
Abstract
Gastric cancer (GC) remains a global clinical challenge due to late diagnosis, high heterogeneity, and poor prognosis. Tumor stemness has emerged as a key factor driving tumor aggressiveness and therapeutic resistance. However, the systematic characterization of high-stemness GC cells and their molecular features remains limited. We integrated single-cell RNA sequencing (scRNA-seq), spatial transcriptomics, and bulk RNA-seq data to identify and characterize high-stemness GC cells. Stemness scores were calculated using CytoTRACE, and malignant cells were classified into high stemness (top 25% CytoTRACE-scored cells, HighStem), dynamic transition stemness (DTStem), and low stemness (LowStem) subpopulations based on the quartile method cutoff. ScPagwas and cell–cell communication profiling were used to explore genomic instability, genetic susceptibility, and microenvironmental interactions. HighStem-specific co-expression modules were identified via high-dimensional WGCNA (hdWGCNA), and features were screened using six machine learning algorithms. A benchmark model was constructed for HighStem prediction and interpreted using SHAP analysis. HighStem GC cells exhibited enhanced intercellular signaling, metabolic reprogramming, and stemness-related pathway activity. Five genes—APMAP, MAPRE1, GLB1, TSPAN6, and CDKN2A—were identified as robust HighStem features. Spatial and bulk transcriptomic validation confirmed their tumor-specific expression and prognostic relevance. The Support Vector Machine (SVM) model incorporating these genes achieved high accuracy (AUC = 0.973) in distinguishing HighStem cells, demonstrating strong clinical utility at the scRNA-seq level. In addition, experimental validation through knockdown of core genes (APMAP, CDKN2A, TSPAN6, MAPRE1, and GLB1) in SGC7901 and HGC-27 gastric cancer cell lines revealed a significant reduction in JAK1-STAT3 pathway activity, supporting their functional involvement in tumor stemness regulation. Furthermore, knockdown of these genes increased the sensitivity of GC cells to chemotherapeutic agents like 5-FU and cisplatin, indicating their potential role in chemoresistance. This study provides a comprehensive molecular and functional characterization of high-stemness GC cells. The identified signature genes and predictive models offer novel insights into GC stemness biology and could guide personalized therapeutic strategies. Furthermore, our findings suggest that the core genes identified in this study may serve as potential biomarkers for predicting treatment outcomes and monitoring therapeutic resistance in GC.
Similar content being viewed by others


Introduction
Gastric cancer (GC) is one of the most common malignant tumors worldwide, ranking fifth in incidence and fourth in cancer-related mortality1,2. Despite advances in treatment, GC remains a major clinical challenge due to its low 5-year survival rate among digestive system malignancies and its high propensity for distant metastasis3,4. Clinically, early-stage GC is often asymptomatic or presents with nonspecific gastrointestinal symptoms, leading to frequent misdiagnosis as gastritis or other benign conditions5,6. As a result, most patients are diagnosed at an advanced stage. GC is characterized by significant heterogeneity7. Pathologically, it includes several subtypes such as adenocarcinoma, squamous cell carcinoma, and carcinoid tumors1. From a molecular perspective, The Cancer Genome Atlas (TCGA) categorizes GC into four major subtypes: Epstein–Barr virus (EBV)-positive, microsatellite instability (MSI), genomically stable (GS), and chromosomal instability (CIN)8,9. Numerous studies have demonstrated that these molecular subtypes exhibit distinct prognoses and responses to immunotherapy or targeted therapies. However, a robust and clinically applicable molecular classification system or predictive model for guiding individualized treatment remains lacking10. Therefore, it is crucial to further elucidate the molecular mechanisms underlying GC and to promote the development of reliable biomarkers and personalized therapeutic strategies, which may significantly enhance diagnostic accuracy and treatment efficacy in GC management.
Cancer stem cells (CSCs) are a distinct subpopulation of cancer cells with unlimited self-renewal and differentiation potential. They play critical roles in tumor initiation, progression, metastasis, and therapeutic resistance11. In GC, beyond the classical CSCs, recent transcriptomic analyses—particularly at the single-cell level—have identified cancer cell subsets with high stemness scores12,13. Although these cells may not fully meet the phenotypic or functional criteria of traditional gastric cancer stem cells (GCSCs), they display pronounced stemness-like features and contribute similarly to tumor aggressiveness, immune evasion, and drug resistance14,15. As a result, high-stemness GC cells have garnered increasing research interest. Given their malignant molecular characteristics and transcriptional profiles, these cells represent a biologically and clinically relevant subpopulation. Investigating their regulatory networks, functional roles, and interactions with the tumor microenvironment (TME) may enhance our understanding of GC heterogeneity and uncover novel therapeutic targets16,17,18.
Based on these insights, our study identified a subset of malignant GC cells with high stemness scores (HighStem) using CytoTRACE analysis of scRNA-seq data. We comprehensively characterized the biological features of these cells and explored their interactions with other cell types within TME. Through high-dimensional WGCNA (hdWGCNA) and multiple machine learning approaches, we further identified five core marker genes of HighStem cells—APMAP, CDKN2A, TSPAN6, MAPRE1, and GLB1. Leveraging these key features, we constructed a robust and highly accurate predictive model for HighStem cell identification using a benchmark machine learning framework. Together, our findings offer novel insights into the molecular landscape of high-stemness GC cells and may provide potential targets and strategies for improving the clinical diagnosis and treatment of gastric cancer.
Results
Identification of cell populations
scRNA-seq analysis revealed 38 transcriptionally distinct clusters with 0.8 resolution in gastric cancer samples via Seurat pipeline (Fig. 1A). Based on canonical marker genes, these clusters were annotated into major cell types including T cells, B cells, NK cells, monocytes, macrophages, dendritic cells, epithelial cells, fibroblasts, endothelial cells, and others (Fig. 1B). Marker gene expression supported accurate cell-type classification (Fig. 1C). For example, CD3D and CD8A marked T cells, NKG7 and KLRD1 marked NK cells, CD68 and CD163 marked macrophages, EPCAM and CDH1 marked epithelial cells, while CD1C, FCER1A, and CLEC9A were enriched in dendritic cells. To visualize gene expression patterns across the UMAP space, gene density plots were generated for selected marker genes (Fig. 1D).

A UMAP plot showing 38 transcriptionally distinct cell clusters identified by Seurat. B Cell type annotation based on canonical marker genes. C Dot plot displaying representative marker gene expression across major cell types. Dot size indicates the percentage of expressing cells; color denotes relative expression level. D Gene density plots show the distribution of selected marker genes in the UMAP space.
Identification and characterization of HighStem malignant gastric cancer cells
Given that GC originates from epithelial cells, we first isolated epithelial subpopulations for CNV analysis (Fig. 2A). Cluster 3 exhibited significantly lower CNV scores compared to clusters 1, 2, and 4 (Fig. 2B), indicating a lack of large-scale chromosomal alterations. Therefore, cluster 3 was annotated as normal epithelial cells, while the remaining epithelial clusters were considered malignant GC cells. To evaluate stemness, we applied CytoTRACE, which estimates cellular differentiation potential based on transcriptional diversity. As shown in Fig. 2C, malignant epithelial cells exhibited a continuum of differentiation states. Based on the distribution of CytoTRACE scores, cells were stratified into three groups: LowStem, DTStem (intermediate), and HighStem, corresponding to the bottom 25%, middle 50%, and top 25% of the score range, respectively (Fig. 2D). UMAP visualization demonstrated distinct spatial separation of these subgroups (Fig. 2E), and HighStem cells displayed significantly elevated CytoTRACE scores compared to the other groups (Fig. 2F).

A Heatmap of CNV signals across epithelial clusters. B Violin plot shows CNV score differences among epithelial clusters. C CytoTRACE plot displaying stemness score distribution in malignant epithelial cells. D Histogram of CytoTRACE score-based subgroup classification: LowStem, DTStem, and HighStem. E UMAP visualization of malignant cells colored by stemness subgroups. F Box plot comparing CytoTRACE scores among subgroups. G UMAP showing scPagwas-derived tumor relevance scores (TRS). J UMAP plots of stemness score, TRS score, and their combined distribution. H Box plot of TRS scores among stemness subgroups. I Correlation between CytoTRACE stemness scores and TRS values.
To further explore the genetic basis underlying stemness, we performed scPagwas analysis. The tumor relevance score (TRS), representing the degree of GWAS signal enrichment at the single-cell level, was visualized across the UMAP space (Fig. 2G). HighStem cells exhibited significantly higher TRS scores (Fig. 2H), suggesting a potential genetic contribution to their stemness phenotype. Consistently, UMAP plots of stemness score, TRS score, and their combination revealed overlapping high-scoring regions (Fig. 2J). Finally, correlation analysis revealed a strong positive relationship between CytoTRACE-based stemness scores and TRS values (Fig. 2I), supporting the hypothesis that stemness in malignant GC cells may be driven by inherited genetic regulatory programs.
HighStem exhibit enhanced intercellular communication, active signaling pathways, and distinct metabolic reprogramming
To explore the functional characteristics of HighStem GC cells, we analyzed intercellular communication patterns using the CellChat framework. Compared with LowStem and DTStem groups, HighStem cells exhibited markedly increased interaction frequency and strength with diverse cell types in TME, particularly with macrophages, endothelial cells, and fibroblasts (Fig. 3A–C). Outgoing and incoming signaling analysis further revealed that HighStem cells were active hubs in multiple signaling pathways, suggesting enhanced crosstalk potential (Fig. 3B).

A, B Cell–cell interaction networks showing total interaction counts and strengths, highlighting HighStem cells. C Incoming vs. outgoing interaction strength across cell types. D, E Increased ligand–receptor interaction pairs involving HighStem cells compared to LowStem. F, G Key ligand–receptor pairs enriched in HighStem-related interactions. H Pathway activity heatmap showing upregulation of oncogenic signals in HighStem cells. I Bubble plots illustrate metabolic pathway enrichment, with broad metabolic activation in HighStem cells.
Further quantitative comparison of ligand–receptor interactions showed that HighStem cells engaged in significantly more communication events than LowStem cells, both as ligand providers and receptor recipients (Fig. 3D, E). Key ligand–receptor pairs enriched in HighStem cells included MIF–CD74, NAMPT–INSR, and MDK–SDC1, indicating their involvement in immune modulation, stress responses, and stemness maintenance (Fig. 3F, G).
Pathway enrichment analysis highlighted that HighStem cells were positively associated with signaling cascades such as PI3K, WNT, TGF-β, and JAK–STAT, all of which are known to promote stemness and tumor progression (Fig. 3H). Moreover, metabolic profiling revealed that HighStem cells exhibited upregulation of a wide range of metabolic pathways, including glutathione metabolism, fatty acid metabolism, steroid biosynthesis, and glycosaminoglycan biosynthesis, indicating active metabolic reprogramming that supports their aggressive phenotype (Fig. 3I).
Identification of HighStem gene co-expression modules via hdWGCNA
To uncover gene expression programs associated with HighStem cells, we performed hdWGCNA. A soft-thresholding power of 6 was selected based on scale-free topology and connectivity criteria (Fig. 4A). Hierarchical clustering identified five distinct co-expression modules, each represented by a unique color (Fig. 4B). Module eigengene (ME) analysis revealed key genes contributing to each module (Fig. 4C). The UMAP projection of module expressions confirmed the modules across malignant cells (Fig. 4D). Among them, the brown, green, yellow, and blue modules showed distinct expression patterns across cells with different stemness states.

A Soft-thresholding power selection based on scale-free topology and connectivity. B Dendrogram showing five gene co-expression modules. C Module eigengene-based bar plots highlighting top-contributing genes. D UMAP projections of module expression across malignant cells. E Correlation matrix among modules. F Dot plot showing enrichment of brown, green, and yellow modules in HighStem cells.
Correlation analysis between modules revealed moderate co-expression relationships, with the turquoise module showing less connectivity to others (Fig. 4E). Importantly, the brown, green and yellow modules were specifically enriched in HighStem cells, both in terms of average expression and proportion of expressing cells (Fig. 4F). The 246 genes included in these modules are shown in Supplementary Table 1.
HighStem signature gene selection
To obtain a robust HighStem gene set, we initially identified the 171 genes showing the strongest positive correlation with the HighStem phenotype from three hdWGCNA modules (brown, green, and yellow) in 3367 HighStem cells using Pearson correlation analysis (Fig. 5A, Supplementary Table 2), collectively defined as the HighStem activity gene set. Spatial transcriptomics analysis demonstrated that HighStem activity scores were predominantly enriched within tumor regions (Fig. 5B). Consistently, RNA-seq data from the TCGA cohort revealed that HighStem activity was significantly elevated in GC tissues compared to adjacent normal tissues (Fig. 5C). All GC samples were subsequently scored for HighStem activity and divided into high- and low-activity groups based on the optimal cutoff values. Kaplan–Meier survival analysis showed that patients with high HighStem activity had significantly shorter overall survival than those with low activity (Fig. 5D), highlighting its prognostic relevance.

A Positively genes (displayed top 100 genes) correlated with the HighStem phenotype across three hdWGCNA modules. B Spatial transcriptomics showing tumor-enriched HighStem activity. C Comparison of HighStem activity between normal and tumor tissues in TCGA. D Kaplan–Meier survival curves stratified by HighStem activity score. E, F Feature importance ranking based on the Boruta algorithm. G Gene importance ranking from Decision Tree analysis. H Feature selection via LASSO regression. I Error rate curve and feature ranking from Random Forest analysis. J Feature importance ranking from GBM analysis. K Feature selection and optimal subset identification using ABESS. L Upset plot showing the intersection of top genes identified by six machine learning algorithms, with five shared hub genes highlighted.
Among them, Boruta and Random Forest were first used to rank gene importance based on their contribution to classification accuracy, with Boruta evaluating feature relevance through comparison with shadow features (Fig. 5E, F, Supplementary Table 3), and DT analysis provided a clear hierarchical view of the gene importance based on node splitting criteria (Fig. 5G, Supplementary Table 4). LASSO regression applied L1 regularization for dimensionality reduction, screening the most valuable predictors while minimizing overfitting (Fig. 5H, Supplementary Table 5). RF analysis further evaluated the importance of each gene based on the mean decrease in accuracy, with a stable decline in model error observed as the number of decision trees increased (Fig. 5I, Supplementary Table 6).GBM, as an integrated boosting method, continuously optimized the residuals of weak learners to improve the model’s accuracy, and prioritized candidate genes based on feature importance (Fig. 5J, Supplementary Table 7). Additionally, ABESS selected the optimal gene subset by evaluating different feature combinations with the lowest loss function, enhancing the reliability of feature screening (Fig. 5K, Supplementary Table 8). By integrating results from all four algorithms, five consistently selected hub genes (APMAP, CDKN2A, TSPAN6, MAPRE1, and GLB1) were identified (Fig. 5L).
Validation of HighStem hub genes and machine learning model performance
To investigate the spatial expression characteristics of the HighStem hub genes, we first visualized their expression density across all single cells. The results showed that APMAP, CDKN2A, TSPAN6, MAPRE1, and GLB1 were broadly expressed in the tumor tissue microenvironment, with relatively high signal intensity in specific cell populations (Fig. 6A). When focusing on malignant epithelial cells, these five genes demonstrated expression patterns that closely overlapped with the distribution of stemness scores, as indicated by CytoTRACE, suggesting that their expression was tightly associated with the high-stemness phenotype (Fig. 6B). Next, we evaluated the predictive performance of each gene in distinguishing malignant from non-malignant cells at the single-cell level. ROC curve analysis showed that all five genes had moderate to strong discriminatory power, with AUCs ranging from 0.709 to 0.832, among which APMAP (AUC = 0.832) and MAPRE1 (AUC = 0.827) showed the highest accuracy (Fig. 6C).

A Expression density plots of five HighStem hub genes in all cells. B Expression density in malignant epithelial cells, showing spatial overlap with stemness scores. C ROC curves show the single-cell classification performance of each gene. D Benchmark comparison of eight machine learning models. E Precision–recall and ROC curves of top-performing models. F ROC and PRAUC of the SVM model in development and test cohorts. G Confusion matrices of the training and test sets. H Decision curve analysis of the final classifier. I SHAP summary plot showing the contribution of each gene. J Predicted probability plots based on normalized gene expression. K SHAP dependency plots illustrate the relationship between feature values and model output.
Furthermore, to develop a reliable classifier for identifying HighStem cells, we systematically compared the performance of 8 machine learning algorithms, including support vector machine (SVM), random forest (ranger), XGBoost, decision tree (rpart), etc. Benchmark analysis showed that the SVM model achieved the highest average AUC in cross-validation folds, showing excellent and stable predictive performance (Fig. 6D, Supplementary Table 9). The SVM model also demonstrated superior precision–recall and ROC curve performance (Fig. 6E), with a final AUC of 0.973 in the independent test set (Fig. 6F). Confusion matrix analysis revealed high classification accuracy in both training and test cohorts, with balanced sensitivity and specificity (Fig. 6G). Decision curve analysis further confirmed the clinical utility of the SVM model, with the proposed classifier showing a significantly higher net benefit across a wide range of thresholds (Fig. 6H). SHAP analysis identified APMAP, MAPRE1, GLB1, TSPAN6, and CDKN2A as the top contributors to model output (Fig. 6I). Predicted probability plots demonstrated a strong positive correlation between normalized gene expression and the predicted HighStem probability (Fig. 6J). Similarly, SHAP dependency plots revealed that increased expression levels of these genes positively contributed to the classification score, further supporting their functional relevance in defining HighStem GC cells (Fig. 6K).
Validation of HighStem hub gene expression and prognostic significance
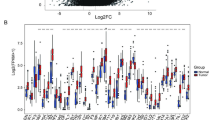
To further validate the HighStem hub genes, we assessed their expression and clinical relevance using bulk RNA-seq data from TCGA. All five genes—APMAP, CDKN2A, TSPAN6, MAPRE1, and GLB1—were significantly upregulated in GC tissues compared to adjacent normal tissues (Fig. 7A). These findings were further confirmed in a paired GC cohort, where consistent overexpression of APMAP, CDKN2A, MAPRE1, and GLB1 was observed, while TSPAN6 showed an upward trend that did not reach statistical significance (Fig. 7B). Time-dependent ROC analysis further supported the diagnostic potential of these genes, with APMAP demonstrating the highest AUC (0.934), followed by TSPAN6 (0.836), MAPRE1 (0.801), CDKN2A (0.750), and GLB1 (0.697) (Fig. 7C). We next explored the prognostic value of these genes. K–M survival analysis revealed that high expression of TSPAN6 (p = 0.018) and MAPRE1 (p = 0.027) was significantly associated with worse overall survival in GC patients (Fig. 7D). GLB1 and APMAP showed borderline significance (p = 0.053 and p = 0.079, respectively), while CDKN2A did not reach statistical significance. These results highlight the potential of the five hub genes as diagnostic and prognostic biomarkers for high-stemness GC cells.

A Expression levels of APMAP, CDKN2A, TSPAN6, MAPRE1, and GLB1 in normal and GC tissues. B Paired expression comparison in matched normal and GC samples. C Time-dependent ROC curves showing the diagnostic performance (AUC) of the five hub genes. D K–M survival curves stratified by gene expression levels in TCGA-GC cohort.
Five core marker genes were positively related with maker of stem cells
To further validate the correlation among the core marker genes (APMAP, CDKN2A, TSPAN6, MAPRE1, and GLB1), we first analyzed their association with tumor stem cell-related markers—specifically components of the JAK1-STAT3 signaling pathway—using the TIMER 2.0 platform. The analysis revealed that APMAP (also known as C200RF3), GLB1, TSPAN6, and MAPRE1 were positively correlated with both JAK1 and STAT3 expression levels. In contrast, CDKN2A showed no significant correlation with either JAK1 or STAT3 (Fig. 8A).

A TIMER 2.0 platform analyzed APMAP (also known as C200RF3), GLB1, TSPAN6, and MAPRE1 correlation with both JAK1 and STAT3 expression levels. B RT-PCR analyzed the expression of highStem signature genes following knockdown or overexpression of the core genes in the SGC7901 and HGC-27 GC cell lines.
In addition, we explored the functional involvement of these core genes in tumor stemness. We assessed the expression of highStem signature genes following knockdown or overexpression of the core genes in the SGC7901 and HGC-27 GC cell lines. The results demonstrated that the mRNA expression levels of highStem signature genes—including JAK1 (a tyrosine kinase that mediates cytokine signaling), STAT3 (a transcription factor crucial for cell proliferation and stemness maintenance), Hippo (a signaling pathway that restricts organ size and regulates stem cell self-renewal), YAP1 (a key effector of the Hippo pathway involved in cell growth and survival), and WNT3A (a ligand in the Wnt signaling pathway essential for stem cell regulation and tumor progression). The highStem signature genes were downregulated upon knockdown of the core genes, whereas their expression was upregulated in cells overexpressing the core genes (Fig. 8B).
Knockdown of core genes suppressed JAK1-STAT3 pathway
The JAK-STAT3 signaling pathway plays a pivotal role in the progression of various cancers by promoting cell proliferation, survival, immune evasion, and the maintenance of cancer stem cell properties. Aberrant activation of this pathway has been closely associated with tumor development and poor clinical outcomes in multiple malignancies, including GC.
Then, we analyzed the protein expression levels of components of the JAK1-STAT3 pathway in SGC7901 and HGC-27 GC cell lines following the knockdown of the core marker genes. The results demonstrated that in both SGC7901 and HGC-27 cell lines, knockdown of the core marker genes—APMAP, CDKN2A, TSPAN6, MAPRE1, and GLB1—led to a marked reduction in the protein expression of JAK1 and STAT3 (Fig. 9A–E), suggesting that these genes may act as upstream regulators of the JAK-STAT3 axis in GC cells.

A–E Western blot analyzed the JAK1-STAT3 pathway in SGC7901 and HGC-27 GC cell lines following the knockdown of the core marker genes.
Knockdown of core genes enhances drug sensitivity in GC cell line
5-Fluorouracil (5-FU) and cisplatin are widely used chemotherapeutic agents in the treatment of GC and other solid tumors. 5-FU functions primarily as a pyrimidine analog that inhibits thymidylate synthase, thereby disrupting DNA synthesis and inducing apoptosis in rapidly dividing cells. Cisplatin exerts its antitumor effect by forming DNA crosslinks, which interfere with DNA replication and transcription, ultimately triggering cell death. However, resistance to these agents remains a major clinical challenge, often associated with tumor stemness and molecular alterations.
Further, we investigated the role of the core marker genes in modulating chemotherapy sensitivity. Upon treatment with 5-FU or cisplatin, knockdown of the core genes enhanced the sensitivity of both SGC7901 and HGC-27 GC cell lines to these agents. Notably, silencing of TSPAN6 and MAPRE1 led to a pronounced inhibition of cell proliferation under chemotherapeutic stress (Fig. 10A–D), suggesting that these genes may contribute to drug resistance mechanisms and could serve as potential therapeutic targets to overcome chemoresistance.

A, B CCK-8 assay was carried to detect the drug sensitivity of 5-FU and cisplatin in HGC-27 GC cell lines after core genes (APMAP, CDKN2A, TSPAN6, MAPRE1, and GLB1) knockdown. C, D CCK-8 assay was carried to detect the drug sensitivity of 5-FU and cisplatin in SGC7901 GC cell lines after core genes (APMAP, CDKN2A, TSPAN6, MAPRE1, and GLB1) knockdown.
Discussion
Tumor cell stemness has attracted increasing attention in recent years. Many cancer cells acquire enhanced proliferative capacity and immune evasion ability through the activation of embryonic development-related gene programs19,20. In 2006, the American Association for Cancer Research (AACR) defined CSCs as a subpopulation of tumor cells with self-renewal ability and the potential to generate heterogeneous tumor progeny. Experts at the time also suggested that CSCs may be inherently resistant to conventional therapies, making tumor stemness one of the emerging hallmarks of cancer21.
To date, extensive efforts have been made to identify markers associated with high-stemness tumor cells22. Surface markers such as CD44, CD24, CD29, CD90, and CD133 have been widely used to isolate CSCs from various cancers and cell lines23,24. With the rapid development of transcriptome-based analytical tools such as CytoTRACE25, mRNAsi (OCLR)26, and StemSC27, numerous potential markers have been further identified. For example, Yibo Fan et al. demonstrated that SOX9 maintains stem-like properties in advanced gastric cancer28, while Xiaoli Liu et al. showed that the activation of the Hippo-YAP1 signaling pathway upregulates FOXP4 to sustain gastric cancer stemness, highlighting FOXP4 as both a biomarker and a therapeutic target29. In addition, several non-coding RNAs such as lncRNA HCP530 and circSLC4A731 have also been implicated in stemness regulation. Despite these advances, few studies have systematically screened for high-stemness markers in GC or comprehensively evaluated their predictive value. To address this gap, we applied CytoTRACE to quantitatively assess stemness in malignant GC cells and further identified five robust HighStem signature genes through hdWGCNA combined with multiple machine learning algorithms. A predictive model with high classification accuracy was then constructed using a benchmark machine learning framework. This study not only provides a novel strategy for identifying high-stemness GC cells but also offers new insights into addressing the current clinical challenges in GC treatment.
In our study, five HighStem hub genes were identified, namely APMAP, MAPRE1, GLB1, TSPAN6, and CDKN2A. APMAP (Adipocyte Plasma Membrane Associated Protein) encodes a protein predominantly localized to the plasma membrane and endoplasmic reticulum of adipocytes32,33,34. Recent studies suggest that APMAP plays a potential regulatory role in promoting tumor progression, particularly through the induction of epithelial-mesenchymal transition (EMT) in certain tumor types35,36,37. Notably, EMT is closely linked to tumor stemness. During EMT, tumor cells lose epithelial traits and acquire mesenchymal features, including enhanced migratory capacity and stem cell-like properties38,39. Conversely, CSCs are often characterized by high expression of EMT-related signaling pathways, such as TGF-β40, Notch41, Wnt42, and Hippo/YAP143. Interestingly, these pathways were also enriched in the HighStem subpopulation in our cell–cell communication analysis. Furthermore, APMAP demonstrated the highest SHAP value among all features, indicating its strongest positive predictive contribution to the HighStem phenotype, supporting its potential role as a key regulator of stemness in GC.
MAPRE1 (Microtubule-Associated Protein RP/EB Family Member 1) encodes a protein that regulates microtubule dynamics and was originally identified through its interaction with the adenomatous polyposis coli (APC) gene44. It plays a critical role in maintaining microtubule organization and chromosomal stability45,46. The aberrant expression of MAPRE1 has been implicated in the pathogenesis of various malignancies by disrupting these essential cellular processes47,48,49. In GC, a study by Ye Feng et al. reported CNV of the MAPRE1 gene, suggesting its potential genomic instability50. These features may contribute to the self-renewal capacity and undifferentiated phenotype observed in HighStem GC cells.
The GLB1 gene encodes β-galactosidase, a lysosomal enzyme responsible for the degradation of specific glycolipids and glycoproteins51. Although GLB1 is widely recognized as a canonical marker of cellular senescence52,53, its high expression in the HighStem cell population appears paradoxical. However, it is important to note that GLB1, beyond serving as a senescence marker, plays broader biological roles in intracellular metabolism, glycolipid catabolism, and lysosomal homeostasis54,55. Interestingly, a study by Monique Bernard et al. highlighted that both cancer stem cells and senescent cells exhibit enhanced stress resistance, suggesting that elevated GLB1 expression may reflect a quiescent or dormant cellular state56. In this context, GLB1 may help HighStem cells reduce proliferation and metabolic burden under adverse conditions, thereby facilitating long-term survival and potentially contributing to tumor recurrence or metastasis at later stages. Moreover, emerging evidence indicates that certain cells in EMT or high-stemness states can concurrently activate senescence-associated signaling pathways, such as p16 and p2157,58, implying that HighStem cells are not constantly in a proliferative state. Instead, they may undergo dynamic transitions between dormancy and reactivation in response to tumor microenvironmental cues. This regulated balance between quiescence and activation could represent a self-protective mechanism by which tumor cells maintain homeostasis and stemness potential.
Similarly, CDKN2A (Cyclin-Dependent Kinase Inhibitor 2A) is a well-established tumor suppressor gene involved in the regulation of the cell cycle, induction of cellular senescence, and inhibition of tumorigenesis59,60,61. While the CDKN2A gene can exert antiproliferative effects when properly transcribed and translated, numerous studies have shown that in many tumor types, it is frequently subject to genetic alterations such as mutations, deletions, or epigenetic modifications—particularly promoter hypermethylation—which result in reduced expression or complete functional loss62,63,64. This downregulation contributes to uncontrolled cell proliferation and tumor progression. In GC, CDKN2A promoter hypermethylation has been associated with the loss of p16^INK4a protein expression65,66, enabling GC cells to escape cell cycle control and acquire a highly proliferative phenotype. Moreover, copy number loss of CDKN2A has been identified as a potential biomarker for predicting hematogenous metastasis in GC patients67. These findings align, to a certain extent, with the biological features of high-stemness GC cells, which are characterized by enhanced proliferative capacity and metastatic potential. It is also important to note that, although our study observed elevated CDKN2A mRNA levels in HighStem cells, this may not directly reflect functional protein expression due to possible post-transcriptional regulatory mechanisms such as RNA methylation, miRNA-mediated repression, or impaired translation. Therefore, when interpreting the role of CDKN2A in the context of tumor stemness, both transcriptomic and proteomic levels should be considered.
TSPAN6 (tetraspanin 6) is a member of the transmembrane 4 superfamily, also known as the tetraspanin family. Current studies have reported conflicting roles of TSPAN6 across different cancer types68,69. For instance, in colorectal cancer, TSPAN6 is considered to function as a tumor suppressor, and its downregulation has been associated with tumor progression70. In contrast, in glioblastoma, high TSPAN6 expression has been linked to malignant progression and poor patient prognosis, suggesting a context-dependent role in tumor biology71. Although TSPAN6 has been relatively understudied in GC, other members of the tetraspanin family, such as TSPAN872, TSPAN173, CD15174, and TSPAN475, have been shown to be highly expressed in GC tissues and are closely associated with enhanced proliferation, migration, and invasiveness of GC cells. Given the functional similarity within the tetraspanin family, it is plausible that TSPAN6 may play a comparable role in GC. However, further experimental validation is required to clarify its function and clinical relevance in GC.
The JAK-STAT3 signaling pathway has long been recognized as a central mediator in cancer stemness, tumor cell proliferation, and anti-apoptosis processes76,77,78. Our data indicate that APMAP, TSPAN6, MAPRE1, and GLB1 are positively correlated with the expression levels of JAK1 and STAT3, suggesting that these genes may promote GC stemness by modulating the JAK-STAT3 axis. Notably, although CDKN2A did not show a direct correlation with either JAK1 or STAT3, its known role in cell cycle regulation may contribute indirectly to the modulation of cell differentiation and proliferation. Thus, these core genes likely interact in a complex molecular network to regulate tumor stemness and promote cancer progression. Further experimental validation demonstrated that knockdown of these core genes resulted in a significant reduction in JAK1 and STAT3 protein expression, underscoring their role as upstream regulators in the JAK-STAT3 pathway. This finding not only enhances our understanding of the involvement of these genes in maintaining tumor stemness but also suggests their potential as therapeutic targets, especially in strategies targeting cancer stemness and overcoming therapeutic resistance. In terms of chemoresistance, knockdown of these genes notably increased the sensitivity of GC cells to common chemotherapy agents such as 5-FU and cisplatin. This suggests that these genes play a crucial role not only in maintaining tumor stemness but also in mediating drug resistance mechanisms. Specifically, the silencing of TSPAN6 and MAPRE1 led to a pronounced inhibition of cell proliferation under chemotherapy stress, further supporting their potential as targets for overcoming chemoresistance.
In summary, the five identified HighStem hub genes may contribute to maintaining stemness in gastric cancer through diverse yet complementary mechanisms. These findings provide a foundation for future studies aimed at validating their roles and exploring their potential as biomarkers or therapeutic targets in GC.
Methods
Data collection and processing
scRNA-seq datasets (GSE1839047, GSE20678579) and stRNA-seq data (GSE25195080) were obtained from GEO. Bulk RNA-seq data were retrieved from TCGA, and GWAS summary statistics for scPagwas analysis were downloaded from the IEU OpenGWAS database. Dataset details are provided in Supplementary Table 10. Cells with >20% mitochondrial content or <200 detected genes were excluded. Genes expressed in ≥3 cells and within 200–7000 counts were retained, yielding 269213 high-quality cells from 88 samples for analysis. Data were processed using Seurat, including normalization, PCA, UMAP, and clustering (resolution = 0.8), with batch correction via Harmony. Cell types were annotated using known markers. For stRNA-seq, SCTransform normalization and unsupervised clustering defined spatial domains, supported by H&E staining and marker gene expression. Spatial patterns were visualized with “SpatialDimPlot” and “SpatialFeaturePlot”.
Inference of copy number variations
Copy number variations (CNVs) were inferred from scRNA-seq data using the inferCNV R package. To reduce technical variability, normalization procedures were applied, and malignant cells were analyzed relative to normal reference cells to detect regions with abnormal expression indicative of genomic instability81,82. A CNV score was then calculated to quantify the extent of deviation in each cell from the reference baseline. Malignant cells were extracted according to CNV-based classification, yielding 13,483 tumor cells for downstream analysis.
Stemness scoring of malignant gastric cancer cells
To evaluate cellular stemness, we applied the CytoTRACE algorithm to malignant GC cells identified through CNV analysis. CytoTRACE is a computational framework that infers the differentiation state of individual cells based on transcriptional diversity, under the premise that less differentiated cells express a broader array of genes. Unlike traditional stemness assessments, CytoTRACE does not depend on predefined gene sets or prior biological assumptions, making it broadly applicable across diverse cell types and tissues25,83. Based on the quartile distribution of CytoTRACE scores, cells were stratified into three groups: high stemness (top 25%), dynamic transition stemness (25–75%), and low stemness (bottom 25%)84.
scPagwas analysis
To explore the genetic underpinnings of cellular stemness and tumor progression, we employed scPagwas, an integrative computational framework implemented in the “scPagwas” R package. This method enables the integration of scRNA-seq data with GWAS summary statistics to uncover trait-associated genetic variants that potentially influence cell fate decisions at single-cell resolution. In this study, we focused on mapping genes associated with stemness scores to GWAS summary data derived from large-scale population studies85. By linking cell-type-specific gene expression patterns to genomic loci associated with cancer-related traits, scPagwas allowed us to identify candidate genetic variants that may drive intercellular heterogeneity in stemness and contribute to tumor evolution.
High-dimensional WGCNA (hdWGCNA) analysis
As CytoTRACE provides a global stemness score for individual cells without pinpointing gene-level expression patterns across subpopulations, we employed high-dimensional weighted gene co-expression network analysis (hdWGCNA) to further elucidate the transcriptional characteristics of malignant GC cells. A weighted co-expression network was constructed by calculating pairwise gene expression correlations, and genes were clustered into distinct co-expression modules86,87. To identify modules associated with tumor progression, we performed module–trait relationship analysis, focusing on stemness and metastatic phenotypes. Modules showing strong correlations with high-stemness or metastatic features were considered functionally relevant. Within these key modules, hub genes were defined based on high intra-module connectivity, representing potential core regulators that may drive tumor heterogeneity and malignant progression in GC.
Cell–cell interaction analysis
To investigate intercellular communication within the tumor microenvironment, we utilized the CellChat R package, which infers ligand–receptor interactions based on scRNA-seq data88. Communication networks were constructed to delineate signaling exchanges among annotated cell populations. Visualization of interaction strength and frequency between specific cell types was performed using the netVisual_circle function, which provides a circular plot representing outgoing and incoming signaling patterns. To further dissect individual signaling pathways, the netVisual_bubble function was applied, generating bubble plots that highlight key ligand–receptor pairs and their associated signaling axes.
Screening of HighStem (top 25% CytoTRACE-scored cells) signature genes
To identify key signature genes of the HighStem subpopulation, we employed an integrated machine learning approach combining six algorithms: random forest, LASSO, Boruta, and decision tree (DT), Adaptive Best Subset Selection (ABESS), and Gradient Boosting Machine (GBM). These methods were chosen for their complementary strengths in feature selection and bias reduction.
Random forest89 and Boruta90 (a random forest-based wrapper algorithm) were used to rank gene importance. LASSO regression91 applied L1 regularization to eliminate redundant features, while decision tree92 analysis provided interpretable hierarchical classification. ABESS93, a recently developed algorithm that performs optimal subset selection with theoretical guarantees, was introduced to further refine the gene set while avoiding overfitting or selection bias. Meanwhile, the GBM algorithm94, an ensemble learning method that builds additive models in a forward stage-wise fashion using decision trees, was applied to capture complex nonlinear relationships between genes and stemness phenotypes.Genes identified by all four methods were considered hub HighStem markers, and their intersection was visualized using a Venn diagram.
Machine learning benchmark models for HighStem features
To identify the most effective predictive model for HighStem features at the single-cell level, we benchmarked nine machine learning algorithms using the “mlr3” R package. The models included k-nearest neighbor (KNN), linear discriminant analysis (LDA), naive Bayes (NB), random forest (Ranger), recursive partitioning and regression trees (RPART), support vector machine (SVM), and extreme gradient boosting (XGBoost). Cells classified as HighStem were used as positive samples, and LowStem cells served as controls. The dataset was randomly split into training (80%) and test (20%) sets. Hyperparameter tuning was conducted using five-fold internal cross-validation (CV), and model generalization was evaluated via ten-fold external CV. The model with the highest average area under the curve (AUC) was selected as the optimal framework for HighStem signature prediction.
SHAP analysis for feature interpretation
To interpret the contributions of individual genes to the HighStem prediction model, we applied SHAP (SHapley Additive exPlanations) analysis95. SHAP is a model-agnostic method that quantifies the impact of each feature on the model’s output, based on cooperative game theory. This approach enables both global and local interpretation of feature importance. We computed SHAP values for all samples to evaluate the contribution of each gene to model predictions. The mean absolute SHAP value (mean_phi) for each gene was used as an indicator of its overall importance, with higher values reflecting greater average influence on classification outcomes. Genes with the highest mean SHAP values were considered the most influential predictors and were selected for further biological interpretation.
Cell culture
The SGC7901 and HGC-27 GC cell lines, originally from the American Type Culture Collection (Manassas, VA), was stored by our laboratory and grown in RPMI 1640 supplemented with 2 mM glutamine, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4, and 10% fetal bovine serum at 37 °C in a 5% CO2 humidified incubator.
Transfection with siRNA
Core genes expression was silenced using siRNA transfection mediated by Lipofectamine 2000 (Invitrogen). Cells were seeded in 6-well plates at 50–70% confluency and transfected the following day. For each well, 5 μL of Core genes-targeting siRNA (final concentration 50 nM) was diluted in 125 μL of Opti-MEM (Gibco), and separately, 5 μL of Lipofectamine 2000 was diluted in another 125 μL of Opti-MEM. After a 5-min incubation at room temperature, the two solutions were combined and incubated for 20 min to form siRNA-lipid complexes. The complexes were then added to the cells in antibiotic-free complete medium. After 4–6 h, the medium was replaced with fresh complete medium. Cells were harvested 24–48 h post-transfection for RNA and protein extraction to evaluate gene silencing efficiency. The information of siRNA sequence are described in the Supplementary Table 11.
Western blot
Cells were transiently transfected or treated with 5-Fluorouracil (5-FU) and cisplatin. The cells were lysed in RIPA buffer. The lysates were subjected to immunoprecipitation with anti-Flag antibody. The lysates and immunoprecipitates were subjected to SDS- 12.5% PAGE, transferred onto PVDF membranes and probed with antibody(s) described. The following antibodies were used: JAK1 (1:1000, Santa Cruz), STAT3 (1:1000, Santa Cruz). Secondary HRP-conjugated Abs were obtained from GE Healthcare Life Sciences,and the light emission was quanti¢ed with a Lumino image analyzer LAS-1000 (FUJI, Japan). Signal quantification was performed by using ImageJ Software.
Realtime PCR
Total RNAwasextractedfromcellsbyusingTrizolreagent (Life Technologies). Taqman probes were used for the detection of miRs (Applied Biosystems), as described by the manufacturer, by using beta-actin as endogenous control. For mRNA-level analysis, cDNA was generated by using reverse transcriptase SuperScript II and poly dT primers (Invitrogen). Realtime PCR was performed by using SYBR Green Master Mix (Invitrogen) and the ABI 7900HT fast real-time PCR System (Applied Biosystems). Primers are described in the Supplemental Information. S18 was used as endogenous control. Primers are described in the Supplementary Table 12. β-actin was used as endogenous control.
Statistical analysis
All data processing, statistical analyses, and visualizations were performed using R software (version 4.1.3). Group comparisons for continuous variables were conducted using either the Wilcoxon rank-sum test or Student’s t test, depending on the distribution. Categorical variables were compared using the chi-squared test or Fisher’s exact test, as appropriate. Multiple testing correction was applied using the False Discovery Rate (FDR) method. Pearson correlation analysis was used to evaluate associations between continuous variables. All statistical tests were two-sided, and a p-value < 0.05 was considered statistically significant.
Data availability
The data that supports the findings of this study are available in the manuscript and supplementary materials. Further inquiries can be directed to the corresponding authors.
References
Smyth, E. C. et al. Gastric cancer. Lancet 396, 635–648 (2020).
Chia, N. Y. & Tan, P. Molecular classification of gastric cancer. Ann. Oncol. 27, 763–769 (2016).
Joshi, S. S. & Badgwell, B. D. Current treatment and recent progress in gastric cancer. CA Cancer J. Clin. 71, 264–279 (2021).
Guan, W. L., He, Y. & Xu, R. H. Gastric cancer treatment: recent progress and future perspectives. J. Hematol. Oncol. 16, 57 (2023).
Machlowska, J. et al. Gastric Cancer: Epidemiology, Risk Factors, Classification, Genomic Characteristics and Treatment Strategies. Int. J. Mol. Sci. 21, 4012 (2020).
Yang, W. J. et al. Updates on global epidemiology, risk and prognostic factors of gastric cancer. World J. Gastroenterol. 29, 2452–2468 (2023).
Kumar, V. et al. Single-Cell Atlas of Lineage States, Tumor Microenvironment, and Subtype-Specific Expression Programs in Gastric Cancer. Cancer Discov. 12, 670–691 (2022).
Gullo, I. et al. Heterogeneity in Gastric Cancer: From Pure Morphology to Molecular Classifications. Pathobiology 85, 50–63 (2018).
Lim, B. et al. Genomic and epigenomic heterogeneity in molecular subtypes of gastric cancer. World J. Gastroenterol. 22, 1190–1201 (2016).
Wang, F. H. et al. The Chinese Society of Clinical Oncology (CSCO): Clinical guidelines for the diagnosis and treatment of gastric cancer, 2023. Cancer Commun. 44, 127–172 (2024).
Loh, J. J. & Ma, S. Hallmarks of cancer stemness. Cell Stem Cell 31, 617–639 (2024).
Pan, G. et al. EMT-associated microRNAs and their roles in cancer stemness and drug resistance. Cancer Commun. 41, 199–217 (2021).
Kim, J. et al. Single-cell analysis of gastric pre-cancerous and cancer lesions reveals cell lineage diversity and intratumoral heterogeneity. NPJ Precis. Oncol. 6, 9 (2022).
Rao, X. et al. Targeting Gastric Cancer Stem Cells to Enhance Treatment Response. Cells 11, 2828 (2022).
Chang, Z. et al. THBS2 promotes gastric cancer progression and stemness via the Notch signaling pathway. Am. J. Cancer Res. 14, 3433–3450 (2024).
Fan, X. et al. YTHDF2-mediated m(6)A modification of ONECUT2 promotes stemness and oxaliplatin resistance in gastric cancer through transcriptionally activating TFPI. Drug Resist Updat 79, 101200 (2025).
Liu, T. et al. FAP(+) gastric cancer mesenchymal stromal cells via paracrining INHBA and remodeling ECM promote tumor progression. Int. Immunopharmacol. 144, 113697 (2025).
He, C. et al. Stem cell landscape aids in tumor microenvironment identification and selection of therapeutic agents in gastric cancer. Cell Signal 113, 110965 (2024).
Marimuthu, S. et al. Mucins reprogram stemness, metabolism and promote chemoresistance during cancer progression. Cancer Metastasis Rev. 40, 575–588 (2021).
Zeng, L. et al. New insights into KLFs and SOXs in cancer pathogenesis, stemness, and therapy. Semin Cancer Biol. 90, 29–44 (2023).
Clarke, M. F. et al. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 66, 9339–9344 (2006).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Medema, J. P. Cancer stem cells: the challenges ahead. Nat. Cell Biol. 15, 338–344 (2013).
Nassar, D. & Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 11, 47–76 (2016).
Gulati, G. S. et al. Profiling cell identity and tissue architecture with single-cell and spatial transcriptomics. Nat. Rev. Mol. Cell Biol. 26, 11–31 (2025).
Chen, D. et al. Integrated Machine Learning and Bioinformatic Analyses Constructed a Novel Stemness-Related Classifier to Predict Prognosis and Immunotherapy Responses for Hepatocellular Carcinoma Patients. Int. J. Biol. Sci. 18, 360–373 (2022).
Zheng, H. et al. StemSC: a cross-dataset human stemness index for single-cell samples. Stem Cell Res. Ther. 13, 115 (2022).
Fan, Y. et al. Epithelial SOX9 drives progression and metastases of gastric adenocarcinoma by promoting immunosuppressive tumour microenvironment. Gut 72, 624–637 (2023).
Liu, X. et al. FOXP4 Is a Direct YAP1 Target That Promotes Gastric Cancer Stemness and Drives Metastasis. Cancer Res. 84, 3574–3588 (2024).
Wu, H. et al. MSC-induced lncRNA HCP5 drove fatty acid oxidation through miR-3619-5p/AMPK/PGC1α/CEBPB axis to promote stemness and chemo-resistance of gastric cancer. Cell Death Dis. 11, 233 (2020).
Hui, Y. et al. circSLC4A7 accelerates stemness and progression of gastric cancer by interacting with HSP90 to activate NOTCH1 signaling pathway. Cell Death Dis. 14, 452 (2023).
Albrektsen, T. et al. Identification of a novel integral plasma membrane protein induced during adipocyte differentiation. Biochem. J. 359, 393–402 (2001).
Ilhan, A. et al. Localization and characterization of the novel protein encoded by C20orf3. Biochem. J. 414, 485–495 (2008).
Mekenkamp, L. J. et al. Chromosome 20p11 gains are associated with liver-specific metastasis in patients with colorectal cancer. Gut 62, 94–101 (2013).
Jiang, S. et al. Cholesterol Induces Epithelial-to-Mesenchymal Transition of Prostate Cancer Cells by Suppressing Degradation of EGFR through APMAP. Cancer Res. 79, 3063–3075 (2019).
Zhu, X. et al. APMAP Promotes Epithelial-Mesenchymal Transition and Metastasis of Cervical Cancer Cells by Activating the Wnt/β-catenin Pathway. J. Cancer 12, 6265–6273 (2021).
Akhter, M. D. Q. et al. An exploratory SWATH plasma proteomics analysis of phyllodes tumor- a type of female breast tumor. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1254, 124508 (2025).
Huang, Y., Hong, W. & Wei, X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J. Hematol. Oncol. 15, 129 (2022).
Pastushenko, I. & Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 29, 212–226 (2019).
Peng, D. et al. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 21, 104 (2022).
Espinoza, I. & Miele, L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 341, 41–45 (2013).
Xue, W. et al. Wnt/β-catenin-driven EMT regulation in human cancers. Cell Mol. Life Sci. 81, 79 (2024).
Wang, H. et al. GABAB receptor inhibits tumor progression and epithelial-mesenchymal transition via the regulation of Hippo/YAP1 pathway in colorectal cancer. Int. J. Biol. Sci. 17, 1953–1962 (2021).
Ladd, J. J. et al. Increased plasma levels of the APC-interacting protein MAPRE1, LRG1, and IGFBP2 preceding a diagnosis of colorectal cancer in women. Cancer Prev. Res. 5, 655–664 (2012).
Su, L. K. & Qi, Y. Characterization of human MAPRE genes and their proteins. Genomics 71, 142–149 (2001).
Islam, F. et al. Protein interactions of FAM134B with EB1 and APC/beta-catenin in vitro in colon carcinoma. Mol. Carcinog. 57, 1480–1491 (2018).
Tang, L. et al. circSETD3 regulates MAPRE1 through miR-615-5p and miR-1538 sponges to promote migration and invasion in nasopharyngeal carcinoma. Oncogene 40, 307–321 (2021).
Chanez, B. et al. EB1 Restricts Breast Cancer Cell Invadopodia Formation and Matrix Proteolysis via FAK. Cells 10, 388 (2021).
Rodrigues-Ferreira, S. et al. Combinatorial expression of microtubule-associated EB1 and ATIP3 biomarkers improves breast cancer prognosis. Breast Cancer Res. Treat. 173, 573–583 (2019).
Feng, Y. et al. Integrated Analysis of DNA Copy Number Changes and Gene Expression Identifies Key Genes in Gastric Cancer. J. Comput. Biol. 27, 877–887 (2020).
Lee, B. Y. et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 5, 187–195 (2006).
Sun, J. et al. A Glb1-2A-mCherry reporter monitors systemic aging and predicts lifespan in middle-aged mice. Nat. Commun. 13, 7028 (2022).
Guerrero, A. et al. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 19, e13133 (2020).
Liu, J. et al. Proteomics-Derived Biomarker Panel Facilitates Distinguishing Primary Lung Adenocarcinomas With Intestinal or Mucinous Differentiation From Lung Metastatic Colorectal Cancer. Mol. Cell Proteom. 23, 100766 (2024).
Mou, L. et al. Construction of a lipid metabolism-related risk model for hepatocellular carcinoma by single cell and machine learning analysis. Front. Immunol. 14, 1036562 (2023).
Bernard, M. et al. Autophagy drives fibroblast senescence through MTORC2 regulation. Autophagy 16, 2004–2016 (2020).
Park, S. S. et al. Cellular senescence is associated with the spatial evolution toward a higher metastatic phenotype in colorectal cancer. Cell Rep. 43, 113912 (2024).
Brambs, C. E. et al. Epithelial-mesenchymal transition (EMT) in vulvar cancer with and without inguinal lymph node involvement. J. Cancer Res. Clin. Oncol. 148, 1183–1193 (2022).
Negrini, S., Gorgoulis, V. G. & Halazonetis, T. D. Genomic instability-an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228 (2010).
Zucman-Rossi, J. et al. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 149, 1226–1239.e4 (2015).
Sherr, C. J. Cancer cell cycles. Science 274, 1672–1677 (1996).
Dilworth, D. et al. Germline CDKN2A mutation implicated in predisposition to multiple myeloma. Blood 95, 1869–1871 (2000).
Tang, V. et al. Loss of p16 expression is a sensitive marker of CDKN2A homozygous deletion in malignant meningiomas. Acta Neuropathol. 145, 497–500 (2023).
Kreuger, I. Z. M. et al. Therapeutic Strategies for Targeting CDKN2A Loss in Melanoma. J. Invest. Dermatol. 143, 18–25.e1 (2023).
Spagnol, L. W. et al. P16 gene promoter methylation is associated with oncogenesis and progression of gastric carcinomas: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 180, 103843 (2022).
Wang, M. et al. The SMAD2/miR-4256/HDAC5/p16(INK4a) signaling axis contributes to gastric cancer progression. Oncol. Res. 31, 515–541 (2023).
Dermawan, J. K. et al. Novel Genomic Risk Stratification Model for Primary Gastrointestinal Stromal Tumors (GIST) in the Adjuvant Therapy Era. Clin. Cancer Res. 29, 3974–3985 (2023).
Qi, Y. et al. Expression and function of transmembrane 4 superfamily proteins in digestive system cancers. Cancer Cell Int. 20, 314 (2020).
Huang, R. et al. The role of tetraspanins pan-cancer. iScience 25, 104777 (2022).
Andrijes, R. et al. Tetraspanin 6 is a regulator of carcinogenesis in colorectal cancer. Proc. Natl Acad. Sci. USA, 118, e2011411118 (2021).
Zhang, C. et al. TSPAN6 reinforces the malignant progression of glioblastoma via interacting with CDK5RAP3 and regulating STAT3 signaling pathway. Int. J. Biol. Sci. 20, 2440–2453 (2024).
Zhang, L. et al. TSPAN8 regulates EGFR/AKT pathway to enhance metastasis in gastric cancer. Mol. Biol. Rep. 50, 7955–7965 (2023).
Lu, Z. et al. Delivery of TSPAN1 siRNA by Novel Th17 Targeted Cationic Liposomes for Gastric Cancer Intervention. J. Pharm. Sci. 109, 2854–2860 (2020).
Deng, Y. et al. Tetraspanins: Novel Molecular Regulators of Gastric Cancer. Front. Oncol. 11, 702510 (2021).
Qi, W. et al. Tetraspanin family identified as the central genes detected in gastric cancer using bioinformatics analysis. Mol. Med. Rep. 18, 3599–3610 (2018).
Johnson, D. E. et al. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 15, 234–248 (2018).
Wang, T. et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 27, 136–150.e5 (2018).
Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition. Cells 9, 217 (2020).
Kang, B. et al. Parallel single-cell and bulk transcriptome analyses reveal key features of the gastric tumor microenvironment. Genome Biol. 23, 265 (2022).
Lee, S. H. et al. Spatial dissection of tumour microenvironments in gastric cancers reveals the immunosuppressive crosstalk between CCL2+ fibroblasts and STAT3-activated macrophages. Gut 74, 714–727 (2024).
Luo, L. et al. Single-cell RNA sequencing identifies molecular biomarkers predicting late progression to CDK4/6 inhibition in patients with HR+/HER2- metastatic breast cancer. Mol. Cancer 24, 48 (2025).
Xin, S. et al. ScRNA-seq revealed an immunosuppression state and tumor microenvironment heterogeneity related to lymph node metastasis in prostate cancer. Exp. Hematol. Oncol. 12, 49 (2023).
Gulati, G. S. et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411 (2020).
Fan, G. et al. Single-cell and spatial analyses revealed the co-location of cancer stem cells and SPP1+ macrophage in hypoxic region that determines the poor prognosis in hepatocellular carcinoma. NPJ Precis Oncol. 8, 75 (2024).
Ye, L. et al. Inferring the genetic relationships between unsupervised deep learning-derived imaging phenotypes and glioblastoma through multi-omics approaches. Brief Bioinform 26, bbaf037 (2024).
Dai, L. et al. Single-cell and spatial transcriptomics reveal a high glycolysis B cell and tumor-associated macrophages cluster correlated with poor prognosis and exhausted immune microenvironment in diffuse large B-cell lymphoma. Biomark. Res. 12, 58 (2024).
Sun, Y. et al. Single-cell hdWGCNA reveals metastatic protective macrophages and development of deep learning model in uveal melanoma. J. Transl. Med. 22, 695 (2024).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Wallace, M. L. et al. Use and misuse of random forest variable importance metrics in medicine: demonstrations through incident stroke prediction. BMC Med. Res. Methodol. 23, 144 (2023).
Yan, F. et al. Association between the stress hyperglycemia ratio and 28-day all-cause mortality in critically ill patients with sepsis: a retrospective cohort study and predictive model establishment based on machine learning. Cardiovasc. Diabetol. 23, 163 (2024).
Kang, J. et al. LASSO-Based Machine Learning Algorithm for Prediction of Lymph Node Metastasis in T1 Colorectal Cancer. Cancer Res. Treat. 53, 773–783 (2021).
Lyu, M. et al. Maxillary sinus floor augmentation: a review of current evidence on anatomical factors and a decision tree. Int. J. Oral. Sci. 15, 41 (2023).
Kong, W. et al. Adaptive best subset selection algorithm and genetic algorithm aided ensemble learning method identified a robust severity score of COVID-19 patients. Imeta 2, e126 (2023).
Liu, X. P. et al. Clinical significance and molecular annotation of cellular morphometric subtypes in lower-grade gliomas discovered by machine learning. Neuro Oncol. 25, 68–81 (2023).
Qi, X. et al. Machine learning and SHAP value interpretation for predicting comorbidity of cardiovascular disease and cancer with dietary antioxidants. Redox Biol. 79, 103470 (2025).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (82072733) and the Natural Science Foundation of Liaoning Province (2020-BS-088).
Author information
Authors and Affiliations
Contributions
W.M. conceived, designed and funded the study. Z.W. prepared and wrote the paper and prepared the figures. X.L. and J.W. performed data analyses. Z.W. and X.L. performed experiments. H.Y. and D.Z. acquired tissues and data. Supervision, manuscript reviewing, and editing was performed by Y.X. and S.Z. All the authors contributed to writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Z., Li, X., Wang, J. et al. Comprehensive molecular characterization of high-stemness gastric cancer cells using single-cell transcriptomics, spatial mapping, and machine learning. npj Precis. Onc. 9, 400 (2025). https://doi.org/10.1038/s41698-025-01177-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01177-0
