
Abstract
While cancer immunotherapy has transformed clinical management for cancer patients, its low response rates remain a critical challenge to be addressed. Tumor immune evasion now extends beyond the tumor microenvironment (TME), as advanced tumors induce extramedullary hematopoiesis (EMH) in the spleen, leading to a substantial expansion of erythroid progenitor cells (EPCs) with potent immunosuppressive capacity. EPCs are typically transient populations in erythroid maturation and differentiation; however, under tumor burden, they undergo profound metabolic reprogramming that exacerbates their immunosuppressive effects. This review examines the role and mechanisms of tumor-hijacked metabolic reprogramming in EPCs and provides strategies for targeting this reprogramming to potentiate cancer immunotherapy. In particular, we synthesize the metabolic interplay between EPCs, tumor cells, and immune cells, integrating EPC metabolic reprogramming with established concepts of tumor cell metabolism and immunometabolism. Furthermore, this review outlines future directions for the field, including multi-modal approaches to decipher the mechanisms of EPC metabolic reprogramming, biomarker development, and metabolism-based targeted therapies, all aimed at improving survival and prognosis for cancer patients.
Similar content being viewed by others

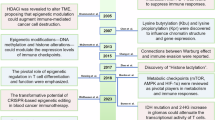

Introduction
Cancer now represents the second leading cause of human mortality after cardiovascular diseases1,2. This epidemic directly impedes life expectancy improvements while imposing a substantial global public health burden, with particularly severe impacts in low- and middle- income countries3. Immunotherapy has revolutionized cancer treatment by activating or enhancing immune system responses to eliminate cancer cells through natural mechanisms. As a representative class of immunotherapies, immune checkpoint inhibitors (ICIs) have significantly advanced cancer treatment and demonstrate broad application potential across multiple cancer types4. ICIs targeting programmed cell death protein 1/programmed death-ligand 1 (PD-1/PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) signaling have been shown to counteract tumor immune evasion and have achieved notable success in clinical practice5,6. Despite these advances, ICI efficacy varies considerably among individuals, with durable and effective disease control observed in only a subset of patients7,8. To address this limitation, researchers aim to enhance ICI therapeutic outcomes by improving response rates and overcoming primary and acquired resistance, ultimately aiming to improve prognosis for cancer patients.
Resistance to ICIs is often associated with immunosuppression within the tumor microenvironment (TME)9,10. The TME contains multiple immunosuppressive cells and inhibitory factors that weaken T-cell responses by suppressing T-cell proliferation or activation, thereby facilitating tumor immune escape11. Various myeloid cells, including tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), are recognized as immunosuppressive cell types that mediate immunosuppression by inhibiting T cells and promote tumor progression. In recent years, novel approaches for cancer treatment have been explored by targeting and inhibiting the metabolism of these myeloid cells. However, immunotherapies targeting these cells appear unable to completely cure tumors. This indicates that there are still deficiencies in research on immunosuppressive cells. Numerous studies have shown that erythroid progenitor cells (EPCs) are a new type of immunosuppressive cell and are at least equal to MDSCs in immunosuppressive activity. Therefore, the metabolism of EPCs may be a key factor affecting the efficacy of immunotherapy, further research into the mechanisms governing EPC-mediated immunosuppression and their interactions with immune cells holds promise for the development of novel therapeutic strategies targeting the immunometabolism.
This review synthesizes the metabolic interactions among EPCs, tumor cells, and immune cells, integrating EPC metabolic reprogramming with tumor cell metabolism and immunometabolism. It further proposes strategies to target EPC metabolic reprogramming for enhancing cancer immunotherapy, offering a novel perspective on improving therapeutic efficacy and providing a framework for the development of targeted interventions. Moreover, a deeper understanding of the mechanisms underlying EPC metabolic reprogramming will inform the design of metabolism-based biomarkers and support the development of synergistic treatment approaches that combine metabolic modulation with cancer immunotherapy (Fig. 1).

During their own maturation and differentiation, EPCs may undergo tumor-induced metabolic reprogramming. Furthermore, via interactions with tumor cells and immune cells, EPCs can subsequently alter the metabolism of both tumor and immune cells, thereby promoting cancer immune evasion. Targeting EPCs metabolic reprogramming can enhance cancer immunotherapy. EPC erythroid progenitor cell, HPSC hematopoietic stem cell, RBC red blood cell, ROS reactive oxygen species, ARG arginase, EDMC erythroid-derived myeloid cell, CCL5 c-c motif chemokine ligand 5.
Development and differentiation of EPCs
As key mediators of immunosuppression derived from the erythroid lineage, EPCs create a self-reinforcing cycle that actively drives tumor progression12. Human EPCs are characterized by the surface markers CD71 and CD235a13, mice EPCs are defined by the surface markers CD71 and Ter119. During normal development, hematopoietic stem and progenitor cells (HSPCs) differentiate into common lymphoid progenitors (CLPs) and common myeloid progenitors (CMPs). The latter further differentiate through the megakaryocyte-erythroid progenitor (MEP) stage into EPCs, ultimately producing mature erythrocytes14. In contrast, MDSCs are defined as a heterogeneous population of myeloid cells originating from altered myelopoiesis in the bone marrow and secondary lymphoid organs, where their maturation is blocked, and from the reprogramming of mature myeloid cells in peripheral tissues15,16. These altered differentiation stages collectively result from dysregulated myelopoiesis and developmental disruption. A fundamental characteristic of these heterogeneous myeloid cells is immunosuppressive capacity under the pathological conditions that drive their expansion17. Beyond cancer, such conditions include chronic inflammation and infection, autoimmunity, and allergy18.
Under conditions that require rapid erythrocyte expansion, such as anemia, pregnancy, or infection, erythropoiesis can occur outside the bone marrow. This extramedullary erythropoiesis takes place in organs including the liver, spleen, and lymph nodes19, which is called extramedullary hematopoiesis (EMH). Cytokines and physical or chemical stresses specific to the TME can induce changes in the function and composition of the spleen through the systemic circulatory system and systemic immune system, thereby causing EMH in the spleen. Tumor-derived regulatory factors and anemia commonly observed in cancer patients promote hematopoiesis, leading to abundant EPCs generation and recruitment into the spleen or infiltration into the TME13,20. Based on surface markers and functional properties, EPCs are categorized into two distinct subsets, CD45⁺ EPCs and CD45⁻ EPCs. The CD45⁺ subset primarily suppresses T cell immune responses21. In mice tumor-bearing models, CD45⁺ EPCs significantly outnumber regulatory T cells (Tregs) and are considered to possess immunosuppressive potency at least comparable to that of MDSCs. Given that their abundance exceeds that of MDSCs by more than threefold, CD45⁺ EPCs are regarded as a potentially more potent immunosuppressive population than MDSCs19. In peripheral blood samples from patients with hepatocellular carcinoma, CD45⁺ EPCs are fewer in number than monocytic MDSCs (M-MDSCs) but more abundant than polymorphonuclear MDSCs (PMN-MDSCs) and Tregs22. Thus, they are considered a stronger immunosuppressive cell population than Tregs and likely play a role in human cancer immune evasion analogous to that of MDSCs. Another population of EPCs differentiates into CD45- EPCs (Ter-cells), which participate in immune regulation by secreting cytokines14. The CD45⁻ subset of EPCs exhibits a distinct transcriptional signature, characterized by the downregulation of genes associated with reactive oxygen species (ROS) pathways19. Within the splenic niche, EPCs can undergo myeloid differentiation to become erythroid-derived myeloid cells (EDMCs), resulting in the generation of cells that inhibit anti-tumor immunity23. Moreover, EPCs significantly contribute to resistance against ICI-based therapies24. Therefore, understanding EPCs-related metabolic mechanisms and developing targeted therapeutic strategies may disrupt this vicious cycle, improve the response rate and long-term efficacy of immunotherapy.
Metabolic regulation of EPCs
EPCs represent a specific subpopulation in erythroid development and differentiation. Their metabolic state during development influences both their differentiation fate and biological functions12. Under pathological conditions, the metabolism of EPCs becomes altered, and such metabolic alterations may subsequently shape the formation and characteristics of the TME (Fig. 2).

EPCs represent a critical subpopulation during erythroid differentiation. They originate in the spleen and participate in EMH under pathological conditions. Furthermore, EPCs can infiltrate the TME and contribute to tumor progression. EPC erythroid progenitor cell, HPSC hematopoietic stem cell, EPO erythropoietin, GM-CSF granulocyte-macrophage colony-stimulating factor, G-CSF granulocyte colony-stimulating factor, PDGF platelet-derived growth factor, TGF-β transforming growth factor beta, CCL2 c-c motif chemokine ligand 2, PD-L1 programmed death-ligand 1, EDMC erythroid-derived myeloid cell, ARG arginase.
Metabolic adaptations during EPCs expansion and mobilization
Under physiological conditions, EPCs reside within the bone marrow niche and ultimately differentiate into oxygen-transporting erythrocytes25. Initially, stem cell factor (SCF) secreted by bone marrow endothelial and stromal cells binds to its receptor c-Kit, inducing EPCs proliferation and the formation of burst-forming unit-erythroid (BFU-E)26. Subsequently, under the influence of EPO, BFU-E further differentiate into colony-forming unit-erythroid (CFU-E)27. Ultimately, multiple cytokines mediate the upregulation of GATA-2 expression, driving EPCs maturation into functional erythrocytes25.
In the tumor-bearing state, tumor-derived cytokines induce rapid expansion and accumulation of hematopoietic stem and progenitor cells (HSPCs). Key cytokines involved include granulocyte colony-stimulating factor (G-CSF), platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β), and SCF. This process promotes EMH21. Specifically, tumor-derived PDGF-BB stimulates EPO production by targeting stromal and perivascular cells that express PDGFR-β28. Tumors can also promote EPO generation through tissue hypoxia and anemia. EPO is essential for the proliferation and survival of EPCs. Upon binding to its receptor, EPO activates Janus kinases (JAKs) and Src family kinases, which leads to STAT protein activation and enhanced expansion of EPCs29. Additionally, EPO promotes erythroid differentiation of HSPCs, giving rise to CD45⁺ EPCs. It also enhances cell survival by upregulating Bcl-2 and Bcl-X and inhibiting apoptosis30. During early tumor progression, serum G-CSF levels rise31. As a potent hematopoietic growth factor, G-CSF plays a key role in tumor-induced hematopoiesis32. By binding to its receptor (G-CSFR), it activates JAK-STAT signaling, primarily through STAT3 and STAT5 activation, to mediate HSPCs expansion and differentiation33,34.
CXCL12 is a critical component of the bone marrow stem cell niche. Under physiological conditions, HSPCs colonize the marrow niche by binding to CXCL12 expressed on perivascular mesenchymal cells, where they undergo further development35,36. In tumor-bearing hosts, G-CSF promotes enzyme release that disrupts CXCR4-CXCL12 interactions between HSPCs and bone marrow stromal cells. Additionally, VEGF binding to VEGFR2 on bone marrow endothelial cells facilitates HSPCs egress. Once mobilized into circulation, HSPCs are recruited to the spleen37,38,39.
Metabolism of EPCs in extramedullary hematopoiesis
EMH serves as a compensatory hematopoietic mechanism that occurs under pathological stress conditions such as hematopoietic insufficiency, hematopoietic disorders, tumors, anemia, or infections40. Tumors can induce EMH and preferentially promote myeloid cell production41. This is driven by the intrinsically hypoxic TME, where tumor cells secrete cytokines including HIF-1α and elevated G-CSF levels21,39. These circulating cytokines act on HSPCs, thereby promoting EMH. However, due to the dense tumor stroma, erythrocytes generated through EMH fail to infiltrate the TME and consequently cannot alleviate hypoxia. This failure perpetuates tumor-induced EMH42. In tumor-bearing states, EMH exhibits a myeloid-biased output due to tumor-derived GM-CSF and G-CSF, which direct HSPCs differentiation toward myeloid lineages41. These myeloid cells critically contribute to establishing an immunosuppressive TME. Substantial evidence correlates elevated myeloid cell levels with poor prognosis in cancer43,44. Recent studies indicate that this tumor-induced myeloid bias in EMH is mechanistically linked to intrinsic metabolic reprogramming of EPCs45.
EMH primarily occurs in the spleen, with HSPCs recruitment requiring multiple regulatory factors46. Studies using orthotopic Hepa1-6 hepatocellular carcinoma mouse models revealed that splenic stromal cells upregulate CCL2 expression, which recruits circulating HSPCs to the spleen via CCR2 binding42. To identify HSPCs colonization sites, immunohistochemical analysis demonstrated that EPCs accumulate within the splenic intrasinusoidal space47. Further investigations of EMH-positive patient spleens showed significantly elevated CXCL12 expression on red pulp sinusoidal endothelial cells48. This facilitates EPCs niche colonization through CXCR4 binding.
The fate of HSPCs is regulated by stromal cell types constituting specialized niches49. Consequently, activation of the splenic niche plays a critical role in EMH. Single-cell RNA sequencing (scRNA-seq) of HSPCs isolated from bone marrow and spleen revealed that splenic HSPCs from tumor-bearing mice exhibit upregulated inflammatory gene signatures and produce tumor necrosis factor-α (TNF-α) compared to those from normal mice. Further studies demonstrated that TNF-α derived from HSPCs binds to Pdgfra⁺/Pdgfrb⁺ adventitial stromal (ABS) cells expressing high levels of TNF-α receptors, inducing elevated expression of vascular cell adhesion molecule-1 (VCAM-1) on ABS cells. This promotes HSPCs colonization in the spleen. TNF-α also sustains CXCL12 secretion by ABS cells, further recruiting HSPCs50. Additionally, TNF-α secreted by CD4⁺ T cells have been shown to induce HSPC proliferation and differentiation, potentially contributing to splenic niche activation51. In summary, HSPCs can activate niche cells via inflammatory factor TNF-α expression, thereby promoting EMH. During EMH, stem cell factor (SCF) expressed by splenic endothelial cells and Tcf21⁺ stromal cells binds to c-Kit on EPCs, inducing receptor dimerization and activation of tyrosine kinases52,53. This initiates multiple signaling pathways that regulate EPCs development. Studies indicate that the c-Kit/SCF and EPO-R/EPO pathways functionally synergize as key regulators of EPCs development, playing essential roles in maintaining EPCs expansion and survival34.
Metabolic regulation of EPCs in the spleen
The metabolic regulation of HSPCs relies on support from the splenic niche. In a breast cancer mouse model, elevated circulating levels of IL-1α were observed. Further studies revealed that tumor-derived IL-1α activates niche cells by inducing TNF-α expression in HSPCs. These activated niche cells promote hematopoiesis by secreting various cytokines, including VCAM-1, CXCL1, and CXCL12. Tumors also expand the splenic niche directly by inducing proliferation of PDGFRα⁺/β⁺ stromal cell populations via leukemia inhibitory factor (LIF)50. IL-1α, which promotes EMH, has been implicated in driving the expansion of EPCs. Notably, IL-1α stimulates niche cells in the spleen to produce CXCL1 and CXCL12, thereby promoting the proliferation of hematopoietic progenitor cells.
EPO promotes EPCs differentiation into mature erythrocytes by inducing expression of GATA family transcription factors54. However, this erythropoietic differentiation mediated by EPO is inhibited upon CD45 expression55. CD45 dephosphorylates Src family kinases (including Fyn, Lck, Lyn, and Hck)56. Lyn kinase plays an important role in EPO signaling and promotes erythroid differentiation57. Under EPO culture conditions, co-incubation with an anti-CD45 antibody reduced Lyn phosphorylation levels and significantly impeded EPO-induced EPCs proliferation and erythroid differentiation55.
Under tumor-bearing conditions, CD45⁺ EPCs undergo myeloid differentiation. Studies in lung cancer mouse models revealed that GM-CSF promotes lineage conversion of EPCs into EDMCs45. Compared to CD45⁺ EPCs from tumor-free mice, those from tumor-bearing mice exhibit high GM-CSF receptor (GM-CSFR) expression on their surface. In vitro induction with GM-CSF led to lineage conversion in over 80% of these cells, demonstrating that tumors induce EPCs differentiation into EDMCs, a process mediated by GM-CSF signaling23.
A CD45⁻ subpopulation of EPCs, termed Ter-cells, also exists. TGF-β mediates its effects through binding to type III TGF-β receptor (TβR-III), which is significant for Ter cell generation58. Administration of TGF-β neutralizing antibodies to tumor-bearing mice inhibited Ter cell generation in the spleen. Similarly, inoculation of TGF-β-knockout hepatocellular carcinoma cells into mice resulted in significantly reduced Ter cell induction. Further investigations showed that TGF-β mediates Ter cell generation through Smad3 phosphorylation20. Additionally, elevated serum TGF-β levels in advanced cancer patients may explain the expansion of Ter cells during late-stage tumors59.
Recombinant human EPO (rhEPO) has been used to treat anemia in cancer patients during chemotherapy or radiotherapy. However, clinical trials have indicated adverse effects of rhEPO in cancer therapy, which are associated with EPO-promoted Ter cell generation60. Injection of EPO into naïve mice resulted in Ter-cell accumulation in the spleen. Further depletion of Ter-cells using Ter119 antibodies in LLC model mice showed that EPO-injected mice exhibited faster tumor growth compared to controls, suggesting that EPO enhances Ter-cell generation and potentially strengthens their tumor-promoting effects61. Gene expression profiling of Ter-cells revealed significant overexpression of artemisinin. Further studies found that serum artemisinin levels were markedly elevated during tumor progression, whereas Smad3-deficient mice inoculated with hepatocellular carcinoma cells exhibited reduced serum artemisinin. Splenectomy in tumor-bearing mice nearly eliminated elevated serum artemisinin, confirming that Ter-cells in the spleen are the primary source of serum artemisinin.
Interplay between EPCs and cancer cell metabolism
EPCs act as collaborators in cancer progression. Beyond their intrinsic metabolic activities, they may directly influence cancer cell metabolism. These metabolic alterations could integrate into the broader landscape of tumor metabolic reprogramming, potentially impacting antitumor immunity and contributing to therapy resistance46. Consequently, targeting EPC-mediated modulation of cancer cell metabolism may represent a critical strategy to enhance immunotherapy efficacy and overcome cancer cell drug resistance.
CCL5/CCR5 axis mediates recruitment of CD45⁺ EPCs into the TME
CD45⁺ EPCs mediate immunosuppression through direct cell contact and paracrine signaling, necessitating their infiltration into the TME to exert functional effects22. CCL5, a critical intercellular communication mediator, is highly expressed in the TME of multiple cancers and promotes various protumor functions by binding to its receptor CCR562. In a recent study using SCC7 model mice, elevated transcriptional levels of CCL5 were detected in tumor cells. RNA sequencing analysis of intratumoral CD45⁺ EPCs revealed upregulated CCR5 transcription, which was further confirmed at the protein level via flow cytometry, demonstrating high CCR5 expression on these cells. Specific blockade of CCR5 significantly reduced the infiltration of CD45⁺ EPCs into tumors46. These findings indicate that CD45⁺ EPCs are recruited into the TME via tumor-secreted CCL5 through upregulation of CCR5 expression.
ROS metabolism of EPCs
ROS is generated through multiple intracellular pathways and participate in redox signaling and oxidative stress homeostasis under physiological conditions63. ROS generally play a promotive role in immune responses, contributing to both innate and adaptive immunity. However, in the high-ROS environment of the TME, ROS can also exert immunosuppressive effects and promote tumorigenesis and angiogenesis. High ROS levels exhibit broad inhibitory effects on immune cells. EPCs regulate ROS generation through the co-localization of galectin-3 (Gal-3) and GARP, a surface TGF-β receptor. Gal-3 expression shows a strong positive correlation with ROS levels. Consequently, Gal-3⁺ EPCs exhibit significantly stronger immunosuppressive properties compared to their Gal-3⁻ counterparts, primarily through the inhibition of CD4⁺ and CD8⁺ T-cell proliferation64.
Arginine metabolism of EPCs
Arginase is a manganese-centered metalloenzyme that catalyzes the conversion of L-arginine to L-ornithine and urea, serving as a key enzyme in the urea cycle. In mammals, two arginase isozymes exist: ARG-1, predominantly expressed in the liver, and ARG-2, mainly found in the kidneys65. ARG overexpression plays a significant immunosuppressive role in the TME and represents a major mechanism through which EPCs exert immunosuppression. EPCs enriched in the placenta and umbilical cord blood show high expression of ARG-1 and PD-L1, and their depletion in mouse models confirmed their immunosuppressive role at the maternal–fetal interface66. Within the TME, mediated by the granulocyte-macrophage colony-stimulating factor receptor (GM-CSFR), EPCs can differentiate into myeloid lineages, yielding EDMCs that produce ARG-1 and mediate immune suppression23. In the multiple myeloma (MM) TME, the proportion of EPCs expressing ARG-1 increases, and overall ARG-1 protein levels are elevated, impairing T cell-mediated immune responses. This impairment is significantly alleviated upon administration of ARG inhibitors67.
Roles of EPCs on immunometabolism
EPCs may influence cancer cell metabolism, thereby contributing to the formation of an immunosuppressive TME. During this process, EPCs can interact with immune cells and alter their metabolic profiles. Such metabolic reprogramming may suppress immune cell recognition and antigen presentation capabilities (Fig. 3), ultimately exacerbating the immunosuppressive properties of the TME12.

EPCs can interact with immune cells and influence their metabolic processes. Alternatively, they may remodel the immune microenvironment by regulating their own metabolism, ultimately impacting antitumor immunity. EPC erythroid progenitor cell, ARG arginase, NK natural killer, PD-L1 programmed death-ligand 1, PD-1 programmed death receptor 1, TGF-β transforming growth factor beta, CTL cytotoxic T lymphocyte, DC dendritic cell, Treg regulatory T cell, TCR T cell receptor, MDSC myeloid-derived suppressor cell, EDMC erythroid-derived myeloid cell.
CD45⁺ EPCs suppress CD8⁺ T cells via ROS metabolism
Similar to MDSCs, which mediate T cell immunosuppression through excessive ROS production, ROS serve as a primary mechanism underlying CD45⁺ EPC-mediated immunosuppression19,68. ROS can combine with nitric oxide (NO) to form bioactive peroxynitrite (ONOO⁻), which downregulates T-cell receptor (TCR) expression and disrupts TCR complex integrity through tyrosine nitration to nitrotyrosine (NT)69,70,71. When CD45⁺ EPCs isolated from tumor-bearing mice were co-cultured with CD8⁺ T cells for 48 h, a significant increase in NT levels was observed in the T cells. This effect was abolished upon addition of the ROS inhibitor uric acid. Further experiments demonstrated that blocking peroxynitrite reversed CD45⁺ EPCs-mediated suppression of CD8⁺ T cells72. These findings indicate that CD45⁺ EPCs inhibit CD8⁺ T cell immune responses via ROS-dependent nitration of the TCR.
EDMCs exhibit high PD-L1 expression
CD45⁺ EPCs can also differentiate into myeloid lineages, giving rise to EDMCs that resemble MDSCs. PD-1 is a co-inhibitory receptor expressed on antigen-stimulated T cells and interacts with two ligands, PD-L1 and PD-L2. PD-L1 is primarily detected on hematopoietic and non-hematopoietic healthy tissue cells, while PD-L2 is mainly expressed on macrophages and mast cells73. The PD-1/PD-L1 axis mediates immune escape; EDMCs upregulate PD-L1 expression, leading to exhaustion of CD8⁺ T cells in the TME23. PD-L1 derived from EDMCs binds to PD-1 on CD8⁺ T cells, inhibiting the co-stimulatory receptor CD28 and suppressing kinase activation around the TCR, thereby inducing T cell exhaustion74. PD-L1 also inhibits the phosphoinositide 3-kinase (PI3K)/Akt pathway, suppresses T cell proliferation, promotes apoptosis, and ultimately depletes CD8⁺ T cells75. Furthermore, PD-L1 impedes the release of inflammatory cytokines (e.g., IFN-γ, IL-2 and TNF-α), further compromising CD8⁺ T cell function. Additionally, PD-L1 promotes the development, maintenance, and function of Tregs, contributing to overall immunosuppression76. In cancer patients experiencing, CD45⁺ EPCs in the spleen are modulated by heparin-binding growth factors derived from CD45⁻ EPCs. These cells migrate into tumors via the CCL5/CCR5 axis and exert immunosuppression through PD-L146. EPCs themselves can also express PD-L1 to mediate immune suppression. During murine pregnancy, high expression of PD-L1 and PD-L2 is observed in placental erythroid cells, maintaining fetal tolerance. Blocking PD-L1 restores immune responsiveness during pregnancy77.
EDMCs inhibit T cells via arginase secretion
L-arginine plays a crucial role in T cell metabolism. Its deficiency reduces T cell survival and suppresses antitumor activity78. Similar to MDSCs, EDMCs secrete ARG-1 to catabolize L-arginine into ornithine, depleting arginine in the TME23. Extensive research demonstrates that L-arginine deficiency downregulates CD3ζ chain expression in T cells, inhibiting their proliferation79. Furthermore, under low arginine conditions, T cells fail to upregulate cyclin D3 expression, causing cell cycle arrest at the G0-G1 phase and consequently blocking proliferation80. Arginine blockade in CD4⁺ and CD8⁺ T cells significantly reduce proliferation, while elevated L-arginine levels promote the serine biosynthesis pathway by upregulating intermediate metabolites. This pathway enhances the tricarboxylic acid (TCA) cycle and subsequent oxidative phosphorylation (OXPHOS), thereby elevating T cell energy metabolism-—essential for biosynthetic processes and immune function78,81. Additionally, L-arginine deficiency impairs T cell differentiation and maturation, substantially suppressing IFN-γ and TNF-β secretion82. Collectively, EDMCs suppress T cell immunity through ARG-1-mediated arginine depletion in the TME.
CD45⁺ EPCs induce CD8⁺ T cell tolerance via antigen presentation
Tumor immunosuppression can be initiated through the induction of CD8⁺ T cell tolerance. GR-1⁺ immature myeloid cells (IMCs) from tumor-bearing mice can uptake soluble proteins, process them, and present antigenic epitopes on their surface, thereby inducing antigen-specific CD8⁺ T cell tolerance83. Similarly, EPCs possess the ability to uptake, process, and present antigens, leading to CD8⁺ T cell tolerance. Following the establishment of a murine tumor model using MC38-OVA cells, the presence of OVA was detected on the surface of CD45⁺ EPCs, indicating that these cells can capture and process tumor-associated antigens. However, this study did not elucidate whether the process is MHC-dependent or occurs through alternative pathways, warranting further investigation72. Specifically, CD45⁺ EPCs loaded with tumor-derived peptides induce antigen-specific CD8⁺ T cell tolerance. This effect is mediated by ROS and peroxynitrite produced by CD45⁺ EPCs, which promote nitration of tyrosine residues within TCR/CD8 molecules, ultimately driving antigen-specific T cell tolerance.
CD45⁻ EPCs mediate immunosuppression via TGF-β secretion
TGF-β signaling plays a dual role in cancer progression, acting as a tumor suppressor in early stages while promoting tumor development in advanced disease. TGF-β signaling contributes to the formation of an immunosuppressive TME and facilitates cancer growth, invasion, metastasis, recurrence, and drug resistance84. Immunofluorescence staining of tumor tissues has revealed upregulated TGF-β expression in EPCs, suggesting their involvement in TGF-β-mediated immunosuppression22. TGF-β exerts broad inhibitory effects on immune cells: it downregulates the expression of NKP30 and NKG2D on natural killer (NK) cells, impairing their cytotoxicity and antitumor responses; it reduces dendritic cell (DC) infiltration into tumors and inhibits their migration to lymph nodes, thereby suppressing antigen presentation; it suppresses the expression of cytotoxic genes (e.g., perforin, granzyme, IFN-γ) in CD8⁺ T lymphocytes and inhibits T cell proliferation85,86. TGF-β derived from CD45⁻ EPCs acts on naïve CD4⁺ T cells, significantly downregulating phosphorylation of Akt and mTOR and inhibiting their proliferation and differentiation into effector T cells. Beyond suppressing effector cells, TGF-β also promotes the generation of suppressive populations by inducing FOXP3 expression in CD4⁺ T cells, promoting their differentiation into immunosuppressive regulatory T cells87. As immunosuppressive cells, Tregs highly express PD-L1, playing a critical role in facilitating tumor immune escape. In summary, CD45⁻ EPCs secrete TGF-β to broadly suppress T cell function and promote the generation of immunosuppressive cells.
Targeting EPCs metabolism to improve cancer immunotherapy
Immunotherapy has significantly advanced cancer treatment, but its clinical translation remains limited by low response rates, particularly in locally advanced patients receiving ICIs88,89. EPCs substantially contribute to ICI resistance; thus, targeting EPCs metabolism holds promise for enhancing therapeutic efficacy and improving patient outcomes (Fig. 4).

Metabolic reprogramming of EPCs can be targeted to reverse immunosuppressive effects, thereby potentiating the efficacy of various cancer treatment strategies, including radiotherapy and immunotherapy. EPC erythroid progenitor cell, HPSC hematopoietic stem cell, ROS reactive oxygen species, ARG arginase, EDMC erythroid-derived myeloid cell, CCL5 c-c motif chemokine ligand 5, CCR5 c-c motif chemokine receptor 5, TME tumor microenvironment.
Reduce EPC abundance
Promoting EPCs to complete terminal erythroid maturation and exit the progenitor pool, thereby reducing the pool of immature, immunosuppressive EPCs that are susceptible to myeloid diversion and minimize the generation of EDMCs. G-CSF and GM-CSF facilitate myeloid differentiation of EPCs and splenic HSPCs, as well as transdifferentiation of erythroid precursors into myeloid lineages. Intervention strategies targeting GM-CSF may reverse the differentiation direction of EPCs and alleviate tumor-induced immunosuppression90. GATA1 is a crucial transcription factor in erythropoiesis, regulating erythroid maturation and function at the transcriptional level. Maintaining GATA1 homeostasis is essential for proper erythroid development. Overexpression of GATA1 can reprogram myeloid progenitors into megakaryocytic or erythroid lineages, demonstrating its ability to balance erythroid commitment. Inflammasomes promote caspase-mediated cleavage of GATA1 in HSPCs, leading to biased myeloid differentiation of EPCs91. TGF-β exerts inhibitory effects on EPCs, impeding their proliferation and maturation while promoting myeloid-skewed differentiation92. Inhibiting TGF-β signaling with Smad inhibitors has been shown to restore T-cell proliferation suppressed by EPCs22. FOXO3 mutation results in hyperactivation of mTOR signaling in immature erythroblasts, causing maturation defects. Rapamycin can effectively alleviate the inhibition of erythroid maturation93.
Inhibiting the recruitment of HSPCs to the spleen effectively attenuates EMH. The CXCL12/CXCR4 axis and adhesion molecules critically regulate HSPC homing and mobilization, where CXCL12/CXCR4 activation promotes HSPC retention. Concurrently, the CCL2/CCR2 signaling pathway plays a pivotal role in selectively recruiting splenic HSPCs and triggering EMH-mediated immunosuppression42. Blockade of the CCL2/CCR2 pathway significantly reduces EMH, with CCR2 inhibitors combined with chemotherapy already demonstrating robust antitumor efficacy. Low-dose sorafenib induces HSPC apoptosis to deplete splenic HSPC reservoirs, thereby weakening EMH-driven immunosuppression. Additionally, Anemia serves as a significant inducer of EMH and affects a considerable proportion of cancer patients. It not only directly reduces the quality of life and survival rates of cancer patients but also promotes the proliferation of EPCs by inducing EMH. Improving anemia can alleviate EMH and suppress EPC generation.
Block recruitment
Suppressing EPC recruitment to the TME represents a viable therapeutic strategy. Our previous work established that splenic CD45⁺ EPCs migrate to tumors via the CCL5–CCR5 chemokine axis, suggesting that targeting this pathway could impede EPC recruitment into the TME. In SCC-7 tumors, where CCR5 protein expression is notably high in CD45⁺ EPCs, CCR5 blockade alone reproduced the effect of splenectomy and reduced intratumoral CD45⁺ EPC infiltration. However, CCR5 inhibition did not substantially alter the frequency of CD45⁺ EPCs in the spleen. Furthermore, a combined strategy of CCR5 blockade plus splenectomy did not significantly further decrease tumor-infiltrating CD45⁺ EPCs compared to CCR5 blockade alone46. Thus, while targeting CCR5 can effectively inhibit the recruitment of CD45⁺ EPCs into the TME, it does not address the underlying cause of their expansion driven by EMH.
Targeted depletion of immunosuppressive EPCs through apoptosis induction via specific biomarkers represents a promising strategy. Since EPCs are potential sources of erythrocytes, non-selective targeting may lead to adverse effects such as anemia, highlighting the importance of precise targeting. Cytotoxic drug-conjugated CD45 antibodies can clear and replace myeloid cells in tissues, and CD71 antibodies can eliminate the immunosuppressive effects of CD71⁺ erythroid cells94,95. Similar approaches may be applied to deplete immunosuppressive EPCs in cancer patients. Radiation therapy promotes EPCs apoptosis by stimulating IFN-γ production, and supplementation with IFN-γ effectively induces apoptosis of EPCs in the spleens of tumor-bearing mice61. Chemotherapeutic agents can also induce EPCs apoptosis, associated with upregulation of Bcl-2 leading to cytochrome c release and formation of apoptotic vesicles. However, due to the broad suppression of bone marrow cells by chemotherapy, more precise drug delivery approaches are needed to minimize adverse effects.
Reverse metabolic reprogramming
Reversing the metabolic reprogramming of EPCs represents a key strategy for targeting EPCs to enhance cancer immunotherapy. In the TME, EPC-mediated arginine depletion impairs OXPHOS in T cells, thereby suppressing their proliferation and effector functions while promoting tumor cell growth and immune evasion96. Therefore, arginine supplementation in the TME may offer a promising approach to alleviate EPC-driven immune tolerance. Notably, however, arginine exerts dual roles in both promoting tumor progression and supporting immune activation, underscoring the need for further preclinical validation of such metabolic intervention78. Additionally, CD45⁺ EPCs abundantly produce ROS, which significantly influence immune escape19. Although tumor cells maintain higher basal ROS levels and more robust antioxidant systems compared to normal cells, T cells are poorly adapted to drastic fluctuations in ROS within the TME97. Excessively high or low ROS levels disrupt normal T cell function98. Consequently, selective scavenging of ROS in the TME can effectively restore T cell activity and improve the efficacy of immunotherapeutic approaches.
Inhibit ROS and ARG-1 pathways
EPCs highly express ARG-1, depleting arginine in the TME14. This high ARG-1 expression is a common feature of nearly all myeloid cells in the TME and severely impairs T-cell immune responses99. Although arginine supplementation was discussed in the previous section as a potentially beneficial strategy for antitumor immunity, it is important to note that tumor cells in the TME also uptake arginine to support their own growth and proliferation100. Therefore, arginine supplementation alone may not achieve satisfactory clinical outcomes. Consequently, targeting ARG-1 in myeloid cells or EPCs represents a more promising therapeutic approach. Indeed, monoclonal antibodies against ARG-1, when combined with ICIs, have been shown to increase CD8⁺ T cell infiltration in the TME and enhance ICI efficacy101. Furthermore, RNA-seq analysis has revealed that CD45⁺CD71⁺ EPCs possess a greater capacity for ROS production compared to their CD45⁻CD71⁺ counterparts19, accompanied by significantly elevated expression of NADPH oxidase 2 (NOX2), a key enzyme responsible for ROS generation102. The regulation of ROS within EPCs is complex, and targeting a single signaling pathway or molecule may therefore yield limited effects. Deeper investigation into the ROS regulatory network will provide a more solid foundation for developing strategies that target ROS metabolism in EPCs.
Conclusion and prospects
Immunotherapy has significantly improved clinical care for cancer patients, particularly those with advanced disease. Enhancing the efficacy of immunotherapy is crucial for prolonging patient survival. EPCs represent a key subpopulation in erythrocyte formation. EPCs originate from bone marrow HSPCs and differentiate into mature erythrocytes under physiological conditions. However, in the tumor-bearing state, erythroid differentiation of EPCs is blocked, leading to reduced erythrocyte production and anemia, which subsequently activates compensatory hematopoietic responses.
Under tumor-bearing conditions, hematopoiesis predominantly occurs outside the bone marrow, a process termed EMH. The spleen is a common site of EMH and has been identified as a major source of EPCs in tumor-bearing hosts. Within the splenic niche, EPCs differentiate into two distinct subpopulations: CD45⁺ EPCs and CD45⁻ EPCs. CD45⁺ EPCs have been extensively studied as immunosuppressive cells that infiltrate the TME to mediate pro-tumor and immunosuppressive responses. In contrast, CD45⁻ EPCs are not recruited to tumors and remain in the spleen. Through metabolic reprogramming, EPCs influence erythroid maturation and contribute to tumor immune escape. Within the TME, CD45⁺ EPCs mediate immunosuppression via ROS and arginine metabolism. The EDMCs are generated through GM-CSF-induced lineage conversion. EDMCs highly express PD-L1 to directly suppress CD8⁺ T cells through contact-dependent mechanisms and secrete ARG-1 to deplete arginine in the TME, thereby inhibiting T cell proliferation and inducing immunosuppression. Furthermore, EDMCs may also induce antigen-specific tolerance in CD8⁺ T cells. EPCs that do not infiltrate the TME differentiate into CD45⁻ EPCs under the influence of TGF-β. CD45⁻ EPCs remotely modulate immune responses by secreting immunosuppressive factors such as artemisinin and TGF-β. By interfering with EPCs metabolism, it may be possible to inhibit their proliferation and differentiation, suppress lineage conversion, and promote apoptosis, thereby restraining cancer progression.
Elucidating the mechanisms underlying EPCs metabolic reprogramming
EPCs abundance positively correlates with tumor progression, including metastasis and poor prognosis. Altered EPCs metabolism or its regulatory processes may lead to new clinical symptoms in cancer patients, yet the relationship between these symptoms and EPC-derived metabolites remains unclear. Although significant progress has been made in understanding EPCs metabolism and its role in immunotherapy, translating these findings into clinical practice faces considerable challenges. Overcoming these obstacles will require deeper investigation into the metabolic regulation of EPCs and its underlying mechanisms. There is an urgent need to elucidate the mechanisms governing EPCs differentiation into erythrocytes, particularly how metabolic reprogramming influences erythroid versus myeloid fate decisions. This understanding is crucial for developing anti-EPCs therapeutics aimed at restoring normal erythropoiesis. Furthermore, although CD45⁺ EPCs mediate immunosuppression through mechanisms similar to MDSCs, with a notable capacity for inhibiting CD8⁺ T cell function, their broader metabolic influence on other immune cells, including NK cells and dendritic cells, remains poorly understood. The differences in immunosuppressive mechanisms between CD45⁺ EPCs and MDSCs also warrant clarification. Additionally, the metabolic processes and alterations driving the conversion of CD45⁺ EPCs to EDMCs within the TME need further exploration.
Applying multi-omics technologies, particularly spatial transcriptomics and single-cell sequencing, offers a promising avenue to uncover the metabolic and immunosuppressive mechanisms underlying EPC function. Profiling erythroid cells at different developmental stages may help delineate metabolic regulation during erythropoiesis and identify how metabolic dysregulation or reprogramming contributes to immune suppression. Furthermore, as immunosuppressive cells, EPCs may act as collaborators with cancer cells to promote tumor progression. However, the role of EPCs in cancer recurrence and metastasis remains unclear. Cancer metastasis includes both lymph node metastasis and distant organ spread103. It represents one of the most critical markers of poor prognosis and is a major cause of cancer related mortality. Interestingly, EPCs and EDMCs are believed to function similarly to MDSCs, potentially with even greater potency. Thus, it is reasonable to hypothesize that EPCs may contribute to malignant progression and metastasis. Notably, metastatic cancers often exhibit stronger resistance to immunotherapy compared to primary tumors and may undergo metabolic reprogramming to evade immune attack and develop therapy resistance. Research in this area remains limited and warrants further exploration.
Advances in this field necessitate the development of more physiologically relevant preclinical models. Currently, murine models remain the primary system for studying EPCs; however, physiological and functional differences between mice and humans represent a major obstacle to clinical translation. A key issue is the divergence in surface markers between murine and human EPCs, which may limit the translatability of metabolism-targeting strategies developed in mice to clinical settings. While organ-on-a-chip systems, including tumor organoids, offer improved in vitro modeling, they often fail to fully recapitulate systemic physiological changes and multi-organ interactions. A more promising approach may involve multi-organ chip platforms that simulate the systemic microenvironment of cancer patients, thereby providing a more comprehensive model for evaluating therapeutic efficacy and biological complexity.
Exploring biomarkers based on EPCs metabolic regulation
The metabolic reprogramming of EPCs profoundly influences their maturation and differentiation, potentially altering the entire erythroid lineage. Metabolic dysregulation may promote myeloid differentiation, thereby contributing to the formation of an immunosuppressive TME and ultimately impairing antitumor immune responses, which could represent a key mechanism of resistance to cancer immunotherapy. Accurately identifying and tracking metabolic alterations and regulatory pathways in EPCs is critical for developing EPC-targeted therapies. This aligns with the molecular mechanisms discussed earlier, as elucidating metabolic processes and discovering associated metabolites or substrates may lead to the identification of molecular signatures indicative of EPC metabolic dysfunction. Developing clinically relevant biomarkers for EPC-driven metabolic reprogramming requires addressing some intertwined methodological and conceptual barriers. Current isolation protocols, predominantly relying on flow cytometric sorting of splenic populations, remain unsuitable for clinical translation and preclude precise lineage tracing and ontogeny studies. Advancing robust, translation-ready isolation strategies is therefore imperative. Equally critical is resolving the causal relationship between EPCs and immune evasion in the TME. Should EPCs function as active drivers of immunosuppression, their recruitment signals or aberrant metabolic byproducts represent strong biomarker candidates. If, however, EPC accumulation is a secondary consequence of established immune escape, then EPC frequency or phenotype itself may serve as a prognostic indicator. Clarifying this causality is essential for rational biomarker design.
The detection of such biomarkers can be achieved through integrated multi-technology strategies, including molecular probes and liquid biopsy, offering novel approaches for precise monitoring. Furthermore, tracing metabolic dynamics and dysregulation in EPCs will deepen our understanding of the underlying reprogramming mechanisms. Detecting these biomarkers could form the basis for metabolically targeted interventions against EPCs, serve as indicators for treatment modulation, aid in screening potential patient populations likely to benefit from therapy, and provide insights into treatment efficacy.
Targeting EPCs metabolism to enhance cancer immunotherapy
Pharmacologic strategies aimed at EPCs offer considerable promise for clinical translation. Antibody-drug conjugates (ADCs), which integrate the high specificity of monoclonal antibodies with potent cytotoxic payloads, exemplify one promising direction. The development of antibodies targeting EPC-specific surface markers, when conjugated with cytotoxic agents such as IFN-γ, facilitates the precise elimination of EPC populations while minimizing off-target effects. However, a significant obstacle lies in the divergent biomarker profiles of EPCs between mice and humans, including structural differences in CD45, which complicate the targeted delivery of ADC-based therapeutics. Preclinical studies conducted in murine models may therefore encounter substantial translational challenges when advancing to clinical trials.
Beyond direct cytotoxicity, metabolic intervention offers a compelling alternative approach. This can be achieved by using inhibitors that target metabolic pathways underlying immunosuppression, including those involved in arginine metabolism and intracellular ROS accumulation, thereby reversing the associated phenotypic dysregulation in EPCs. However, many conventional metabolic modulators lack cellular specificity, necessitating advanced delivery systems. Nanomedicine platforms facilitate targeted drug delivery to EPCs or the splenic niche, thereby supporting erythroid differentiation, suppressing aberrant myelopoiesis, and potentially ameliorating anemia associated with EMH or tumor progression. ICIs have revolutionized cancer therapy by countering immune evasion mechanisms, yet their efficacy is often limited by compensatory immunosuppressive pathways, some of which may be mediated by EPCs metabolism. Current research on targeting the metabolism of tumor-associated EPCs is still in its early stages but holds promise for providing new strategies to enhance immunotherapy. In a murine model of malignant melanoma, neutralization of EPO with specific antibodies significantly suppressed tumor growth by inhibiting EPCs proliferation104. Thus, anti-TGF-β therapies may improve anemia and suppress tumors by modulating EPCs metabolism. Treatment with CD45 antibodies promotes erythroid differentiation of EPCs and inhibits the generation of immunosuppressive EDMCs. Similarly, administration of IFN-γ markedly induces apoptosis of EPCs in the spleens of tumor-bearing mice and similarly attenuates cancer progression. Therefore, targeting EPCs metabolism represents a promising potential strategy for cancer therapy and immunotherapy. Notably, combined inhibition of TGF-β and immune checkpoints has demonstrated enhanced antitumor responses. Strategic modulation of EPCs metabolism may thus serve as a synergistic approach to improve the efficacy of ICIs. Ongoing research into EPC-directed metabolic therapeutics is expected to open new avenues in combinatorial immunotherapy and bring transformative advances to cancer treatment.
In summary, EPCs are increasingly recognized as novel immunosuppressive cells within the scientific community. Metabolic targeting strategies show promise for enhancing immunotherapy efficacy (Fig. 5). Current research has identified multiple molecular targets and regulatory mechanisms in EPCs metabolism. Nevertheless, these advances remain largely confined to preclinical studies, with substantial hurdles impeding clinical translation. Future work must elucidate EPCs metabolic pathways, improve drug specificity, and develop clinically viable approaches to ultimately enhance cancer patient care and survival outcomes.

A deeper understanding of the metabolic mechanisms of EPCs and their influence on the metabolism of other cells may enable the development of metabolism-based biomarkers for guiding clinical therapy. Additionally, targeting EPCs metabolism could enhance the efficacy of cancer immunotherapy, ultimately improving patient outcomes. EPC erythroid progenitor cell, EDMC erythroid-derived myeloid cell, ADC antibody-drug conjugate.
Data availability
No data was used for the research described in the article.
References
Siegel, R. L., Kratzer, T. B., Giaquinto, A. N., Sung, H. & Jemal, A. Cancer statistics, 2025. CA Cancer J. Clin. 1, 10–45 (2025).
Zeng, H. et al. Cancer survival statistics in China 2019-2021: a multicenter, population-based study. J. Natl. Cancer Cent. 3, 203–213 (2024).
Saka, A. H. et al. Cancer statistics for African American and black people, 2025. CA Cancer J. Clin. 2, 111–140 (2025).
Riley, R. S., June, C. H., Langer, R. & Mitchell, M. J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 3, 175–196 (2019).
Mc Neil, V. & Lee, S. W. Advancing cancer treatment: A review of immune checkpoint inhibitors and combination strategies. Cancers (Basel) 17, 1408 (2025).
Garner, H. & de Visser, K. E. Immune crosstalk in cancer progression and metastatic spread: A complex conversation. Nat. Rev. Immunol. 8, 483–497 (2020).
Morad, G., Helmink, B. A., Sharma, P. & Wargo, J. A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 185, 576 (2022).
Kalbasi, A. & Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 20, 25–39 (2020).
Cao, L. M. et al. Extracellular vesicles: Hermes between cancers and lymph nodes. Cancer Lett. 623, 217735 (2025).
Bader, J. E., Voss, K. & Rathmell, J. C. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol. Cell 6, 1019–1033 (2020).
Li, Z. Z., He, J. Y., Wu, Q., Liu, B. & Bu, L. L. Recent advances in targeting myeloid-derived suppressor cells and their applications to radiotherapy. Int Rev. Cell Mol. Biol. 378, 233–264 (2023).
Mo, W. T., Huang, C. F. & Sun, Z. J. Erythroid progenitor cell modulates cancer immunity: Insights and implications. Biochim Biophys. Acta Rev. Cancer 1879, 189209 (2024).
Bozorgmehr, N. et al. CD71(+) erythroid cells suppress T-cell effector functions and predict immunotherapy outcomes in patients with virus-associated solid tumors. J. Immunother. Cancer 11, e006595 (2023).
Li, S. R., Wu, Z. Z., Yu, H. J. & Sun, Z. J. Targeting erythroid progenitor cells for cancer immunotherapy. Int J. Cancer 11, 1928–1938 (2024).
Lasser, S. A., Ozbay Kurt, F. G., Arkhypov, I., Utikal, J. & Umansky, V. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat. Rev. Clin. Oncol. 2, 147–164 (2024).
He, S., Zheng, L. & Qi, C. Myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment and their targeting in cancer therapy. Mol. Cancer 24, 5 (2025).
Raskov, H., Orhan, A., Gaggar, S. & Gogenur, I. Neutrophils and polymorphonuclear myeloid-derived suppressor cells: An emerging battleground in cancer therapy. Oncogenesis 11, 22 (2022).
Tang, H., Li, H. & Sun, Z. Targeting myeloid-derived suppressor cells for cancer therapy. Cancer Biol. Med 4, 992–1009 (2021).
Zhao, L. et al. Late-stage tumors induce anemia and immunosuppressive extramedullary erythroid progenitor cells. Nat. Med 10, 1536–1544 (2018).
Han, Y. et al. Tumor-induced generation of splenic erythroblast-like Ter-cells promotes tumor progression. Cell 184, 1392 (2021).
Wang, Y. Y., Wu, Z. Z., Huang, C. F. & Sun, Z. J. Tumor-host colluding through erythroid progenitor cells: Mechanisms and opportunities. Cancer Lett. 563, 216193 (2023).
Chen, J. et al. Intratumoral CD45(+)CD71(+) erythroid cells induce immune tolerance and predict tumor recurrence in hepatocellular carcinoma. Cancer Lett. 499, 85–98 (2021).
Long, H. et al. Tumor-induced erythroid precursor-differentiated myeloid cells mediate immunosuppression and curtail anti-PD-1/PD-L1 treatment efficacy. Cancer Cell 40, 674–693 e7 (2022).
Cheng, X., Wang, H., Wang, Z., Zhu, B. & Long, H. Tumor-associated myeloid cells in cancer immunotherapy. J. Hematol. Oncol. 16, 71 (2023).
Zhang, H., Wan, G. Z., Wang, Y. Y., Chen, W. & Guan, J. Z. The role of erythrocytes and erythroid progenitor cells in tumors. Open Life Sci. 1, 1641–1656 (2022).
Jafari, M., Ghadami, E., Dadkhah, T. & Akhavan-Niaki, H. PI3k/AKT signaling pathway: Erythropoiesis and beyond. J. Cell Physiol. 3, 2373–2385 (2019).
Wu, H., Klingmuller, U., Acurio, A., Hsiao, J. G. & Lodish, H. F. Functional interaction of erythropoietin and stem cell factor receptors is essential for erythroid colony formation. Proc. Natl. Acad. Sci. USA 5, 1806–1810 (1997).
Xue, Y. et al. PDGF-BB modulates hematopoiesis and tumor angiogenesis by inducing erythropoietin production in stromal cells. Nat. Med 1, 100–110 (2011).
Rane, S. G. & Reddy, E. P. JAKs, STATs and Src kinases in hematopoiesis. Oncogene 21, 3334–3358 (2002).
Silva, M. et al. Erythropoietin can promote erythroid progenitor survival by repressing apoptosis through Bcl-XL and Bcl-2. Blood 5, 1576–1582 (1996).
Casbon, A. J. et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl. Acad. Sci. USA 6, E566–E575 (2015).
Seymour, J. F. et al. Mice lacking both granulocyte colony-stimulating factor (CSF) and granulocyte-macrophage CSF have impaired reproductive capacity, perturbed neonatal granulopoiesis, lung disease, amyloidosis, and reduced long-term survival. Blood 8, 3037–3049 (1997).
Dong, F. et al. Stimulation of Stat5 by granulocyte colony-stimulating factor (G-CSF) is modulated by two distinct cytoplasmic regions of the G-CSF receptor. J. Immunol. 12, 6503–6509 (1998).
Barisas, D. A. G. & Choi, K. Extramedullary hematopoiesis in cancer. Exp. Mol. Med 3, 549–558 (2024).
Ara, T. et al. Long-term hematopoietic stem cells require stromal cell-derived factor-1 for colonizing bone marrow during ontogeny. Immunity 2, 257–267 (2003).
Sugiyama, T., Kohara, H., Noda, M. & Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 6, 977–988 (2006).
Lapidot, T. & Kollet, O. The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homing and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2m(null) mice. Leukemia 10, 1992–2003 (2002).
Dale, D. C. Editorial: The mysteries of the spleen. J. Leukoc. Biol. 2, 249–251 (2016).
Steenbrugge, J., De Jaeghere, E. A., Meyer, E., Denys, H. & De Wever, O. Splenic hematopoietic and stromal cells in cancer progression. Cancer Res 1, 27–34 (2021).
Yang, X., Chen, D., Long, H. & Zhu, B. The mechanisms of pathological extramedullary hematopoiesis in diseases. Cell Mol. Life Sci. 14, 2723–2738 (2020).
Wu, W. C. et al. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc. Natl. Acad. Sci. USA 11, 4221–4226 (2014).
Wu, C. et al. Spleen mediates a distinct hematopoietic progenitor response supporting tumor-promoting myelopoiesis. J. Clin. Invest 8, 3425–3438 (2018).
Hori, T. et al. Usefulness of palliative prognostic index, objective prognostic score, and neutrophil-lymphocyte ratio/albumin ratio as prognostic indicators for patients without cancer receiving home-visit palliative care: A pilot study at a community general hospital. Palliat. Med. Rep. 1, 142–149 (2024).
Iwai, N. et al. Neutrophil to lymphocyte ratio predicts prognosis in unresectable pancreatic cancer. Sci. Rep. 10, 18758 (2020).
Mandula, J. K. & Rodriguez, P. C. Tumor-directed dysregulation of erythroid progenitors drives immunosuppressive myeloid cells. Cancer Cell 6, 597–599 (2022).
Wu, Z. Z. et al. Erythroid progenitor cell-mediated spleen-tumor interaction deteriorates cancer immunity. Proc. Natl. Acad. Sci. USA 9, e2417473122 (2025).
Miwa, Y. et al. Up-regulated expression of CXCL12 in human spleens with extramedullary haematopoiesis. Pathology 4, 408–416 (2013).
Peled, A. et al. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34(+) cells on vascular endothelium under shear flow. J. Clin. Invest 9, 1199–1211 (1999).
Sugiyama, T., Omatsu, Y. & Nagasawa, T. Niches for hematopoietic stem cells and immune cell progenitors. Int Immunol. 1, 5–11 (2019).
Barisas, D. A. G. et al. Tumor-derived interleukin-1alpha and leukemia inhibitory factor promote extramedullary hematopoiesis. PLoS Biol. 21, e3001746 (2023).
Al Sayed, M. F. et al. T-cell-secreted TNFalpha induces wmergency myelopoiesis and myeloid-derived suppressor cell differentiation in cancer. Cancer Res. 2, 346–359 (2019).
Inra, C. N. et al. A perisinusoidal niche for extramedullary haematopoiesis in the spleen. Nature 7579, 466–471 (2015).
Munugalavadla, V. & Kapur, R. Role of c-Kit and erythropoietin receptor in erythropoiesis. Crit. Rev. Oncol. Hematol. 1, 63–75 (2005).
De Maria, R. et al. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 6752, 489–493 (1999).
Harashima, A. et al. CD45 tyrosine phosphatase inhibits erythroid differentiation of umbilical cord blood CD34+ cells associated with selective inactivation of Lyn. Blood 13, 4440–4445 (2002).
Thomas, M. L. & Brown, E. J. Positive and negative regulation of Src-family membrane kinases by CD45. Immunol. Today 9, 406–411 (1999).
Karur, V. G., Lowell, C. A., Besmer, P., Agosti, V. & Wojchowski, D. M. Lyn kinase promotes erythroblast expansion and late-stage development. Blood 5, 1524–1532 (2006).
Gao, X. et al. TGF-beta inhibitors stimulate red blood cell production by enhancing self-renewal of BFU-E erythroid progenitors. Blood 23, 2637–2641 (2016).
Matsuzaki, K. et al. Autocrine stimulatory mechanism by transforming growth factor beta in human hepatocellular carcinoma. Cancer Res 5, 1394–1402 (2000).
Henke, M. et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 9392, 1255–1260 (2003).
Hou, Y. et al. Radiotherapy and immunotherapy converge on elimination of tumor-promoting erythroid progenitor cells through adaptive immunity. Sci. Transl. Med 13, eabb0130 (2021).
Hu, W. T. et al. A silence catalyst: CCL5-mediated intercellular communication in cancer. Arch. Toxicol. 7, 2699–2712 (2025).
Sies, H. et al. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 7, 499–515 (2022).
Elahi, S. et al. Galectin-3 regulates erythropoiesis and enhances the immunoregulatory properties of CD71+ erythroid cells across developmental stages. J. Immunol. 6, 1202–1218 (2025).
Niu, F. et al. Arginase: An emerging and promising therapeutic target for cancer treatment. Biomed. Pharmacother. 149, 112840 (2022).
Delyea, C. et al. CD71(+) erythroid suppressor cells promote fetomaternal tolerance through arginase-2 and PDL-1. J. Immunol. 12, 4044–4058 (2018).
Czubak, K. et al. CD71(+) erythroid cells promote multiple myeloma progression and impair anti-bacterial immune response. Br. J. Haematol. 2, 478–483 (2025).
Ohl, K. & Tenbrock, K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front Immunol. 9, 2499 (2018).
Dubaniewicz, A. Mycobacterial heat shock proteins in sarcoidosis and tuberculosis. Int J. Mol. Sci. 24, 5084 (2023).
Rotem, S. et al. A novel approach to vaccine development: Concomitant pathogen inactivation and host immune stimulation by peroxynitrite. Vaccines (Basel) 10, 1593 (2022).
Nagaraj, S., Schrum, A. G., Cho, H. I., Celis, E. & Gabrilovich, D. I. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J. Immunol. 6, 3106–3116 (2010).
Fan, X. et al. Tumor-associated CD8(+)T cell tolerance induced by erythroid progenitor cells. Front Immunol. 15, 1381919 (2024).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med 26, 2455–2465 (2012).
Boucher, J. C. et al. CD28 costimulatory domain-targeted mutations enhance chimeric antigen receptor T-cell function. Cancer Immunol. Res. 1, 62–74 (2021).
Zhao, R. et al. PD-1/PD-L1 blockade rescue exhausted CD8+ T cells in gastrointestinal stromal tumours via the PI3K/Akt/mTOR signalling pathway. Cell Prolif. 52, e12571 (2019).
Francisco, L. M. et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med 13, 3015–3029 (2009).
Nazarov, K. et al. Murine placental erythroid cells are mainly represented by CD45(+) immunosuppressive erythroid cells and secrete CXCL1, CCL2, CCL3 and CCL4 chemokines. Int J. Mol. Sci. 24, 8130 (2023).
Geiger, R. et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 167, 829–842.e13 (2016).
Rodriguez, P. C. et al. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J. Biol. Chem. 24, 21123–21129 (2002).
Rodriguez, P. C., Quiceno, D. G. & Ochoa, A. C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 4, 1568–1573 (2007).
Unterlass, J. E. et al. Validating and enabling phosphoglycerate dehydrogenase (PHGDH) as a target for fragment-based drug discovery in PHGDH-amplified breast cancer. Oncotarget 17, 13139–13153 (2018).
Munder, M. et al. Suppression of T-cell functions by human granulocyte arginase. Blood 5, 1627–1634 (2006).
Kusmartsev, S., Nagaraj, S. & Gabrilovich, D. I. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J. Immunol. 7, 4583–4592 (2005).
Shi, X. et al. TGF-beta signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 15, 135 (2022).
Flavell, R. A., Sanjabi, S., Wrzesinski, S. H. & Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 8, 554–567 (2010).
Weber, F. et al. Transforming growth factor-beta1 immobilises dendritic cells within skin tumours and facilitates tumour escape from the immune system. Cancer Immunol. Immunother. 9, 898–906 (2005).
Shahbaz, S. et al. CD71+VISTA+ erythroid cells promote the development and function of regulatory T cells through TGF-beta. PLoS Biol. 16, e2006649 (2018).
O’Donnell, J. S., Teng, M. W. L. & Smyth, M. J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 3, 151–167 (2019).
Wang, C. et al. Tumor-associated myeloid cells selective delivery of a therapeutic tumor nano-vaccine for overcoming immune barriers for effective and long-term cancer immunotherapy. Adv. Health. Mater. 13, e2401416 (2024).
Gargett, T. et al. GM-CSF signalling blockade and chemotherapeutic agents act in concert to inhibit the function of myeloid-derived suppressor cells in vitro. Clin. Transl. Immunol. 5, e119 (2016).
Gutierrez, L., Caballero, N., Fernandez-Calleja, L., Karkoulia, E. & Strouboulis, J. Regulation of GATA1 levels in erythropoiesis. IUBMB Life 1, 89–105 (2020).
Bataller, A., Montalban-Bravo, G., Soltysiak, K. A. & Garcia-Manero, G. The role of TGFbeta in hematopoiesis and myeloid disorders. Leukemia 5, 1076–1089 (2019).
Zhang, X. et al. FOXO3-mTOR metabolic cooperation in the regulation of erythroid cell maturation and homeostasis. Am. J. Hematol. 10, 954–963 (2014).
Gustafsson, K. et al. Clearing and replacing tissue-resident myeloid cells with an anti-CD45 antibody-drug conjugate. Blood Adv. 22, 6964–6973 (2023).
Yang, L. et al. Regulation of bile duct epithelial injury by hepatic CD71+ erythroid cells. JCI Insight 5, e135751 (2020).
Bai, D., Zhou, Y., Jing, L., Guo, C. & Yang, Q. Arginine metabolism in cancer biology and immunotherapy. Immune Netw. 25, e30 (2025).
Cheung, E. C. & Vousden, K. H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 5, 280–297 (2022).
Ren, Y. et al. Multifaceted role of redox pattern in the tumor immune microenvironment regarding autophagy and apoptosis. Mol. Cancer 22, 130 (2023).
Grzywa, T. M. et al. Myeloid cell-derived arginase in cancer immune response. Front Immunol. 11, 938 (2020).
Zhang, Y., Lyu, J. & Fang, H. Rewiring amino acids in cancer. Biochim Biophys. Acta Rev. Cancer 1880, 189465 (2025).
Cane, S. et al. Neutralization of NET-associated human ARG1 enhances cancer immunotherapy. Sci. Transl. Med 15, eabq6221 (2023).
Elahi, S. Neglected cells: Immunomodulatory roles of CD71(+) erythroid cells. Trends Immunol. 40, 181–185 (2019).
Li, Z. Z., Zhou, K., Wu, Q., Liu, B. & Bu, L. L. Lymph node metastasis in cancer: Clearing the clouds to see the dawn. Crit. Rev. Oncol. Hematol. 204, 104536 (2024).
Sano, Y. et al. Multiorgan signaling mobilizes tumor-associated erythroid cells expressing immune checkpoint molecules. Mol. Cancer Res. 3, 507–515 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82273202); the Fundamental Research Funds for the Central Universities (2042022dx0003).
Author information
Authors and Affiliations
Contributions
Zi-Zhan Li: writing—review ānd editing, writing—original draft, visualization, validation, software, resources, investigation. Hai-Liang Huang and Yu-Cheng Li: writing—original draft, visualization, software. Wen-Da Wang: visualization, validation, software. Zhi-Jun Sun: writing—review and editing, supervision, investigation, funding acquisition, conceptualization.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, ZZ., Huang, HL., Li, YC. et al. Targeting erythroid progenitor cell metabolism to enhance cancer immunotherapy. npj Precis. Onc. 10, 163 (2026). https://doi.org/10.1038/s41698-026-01362-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-026-01362-9
