
Abstract
The response to neoadjuvant chemoradiotherapy (neoCRT) in patients with locally advanced colorectal cancer is variable. However, the detailed mechanisms underlying the poor response to neoCRT are elusive. Here, we found that the KRAS mutation significantly influences the therapeutic response to neoCRT and tumor lapses through evasion of immune surveillance. We found that oncogenic KRAS activation led to the upregulation of DNMT3b expression, which suppressed STING expression and attenuated radiotherapy (RT)-induced antitumor immunity. Oncogenic KRAS activation significantly decreased miR-29b-3p expression, resulting in DNMT3b upregulation, which in turn decreased RT-induced STING signaling for type I interferon (IFN) production. We found that supplementation with colorectal cancer-specific miR-29b-3p markedly increased the response to radiotherapy in KRAS-mutated CRC tumors in vivo and significantly reshaped the tumor microenvironment to promote immune cell infiltration. Furthermore, supplementation with miR-29b-3p also enhanced the response to immune checkpoint blockade as well as radiotherapy. Taken together, these results suggest a therapeutic approach in which tumor-specific miR-29b-3p increases the benefits of RT by overcoming defective STING in KRAS-mutated CRC patients.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is a major cause of cancer mortality, and the prognosis of patients with metastatic CRC remains poor1. Targeting T-cell immune checkpoint molecules such as PD-1/PD-L1 induces T-cell-mediated antitumor immunity with dramatic efficacy in hypermutated tumors with microsatellite instability-high (MSI-H) or DNA mismatch repair deficiency (dMMR)2,3. Nonetheless, this comprises a minor subset of CRC (~5–10%)4, highlighting the need to improve the response to ICB in CRC.
Mutant KRAS represents one of the most common tumor driver mutations, and its involvement in multistep tumorigenesis and tumor progression is well recognized5. Activating KRAS mutations (~45%) are drivers of CRC. Oncogenic KRAS correlates positively with aggressive tumor behaviors such as dysregulated tumor growth, distant metastasis, and resistance to EGFR-targeted therapy in metastatic CRC6. KRAS mutations not only confer resistance to targeted therapy but have also been linked to cellular and clinical radioresistance in several malignancies, such as lung adenocarcinoma7,8 and rectal adenocarcinoma9. However, the molecular mechanisms of cellular radioresistance are highly variable and unsolved in KRAS-mutated CRC. According to the consensus molecular subtype (CMS) for CRC10, KRAS mutation is associated with the CMS3 subtype with poor immune infiltration. Mutant KRAS is associated with reduced infiltration of lymphocytes in the Cancer Genome Atlas (TCGA) and KFSYSCC CRC datasets11. Additionally, oncogenic KRAS also induces aberrant DNA methylation via DNA methyltransferase I (DNMT1) activity to suppress the expression of tumor suppressors as well as immunostimulatory proteins such as STING12, resulting in decreased type I interferon (IFN) production13,14,15. Moreover, oncogenic KRAS represses IRF2 levels to repress CXCL3 signaling in the immunosuppressive TME in CRC16. Mice with KrasG12D mutant CRC exhibit immunosuppression and resistance to immune checkpoint blockade (ICB). These findings highlight the role of mutant KRAS in driving CRC progression by suppressing antitumor immunity17. However, the role of oncogenic KRAS in radiotherapy-induced antitumor immunity has not been fully elucidated.
Radiotherapy-induced DNA damage contributes to the accumulation of cytosolic double-stranded DNA (dsDNA) fragments that are recognized by cGAS, triggering STING-mediated type I IFN production for antitumor immunity18,19. However, defects in cGAG/STING induced by epigenetic control prevent immune surveillance and attenuate the antitumor immune response by decreasing antigenicity and T-cell-mediated cytotoxicity in several malignancies, such as colorectal cancer and lung cancer12,20,21,22. Suppression of STING by oncogenic mutations such as Myc and KRAS promoted immune evasion and resistance to ICBs12,23. Recently, we reported that DNMT1 directly inhibited tumor PD-L1 expression to decrease the response to radiotherapy and ICBs24. Administration of mesenchymal stem cells (MSCs) engineered with IFNβ and sTRAIL can overcome STING deficiency to increase the therapeutic efficacy of radiotherapy and ICB25, suggesting that the activity of the cancer cell-intrinsic STING pathway defines the immunogenicity and efficacy of ICB. These findings suggest that re-engagement of STING signaling might represent a promising strategy to restore immunogenicity to increase the response to radiotherapy.
The regulation of STING expression by oncogenic mutations has been reported in many malignancies12,20,22. We found that oncogenic KRAS decreased the therapeutic efficacy of radiotherapy by decreasing STING-mediated antitumor immunity. Oncogenic KRAS significantly inhibits microRNA-29b-3p (miR-29b-3p) expression, which in turn upregulates DNMT3b to suppress STING expression via epigenetic control, leading to less systemic antitumor immunity in response to radiotherapy. Knocking down oncogenic KRAS significantly enhanced the response to radiotherapy and inhibited distant tumor growth (abscopal effect) by promoting cytotoxic T-cell-mediated immune responses. Moreover, we developed an adenovirus-associated virus (AAV) expressing miR-29b-3p under the control of the colorectal cancer-specific CEA promoter. Administration of tumor-specific AAV-miR-29b-3p increased the response to radiotherapy in a KRAS-mutated CRC animal model. Moreover, AAV-miR-29b-3p remodeled the tumor microenvironment and enhanced the eradication of distant nonirradiated tumor cells. Taken together, these results suggest a therapeutic approach in which tumor-specific AAV-miR-29b-3p increases the benefits of RT by overcoming defective STING in KRAS-mutated CRC patients.
Results
KRAS mutations decreased RT-induced antitumor immunity by inhibiting STING-mediated type I IFN signaling
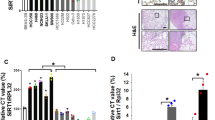
Previous studies have shown that oncogenic KRAS leads to reduced T-cell infiltration in colorectal cancer11. To identify gene expression signatures enriched specifically in oncogenic KRAS colorectal cancer relative to wild-type KRAS colorectal cancer, we analyzed colon adenocarcinoma (COAD) from The Cancer Genome Atlas (TCGA) via gene set enrichment analysis (GSEA). As expected, this analysis revealed oncogenic KRAS-specific downregulation of signatures associated with the CD8 signaling pathway and the IL12/STAT4 signaling pathway (activated CD8+ T cells, q value < 0.001; Fig. S1A and 1A). Consistent results of a low CD8A signature in oncogenic KRAS were found in rectal adenocarcinoma (READ, Fig. S1B). READ patients carrying mutant KRAS had fewer CD8A signatures [CD8+ T cells (Quantizeq, p = 0.052; CD8+ T cells (MCPcounter, p = 0.093), naïve CD8+ T cells (p = 0.035) and CD8+ TCM cells (p = 0.021)] within tumors according to the TIMER 2.0 website [http://timer.cistrome.org/, Fig. S1B]. Moreover, type I IFN signaling (q value < 0.001) and IFNγ signaling were among the most significantly downregulated pathway signatures in COAD patients with oncogenic KRAS (Fig. S1C). Cancer cell-autonomous type I IFN responses are typically associated with immunogenicity in response to radiotherapy24,25. We then evaluated the density of tumor-infiltrating CD8+ T cells before and after radiotherapy in patients with advanced CRC (Fig. 1B). We found a greater density of tumor-infiltrating CD8+ T cells in the KRASWT patients than in the KRASMut patients after nCRT (post-neoCRT, Fig. 1B, p = 0.0385, n = 94). However, there was no significant difference in pre-neoCRT biopsies, suggesting that oncogenic KRAS may inhibit type I IFN to decrease cancer immunogenicity to reduce T-cell infiltration and decrease the therapeutic efficacy of neoCRT.

A The gene expression data were downloaded from TCGA database for GSEA analysis (KRASWT-CRC patients (n = 150) and KRASG12/13*-CRC patients (n = 150). B The association between KRAS status and CD8+ TILs in pre-neoCRT biopsies and post-neoCRT surgical tissues was analyzed in advanced CRC patients (Unpaired t test, n = 94, p = 0.0385). C HT29-KRASWT (endogenous wild-type KRAS) and HT29-KRASG12D cells were irradiated (5 Gy) and labeled with CFSE. These cells were individually co-cultured with immature dendritic cells (THP1-iDCs) for 24 hr and Jurkat T cells for 18 hr. The level of surface CD80 (THP1-iDC) and intracellular IFNγ (Jurkat) was evaluated by flow cytometry (n = 3). One-way ANOVA t test. D CT26shNC and CT26shKRAS cells were generated by infecting with lentivirus carrying shNC and shKRAS. Cells were inoculated into right hind leg (2 × 105 cells) and left back (1 × 105 cells) by subcutaneous injection for 14 day. On days 14 and 18, local tumors in right hind leg were irradiated (5 Gy). The tumor volume was measured every 3 days (n = 4–6). The resected tumors on Day 28 were weighted (n = 4-6). One-way ANOVA and two-way ANOVA t test. *p < 0.05, **p < 0.01 and ***p < 0.001. E The subset of tumor-infiltrating CD4+ cells within primary tumors was analyzed by flow cytometric analysis (n = 3–4). One-way ANOVA t test. *p < 0.05. F The subset of tumor-infiltrating CD8+ cells within primary tumors was analyzed by flow cytometric analysis (n = 3–4). One-way ANOVA t test. *p < 0.05. G The subset of tumor-infiltrating MDSCs (Gr1+CD11b+) within primary tumors was analyzed by flow cytometric analysis (n = 3-4). One-way ANOVA t test. H. The subset of tumor-infiltrating regulatory T lymphocytes (CD4+CD25+Foxp3+, Treg) within primary tumors was analyzed by flow cytometric analysis (n = 3–4). One-way ANOVA t test. *p < 0.05.
We then examined T-cell-mediated cytotoxicity to HT29 (endogenous KRASWT) and HT29-KRASG12D cells in response to radiotherapy (Fig. 1C). By coculture with THP-1-iDCs (THP-1-immature dendritic cells), we found that irradiated HT29-KRASG12D cells exhibited decreased expression of activated DC markers (CD80) on the THP-1-iDCs (Fig. 1C). Moreover, the level of intracellular IFNγ in Jurkat T cells, which represented to T-cell-mediated cytotoxicity, was also significantly lower in HT29-KRASG12D cells than HT29-KRASWT cells (Fig. 1C). We then implanted BALB/c mice with subcutaneous CT26shNC (endogenous KRASG12D) and CT26shKRAS tumors bilaterally and treated the ipsilateral tumors with or without local irradiation. Compared with CT26shNC, local RT significantly improved tumor growth at irradiated sites in BALB/c mice bearing CT26shKRAS tumors (Fig. 1D). We observed significantly higher complete response rates (CR, 40%) in CT26shKRAS-bearing mice treated with RT than in CT26shNC-bearing mice receiving RT (20%, Fig. 1D). Furthermore, subcutaneous tumor rechallenge was performed in mice with CR on Day 35, and the rechallenged tumors were rejected in these tumor-free mice, indicating the generation of antitumor immune memory. To validate this finding, we harvested subcutaneous tumors for flow cytometry analysis (Fig. S2A and S2B). Compared with those in the CT26shNC controls, significant increases in CD4+ and CD8+ T cells were observed in the irradiated CT26shKRAS tumor cells (Figs. 1E and Fig. 1F). There was no significant difference in the number of MDSCs (Gr1+CD11b+) in CT26shKRAS tumors (Fig. 1G). Furthermore, we detected decreased recruitment of Foxp3+ Tregs (Foxp+CD25+CD4+) in CT26shKRAS tumors (Fig. 1H). Additionally, the number of cytotoxic IFNγ+CD8+ T cells was also increased within the tumor microenvironment (Fig. 2A). The ratio of IFNγ+CD8+ T/Foxp3+ Tregs was also changed, suggesting that KRAS mutation may decrease the therapeutic efficacy of RT through its immunological effects. The cancer immunogenicity of resected tumors was also evaluated via qRT‒PCR, which revealed that type I IFN signatures, which constitute the main RT-induced antitumor immune system, were significantly increased, including Ifnβ1, Cxcl10, Irf7 and Irf9 (Fig. 2B). Immunohistochemical analysis revealed that the expression of the proliferating marker Ki67 in resected tumors was markedly decreased in resected CT26shKRAS tumors that received RT (Fig. 2C). Additionally, the levels of tumor STING, the cytotoxic T-cell marker GzmB and the dendritic cell marker CD11c were elevated in resected CT26shKRAS/RT tumors (Fig. 2C and Fig. 2D). The level of the apoptotic cell marker cleaved caspase-3 (c. Casp.3) was significantly increased in the CT26shKRAS/RT subgroup (Fig. 2E). These results indicate that oncogenic KRAS might attenuate RT-induced cancer immunogenicity by inhibiting STING, leading to poor therapeutic efficacy.

A The subset of tumor-infiltrating cytotoxic IFNγ+CD8+ cells within primary tumors was examined by flow cytometric analysis. The ratio of IFNγ+CD8+/Foxp3+ Treg was shown (n = 3-4). One-way ANOVA t test. *p < 0.05. B The resected primary tumors were homogenized to evaluate type I signatures by qRT-PCR (n = 3-4). One-way ANOVA t test. *p < 0.05 and **p < 0.01. C The proliferation marker Ki67, tumor STING, GzmB+ immune cells and CD11c+ DCs were evaluated by immunohistochemistry (n = 3). D The quantification of tumor STING (H-score), GzmB+ immune cells and CD11c+ DCs (n = 3). One-way ANOVA t test. *p < 0.05 and **p < 0.01. E The resected primary tumors were homogenized for immunoblotting analysis (n = 3). One-way ANOVA t test. **p < 0.01. F The non-irradiated distant tumor volume (abscopal tumor) was measured every 3 days (n = 4–6). The resected abscopal tumors on Day 28 were weighted (n = 4-6). One-way ANOVA and two-way ANOVA t test. *p < 0.05, **p < 0.01 and ***p < 0.001. G The subset of tumor-infiltrating CD4+ cells, CD8+ cells, MDSCs (Gr1+CD11b+) and Treg (CD4+CD25+Foxp3+) within abscopal tumors was evaluated by flow cytometric analysis (n = 3-4). One-way ANOVA t test. *p < 0.05. H The proliferation marker Ki67 and CD8+ immune cells were evaluated by immunohistochemistry (n = 3). I The quantification of Ki67+ cells was shown (n = 4). One-way ANOVA t test. *p < 0.05. J The quantification of CD8+ immune cells was shown (n = 4). One-way ANOVA t test. *p < 0.05. K The resected abscopal tumors were homogenized to evaluate Ifnb1 mRNA by qRT-PCR (n = 3-4). One-way ANOVA t test. *p < 0.05 and **p < 0.01. L The resected abscopal tumors were homogenized to evaluate Cxcl10 mRNA by qRT-PCR (n = 3-4). One-way ANOVA t test. *p < 0.05 and **p < 0.01.
Moreover, the tumor volume of nonirradiated abscopal tumors in BALB/c mice bearing CT26shKRAS tumors was significantly decreased after RT compared with that of CT26shNC tumors (Fig. 2F). cGAS/STING promoted radiation-induced type I IFN-dependent antitumor immunity for abscopal effects18,19. Therefore, we evaluated whether immune cell profiles were reshaped in abscopal tumors in CT26shKRAS tumor-bearing mice in response to radiation (Fig. 2G). We detected a greater density of CD8+ T cells in the CT26shKRAS abscopal tumors than in the CT26shNC abscopal tumors (Fig. 2G). Additionally, increased infiltration of CD8+ T cells was observed in the abscopal tumors of the CT26shKRAS/RT mice. The infiltration of MDSCs and Foxp3+ Tregs was not induced in the abscopal tumors of the CT26shKRAS/RT mice. By immunohistochemical analysis, we observed fewer Ki67+ tumor cells and more CD8+ T cells in the abscopal tumors from the CT26shKRAS/RT mice (Fig. 2H–J). Compared with those in CT26shNC/RT mice, the levels of Ifnβ1 and Cxcl10 in abscopal tumors from CT26shKRAS/RT mice were also greater (Fig. 2K and L). These results suggest that KRAS mutations may inhibit STING expression to decrease radiotherapy-induced type I IFN-dependent antitumor immunity to achieve an abscopal effect.
Loss of STING expression by DNMT1 and DNMT3b in KRAS-mutated cells decreased systemic antitumor immunity to eradicate distant abscopal tumors
When KRASG12D was ectopically expressed in WiDr and CoLo320 (endogenous wild-type KRAS) cells, the mRNA levels of STING (TMEM173), IFNβ1, CXCL10 and IFI27 were not increased by RT compared with those in parental cells (Fig. 3A and B). Knockdown of KRASG12D in HCT116 and SW620 (KRAS-mutant) cells significantly increased STING expression and augmented RT-induced type I IFN signatures (Fig. 3C and D).

A WiDr cells were infected with lentivirus carrying pBabe-puro-vector (Vec.) or pBabe-puro-KRASG12D and selected for three days. WiDr-Vec. and WiDr-KRASG12D cells were irradiated (5 Gy), and then harvested for qRT-PCR analysis after 24 hr (n = 3). One-way ANOVA t test. *p < 0.05. B CoLo320 cells were infected with lentivirus carrying pBabe-puro-vector (Vec.) or pBabe-puro-KRASG12D and selected for three days. CoLo320-Vec. and CoLo320-KRASG12D cells were irradiated (5 Gy), and then harvested for qRT-PCR analysis after 24 hr (n = 3). One-way ANOVA t test. *p < 0.05. C HCT116 cells were infected with lentivirus carrying pLKO-scramble shRNA (shNC) or pLKO-shKRAS (shKRAS) and selected for three days. HCT116shNC and HCT116shKRAS cells were irradiated (5 Gy), and then harvested for qRT-PCR analysis after 24 hr (n = 3). One-way ANOVA t test. *p < 0.05 and **p < 0.01. D SW620shNC and SW620shKRAS cells were irradiated (5 Gy), and then harvested for qRT-PCR analysis after 24 hr (n = 3). One-way ANOVA t test. *p < 0.05 and **p < 0.01. E WiDr and Colo320DM (endogenous wild-type KRAS) cells were individually infected with vector (Vec.) and KRASG12D. Cells were selected by puromycin for three days. The level of DNMT1, DNMT3a and DNMT3b was evaluated by immunoblotting (n = 3). One-way ANOVA t test. **p < 0.01. F CT26, HCT116 and SW620 (endogenous mutant KRAS) cells individually infected with lentivirus carry shNC and shKRAS. Cells were selected by puromycin for three days. The level of DNMT1, DNMT3a and DNMT3b was evaluated by immunoblotting (n = 3). One-way ANOVA t test. **p < 0.01. G KRASG12D-tranduced WiDr (endogenous wild-type KRAS) cells were treated with 2.5 μmol/L and 5.0 μmol/L AZA for two days. The level of STING was evaluated by immunoblotting (n = 3). One-way ANOVA t test. *p < 0.05 and **p < 0.01. H. KRASG12D-tranduced CoLo320 (endogenous wild-type KRAS) cells were treated with 2.5 μmol/L and 5.0 μmol/L AZA for two days. The level of STING was evaluated by immunoblotting (n = 3). One-way ANOVA t test. *p < 0.05 and **p < 0.01. I HCT116 and SW620 (endogenous mutant KRAS) cells individually infected with lentivirus carry shDNMT1, shDNMT3a and shDNMT3b. Cells were selected by puromycin for three days. The mRNA level of STING was evaluated by qRT-PCR (n = 3). One-way ANOVA t test. *p < 0.05. J The protein level of STING was evaluated by immunoblotting (n = 3).
Loss of STING expression caused by hypermethylation has been reported20,21. We therefore explored whether the level of the DNMT family might be related to this phenomenon. Indeed, the overexpression of oncogenic KRAS resulted in decreased STING expression and high expression levels of DNMTs, including DNMT1, DNMT3a and DNMT3b (Fig. 3E). In contrast, knockdown of KRAS restored STING expression and decreased DNMT expression in KRAS-mutated cell lines (CT26, HCT116 and SW620), suggesting that KRAS mutation led to global hyperactivation of DNMTs to suppress STING expression (Fig. 3F). Indeed, treatment with 5-azacytidine (AZA), an FDA-approved DNMT inhibitor, in KRASG12D-transduced WiDr and CoLo320 (endogenous wild-type KRAS) cells restored the STING level to a lower level than that in WiDr and Colo320DM parental cells (Fig. 3G and H). To identify which DNMT is involved in oncogenic KRAS-mediated STING suppression, we generated DNMT1-, DNMT3a- and DNMT3b-silenced HCT116 and SW620 cell lines (Fig. 3I). We found that knockdown of DNMT3b markedly restored STING expression in two KRAS-mutated cell lines, which was verified by qRT‒PCR and immunoblotting (Fig. 3I and J). Furthermore, the level of DNMT3b was negatively correlated with the expression of STING in the TCGA-COAD dataset (Fig. S3). These results indicate that DNMT3b-mediated STING suppression in KRAS-mutated colorectal cancer cells.
By analyzing DNA methylation of the STING promoter in the CCLE database, three methylation loci were found to be located peripheral to the CpG-dense area of the STING promoter (P1, 5:138861649; P2, 5:138861807 and P3, 5:138862442.). Significant changes in methylation status were observed at the STING promoters P1 and P2. Moreover, the methylation status of the STING promoter P1 region was significantly associated with the KRAS mutation (Fig. 4A p = 0.0017). To further analyze whether STING promoter methylation influences the response to RT in advanced CRC patients, we extracted genomic DNA from post-neoCRT surgical tissues from CRC patients and examined the extent of methylation of the TMEM173 (STING) promoter P1 and P2 regions (Figs. 4B and S4A). Hypermethylation in the P1 region was found in KRAS-mutated patients but not in KRAS-WT patients (Fig. 4C). There were no significant changes in P2 regions, including P2-1, P2-2-1 and P2-2-2 (Fig. 4C and D). Moreover, pair-matched tissue analysis revealed that RT significantly increased STING expression in tumors (Fig. S4B). Additionally, we found that the level of tumor STING expression was significantly increased in KRAS-WT CRC patients after neoCRT treatment (Figs. 4E and F, p= 0.024). The level of tumor STING was positively correlated with the density of infiltrating CD8+ T cells in KRAS-WT CRC patients after neoCRT treatment (Fig. 4F, n= 42, p = 0.03). However, there was no significant association between the STING level and CD8+ T-cell infiltration in KRAS-Mut CRC patients after neoCRT treatment (Fig. 4G). Additionally, the level of tumor STING expression was significantly associated with favorable survival outcomes in KRAS-WT CRC patients after neoCRT treatment (Fig. 4H, n = 41, p = 0.0366). These results indicate that methylation of the STING promoter P1 region in KRAS-mutated patients may suppress tumor STING expression to attenuate radiotherapy-induced antitumor immunity, leading to poor survival outcomes.

A Dot plot heatmaps showing DNA methylation levels in the STING promoter in CCLE (red = hypermethylation). The location of each position is as follows: P1, 5:138861649; P2, 5:138861807 and P3, 5:138862442. One-way ANOVA t test. ***p < 0.001. B The DNA methylation levels in the TMEM173(STING) promoter was evaluated in the post-neoCRT surgical tissues. P1, 5:138861649 and P2, 5:138861807. C The relationship between STING promoter P1/P2-1 methylation and KRAS mutations (G12/G13 mutations) was evaluated by Pyrosequencing. One-way ANOVA t test. *p < 0.05. D The relationship between STING promoter P2-2-1/P2-2-2 methylation and KRAS mutations (G12/G13 mutations) was evaluated by Pyrosequencing. One-way ANOVA t test. E The level of tumor STING in KRASWT pair-matched pre-neoCRT biopsies and post-neoCRT surgical tissues. F The level of tumor STING after neoCRT treatment was evaluated in KRASWT-CRC patients (p = 0.03, n = 42). One-way ANOVA t test. G The level of tumor STING in KRASMut pair-matched pre-neoCRT biopsies and post-neoCRT surgical tissues. The level of tumor STING after neoCRT treatment was evaluated in KRASMut-CRC patients (p = 0.15, n = 17). One-way ANOVA t test. H KM analysis on tumor STING in KRASWT-CRC patients (p = 0.0366, n = 41). Log-rank p test.
KRAS mutation suppressed miR-29b-3p expression to upregulate DNMT3b for epigenetic control of STING expression
Since the levels of DNMT3b are regulated by miRNAs26,27,28, we aimed to explore the potential miRNAs that are suppressed by KRAS mutations for DNMT3b upregulation in colorectal cancer. To identify oncogenic KRAS-related miRNA candidates, we performed miRNA sequencing of CoLo320-KRASWT and CoLo320-KRASG12D (Fig. 5A). A total of 1590 miRNAs were differentially expressed in response to oncogenic KRASG12D, and 35 of those miRNAs were significantly downregulated (log2FC < −0.44 and FDR < 0.001). We further prioritized potential miRNAs that potentially targeted DNMT3b based on two criteria: (i) increased expression of oncogenic KRAS in the CCLE dataset and (ii) the presence of high-confidence conserved miRNA binding sites on DNMT3b predicted by at least three different algorithms (Fig. 5A). We determined that miR-29 is a potential target of DNMT3b, which has been reported to target DNMT3b26,27,28. Analysis of the CCLE-CRC and COAD-CRC datasets revealed that high expression of miR-29b was associated with KRAS mutation (Fig. 5B). Two other miRNA members from the miR-29 family, miR-29a and miR-29c, were not associated with KRAS mutation in the CCLE-CRC database (Figure. S5A). Transduction of oncogenic KRAS directly downregulated the level of miR-29b-3p (Fig. 5C). However, the knockdown of oncogenic KRAS significantly restored the level of miR-29b-3p (Fig. 5D). There was no significant difference in the level of miR-29c in response to either transduction of oncogenic KRAS or knockdown of oncogenic KRAS (Fig. S5B and S5C). Reconstitution of the miR-29b-3p level in HT29-KRASG12D cells by miR-29b-3p miRNA mimics decreased the expression of DNMT3b and increased the levels of STING and IFNβ1 in response to radiotherapy (Fig. 5E). However, the inhibition of endogenous miR-29b-3p with hairpin inhibitors in SW620shKRAS cells reduced the expression of STING and IFNβ1 in response to RT (Fig. 5F). The protein level of DNMT3b was also verified by immunoblotting (Fig. 5G and H). Moreover, the protein level of STING was significantly decreased as treatment with miR-29b-3p antagomir (Fig. 5H). To demonstrate the clinical relevance of miR-29b-3p, DNMT3b and STING, we analyze their expression in colorectal cancer patients and stratified based on the KRAS status (KRAS-WT, n = 143; and KRAS-Mut, n = 157, Fig. 6A). We found that decreased miR-29b-3p, increased DNMT3b as well as decreased STING was found in KRAS-Mut CRC patients, compared to KRAS-WT CRC patients. Moreover, the level of miR-29b-3p was negatively correlated with the expression of DNMT3b in KRAS-Mut CRC patients (Fig. 6B, n = 157, p = 0.032). Patients with high miR-29b-3p were associated with favorable survival outcome in KRAS-Mut CRC patients (Fig. 6C, n = 157, p = 0.0459). Consistent with our results, reduced STING and miR-29b was observed in KRAS-Mut patients in TCGA-COAD database (Fig. S5D and S5E). But we did not find significant association in specific KRAS variant due to limited number in each subtype (Fig. S5D and S5E). These results indicated that the KRAS mutation directly decreased miR-29b-3p expression, leading to the upregulation of DNMT3b expression to reduce STING expression and suppress RT-induced antitumor immunity.

A CoLo320-Vec. and CoLo320-KRASG12D cells were harvested for miRNA-seq and identified miR-29b-3p as the potential candidate to regulate DNMT3b expression. B The level of miR-29b-3p was evaluated in KRAS-mutated cell lines in CCLE and TCGA-COAD database. One-way ANOVA t test. C The level of miR29b-3p was evaluated in HT29-KRASG12D and CoLo320-KRASG12D cells, compared to their parental KRASWT cells. One-way ANOVA t test. *p < 0.05. D The level of miR-29b-3p was evaluated in SW620shKRAS and HCT116shKRAS cells, compared to their parental KRASMut cells. One-way ANOVA t test. *p < 0.05 and ***p < 0.001. E HT29-KRASG12D cells were transfected with miR-29b mimetic (20 nM) for 24 hr and then irradiated (5 Gy). After 24 hr, cells were harvested for qRT-PCR (n = 3). One-way ANOVA t test. *p < 0.05 and **p < 0.01. F SW620shKRAS cells were transfected with miR-29b-3p antagomir (20 nM) for 24 hr and then irradiated (5 Gy). After 24 hr, cells were harvested for qRT-PCR (n = 3). One-way ANOVA t test. *p < 0.05 and **p < 0.01. G HT29-KRASG12D cells were transfected with miR-29b-3p mimetic (20 nM) for 24 hr and then irradiated (5 Gy). After 24 hr, cells were harvested for immunoblotting (n = 3). One-way ANOVA t test. **p < 0.01. H SW620shKRAS cells were transfected with miR-29b-3p antagomir (20 nM) for 24 hr and then irradiated (5 Gy). After 24 hr, cells were harvested for immunoblotting (n = 3). One-way ANOVA t test. **p < 0.01.

A The representative images of miR-29b-3p, DNMT3b and STING in colorectal cancer patients. B The correlation between miR-29b-3p and DNMT3b in KRASMut-CRC patients (n = 157, p = 0.032). C KM analysis on the survival outcome of KRASMut-CRC patients with high miR-29b-3p (n = 157, Log-rank p = 0.0459). D Schematic depiction of CRC-specific AAV-miR-29b-3p cassette. E CT26 cells were inoculated into right hind leg (2 × 105 cells) and left back (1 × 105 cells) by subcutaneous injection. On Day 9 and 10, local tumors in right hind leg were irradiated with 5 Gy. AAV-Vec. and AAV-pre-mmu-miR-29b-3p (1 × 108 vg/mouse) was intramuscular injected on Days 4, 9, 14 and 18. The tumor volume was measured every 3 days (n = 4-6). Two-way ANOVA t test. *p < 0.05, **p < 0.01 and ***p < 0.001. F The resected primary tumors on Day 28 were weighted (n = 3). One-way ANOVA t test. *p < 0.05 and ***p < 0.001. G The non-irradiated distant tumor volume (abscopal tumor) was measured every 3 days (n = 4-6). Two-way ANOVA t test. **p < 0.01 and ***p < 0.001. H The resected abscopal tumors on Day 28 were weighted (n = 3). One-way ANOVA t test. *p < 0.05 and ***p < 0.001. I The representative images of CD3 and GzmB+ immune cells were evaluated by immunohistochemistry (n = 3). J The quantification of CD3 immune cells (n = 3). One-way ANOVA t test. *p < 0.05 and **p < 0.01. K The quantification of GzmB+ immune cells (n = 3). One-way ANOVA t test. *p < 0.05 and **p < 0.01. L The resected primary tumors were homogenized for immunoblotting analysis (n = 3). M. The resected primary tumors were homogenized for qRT-PCR analysis (n = 3). One-way ANOVA t test. *p < 0.05 and **p < 0.01. N The isolated splenocytes were co-incubated with CT26 cells (5000 cells) in different ratio. The cell viability was evaluated by CCK8 assay (n = 3). Two-way ANOVA t test. *p < 0.05.
miR-29b-3p reconstitution restores STING expression to enhance RT-induced antitumor immunity for abscopal effects in KRAS–mutated cells
The therapeutic efficacy of FDA-approved small-molecule KRASG12C inhibitors in colorectal cancer is unexpected29. Currently, no small-molecule KRASG12D inhibitor was approved by FDA in colorectal cancer. Therefore, we engineered an adenovirus-associated virus (AAV) with mmu-miR-29b-3p under the control of the CRC-specific CEA promoter, which was previous established in our lab (Fig. 6D)25,30,31. After intramuscular injection of AAV-pCEA-miR-29b-3p into tumor-bearing mice, we analyzed the distribution AAV genome (ITR, inverted terminal repeat) and miR-29b-3p RNA level (Figs. S5F and S5G). We found that AAV genome was broadly expressed in spleen, colon, tumor and brain tissue (Fig. S5F). But miR-29b-3p was highly specific expressed in tumor tissues, compared with other organs (Fig. S5G). Infection of AAV-pCEA-miR-29b-3p in CT26 significantly decreased the DNMT3b and upregulated STING expression (Fig. S5H). Overexpression of DNMT3b reversed these results, demonstrating that the specificity of miR-29b-3p on DNMT3b (Fig. S5H). We then evaluated whether supplementation with cancer-intrinsic miR-29b-3p restored the therapeutic efficacy of radiotherapy in KRASG12D-CRC cells and elicited a better systemic antitumor immune response to eradicate distant tumors (Fig. 6D). Two days after the subcutaneous inoculation of CT26 cells into the right leg of the BALB/c mice, the CT26 cells were inoculated into the left back of the BALB/c mice. AAV-miR-29b-3p was intramuscularly injected at 5-day intervals. Local radiotherapy (5 Gy) was administered on Days 9 and 10. As shown in Fig. 6E, the primary tumor volume was substantially reduced when AAV-miR-29b-3p was administered. Moreover, a high complete response rate was observed for AAV-miR-29b-3p/RT (Fig. 6E and F). Additionally, considerable tumor regression was observed in the abscopal tumors in the AAV-miR-29b-3p/RT group (Fig. 6G and 6H). The resected tumor weight was also reduced in the AAV-miR-29b-3p /RT group on Day 28 (Fig. 6H). The numbers of CD3+ and GzmB+ cells were markedly increased in the AAV-miR-29b-3p/RT group (Fig. 6I‒K). The level of DNMT3b was significantly decreased, and the level of the apoptotic marker cleaved caspase-3 was markedly increased in the resected tumors from the AAV-miR-29b-3p/RT group (Fig. 6L). Proinflammatory cytokines Ifnβ1 and Cxcl10 mRNA levels were markedly increased in tumors resected from the AAV-miR-29b-3p/RT group (Fig. 6M). The cytotoxic ability of the isolated splenic T cells from the AAV-miR-29b-3p/RT group was increased (Fig. 6N). Taken together, these results showed that the administration of CRC-specific miR-29b-3p synergistically increased the therapeutic efficacy of RT to eradicate abscopal tumors by overcoming oncogenic KRAS-mediated STING suppression.
To further investigate the immune profiles within tumors, we repeated this animal experiment to isolate tumor-infiltrating lymphocytes for flow cytometric analysis. The gating strategies was shown in Fig. S6. As shown in Figs. 7A and B, the levels of CD86 and CD80 in tumor-infiltrating DCs (CD11c+MHCII+CD45+ DCs) were significantly increased in the AAV-miR-29b-3p and AAV-Vec/RT groups. Moreover, these DC maturation markers were markedly increased in the AAV-miR-29b-3p/RT group (Figs. 7A, B and S6A). The infiltration of CD4+ and CD8+ T cells (CD4+CD3+CD45+7-AAD- and CD8+CD3+CD45+7-AAD-) was also significantly greater in the AAV-miR-29b-3p/RT group than in the other groups (Fig. 7C). Additionally, the density of effector/memory CD4+ and CD8+ T cells (CD44+CD62L-CD4+CD3+CD45+7-AAD- and CD44+CD62L-CD8+CD3+CD45+7-AAD- TEM) was greater in the AAV-miR-29b-3p/RT group (Figs. 7D, 7E and. S6B). The number of cytotoxic IFNγ+CD8+ (IFNγ+CD8+CD3+CD45+7-AAD-) T cells was significantly increased in the AAV-miR-29b-3p/RT group (Figs. 7F and S6C). Taken together, these results demonstrated that the administration of CRC-specific miR-29b-3p remarkably reinvigorated the immunosuppressive TME in KRAS-mutated CRC, leading to a better therapeutic response to RT.

A The subset of tumor-infiltrating CD86+ DC cells (CD86+CD11c+MHCII+ CD45+7AAD-) within primary tumors was analyzed by flow cytometric analysis (n = 3). One-way ANOVA t test. *p < 0.05. B The subset of tumor-infiltrating CD80+ DC cells (CD80+CD11c+MHCII+ CD45+7AAD-) within primary tumors was analyzed by flow cytometric analysis (n = 3). One-way ANOVA t test. *p < 0.05. C The subset of tumor-infiltrating CD4+ cells (CD4+CD3+CD45+7-AAD-) and CD8+ cells (CD8+CD3+CD45+7-AAD-) within primary tumors was analyzed by flow cytometric analysis (n = 3). One-way ANOVA t test. *p < 0.05. D The subset of tumor-infiltrating effector/memory CD4 TEM cells (CD44+CD62L-CD4+CD3+CD45+7-AAD-) within primary tumors was analyzed by flow cytometric analysis (n = 3). One-way ANOVA t test. *p < 0.05. E The subset of tumor-infiltrating effector/memory CD8 TEM cells (CD44+CD62L-CD8+CD3+CD45+7-AAD-) within primary tumors was analyzed by flow cytometric analysis (n = 3). One-way ANOVA t test. *p < 0.05. F The subset of tumor-infiltrating IFNγ+CD8+ lymphocytes (IFNγ+CD8+CD3+CD45+7-AAD-) within primary tumors was analyzed by flow cytometric analysis (n = 3). One-way ANOVA t test. *p < 0.05. G CT26 cells were inoculated into right hind leg (2 × 105 cells) by subcutaneous injection. On Day 9 and 10, local tumors in right hind leg were irradiated with 5 Gy. AAV-Vec. and AAV-pre-mmu-miR29b (1 × 108 vg/mouse) was intramuscular injected on Day 4, 9, 14 and 18. Anti-mouse CD8 or PD-1 antibodies were intraperitoneal injected on Days 10, 15, 20 and 25. The tumor volume was measured every 3 days (n = 5-6). Two-way ANOVA t test. *p < 0.05, **p < 0.01 and ***p < 0.001. H The proposed mechanism of oncogenic KRAS-mediated miR-29b-3p/DNMT3b/STING pathway to attenuate RT response in KRAS-mutated CRC.
Supplementation with CRC-specific miR-29b-3p significantly increased the therapeutic response to radiotherapy and ICBs in KRAS-mutated CRC
Since oncogenic KRAS promotes an immunosuppressive TME in CRC and is unresponsive to ICBs16,32, we next tested whether the effect of CRC-specific miR-29b-3p enhances the therapeutic response to ICBs in combination with RT in vivo. Therefore, we subcutaneously inoculated the mouse colorectal cancer cell line CT26 (endogenous KRASG12D) into BALB/c mice and then administered AAV-Vec. and AAV-miR-29b-3p. Local radiotherapy (5 Gy ×2) and anti-mouse CD8a and anti-mouse PD1 antibodies were given on the indicated days (Fig. 7G). We found that the AAV-miR-29b-3p/RT regimen significantly delayed tumor growth and prolonged survival (66.7% survival rate). In combination with ICB, the survival rate significantly increased (83.3% survival rate, Fig. 7G). However, administration of anti-mouse CD8a antibodies markedly decreased the survival rate (16.7% survival rate). Taken together, these results showed that supplementation with CRC-specific miR-29b-3p significantly increased RT-induced antitumor immunity to enhance the response to ICBs via the CD8+ cytotoxic T-cell response.
Discussion
Radiotherapy is highly effective in controlling localized colorectal cancer and has been utilized as a standard neoadjuvant treatment modality. However, failure at distant, nonirradiated sites remains a major driver of progression in up to 30% of patients, even after aggressive multimodal interventions33. While the role of the immune system in mediating the local and abscopal antitumor activity of radiotherapy has been appreciated for decades, the underlying mechanisms governing the efficiency of immune priming in this context remain poorly defined. Here, we report that the cell-intrinsic role of mutant KRAS is to suppress cancer immunogenicity and attenuate radiotherapy-induced dendritic cell maturation and cytotoxic CD8+ T-cell tumor infiltration; thus, mutated KRAS-expressing tumors have a poor response to radiotherapy. Mechanistic investigations revealed that a reduction in mutant KRAS-driven miR-29b-3p increased the level of DNMT3b to suppress STING levels via hypermethylation, which inhibited radiotherapy-induced cancer immunogenicity by decreasing type I IFN production for the recruitment of intratumoral dendritic cells and cytotoxic CD8+ T cells (Fig. 7H). The rational combination of cancer-specific miR-29b-3p and radiotherapy was sufficient to reestablish a highly immunogenic TME in KRAS-mutant tumors, enhancing the systematic antitumor immune response to eradicate abscopal tumors.
Although the critical role of oncogenic KRAS is involved in cancer progression and drug resistance via cell-intrinsic mechanisms, recent research has indicated that KRAS mutation is associated with several immune cell types involved in immune escape within the TME34,35,36,37. In addition to resistance to anti-EGFR therapy, KRAS mutations act in a cell-intrinsic fashion to sustain its protumoral activities and are associated with a poor response to chemotherapy and radiotherapy in multiple malignancies, including colorectal and lung cancer8,38,39. Hong et al. reported that the presence of a KRAS mutation was associated with an increased risk of tumor recurrence after high-dose radiation in patients with colorectal cancer metastases40. KRAS mutations are associated with increased survival of cancer cells from oxidative and other genotoxic stresses, possibly by promoting direct removal of oxidized nucleotides, DNA base excision repair (BER) and upregulation of the NRF2-53BP1-mediated nonhomologous end-joining (NHEJ) pathway41,42. Here, we revealed that oncogenic KRAS may drive resistance to radiotherapy by reducing cancer immunogenicity to avoid immune surveillance. We found that oncogenic KRAS significantly decreased the level of miR-29b-3p, which has been reported to act as a tumor suppressor microRNA to inhibit cancer progression via epigenetic regulation43. Our results indicated that miR-29b-3p directly targets DNMT3b to suppress STING via epigenetic regulation, which attenuates radiotherapy-induced type I IFN production for cancer immunogenicity. In support of our results, miR-29b-3p has been proven to be an epi-miRNA that targets DNMTs and regulates DNA demethylation pathway members, leading to the downregulation of global DNA methylation in malignant cells44. MiR-29b either directly targets DNMT1, DNMT3a and DNMT3b, resulting in a marked reduction in their expression, or indirectly downregulates DNMT1 by targeting Sp1, a transactivator of the DNMT1 gene, which in turn re-expresses two tumor suppressors, p15 (INK4b) and ESR144. Moreover, miR-29b-3p is also involved in the production of the proinflammatory cytokine interleukin-1545 and increases sensitivity to radiotherapy by modulating the oncogene axis46. These results indicate that oncogenic KRAS decreases the level of the tumor suppressor miR-29b-3p, which in turn increases DNMT3b to suppress STING expression and weaken immunogenicity, leading to RT-induced type I IFN production for dendritic cell maturation and cytotoxic T-cell infiltration within the TME. However, we cannot exclude the possibility that miR-29b targeting other oncogenes to reverse the immunosuppressive TME in KRAS-Mut CRC. These still need to further investigate.
Mutated KRAS has recently emerged as a player in engineering a permissive microenvironment for CRC progression11. Lal et al. indicated that KRAS mutation is associated with the suppression of Th1 cell and cytotoxic cell immunity via transcriptional analysis11,47. Furthermore, Liao et al. demonstrated that KRAS mutation inhibits IRF2 expression, resulting in high expression of CXCL3, which binds to CXCR2 on myeloid-derived suppressor cells (MDSCs) and attracts them into the TME16. Consistent with these studies, we observed that KRAS mutations may suppress RT-eliciting cancer immunogenicity, leading to less recruitment of CD8+TILs by suppressing STING. This work further proposed a previously undescribed association between oncogenic KRAS and cancer immunology17,48. In addition, we found that cytotoxic CD8+ T-cell tumor infiltration was unlikely to correlate with KRAS mutation after RT, which suggests that tumor immune evasion might be a general feature of KRAS-mutated CRC. Furthermore, due to epigenetically silenced of STING in colorectal cancer, especially in KRAS-mutated CRC12,20, recent studies demonstrated that recovering STING expression by demethylating agents49 and then combined with genotoxic therapies such as radiotherapy and cytotoxic DNA-damaging agents23,50, triggering cGAS/STING activation for antitumor immunity. The elegant study from Kitajima group23 also indicated that upregulation of STING by demethylating agent DNMT inhibitor and combined with monopolar kinase inhibitor (MPS1, a critical regulator of the spindle assembly checkpoint (SAC), triggering profound STING-mediated type I IFN for antitumor immunity. Therefore, identifying potential detailed mechanisms to direct upregulation of STING to enhance RT-induced antitumor immunity in KRAS-mutated CRC patients is critical for clinical relevance. The present findings improve the understanding of the link between oncogenic KRAS and the immune TME. However, we observed that demethylating agent did not complete rescue STING expression in KRASG12D-overexpressing cells, suggesting that other possible mechanisms may participate in regulating STING expression in KRAS-mutated cancers, such as ubiquitin-dependent degradation51. Further experiments for understanding these detailed mechanisms of STING expression in KRAS-mutated cancers are needed in future.
Radiation-induced systemic antitumor effects are associated with the activation of systemic immunity. However, thus far, abscopal responses in the clinic remain relatively rare52, and despite continuing preclinical investigations, it has proven difficult to reproducibly achieve abscopal responses in the clinic. The present study improves the current understanding of the radiation-induced immune response deployed by KRAS-mutated colorectal cancer patients. Our results demonstrated that KRAS-mutated CRC exploits hypermethylation mechanisms to suppress STING, which decreases type I IFN production in response to radiation. Given the complete remission of both irradiated and abscopal tumors and prolonged survival in our murine KRAS-mutated CRC models, we believe that this approach has promising translational potential to significantly increase its therapeutic benefit in KRAS-mutated CRC patients.
Methods
Cell lines and plasmids
Three human colorectal cancer cell lines, WiDr, CoLo320DM, SW480 and HT29, and the mouse colon cancer cell line CT26 were individually cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (HyClone, MA, USA) at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. The retroviral pBabe-puro vector (#1764) and pBabe-puro-KRASG12D (#58902) were purchased from Addgene.
Evaluation of STING and CD8+ immune cells in advanced CRC patients before and after neoadjuvant chemoradiotherapy (neoCRT) treatment
A total of 94 patients with locally advanced rectal cancer who received neoadjuvant chemoradiotherapy (neoCRT) followed by surgery at our hospital from 2006-2016 were enrolled. This study was reviewed and approved by the Institutional Review Board (IRB) of China Medical University Hospital [Protocol number: CMUH105-REC2-072]. Patients with informed consent and biopsy-proven locally advanced rectal cancer [cT3-4 or cN+ by endorectal ultrasonography (EUS), computed tomography (CT), or magnetic resonance imaging (MRI)] who were treated with preoperative chemoradiotherapy followed by radical resection at China Medical University Hospital composed the study cohort. Tumors were staged based on the American Joint Committee on Cancer (AJCC) staging system. EUS, MRI or CT was used to assess the pretreatment clinical stage, and pretreatment biopsies were reviewed by pathologists as previously described4,53,54,55.
Tissue microarrays were constructed from 94 pair-matched preneoCRT biopsies and postneoCRT surgical tissues from rectal cancer patients, and other samples were not available (data not suitable for IHC) as previously described. Areas of tumor cells were marked on hematoxylin and eosin (HE)-stained slides. The corresponding area on the matching paraffin block (donor block) was then identified and marked. We used the AutoTiss 10 C system (EverBio Technology Inc., Taipei, Taiwan) to remove the tissue core from these areas of the donor blocks into the recipient block in a precise, arrayed fashion. The punches were 2 mm in diameter, and a maximum of 60 punches were placed on a single block. Sample sections cut on a microtome were then mounted on capillary gap slides (Dako, Hamburg, Germany).
Immunohistochemistry (IHC) was performed on 3-μm-thick histological TMA sections as previously described53,56,57,58,59. TMA slides were stained individually with horseradish peroxidase-conjugated avidin biotin complex (ABC) via the Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA) and DAB (Vector Laboratories) and counterstained with hematoxylin. The following antibodies were used in this study: anti-CD8 (ab4055, Abcam), anti-STING (ab239074, Abcam) and anti-DNMT3b (NB300-516, Novus Bio.) antibodies. The stained tissue sections were scored separately by two pathologists blinded to the clinicopathological parameters. Intratumoral T lymphocyte staining was positive when detected in the cytoplasm or at the cell membrane of tumor-infiltrating lymphocytes (TILs) and was evaluated via microscopy (OLYMPUS BX53, Tokyo, Japan) according to the intensity of the TILs. Two pathologists who were blinded to all the sample information evaluated the TILs. For the detection of TILs, the tissue was reviewed at 40× magnification, and the area with the highest density adjacent to malignant cells was counted at 400× magnification (no. of TILs/high-power field). The average number of CD8+ TILs in five high-power fields was included in the evaluation. For TILs, the cutoff of criteria for high density was >15 TILs/mm2.
The following antibodies were used in this study: anti-CD8 (ab4055 and ab209775, Abcam), anti-STING (ab239074, Abcam, Cambridge, UK), anti-DNMT3b (NB300-516, Novus Bio.), anti-granzyme B (ab4059), anti-CD3 (ab16669, Abcam), anti-CD11c (ab219799, Abcam) and anti-Ki67 (ab15580, Abcam) antibodies.
Sample preparation and KRAS testing
The pretreated biopsies were formalin fixed, paraffin embedded, and placed on slides that were marked by a pathologist and manually microdissected for tumor tissue. Genomic DNA was extracted via QIAamp DNA FFPE Tissue kits (Qiagen Inc., Valencia, CA). KRAS mutations were originally determined by standard polymerase chain reaction (PCR) followed by Sanger sequencing of exons 2 and 3 of KRAS.
DNA extraction, sodium bisulfite treatment and pyrosequencing
Genomic DNA from nontumor and tumor tissues of rectal cancer patients was extracted from 3–5 (5 μm thick) sequential sections of FFPE slides via a QuickExtract™ FFPE DNA Extraction Kit (QEF81050, Epicenter, WI, USA) according to the manufacturer’s instructions. All genomic DNA samples, as well as synthetic controls for methylated and unmethylated status, underwent bisulfite modification via the Epitect Bisulfite Kit (Qiagen)60.
Pyrosequencing assays were performed for all the study samples both on a PyroMark Q24 MDx and on a PyroMarkQ96 ID using PyroMark Gold reagents (Qiagen). Primers for STING, which target three CpGs (TMEM173_1 (rs11554775), TMEM173_2-1 (rs574732324) and TMEM173_2-2 (rs1751900750, rs1329356200)) in the gene promoter, were chosen according to the literature12 and designed according to PyroMark Assay Design software version 2.0 (Qiagen). PCR analyses were performed in a total volume of 35 μl containing 1X buffer (KCl), 2 mM MgCl2, 200 μM dNTPs, 0.5 μM of each primer, 1.75 U Taq polymerase and 6 μl of bisulfite-modified DNA with the following cycling profile: 95 °C for 1 min followed by 45 cycles of denaturation at 95 °C for 1 min, annealing at the specific temperature for 1 min, and extension at 72 °C for 1 min. Extension at 72 °C for 10 min was finally performed. Amplicons were analyzed via gel electrophoresis on a 2% agarose gel stained with SafeView and visualized via ultraviolet transillumination. The residual PCR product (28 μl for the AQ mode, 20 μl for the CpG mode) was added to distilled water (12 μl for the AQ mode, 18 μl for the CpG mode) and incubated under shaking with binding buffer (pH 7.6; 37 μl for the AQ mode, 40 μl for the CpG mode) containing 10 mM Tris-HCl, 2 M NaCl, 1 mM EDTA, and 0.1% Tween 20, and Sepharose beads (3 μl for the AQ mode, 2 μl for the CpG mode) were covered with streptavidin. The PCR products were washed with 70% ethanol, denatured with 0.2 M and rewashed with 10 mM Tris-acetate, pH 7.6. The pyrosequencing reaction was performed in AQ mode in a total volume of 45 μl, including 44.82 μl of 20 mM Tris-acetate and 5 mM MgAc2, and 0.18 μl of 50 μM sequencing primer (final concentration of 0.3 μM); in CpG mode, it was performed in a total of 25 μl, including 24.85 μl of 20 mM Tris-acetate, 5 mM MgAc2 and 0.15 μl of 50 μM sequencing primer (final concentration of 0.3 μM). Assays were performed according to the manufacturer’s instructions. The nucleotide dispensation order was outlined by Q24 2.0 software.
Evaluation of immune cell infiltration in KRAS-silenced tumors in vivo
A total of 3 × 105 (right leg) and 2×105 (left back) CT26shNC and CT26shKRAS cells were inoculated into BALB/c mice via subcutaneous injection. After 12 days, the local tumors in the right leg were irradiated (5 Gy) two times at 4-day intervals. The tumor volumes at the irradiated site (right leg) and nonirradiated site (left back) were measured every 3 days. Tumors were isolated from the mice, weighed, and then placed in Petri dishes containing blank RPMI media at room temperature to prevent dehydration. The tumors were minced into small pieces (1–2 mm) with a beaver blade, filtered through a 70 μm strainer, spun down, and then resuspended in blank RPMI media. Thereafter, the cell suspensions were layered over Ficoll-Paque media, centrifuged at 1025 × g for 20 min, the layer of mononuclear cells was transferred into a conical tube, 20 ml of complete RPMI media was added, and the mixture was gently mixed and centrifuged at 650 × g for 10 min twice. Finally, the supernatant was removed, and the TILs were resuspended in complete RPMI media61.
TILs were then resuspended in 500 μL of staining buffer (2% BSA and 0.1% NaN3 in PBS). The cells were stained with a surface marker panel and intracellular cytokines by different panels1: CD8/MDSC panel: PerCP/Cy5 anti-mouse CD8a (clone 53-6.7, Cat#100732, BioLegend, CA, USA), FITC anti-human/mouse CD11b (clone M1/70, Cat#101206, BioLegend), PE/Cy7 anti-mouse CD45 (clone 30-F11, Cat#103114, BioLegend), APC anti-mouse Gr-1 (clone RB6-8C5, Cat#108412, BioLegend) and PE anti-mouse CD44 (Clone QA19A43, Cat#163610, BioLegend)2; Foxp+ Treg panel: APC anti-mouse CD25 (Clone PC61, Cat#102012, BioLegend), APC/Cy7 anti-mouse CD4 (Clone RM4-5, Cat#100526, BioLegend), PerCP/Cy5 anti-mouse CD8a (clone 53-6.7, Cat#100732, BioLegend) and PE anti-mouse FOXP3 (Clone QA20A67, Cat#118904, BioLegend)3; IFNγ+CD8+ T-cell panel: FITC anti-mouse IFNγ (Clone XMG1.2, Cat#505806, BioLegend), PerCP/Cy5 anti-mouse CD8a (clone 53-6.7, Cat#100732, BioLegend) and PE/Cy7 anti-mouse CD45 (clone 30-F11, Cat#103114, BioLegend), and their isotypes (BioLegend). The intracellular marker Foxp3 was fixed and permeabilized with a FOXP3 Fix/Perm buffer before staining (Cat# 421403, Biolegend). The intracellular marker INFγ was activated by the protein transport inhibitor brefeldin A for 3 h before staining. These samples were analyzed via a Guava® easyCyte™ flow cytometer (Luminex, CA, USA) and FlowJo v10.0.7 software (Ashland, USA).
Western blot analysis
Total lysates (30 μg) were resolved on an SDS–PAGE gel and transferred onto PVDF membranes (Millipore, MA, USA) [[28,2962 for immunoblot analyses with the indicated antibodies overnight at 4 °C. The membranes were then probed with HRP-conjugated secondary antibodies for 2 h at room temperature. All the antibodies were diluted in T-Pro protein-free blocking buffer (BioLion Tech., Taipei, Taiwan). The membrane was then incubated with Immobilon Western Chemiluminescent HRP Substrate (Millipore, CA, USA), visualized via an ImageQuant™ LAS 4000 biomolecular imager (GE Healthcare, Amersham, UK), processed via Adobe Photoshop, and quantified via ImageJ software (NIH, MD, USA). Each blot was stripped with immunoblotting stripping buffer (BioLion Tech.) before incubation with the other antibodies. The following antibodies were used: phospho-STING-S365 (#19781, Cell Signaling Technology), STING (#13647, Cell Signaling Technology), cleaved caspase-3 (#9661, Cell Signaling Technology and IR96–401, iReal Biotech.), calreticulin (#12238, Cell Signaling Technology), beta-actin (Ab8227, Abcam), DNMT1 (#5032, Cell Signaling Technology), DNMT3a (#49768, Cell Signaling Technology), DNMT3b (#57868, Cell Signaling Technology), KRAS (A1190, ABclonal) and GAPDH (IR3-8, iReal Biotech.)
qRT‒PCR
Total RNA was extracted from the cell lines with TRIzol (Invitrogen, CA, USA), quantified by measuring the absorbance at 260 nm, and then reverse-transcribed into cDNA using iScript™ Reverse Transcription Supermix (Bio-Rad, CA, USA) according to the manufacturer’s instructions31. Primers were designed via the Primer Design Tool (NCBI, USA) according to sequence information from the NCBI database. qRT‒PCR was performed in a final volume of 20 μL with iQ™ SYBR® Green Supermix (Bio-Rad, CA, USA) via the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). All reactions were performed in triplicate for each sample, and GAPDH was used as a reference gene for normalization. The relative gene expression levels were calculated via the 2 − ΔΔCt method. Gene expression levels were compared via t tests.
miRNA sequencing (miRNA-seq) and data analysis
Total RNA was extracted from HT29-KRASWT and HT29-KRASG12D cells via TRIzol reagent. RNA quality was checked via an Agilent 2100 Bioanalyzer System (Santa Clara, CA, USA). mRNA was extracted from total RNA via oligo (dT) magnetic beads and sheared into short fragments of approximately 200 bases. These fragmented mRNAs were then used as templates for cDNA synthesis. The cDNAs were subsequently amplified to prepare a complete library via PCR, and the cDNA library was subjected to paired-end sequencing via the Illumina HiSeqTM 2500 platform24,63.
Clean reads were obtained by removing low-quality reads and/or trimming the adaptor sequences from the raw reads. The clean reads were employed for gene expression analysis. The mapped reads were used for further transcript annotation and for calculating expression levels via the fragments per kilobase per million reads (FPKM) method and the HTseq software package (version 0.6.1). DEGs were identified with the DESeq software package (version 1.12.0). The Benjamini‒Hochberg false discovery rate was employed to correct the p values. Only genes with a p value < .05 were considered to be differentially expressed. The k-means method was used to cluster the DEGs based on their relative expression levels.
Colorectal cancer-specific AAV2 vector generation and recombinant AAV2 virus purification
The murine premmu-miR-29b sequence was generated via PCR and subcloned and inserted into a pAAV2-CEA vector, which contained the CEA promoter25. All the viruses were produced via triple transfection with AAV2-miR-29b and the helper plasmids pRC2-miR-342 and pHelper in 293 T cells. Seventy-two hours after transfection, the cells were collected via centrifugation, and recombinant AAV2 vectors were produced and purified via an AAVpro® purification kit (6232, Takara, Japan). AAV2 titration was performed via quantitative polymerase chain reaction (qPCR) of the vector genomes.
Combined treatment with CRC-specific AAV2-miR-29b-3p and radiotherapy in a syngeneic KRASG12D murine CRC model
BALB/c mice (female, 4 weeks old) were maintained according to the institutional guidelines approved by the China Medical University Institutional Animal Care and Use Committee [Protocol No. CMUIACUC-2018–167]. Before tumor cell inoculation, the mice were randomized into different groups (five in each group). Briefly, CT26 cells (2 × 105 cells/mouse) were suspended in 100 μL of 50% Matrigel and inoculated subcutaneously into the right leg of each mouse. Two days later, CT26 cells (1 × 105 cells/mouse) were inoculated subcutaneously into the left flank of each mouse. AAV2 particles (1× 108 vg) were intramuscularly injected on Days 4, 91, 14 and 19. Local irradiation (5 Gy) was performed on Days 10 and 13. The tumor volume was measured every 3 days throughout the study. The longest and shortest diameters (L and W, respectively) of the tumors were measured via Vernier calipers (Sata, Shanghai, China) every 3 days, and the tumor volume (V) was calculated via the following formula: V = (L × W2)/2. The mice were sacrificed at the termination of the experiments, and the tumor tissues were collected for lysis and subjected to immunoblot analysis and immunohistochemical staining64,65.
Transient transfection with miRNA mimics/inhibitors
miR-29b-3p mimics, inhibitors, and negative control sequences were designed and purchased from TopGen Bio. (Taiwan). Human colorectal cancer cell lines were seeded in a 6-well plate at a density of 2 × 105 cells/well and incubated for 24 h. The cells were transfected with mimics, inhibitors, or negative control sequences via JetPrime (Polypus, France). After transfection for 24 h, the cells were irradiated with 5 Gy and harvested 24 h later for analysis of mRNA expression and 48 h later for western blot analysis of protein expression.
RNA in situ hybridization (ISH)
In situ hybridization (ISH) was performed on 3-μm-thick histological colorectal cancer TMA sections as previously described53,56,57,58. The TMA slides were individually stained with biotin-labeled probes according to the manufacturer’s manual (#TASH02D, BioSpot ISH Kit for Biotin Probes). Briefly, tissue slides were washed three times with phosphate buffer saline (PBS) and subjected to prehybridization.
buffer (2× saline sodium citrate, 10% formamide). The ISH probes for miR-29b-3p (TCCATACCAACTTCGTAGAAA-biotin, Qiagen) were resuspended in hybridization buffer (2× saline sodium citrate, 10% formamide, and 10% dextran sulfate) to a final concentration of 250 nM per probe set. Hybridization was carried out in a humidified chamber at 37 °C for 16 h. After incubation with the ISH probe, the cells were washed three times with PBS, treated with avidin-conjugated HRP for 0.5 h at RT, and then counterstained with hematoxylin. The stained TMA slides were scanned and evaluated.
Statistical analysis
IBM SPSS statistical software version 26 was used to perform the statistical analysis. All tests reported a two-sided p value with a significance level set at 0.05. Student’s t test, the Pearson chi-square test and Fisher’s exact test were used for group comparisons. Cox regression analysis was used to estimate the hazard ratios (HRs) and 95% confidence intervals (CIs) for the univariate and multivariate models. The Kaplan‒Meier method was used to estimate five-year disease-free survival (DFS). Survival time was defined as the time from surgery until the event, including local recurrence, distant metastasis and death. Univariate comparisons were performed via the log-rank test.
Data availability
The original data of western blots has been stored in the supplementary materials. The cell line dataset was download from CCLE database and other dataset can be provided according to reasonable requirements.
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Diaz, L. A. Jr & Le, D. T. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med 373, 1979 (2015).
Roulleaux Dugage, M. et al. Biomarkers of response to immunotherapy in early stage non-small cell lung cancer. Eur. J. Cancer 184, 179–196 (2023).
Huang, C. Y. et al. Clinical significance of programmed death 1 ligand-1 (CD274/PD-L1) and intra-tumoral CD8+ T-cell infiltration in stage II-III colorectal cancer. Sci. Rep. 8, 15658 (2018).
Hofmann, M. H., Gerlach, D., Misale, S., Petronczki, M. & Kraut, N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov. 12, 924–937 (2022).
Formica, V. et al. KRAS and BRAF Mutations in Stage II and III Colon Cancer: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 114, 517–527 (2022).
Wang, M. et al. Radiation Resistance in KRAS-Mutated Lung Cancer Is Enabled by Stem-like Properties Mediated by an Osteopontin-EGFR Pathway. Cancer Res. 77, 2018–2028 (2017).
Mak, R. H. et al. Outcomes by tumor histology and KRAS mutation status after lung stereotactic body radiation therapy for early-stage non-small-cell lung cancer. Clin. Lung Cancer 16, 24–32 (2015).
Chow, O. S. et al. KRAS and Combined KRAS/TP53 Mutations in Locally Advanced Rectal Cancer are Independently Associated with Decreased Response to Neoadjuvant Therapy. Ann. Surg. Oncol. 23, 2548–2555 (2016).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356 (2015).
Lal, N. et al. KRAS Mutation and Consensus Molecular Subtypes 2 and 3 Are Independently Associated with Reduced Immune Infiltration and Reactivity in Colorectal Cancer. Clin. Cancer Res. 24, 224–233 (2018).
Kitajima, S. et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 9, 34–45 (2019).
Muthalagu, N. et al. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 10, 872–887 (2020).
Chu, P. C. et al. Mutant KRAS promotes liver metastasis of colorectal cancer, in part, by upregulating the MEK-Sp1-DNMT1-miR-137-YB-1-IGF-IR signaling pathway. Oncogene 37, 3440–3455 (2018).
Tew, B. Y. et al. Genome-wide DNA methylation analysis of KRAS mutant cell lines. Sci. Rep. 10, 10149 (2020).
Liao, W. et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 35, 559–72 e7 (2019).
Hamarsheh, S., Gross, O., Brummer, T. & Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 11, 5439 (2020).
Deng, L. et al. STING-Dependent Cytosolic DNA sensing promotes radiation-induced Type I Interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852 (2014).
Mackenzie, K. J. et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017).
Xia, T., Konno, H., Ahn, J. & Barber, G. N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 14, 282–297 (2016).
Konno, H. et al. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 37, 2037–2051 (2018).
Falahat, R. et al. Epigenetic reprogramming of tumor cell-intrinsic STING function sculpts antigenicity and T cell recognition of melanoma. Proc. Natl. Acad. Sci. USA. 118, e2013598118 (2021).
Kitajima, S. et al. MPS1 inhibition primes immunogenicity of KRAS-LKB1 mutant lung cancer. Cancer Cell 40, 1128–44 e8 (2022).
Huang, K. C. et al. DNMT1 constrains IFNbeta-mediated anti-tumor immunity and PD-L1 expression to reduce the efficacy of radiotherapy and immunotherapy. Oncoimmunology 10, 1989790 (2021).
Huang, K. C. et al. Engineered sTRAIL-armed MSCs overcome STING deficiency to enhance the therapeutic efficacy of radiotherapy for immune checkpoint blockade. Cell Death Dis. 13, 610 (2022).
Kwon, J. J., Factora, T. D., Dey, S. & Kota, J. A Systematic Review of miR-29 in Cancer. Mol. Ther. Oncolytics 12, 173–194 (2019).
Vera, O. et al. A MAPK/miR-29 Axis Suppresses Melanoma by Targeting MAFG and MYBL2. Cancers. 13, 1408 (2021).
Nguyen, T. et al. Downregulation of microRNA-29c is associated with hypermethylation of tumor-related genes and disease outcome in cutaneous melanoma. Epigenetics 6, 388–394 (2011).
Desai, J. et al. Divarasib plus cetuximab in KRAS G12C-positive colorectal cancer: a phase 1b trial. Nat. Med. 30, 271–278 (2024).
Huang, K. C. et al. A Novel Engineered AAV-Based Neoantigen vaccine in combination with radiotherapy eradicates tumors. Cancer Immunol. Res 11, 123–136 (2023).
Huang, K. C. et al. Colorectal cancer-specific IFNbeta delivery overcomes dysfunctional dsRNA-mediated type I interferon signaling to increase the abscopal effect of radiotherapy. J. Immunother. Cancer 12, e008515 (2024).
Hu, H. et al. Oncogenic KRAS signaling drives evasion of innate immune surveillance in lung adenocarcinoma by activating CD47. J. Clin. Invest. 133, e153470 (2023).
Ogura, A. et al. Neoadjuvant (Chemo)radiotherapy With Total Mesorectal Excision Only Is Not Sufficient to Prevent Lateral Local Recurrence in Enlarged Nodes: Results of the Multicenter Lateral Node Study of Patients With Low cT3/4 Rectal Cancer. J. Clin. Oncol. 37, 33–43 (2019).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Hallin, J. et al. The KRAS(G12C) Inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10, 54–71 (2020).
Boutin, A. T. et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev. 31, 370–382 (2017).
Moon B. S. et al. Role of oncogenic K-Ras in cancer stem cell activation by aberrant Wnt/beta-catenin signaling. J Natl Cancer Inst. 106:djt373. (2014).
Yoon, H. H. et al. KRAS codon 12 and 13 mutations in relation to disease-free survival in BRAF-wild-type stage III colon cancers from an adjuvant chemotherapy trial (N0147 alliance). Clin. Cancer Res. 20, 3033–3043 (2014).
Russo, A. L. et al. Mutational and clinical predictors of pathologic complete response in the treatment of locally advanced rectal cancer. J. Gastrointest. Cancer 45, 34–39 (2014).
Hong, T. S. et al. Phase II study of proton-based stereotactic body radiation therapy for liver metastases: importance of tumor genotype. J. Natl. Cancer Inst. 109, djx031 (2017).
Patel, A. et al. MutT Homolog 1 (MTH1) maintains multiple KRAS-driven pro-malignant pathways. Oncogene 34, 2586–2596 (2015).
Yang, L. et al. Oncogenic KRAS drives radioresistance through upregulation of NRF2-53BP1-mediated non-homologous end-joining repair. Nucleic Acids Res. 49, 11067–11082 (2021).
Yan, B. et al. The role of miR-29b in cancer: regulation, function, and signaling. Onco Targets Ther. 8, 539–548 (2015).
Garzon, R. et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 113, 6411–6418 (2009).
Mishra, A. et al. Aberrant overexpression of IL-15 initiates large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell 22, 645–655 (2012).
Pan, D. et al. miR-29b-3p increases radiosensitivity in stemness cancer cells via modulating oncogenes axis. Front Cell Dev. Biol. 9, 741074 (2021).
Lal, N., Beggs, A. D., Willcox, B. E. & Middleton, G. W. An immunogenomic stratification of colorectal cancer: Implications for development of targeted immunotherapy. Oncoimmunology 4, e976052 (2015).
Tsai, Y. Y. et al. Targeting DRP1 promotes radiotherapy-induced antitumor immunity via mitochondrial DNA-Mediated cGAS-STING signaling in KRAS-mutated colorectal cancer. Cancer Lett 637, 218132 (2026).
Tu, Y. et al. DNMT inhibition epigenetically restores the cGAS-STING pathway and activates RIG-I/MDA5-MAVS to enhance antitumor immunity. Acta Pharmacol. Sin. 47, 197–208 (2025).
Ma, J., Xin, Y., Wang, Q. & Ding, L. Roles of cGAS-STING pathway in radiotherapy combined with immunotherapy for hepatocellular carcinoma. Mol. Cancer Ther. 23, 447–453 (2024).
Kim, D. S., Park, Y., Tsokos, G. C., Cho, M. L. & Kwok, S. K. The ubiquitin E3 ligase TRIM21 suppresses type I interferon signaling via STING degradation and ameliorates systemic autoimmunity. Exp. Mol. Med. 57, 1555–1566 (2025).
Golden, E. B. et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol. 16, 795–803 (2015).
Huang, C. Y. et al. Cytosolic high-mobility group box protein 1 (HMGB1) and/or PD-1+ TILs in the tumor microenvironment may be contributing prognostic biomarkers for patients with locally advanced rectal cancer who have undergone neoadjuvant chemoradiotherapy. Cancer Immunol. Immunother. 67, 551–562 (2018).
Chen, T. W. et al. Prognostic relevance of programmed cell death-ligand 1 expression and CD8+ TILs in rectal cancer patients before and after neoadjuvant chemoradiotherapy. J. Cancer Res Clin. Oncol. 145, 1043–1053 (2019).
Huang, K. C. et al. Decitabine augments chemotherapy-induced PD-L1 upregulation for PD-L1 blockade in colorectal cancer. Cancers 12, 462 (2020).
Huang, C. Y. et al. HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Dis. 9, 1004 (2018).
Lin, T. Y., Fan, C. W., Maa, M. C. & Leu, T. H. Lipopolysaccharide-promoted proliferation of Caco-2 cells is mediated by c-Src induction and ERK activation. Biomedicine 5, 5 (2015).
Wang, X. et al. RSF-1 overexpression determines cancer progression and drug resistance in cervical cancer. Biomedicine 8, 4 (2018).
Ke, T. W. et al. Targeting B7-H3 enhances the efficacy of neoantigen-based cancer vaccine in combination with radiotherapy. NPJ Vaccines 10, 80 (2025).
Huang, K. C. et al. ATAD3A stabilizes GRP78 to suppress ER stress for acquired chemoresistance in colorectal cancer. J. Cell Physiol. 236, 6481–6495 (2021).
Chen, J. Y. et al. CHK1 inhibition increases the therapeutic response to radiotherapy via antitumor immunity in ARID1A-deficient colorectal cancer. Cell Death Dis. 16, 584 (2025).
Huang, K. C. et al. Inhibition of DNMTs increases neoantigen-reactive T-cell toxicity against microsatellite-stable colorectal cancer in combination with radiotherapy. Biomed. Pharmacother. 177, 116958 (2024).
Huang, K. C. et al. Neoantigen-augmented iPSC cancer vaccine combined with radiotherapy promotes antitumor immunity in poorly immunogenic cancers. NPJ Vaccines 9, 95 (2024).
Lin, Y. S. et al. Targeting CD73 increases therapeutic response to immunogenic chemotherapy by promoting dendritic cell maturation. Cancer Immunol. Immunother. 72, 2283–2297 (2023).
Huang, K. C. et al. TNFalpha modulates PANX1 activation to promote ATP release and enhance P2RX7-mediated antitumor immune responses after chemotherapy in colorectal cancer. Cell Death Dis. 15, 24 (2024).
Acknowledgements
This study was supported in part by China Medical University Hospital (DMR-114-168), and the Ministry of Science and Technology (MOST110-2628-B-039-005, MOST110-2314-B-039-032, and MOST 111-2823-8-039-001, Taiwan). Experiments and data analysis were performed in part using the Medical Research Core Facilities Center, Office of Research & Development at China Medical University, Taichung, Taiwan, R. O. C.
Author information
Authors and Affiliations
Contributions
H.Y.C., J.Y.C., Y.W.J., W.Z.H., S.F.C. and K.C.Y.H. conducted and performed the experiments; Y.Y.T., Y.S.L., T.H.C. and T.W.K. enrolled patients and performed the IHC evaluation; K.C.Y.H. and K.S.C.C. supervised the study; C.H.H. assisted in the design of the radiation experiments; and K.C.Y.H. and K.S.C.C. analyzed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chang, HY., Chen, JY., Ke, TW. et al. Loss of MicroRNA-29b promotes DNMT3b-mediated STING downregulation to attenuate radiotherapy-induced antitumor immunity in KRAS-mutated colorectal cancer. npj Precis. Onc. 10, 93 (2026). https://doi.org/10.1038/s41698-026-01290-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-026-01290-8
