (2006).
An, Y. & Duan, H. The role of m6A RNA methylation in cancer metabolism. Mol. Cancer 21, 14 (2022).
Sun, L., Guo, S., Xie, Y. & Yao, Y. The characteristics and the multiple functions of integrin á1 in human cancers. J. Transl. Med 21, 787 (2023).
Wang, X., Wu, X., Hong, N. & Jin, W. Progress in single-cell multimodal sequencing and multi-omics data integration. Biophys. Rev. 16, 13–28 (2023).
Liu, Z. et al. N6-methyladenosine-modified circIGF2BP3 inhibits CD8+ T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of PD-L1 in non-small cell lung cancer. Mol. Cancer 20, 105 (2021).
Pan, J., Huang, T., Deng, Z. & Zou, C. Roles and therapeutic implications of m6A modification in cancer immunotherapy. Front Immunol. 14, 1132601 (2023).
Cordero, A. B., Kwon, Y., Hua, X. & Godwin, A. K. et al. In vivo imaging and therapeutic treatments in an orthotopic mouse model of ovarian cancer. JoVE. 19, 2125 (2010).
Cho, S. et al. Characterization and evaluation of pre-clinical suitability of a syngeneic orthotopic mouse ovarian cancer model. Anticancer Res. 33, 1317–1324 (2013).
Liu, T. et al. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 48, 3816–3831 (2020).
Tsang, S. I., Hassan, A. A., To, S. K. Y. & Wong, A. S. T. Experimental models for ovarian cancer research. Exp. Cell Res. 416, 113150 (2022).
Keefe, F., Monzón-Sandoval, J., Rosser, A. E., Webber, C. & Li, M. Single-cell transcriptomics reveals conserved regulatory networks in human and mouse interneuron development. IJMS 24, 8122 (2023).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Ma, L. et al. Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell 36, 418–430.e6 (2019).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015).
Arunachalam, D. et al. Expression of immune response genes in human corneal epithelial cells interacting with Aspergillus flavus conidia. BMC Genomics 23, 5 (2022).
Linkner, T. R. et al. Cellular proteo-transcriptomic changes in the immediate early-phase of lentiviral transduction. Microorganisms 9, 2207 (2021).
Li, K. P., Yu, T., Hwang, J. D., Yeah, K. S. & Winefordner, J. D. Estimation of rate constants using statistical moments of spatially resolved signal profiles for the elucidation of analyte transformation mechanisms in an inductively coupled plasma. Anal. Chem. 60, 1599–1605 (1988).
Racle, J. & Gfeller, D. EPIC: ATool to estimate the proportions of different cell types from bulk gene expression data. Method. Mol. Biol. 2120, 233–248 (2020).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Lyu, Z. J. et al. Recurrent ZNF83-E293V mutation promotes bladder cancer progression through the NF-κB pathway via transcriptional dysregulation of S100A8. Mol. Ther. 29, 275–290 (2021).
Zhu, S. et al. Preclinical models for bladder cancer research. Hematol./Oncol. Clin. North Am. 35, 613–632 (2021).
Zhang, X., Zhao, Y., Kong, P., Han, M. & Li, B. Expression of circZNF609 is Down-regulated in colorectal cancer tissue and promotes apoptosis in colorectal cancer cells by upregulating p53. Med. Sci. Monit. 25, 5977–5985 (2019).
Qiu, X. et al. M6A demethylase ALKBH5 regulates PD-L1 expression and tumor immunoenvironment in intrahepatic cholangiocarcinoma. Cancer Res. 81, 4778–4793 (2021).
Yan, R. et al. Knockdown of augmenter of liver regeneration in HK-2 cells inhibits inflammation response via the mitogen-activated protein kinase signaling pathway. Inflamm. Res. 64, 453–462 (2015).
Luo, Z. W. et al. Extracellular vesicles from akkermansia muciniphila elicit antitumor immunity against prostate cancer via modulation of CD8+ T cells and macrophages. IJN ume 16, 2949–2963 (2021).
You, Y. et al. ALKBH5/MAP3K8 axis regulates PD-L1+ macrophage infiltration and promotes hepatocellular carcinoma progression. Int J. Biol. Sci. 18, 5001–5018 (2022).
Baharuddin, A. A., Roosli, R. A. J., Zakaria, Z. A. & Md. Tohid, S. F. Dicranopteris linearis extract inhibits the proliferation of human breast cancer cell line (MDA-MB-231) via induction of S-phase arrest and apoptosis. Pharm. Biol. 56, 422–432 (2018).
Tong, D. et al. Circ-Usp10 promotes microglial activation and induces neuronal death by targeting miRNA-152-5p/CD84. Bioengineered 12, 10812–10822 (2021).
Zhou, K. X. et al. Increased nuclear transporter KPNA2 contributes to tumor immune evasion by enhancing PD-L1 expression in PDAC. J. Immunol. Res. 2021, 1–13 (2021).
Zhang, Z., Li, W., Jiang, D., Liu, C. & Lai, Z. MicroRNA-139-5p inhibits cell viability, migration and invasion and suppresses tumor growth by targeting HDGF in non-small cell lung cancer. Oncol. Lett. 19, 1806–1814 (2020).
Guo, S. C. et al. Exosomes derived from platelet-rich plasma promote the re-epithelization of chronic cutaneous wounds via activation of YAP in a diabetic rat model. Theranostics 7, 81–96 (2017).
An, X. et al. Effects of miR-101-3p on goat granulosa cells in vitro and ovarian development in vivo via STC1. J. Anim. Sci. Biotechnol. 11, 102 (2020).
Ning, H., Albersen, M., Lin, G., Lue, T. F. & Lin, C. S. Effects of EdU labeling on mesenchymal stem cells. Cytotherapy 15, 57–63 (2013).
Li, H. et al. Mex3a promotes oncogenesis through the RAP1/MAPK signaling pathway in colorectal cancer and is inhibited by hsa-miR-6887-3p. Cancer Commun. 41, 472–491 (2021).
Wu, Y. et al. Long non-coding RNA SNHG1 stimulates ovarian cancer progression by modulating expression of miR-454 and ZEB1. Mol. Oncol. 15, 1584–1596 (2021).
Tang, Z. et al. KDM1A-mediated upregulation of METTL3 ameliorates Alzheimer’s disease via enhancing autophagic clearance of p-Tau through m6A-dependent regulation of STUB1. Free Radic. Biol. Med. 195, 343–358 (2023).
Wang, C., Zhang, X., Chen, R., Zhu, X. & Lian, N. EGR1 mediates METTL3/m6A/CHI3L1 to promote osteoclastogenesis in osteoporosis. Genomics 115, 110696 (2023).
Su, X. et al. miR-33a-3p regulates METTL3-mediated AREG stability and alters EMT to inhibit pancreatic cancer invasion and metastasis. Sci. Rep. 13, 13587 (2023).
Yang, M., Chen, C., Wang, K., Chen, Y. & Xia, J. Astilbin influences the progression of osteoarthritis in rats by down-regulation of PGE-2 expression via the NF-κB pathway. Ann. Transl. Med 8, 766–766 (2020).
Wang, X. & Wu, Y. Protective effects of autophagy inhibitor 3-methyladenine on ischemia–reperfusion-induced retinal injury. Int. Ophthalmol. 40, 1095–1101 (2020).
Garikipati, V. N. S. et al. Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat. Commun. 10, 4317 (2019).
Wu, Q. & Yi, X. Down-regulation of long noncoding RNA MALAT1 protects hippocampal neurons against excessive autophagy and apoptosis via the PI3K/Akt signaling pathway in rats with epilepsy. J. Mol. Neurosci. 65, 234–245 (2018).
Luo, A. et al. Exosome-derived miR-339-5p mediates radiosensitivity by targeting Cdc25A in locally advanced esophageal squamous cell carcinoma. Oncogene 38, 4990–5006 (2019).
Lu, J. et al. Exosomal miR-9 inhibits angiogenesis by targeting MDK and regulating PDK/AKT pathway in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 37, 147 (2018).
Author information
Authors and Affiliations
Contributions
Z.H., D.W., and W.Z. contributed equally to this work. Z.H. and D.W. performed the majority of the experiments and data analysis. W.Z. assisted with bioinformatics analysis and database validation. M.M. supervised the project, provided critical revisions, and contributed to experimental design. C.D. conceived the study, oversaw the research process, and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The author declares no Competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, Z., Wang, D., Zhao, W. et al. ALKBH5-Mediated ITGB1 m6A Modification in Ovarian Cancer Progression and Immune Evasion. npj Precis. Onc. 9, 399 (2025). https://doi.org/10.1038/s41698-025-01127-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01127-w
