
Abstract
Understanding and controlling native defects is essential for unlocking the full potential of two-dimensional magnetic semiconductors. Here, angle-resolved photoemission spectroscopy (ARPES) and first-principles calculations are used to explore the electronic properties of bulk CrSBr. ARPES measurements reveal clear signatures of conduction band filling in as-grown crystals, indicative of unintentional doping. An analysis of intrinsic defects based on density functional theory (DFT) identifies chromium interstitials (Cri) stabilized between CrSBr layers as the most favorable shallow donors. Bromine-on-sulfur antisites (BrS) and bromine vacancies (VBr) are also found to act as potential donors, albeit with deeper ionization energies. Our results shed light on the origin of unintentional n-type doping of CrSBr and pave the way for new strategies for defect control and electronic property tuning in this van der Waals magnet.
Similar content being viewed by others
Introduction
Two-dimensional (2D) magnetic semiconductors are promising for spintronic and quantum information technologies due to their unique electronic, magnetic, and optical properties. Among these materials, chromium sulfur bromide (CrSBr)1 stands out for its bulk’s robust air stability, high magnetic ordering temperature (TN ≈ 132 K)2,3, and sizable direct band gap of 1.5–2 eV4,5,6, making it particularly suitable for practical device integration.
Despite immense research fueled by these favorable attributes, the intrinsic electrical conductivity of CrSBr remains poorly understood. In nominally undoped semiconductors, intrinsic defects are typically the prime source of electronic carriers (electrons and holes). Prior studies have noted that vapor-grown CrSBr can harbor high defect concentrations, with bromine vacancies (VBr) believed to be the most abundant species7 and suspected to act as the origin of n-type doping8. Additionally, electron transport experiments highlight the interplay between defects and charge carriers in bulk CrSBr9. However, a comprehensive and systematic investigation of native defects and their doping behavior has been lacking.
In this work, we combine first-principles density functional theory (DFT) calculations with angle-resolved photoemission spectroscopy (ARPES). In particular, we systematically investigate the thermodynamic and electronic properties of native defects in CrSBr, including vacancies, self-interstitials, and antisites. To qualify as a favorable n-type dopant, a defect must have a low formation energy when the Fermi level lies near the conduction band minimum (CBM) and possess stable positive charge states over a significant fraction of the bandgap. In contrast to previous assumptions, our calculations suggest chromium interstitials (Cri) as energetically favorable defects under n-type conditions and capable of acting as shallow donors. Two bromine-related defects, VBr and bromine antisites (BrS), are also identified as potential donors but with larger ionization energies. These findings provide a microscopic explanation of unintentional n-type doping in CrSBr and offer guidance for future defect engineering on this promising material.
Results
ARPES measurements
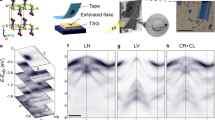
Figure 1 presents our ARPES measurements on bulk CrSBr crystals at room temperature (see Methods). In Fig. 1a, the ARPES intensity below the valence band maximum (VBM) closely reproduces earlier reports4,6. The corresponding energy distribution curve (EDC), shown on the right, reveals a weak but distinct spectral feature above the VBM, highlighted in the inset. We extract an energy separation of (1.48 ± 0.09) eV between the VBM and the onset of this additional spectral weight by linearly extrapolating the EDC and fitting the unoccupied tail with a Gaussian. Due to low signal strength, the overlaid data above -0.85 eV was acquired with 25 times longer acquisition time.

a ARPES intensity map of bulk CrSBr at room temperature measured along high symmetry points. Above −0.85 eV, the acquisition time is increased 25-fold and the contrast is increased tenfold. The corresponding energy distribution curve (EDC), shown on the right, displays a weak spectral feature above the valence band maximum (VBM), highlighted in the inset (intensity multiplied by 100 for clarity). The grey dotted line indicates the linear fit used to extrapolate the VBM position. b Momentum map integrated over the energy range –1.6 eV to –1.3 eV, corresponding to the energy window of the conduction band–like feature. The white dotted line marks the boundary of the first Brillouin zone, and the grey solid line marks the path displayed in (a).
The observed intensity might stem from mid-gap states created by defects at the surface. J. Klein et al. observed an increased density of VBr at the surface using STM7, and M. Weile et al. also found a plethora of defect complexes in CrSBr bilayer with VBr being the most abundant10. However, DFT calculations revealed rather uniform and flat dispersion of these defect states10 in stark contrast to the conduction band asymmetry and intensity distribution observed in Fig. 1a.
Further insight is obtained from the isoenergetic contour along the in-plane momenta (also known as “momentum map”) shown in Fig. 1b, recorded over the energy interval [–1.6, –1.3] eV, corresponding to the region of the additional spectral weight above the VBM. The spectral intensity is highly anisotropic and sharply localized along the \(\bar{\Gamma }\)-\(\bar{{\rm{X}}}\) direction. The resulting contour exhibits a conduction band–like topology, consistent with prior observations in ultrathin CrSBr flakes on metallic substrates11. Together with the ~1.5 eV bandgap, these features provide direct spectroscopic evidence of partial conduction band occupation.
Crucially, while previous ARPES and transport studies reported similar conduction band signatures, they were attributed to substrate-induced charge transfer in exfoliated flakes11 or Li doping12. In contrast, our measurements were performed on bulk, millimeter-thick crystals, ruling out substrate effects. This strongly supports an intrinsic origin of the conduction band filling, most likely due to native donor-type defects in as-grown CrSBr. This is an important step toward validating the microscopic doping mechanism and establishes a foundation for scalable device integration of CrSBr in substrate-free environments.
DFT modeling of native point defects in bulk CrSBr
To identify the intrinsic point defects responsible for the observed conduction band population - and thereby the unintentional n-type conductivity - we performed first-principles calculations within the framework of DFT. Native defect formation energies depend critically on the atomic chemical potentials of Cr, S, and Br (μCr, μS, μBr), which are bounded by the thermodynamic stability region of CrSBr relative to decomposition into competing binary and ternary phases. Therefore, we began our analysis with a thermodynamic assessment of bulk CrSBr. The chemical potentials of the constituting elements are defined as μi = μi⁰ + Δμi, where μi⁰ is the energy per atom of element i in its reference state (Cr in pure bcc chromium crystal, S in α-S8 crystal, and Br in orthorhombic bromine crystal), and Δμi ≤ 0. The thermodynamic stability of CrSBr is defined by the equality:
where ΔHf (CrSBr) is the formation energy of CrSBr per formula unit.
To ensure CrSBr is the most stable phase under given conditions, its formation must be favored over all possible competing phases. Based on our DFT total energy calculations for known entries in the OQMD database13, we identified Cr₂S₃, Cr₃S₄, and CrBr₂ as the relevant competing phases that constrain the chemical potentials and define the boundaries of the CrSBr stability region. The thermodynamic conditions that prevent decomposition into these phases impose the following inequalities:
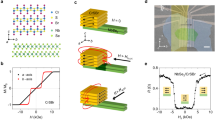
We calculate ΔHf (CrSBr) = –6.09 eV, ΔHf (Cr2S3) = −9.89 eV, ΔHf (Cr3S4) = −14.67 eV, and ΔHf (CrBr2) = −6.50 eV per formula unit. All total energy calculations were performed using the PBE + U approach14,15 (see Methods) that provides reliable formation energies and magnetic ground states, consistent with prior studies of other 2D magnetic semiconductors such as CrI₃ and CrBr316,17,18. Accordingly, from the constraints (1–4), we calculated the stability diagram, which is plotted in Fig. 2a in the (ΔμS, ΔμBr) space (with ΔμCr determined from the stoichiometry constraint).

a DFT-calculated range of chemical potentials (in eV) of the elements in CrSBr. The points A, B, and C mark the representative corners of the stability region of bulk CrSBr (grey area). The inset sketches the atomic structure of CrSBr, with yellow and blue isosurfaces signifying positive and negative magnetization density of the AFM ground state. b Formation energies of native point defects in CrSBr in their neutral charge states calculated for the representative points marked in a. c DFT-optimized atomic structures of the most energetically favorable native defects: Cri, VBr, and BrS. For VBr, the depicted interatomic distances are measured from the nominal lattice site.
The corners (A, B, C) and the nominal midpoint of the CrSBr stability region marked in Fig. 2a define the range of accessible synthesis conditions. These representative points were, therefore, selected for subsequent defect calculations. Point A corresponds to the Cr-rich conditions (ΔμCr = −3.1 eV, ΔμS = − 1.2 eV, ΔμBr = −1.6 eV), point B to the S-rich conditions (ΔμCr = −4.9 eV, ΔμS = 0.0 eV, ΔμBr = −1.1 eV), and point C to the S-rich and Br-rich conditions (ΔμCr = −5.7 eV, ΔμS = 0.0 eV, ΔμBr = −0.4 eV).
We investigated eight native defects that we expect to be most relevant: three vacancies (VCr, VS, VBr), three self-interstitials (Cri, Si, Bri), and two antisites (SBr, BrS). Defect calculations were performed in a 4×3×2 supercell (two CrSBr layers) with antiferromagnetic ordering preserved. First, we computed the formation energies, ΔHf [D⁰], of the defects in their neutral charge states:
where Etot[CrSBr:D⁰] and Etot[CrSBr] are the total energies of the defective and pristine supercells, respectively, ni is the number of atoms of species i added (positive) or removed (negative) to form the defect, and μi is the chemical potential of the corresponding species as defined earlier.
The results are presented in Fig. 2b, showing the variation of neutral defect formation energies across the selected chemical environments. The atomic configurations of the most relevant defects are shown in Fig. 2c. Among the self-interstitials, Cri consistently exhibit favorably low formation energies, particularly under Cr-rich conditions (Point A), where the formation energy falls well below that of other defect types. The relaxed structure of Cri, shown in Fig. 2c, reveals that the most favorable incorporation site is located in the van der Waals gap between the CrSBr layers. The interstitial Cr atom adopts a quasi-octahedral coordination with surrounding atoms, forming bonds of 2.57–2.82 Å to nearby sulfur and bromine atoms. This configuration results in minimal distortion to the adjacent CrSBr layers, preserving the host lattice integrity. Notably, the sulfur and bromine interstitials (Si and Bri) are also stabilized at interlayer sites, but with much higher formation energies.
Previously, Cri has been proposed as a structurally and magnetically significant defect in He+ ion-irradiated CrSBr crystals, where it was found to bridge neighboring layers and stabilize interlayer ferromagnetic coupling19. Our calculations show that this defect can be favorable as well in as-grown CrSBr under a wide range of growth conditions.
At the same time, Fig. 2b illustrates that Cri does not dominate at all corners of the phase space. Specifically, two vacancies, VBr and VS, show low formation energies in Cr-poor environments. Our results indicate that VBr is expected to be the most favorable defect under S-rich conditions (Point B of the phase diagram), which is consistent with the observations of high concentration of this defect in previous experimental works7,8. Its incorporation causes a relatively small local perturbation with slight inward relaxations of neighboring atoms (as shown for VBr in Fig. 2c).
In contrast to VBr and VS, Cr vacancies (VCr) remain the least favorable of the studied defects, with formation energies consistently around 5–7 eV. At the same time, VCr remains a noteworthy species since a recent study of defect complexes in CrSBr pointed out its propensity to form Frenkel pairs with VCr10. We calculated the formation energy of the most favorable configuration of VCr-Cri revealed in ref. 10 (dubbed VCr-Criδ2) using the same settings as for other point defects in Fig. 2, and found it to be of 3.69 eV – approximately halfway between the individual VCr and Cri. It should be noted that this does not exclude the appearance of this Frenkel defect pair under non-equilibrium conditions (due to kinetic effects), consistent with microscopy observations10.
Another noteworthy defect is bromine-on-sulfur antisite, BrS, which becomes the dominant defect in the Br-rich corner of the CrSBr stability region (Point C in Fig. 2). Positioned at the sulfur crystallographic site (Fig. 2c), the bromine atom of BrS exhibits modest bond length changes relative to the pristine lattice, with the distance to nearest-neighbor Br atoms increasing from 2.42 Å to 2.55 Å within the same Br sublayer, and from 2.40 Å to 2.72 Å to Br atoms in the adjacent sublayer. These results suggest that bromine atoms introduced, for instance, upon the formation of bromine vacancies are more likely to substitute sulfur atoms within the CrSBr lattice rather than occupy interstitial positions. In contrast to BrS, the second considered antisite defect – sulfur-on-bromine, SBr – exhibits a significantly higher formation energy, although still consistently lower than that of sulfur interstitials (Si) across most of the CrSBr stability region.
The neutral charge state formation energy calculations thus identify two bromine-related defects, VBr and BrS, and the Cr interstitial as the most thermodynamically accessible intrinsic point defects in CrSBr. To further evaluate which defects are electronically active and capable of contributing to n-type conductivity, we then examined the charge transition levels (CTLs) of all intrinsic point defects. We used the Slater-Janak (SJ) method20, where the charge transition levels are estimated from the Kohn–Sham eigenvalues at fractional occupations. The results of the SJ approach are shown to be in good agreement with those obtained from total energy differences, while avoiding the difficulties associated with electrostatic corrections21,22. This approach allows for the calculation of CTLs without comparing the total energies of differently charged supercells. While the PBE + U functional is suitable for structural relaxation and estimating formation energies, it significantly underestimates the band gap of CrSBr (PBE + U: 0.62 eV). Therefore, single-point SCF calculations with more accurate exchange-correlation functionals were employed to evaluate CTLs on PBE + U-relaxed geometries. Specifically, we tested the HSE06 hybrid functional23 and the meta-GGA SCAN functional24.
It should be noted that the precise value of the band gap in CrSBr remains under debate. Scanning tunneling spectroscopy (STS) and microscopy measurements have reported a gap of approximately 1.5 eV2,5, whereas earlier ARPES studies suggested larger values. Reported VBM lie 1.6-1.8 eV4, 1.5 eV6, and 1.84 eV25 below the Fermi level. These earlier investigations, conducted on bulk crystals similar to those used in our study, did not report any spectral signature associated with the conduction band minimum. In contrast, our measurements reveal a faint spectral feature approximately 1.5 eV above the valence band, whose dispersion is consistent with a partially occupied CBM. The spread in reported VBM-Fermi separations may arise from band bending at the surface that shifts the observed valence band position depending on n-type doping level, photon energy, and photon fluence26,27, thus influencing the VBM position and the appearance of a conduction-band signal in the ARPES spectra26,28.
Beyond the magnitude of the band gap, its character is also contested. Although CrSBr is commonly regarded as a direct-gap semiconductor, two ARPES studies have reported an indirect band gap, one following K dosing25 and another attributed to substrate-induced charge transfer in exfoliated flakes11. In our measurements, the marked \(\bar{\Gamma }\) point of the first Brillouin zone (BZ) shows weak intensity in Fig. 1, whereas neighboring BZs \({\bar{\Gamma }}_{1}\) displays strong spectral weight. This effect was previously noted by M. Bianchi et al.6 and is attributed to photoemission matrix element variations. We therefore compared multiple BZs carefully and found no consistent evidence for an indirect gap with the CBM at the \(\bar{{\rm{X}}}\) point.
In our DFT calculations, the HSE06 hybrid functional yields a band gap of 2.21 eV, while the SCAN meta-GGA functional gives 1.59 eV. Given that the SCAN result closely matches recent transport and STS measurements – as well as the gap inferred from our ARPES data (see also Figure S1) – we adopted SCAN for computing CTLs in our defect analysis.
The calculated CTLs are shown in Fig. 3 for the midpoint of the CrSBr stability region and the Cr-poor, S-rich, Br-rich limit (Point C in Fig. 2a). The total spin of each energetically favorable charge state is listed in Table S1. Our results suggest that among the studied defects, Cri emerges as a potent shallow donor. It is stable only in positive charge states (+3, +2, and +1) throughout the entire band gap, with the (+1/0) transition level lying above the CBM, indicating a stable +1 state under n-type conditions. Its formation energy at n-type conditions is favorable over other studied defects across the majority of the CrSBr stability region (as represented by the midpoint data in Fig. 3), save for the Cr-poor conditions (such as Point C).

Other potential donor candidates include VBr and BrS: their formation energy curves indicate positive charge states are favored at low Fermi levels, with transitions to the neutral charge state occurring high in the band gap. The bromine vacancy – a defect whose high concentration in CrSBr is reported in several experimental works7,8,10 – is stable in +1 and 0 charge states. However, its (+1/0) transition level lies ~0.89 eV below the CBM, implying high ionization energy and classifying it as a deep donor. In contrast to VBr, the BrS antisite has a relatively shallow donor level located ~0.27 eV below the CBM. Moreover, Fig. 3 shows that the computed formation energy of BrS is lower than that of Cri in the Cr-poor region under n-type conditions (i.e., when the Fermi level is close to the CBM). These observations highlight BrS and Cri as the most likely contributors to unintentional n-type doping and the observed conduction-band filling in ARPES, with VBr being the next best candidate.
XPS measurements and DFT analysis
Finally, we analyzed the consistency of the predicted defects with experimental X-ray photoelectron spectroscopy (XPS) data. For this, we computed the Cr 2p3/2 core-level binding energies for both defect-containing and bulk supercells, referenced to the energy of the highest occupied state. The excited Cr atoms were described by a pseudopotential generated with a 2p core hole. Accordingly, the binding energies were obtained as the total energy differences between the core-hole state and the ground state of the (N−1)-electron system29. To account for the pseudopotential formalism used in our calculations, the computed binding energies were supplemented by an additive correction defined as the difference of all-electron and pseudopotential binding energies of the isolated Cr atom (for details, see refs. 30,31). This yields a binding energy of 577.1 eV for Cr atoms in bulk CrSBr. In the supercell containing Cri (in the +1-charge state), the core-level binding energies of bulk atoms remained consistent with this value, while the interstitial Cr atom exhibits a binding energy shifted by about 1 eV, to 576.1 eV.
For comparison, we have also computed the 2p3/2 core-level binding energy of the Cri atom in other relevant charge states. The calculated values are 576.31 eV and 576.32 eV for Cri2+ and Cri3+, respectively, comparable with the +1-charge state of Cri and shifted by about 0.8 eV from the bulk value. Notably, the 2p3/2 binding energies of Cr atoms near other energetically favorable defects, VBr0 and BrS0, are much closer to those of bulk atoms (shifted by 0.2–0.3 eV): for VBr0, the calculated binging energy of the nearest-neighbor chromium is 576.8 eV, and for BrS0, it is 576.9 eV.
The experimental XSP spectrum of the Cr 2p3/2 core level is shown in Fig. 4. In addition to the spin-orbit splitting of the 2p1/2 and 2p3/2 levels, additional multiplet splitting arises from the coupling between the remaining 2p3/2 core electron and the 3d valence electrons32. Even though a multiplet structure is evident by eye in the experimental data, previous studies on CrSBr have neglected this effect32,33, potentially leading to misassignment of the Cr binding energy34,35. In our analysis, we fit the Cr3+ component using a multiplet of five pseudo-Voight functions and include an additional pseudo-Voight peak to account for the Cr1+ interstitials. We applied a standard Shirley background and all resulting fit parameters are summarized in Table S2. The fit reveals a composition of 90.1% Cr3+ and 9.9% Cr1+. We determine the Cr3+ binding energy to be 575.97(04) eV by calculating an area-weighted average of the five multiplet components, while the Cr1+ peak is centered at 574.90(04) eV, yielding a splitting of 1.07(08) eV. The Br 3d and S 2p core level spectra are shown in Figure S2 and support a singular charge state for both elements. Although the absolute values of the Cr peaks differ slightly from our DFT results, the agreement in the relative binding energy shift between the two oxidation states aligns with the presence of interstitial Cr atoms in the +1-charge state. While XPS cannot by itself unambiguously identify the defect species, this close correspondence between theory and experiment suggests that the Cri defects predicted by our DFT analysis are indeed realized in as-grown CrSBr.

The Cr3+ contribution from bulk CrSBr is modeled as a multiplet comprising five pseudo-Voigt components (blue), while the Cr1+ interstitial is fitted with a single pseudo-Voight peak (purple). A standard Shirley background is included in the overall fit. The combined fit reproduces the experimental data with a flat residual. The main Cr3+ peak position is determined via an area-weighted average of the five multiplet components, resulting in a binding energy difference of 1.07(08) eV relative to the Cr1+ state (compare dashed lines).
Discussion
Our combined experimental and DFT results underscore native point defects as possible sources of intrinsic electron doping in CrSBr. The chromium self-interstitial, Cri, bromine-on-sulfur antisites, BrS, and bromine vacancies, VBr, are identified as the most likely intrinsic shallow donor. Importantly, our defect formation energy analysis provides guidance for growth optimization: Cr-poor, Br-rich, and S-rich conditions are expected to suppress the formation of these donor defects and promote intrinsic behavior. This identification and the comprehensive analysis of defect chemistry in CrSBr presented here open new avenues for controlling defect formation and achieving truly intrinsic or intentionally doped states. Establishing control over native doping is not only vital for fundamental studies of CrSBr’s coupled electronic and magnetic phenomena, but also for optimizing this 2D magnet in future applications. In addition, our ARPES measurements provide direct spectroscopic evidence that CrSBr is a direct band gap semiconductor, further confirming its suitability for optoelectronic and spintronic applications.
Methods
Sample preparation
We used commercial CrSBr crystals from HQ Graphene, which we glued to copper sample plates using UHV-compatible silver epoxy (EPO-TEK H21D) to ensure electrical grounding. After transfer into the UHV system, the crystals were exfoliated in situ using Scotch tape to obtain a clean surface. Exfoliation was carried out at a base pressure better than 3 × 10-7 mbar for ARPES measurements and > 5 × 10−8 mbar for XPS to expose a clean surface.
ARPES
We performed μ-ARPES using a Kreios 150 MM momentum microscope (Specs GmbH) under UHV conditions (<3×10-10 mbar)36. This microscope allows to acquire two-dimensional isoenergetic maps at a constant kinetic energy, with a momentum field of view spanning approximately kx, ky ∈ [–2.0, +2.0] Å-1. Photoelectrons were excited using the HeIα resonance line (21.2 eV) from a Helium discharge lamp equipped with a monochromator, providing a spot size of approximately 200 μm in diameter. This configuration allows probing of locally flat regions, suitable for μ-ARPES measurements. All spectra were acquired at room temperature with an energy resolution of 90 meV.
XPS
XPS spectra were acquired at room temperature with a Phoibos 150 hemispherical analyzer (Specs GmbH) under UHV conditions. A monochromatized Al Kα X-ray source with 1486.8 eV photon energy facilitated photoemission at 430 meV energy resolution. The binding energy scale was calibrated using the Fermi edge of a clean Au(111) crystal. Spectral analysis was conducted using the XPST package for Igor Pro37, employing a Gauss-Lorentzian sum function to approximate a Voigt profile and a standard Shirley background for inelastic scattering correction.
DFT Calculations
All calculations were performed within the spin-polarized DFT formalism using the Quantum ESPRESSO program38,39. Geometry optimization and total energy calculations were conducted with the PBE + U functional14,15, where the Coulomb energy U = 5.1 eV defined self-consistently40 was applied to Cr 3d orbitals to correct for strong on-site correlations. To obtain the band structure and CTLs, we employed the meta-GGA SCAN functional21 (see the main text). We used norm-conserving pseudopotentials with the kinetic energy cutoff of 1100 eV imposed to achieve converged results.
Declaration of generative AI and AI-assisted technologies
During the preparation of this work, the authors used ChatGPT to enhance the quality of writing by improving grammar, style, and clarity. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Data availability
The data supporting the findings of this study are available within the article. Additional data related to this manuscript may be requested from the authors.
References
Ziebel, M. E. et al. CrSBr: an air-stable, two-dimensional magnetic semiconductor. Nano Lett. 24, 4319 (2024).
Telford, E. J. et al. Layered antiferromagnetism induces large negative magnetoresistance in the van der Waals semiconductor CrSBr. Adv. Mater. 32, 2003240 (2020).
Göser, O., Paul, W. & Kahle, H. G. Magnetic properties of CrSBr. J. Magn. Magn. Mater. 92, 129 (1990).
Watson, M. D. et al. Giant exchange splitting in the electronic structure of A-type 2D antiferromagnet CrSBr. npj 2D Mater. Appl. 8, 54 (2024).
Klein, J. et al. The bulk van der Waals layered magnet CrSBr is a quasi-1D material. ACS Nano 17, 5316 (2023).
Bianchi, M. et al. Paramagnetic electronic structure of CrSBr: Comparison between ab initio GW theory and angle-resolved photoemission spectroscopy. Phys. Rev. B 107, 235107 (2023).
Klein, J. et al. Sensing the local magnetic environment through optically active defects in a layered magnetic semiconductor. ACS Nano 17, 288 (2022).
Torres, K. et al. Probing defects and spin-phonon coupling in CrSBr via resonant Raman scattering. Adv. Funct. Mater. 33, 2211366 (2023).
Telford, E. J. et al. Coupling between magnetic order and charge transport in a two-dimensional magnetic semiconductor. Nat. Mater. 21, 754 (2022).
Weile, M. et al. Defect complexes in CrSBr revealed through electron microscopy and deep learning. Phys. Rev. X 15, 021080 (2025).
Bianchi, M. et al. Charge transfer induced Lifshitz transition and magnetic symmetry breaking in ultrathin CrSBr crystals. Phys. Rev. B 108, 195410 (2023).
Wu, F. et al. Mott insulating phase and coherent-incoherent crossover across magnetic phase transition in 2D antiferromagnetic CrSBr. Sci. China Phys. Mech. Astron. 68, 267411 (2025).
Saal, J. E., Kirklin, S., Aykol, M., Meredig, B. & Wolverton, C. Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM 65, 1501 (2013).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+ U study. Phys. Rev. B 57, 1505 (1998).
Wines, D., Choudhary, K. & Tavazza, F. Systematic DFT+ U and quantum Monte Carlo benchmark of magnetic two-dimensional (2D) CrX₃ (X = I, Br, Cl, F). J. Phys. Chem. C. 127, 1176 (2023).
Staros, D. et al. A combined first principles study of the structural, magnetic, and phonon properties of monolayer CrI. J. Chem. Phys. 156, 014702 (2022).
Kumar Gudelli, V. & Guo, G. Y. Magnetism and magneto-optical effects in bulk and few-layer CrI₃: a theoretical GGA+ U study. N. J. Phys. 21, 053012 (2019).
Long, F. et al. Ferromagnetic interlayer coupling in CrSBr crystals irradiated by ions. Nano Lett. 23, 8468 (2023).
Sanna, S., Frauenheim, T. & Gerstmann, U. Validity of the Slater-Janak transition-state model within the LDA+U approach. Phys. Rev. B 78, 085201 (2008).
Chakrabarty, A. & Patterson, C. H. Transition levels of defects in ZnO: Total energy and Janak’s theorem methods. J. Chem. Phys. 137, 054709 (2012).
Bertoldo, F., Ali, S., Manti, S. & Thygesen, K. S. Quantum point defects in 2D materials-the QPOD database. npj Comp. Mater. 8, 56 (2022).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 118, 8207 (2003).
Sun, J., Ruzsinszky, A. & Perdew, J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 115, 036402 (2015).
Smolenski, S. et al. Large exciton binding energy in a bulk van der Waals magnet from quasi-1D electronic localization. Nat. Comm. 16, 1134 (2025).
Lev, L. L. et al. Impact of band-bending on the k-resolved electronic structure of Si-doped GaN. Phys. Rev. Res. 4, 013183 (2022).
Roth, F. et al. Dynamical nonlinear inversion of the surface photovoltage at Si (100). Phys. Rev. Lett. 132, 146201 (2024).
Yu, T. et al. Momentum-resolved electronic structure and band offsets in an epitaxial NbN/GaN superconductor/semiconductor heterojunction. Sci. Adv. 7, eabi5833 (2021).
Kahk, J. M., Michelitsch, G. S., Maurer, R. J., Reuter, K. & Lischner, J. Core electron binding energies in solids from periodic all-electron Δ-self-consistent-field calculations. J. Phys. Chem. Lett. 12, 9353 (2021).
Pehlke, E. & Scheffler, M. Evidence for site-sensitive screening of core holes at the Si and Ge (001) surface. Phys. Rev. Lett. 71, 2338 (1993).
Bianchettin, L. et al. Geometric and electronic structure of the N∕Rh(100) system by core-level photoelectron spectroscopy: Experiment and theory. Phys. Rev. B 74, 045430 (2006).
Biesinger, M. C., Brown, C., Mycroft, J. R., Davidson, R. D. & McIntyre, N. S. X-ray photoelectron spectroscopy studies of chromium compounds. Surf. Interface Anal. 36, 1550 (2004).
Shi, W. et al. Controllable antiferromagnetic to ferromagnetic transition of CrSBr by organic cation intercalation. Adv. Electron. Mater 11, e2500125 (2025).
Biesinger, M. C. et al. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 257, 2717 (2011).
Isaacs, M. A., Graf, A. & Morgan, D. J. XPS Insight Note: Multiplet splitting in X-ray photoelectron spectra. Surf. Interface Anal. 57, 285–290 (2025).
Schiller, K. J. et al. Time-resolved momentum microscopy with fs-XUV photons at high repetition rates with flexible energy and time resolution. Sci. Rep. 15, 3611 (2025).
Schmid, M. & Steinrück, H. & Gottfried. J. M. Surf. Interface Anal. 46, 505 (2014).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Giannozzi, P. et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 29, 465901 (2017).
Timrov, I., Marzari, N. & Cococcioni, M. Self-consistent Hubbard parameters from density-functional perturbation theory in the ultrasoft and projector-augmented wave formulations. Phys. Rev. B 103, 045141 (2021).
Acknowledgements
The Momentum Microscope has been financed by the Deutsche Forschungsgemeinschaft (DFG) through the project INST 212/409 and by the “Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfalen”. We acknowledge the Paderborn Center for Parallel Computing (PC2) for the provided computational resources. We acknowledge financial support by the DFG through project 231447078 (TRR 142/3 – projects A08 and B07) and from the European Union’s Horizon 2020 Research and Innovation Programme under Project SINFONIA, grant 964396.
Author information
Authors and Affiliations
Contributions
T.B., W.G.S., K.J.S., A.I., and M.Ci. conceptualized and planned the project and contributed to the writing of the manuscript. T.B. performed DFT calculations. K.J.S., J.E.N., L.S., and G.Z. conducted ARPES measurements. K.J.S., M.S.A., and M.Ca. Conducted XPS measurements. W.G.S. and M.Ci. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Biktagirov, T., Schmidt, W.G., Schiller, K.J. et al. Intrinsic defects as a source of n-type conductivity in CrSBr. npj 2D Mater Appl 9, 106 (2025). https://doi.org/10.1038/s41699-025-00640-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41699-025-00640-7
This article is cited by
-
The two conduction bands of monolayer CrSBr on Au
npj 2D Materials and Applications (2026)
