
Abstract
Long-lived hot carriers in transition metal dichalcogenides (TMDCs) are essential for efficient light–matter interactions, underpinning applications in optoelectronics, photodetection, and quantum technologies. Vanadium doping introduces tunable defect states that significantly modify the electronic structure and carrier dynamics, yet their precise role in electron–hole recombination remains unresolved. Here, we employ mid-infra-red (mid-IR) transient absorption spectroscopy to probe carrier trapping mechanisms in pristine and Vanadium-doped MoS2 monolayers. Under near-resonant excitation of A and B excitons, followed by mid-IR probing at 0.31 and 0.62 eV, we observe distinct non-radiative decay pathways. While pristine MoS2 exhibits fluence-dependent defect state saturation, Vanadium doping introduces additional relaxation channels that suppress this effect, leading to nearly pump fluence- and energy-independent carrier lifetimes. Our results provide direct experimental evidence of doping-mediated suppression of defect state saturation, offering new insight into electron–phonon interactions and defect-assisted recombination in TMDCs. These findings establish mid-IR transient spectroscopy as a powerful tool for quantifying and engineering defect states, paving the way for optimized doping strategies in next-generation TMDC-based optoelectronic devices.

Similar content being viewed by others

Introduction
Transition metal dichalcogenides (TMDCs) have garnered considerable attention owing to their distinctive electronic, optical, and mechanical properties1,2. These materials comprise a transition metal atom (e.g., Mo, W) positioned between two chalcogen atoms (S, Se, or Te)2,3, exhibiting a wide range of band gaps from semiconducting to metallic4. The direct bandgap in monolayer TMDCs enables strong light–matter interactions, making them highly suitable for optoelectronic applications such as photodetectors, light-emitting diodes (LEDs), and solar cells5,6,7. Additionally, their high carrier mobility and mechanical flexibility make them excellent candidates for next-generation flexible electronics8,9 and sensors10. Advances in synthesis techniques, including chemical vapour deposition (CVD) and mechanical exfoliation, have enabled the fabrication of high-quality TMDC monolayers and heterostructures, further expanding their potential applications.
Doping TMDCs has been extensively studied as a means to develop high-performance devices across various fields, including optoelectronics11, neuromorphic computing12,13, and flexible electronics14,15. For instance, chemical doping has been employed to modulate the energy bands of TMDCs, leading to enhanced performance in photodetectors, complementary circuits, and neuromorphic devices16,17. Surface charge-transfer doping has been shown to improve photo-response performance18. Emerging applications such as neuromorphic devices have also benefited from doping processes, where modulation of Schottky barriers and phase transitions in TMDCs facilitated synaptic behaviour12. Functionalisation with chemical dopants, including polymers and self-assembled monolayers, has been utilized to control the band structure and Schottky barriers of TMDCs19,20. Furthermore, annealing processes combined with chemical doping have been explored to enhance the electrical characteristics of TMDC-based devices15,21,22. These advancements underscore the versatility of doping strategies at the application level in TMDCs.
In monolayer MoS2, strong spin–orbit coupling (SOC) and reduced dielectric screening lead to multi-excitonic states bound by Coulomb interactions23,24,25. SOC induces a non-negligible splitting (~10 meV) in the conduction band, which is an order of magnitude smaller than that in the valence band26,27,28. The strong spatial confinement and weak dielectric screening result in tightly bound electron–hole pairs with large binding energy, enabling excitons to interact strongly with light even at room temperature29,30.
The performance and intrinsic properties of the atomically thin 2D semiconductors are critically influenced by the presence of native and extrinsic defects31,32,33. Among various types of defects, substitutional defects are the most significant for preferentially modifying material properties34. These substitutions can occur at either the transition metal site or the chalcogen site, introducing localized electronic states that can fundamentally alter the behaviour of the materials. Precise control of these defect states in 2D TMDCs enables powerful defect-engineering strategies. Introducing dopants, either p-type or n-type, allows for tunable optoelectronic11 and magnetic properties35,36,37, improved stability38, and enhanced charge carrier mobility39. Specifically, Vanadium (V) doping significantly impacts electron decay dynamics by altering the electronic structure40,41 and introducing new recombination pathways, making it possible to design materials for specific applications42. Vanadium acts as a p-type dopant in monolayer MoS2, introducing energy levels near the valence band. However, doping can also create energy levels close to the conduction band, as demonstrated43, introducing additional non-radiative recombination pathways that govern the carrier lifetime. Furthermore, the material can exhibit degenerate semiconductor behaviour at higher concentrations of Vanadium doping44. Additionally, the presence of Vanadium atoms can enhance valley scattering, which is particularly relevant for valleytronic applications, an emerging field that exploits the valley degree of freedom in 2D materials41,45.
For most device-level applications, carrier lifetime plays a crucial role, as it directly influences their performance. However, non-radiative recombination pathways significantly reduce hot carrier lifetimes, necessitating a detailed investigation of these decay mechanisms. Hot carriers in TMDCs can undergo electron–phonon scattering46 to form band-edge excitons that can become trapped in defect states. These processes can only be effectively studied using mid-infra-red (mid-IR) and near-infra-red (near-IR) spectroscopy47,48,49,50. Excitonic dynamics in TMDCs can be explored using probe energies lower than the bandgap energy, which allows for the study of intraexcitonic transitions48, phonon-mediated thermalization49,51,52, Auger recombination53, and exciton trapping at defect levels54. In monolayer MoS2, intraexcitonic transitions within the conduction band are observable due to the high exciton binding energy, which exhibits strong temperature dependence48.
In our previous work, we reported anisotropic non-radiative decay dynamics in few-layer MoS255 using a Mid-IR probe. In this study, we focus on monolayer 1H-MoS2 and lightly Vanadium-doped MoS2 (V-MoS2, 1.8 atomic% V doping) synthesized using the CVD method. The substitutional defects introduced by Vanadium doping create additional non-radiative pathways56, thereby altering the overall carrier dynamics43. Here, we investigate the hot carrier defect-assisted trapping dynamics in monolayer MoS2 and V-MoS2 by pumping at the exciton resonance energies. Selective excitation of A and B excitons enables us to probe exciton formation dynamics. Furthermore, by choosing two probe energies above and below the excitonic binding energy, we selectively probe the intra-excitonic and intra-band transitions. In particular, mid-IR probing allows us to identify the defect-mediated non-radiative decay pathways and compare how these processes differ between pristine MoS2 defects and V-induced defect states in V-MoS2.
A systematic study of defect-related emission peaks in V-MoS2, combining low-temperature photoluminescence (PL) measurements at 77 K and density functional theory (DFT) calculations, has been reported44. Our investigation reveals that Vanadium doping gives rise to a new emission peak around ~850 nm in the low-temperature PL spectra. This feature is consistent with DFT results, which show that Vanadium induces acceptor-like defect bands near the valence band. In addition, sulfur vacancy-related defect bands appear around 750 nm in both pristine and V-MoS244, corresponding to single and double sulfur vacancies57. Vanadium doping can also introduce defect levels ~0.13 eV below the lowest excitonic level, which is the energy difference between defect-induced bleaching and lower excitonic levels as reported experimentally by visible pump-probe spectroscopy43. These conduction band and valence band proximal defect states play a crucial role in enabling non-radiative relaxation pathways.
Results
Exciton selective pumping and probing with mid-IR
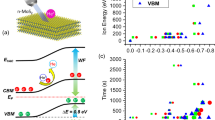
The mid-IR transient spectroscopy setup55, utilizing a femtosecond Ti:Sapphire laser and the detection of the transient signal58, has been detailed in our previous works. A schematic of the home-built pump-probe setup is shown in Fig. S1. To investigate the exciton formation dynamics of A and B excitons, transient absorption spectra were recorded using resonant pump excitation at 1.85 and 2 eV, corresponding to the A and B exciton energies, respectively. These pump energies create stable bound electron-hole pairs at the K and K′ points of monolayer MoS2. Our experiments in this work revealed that the exciton formation time (τᵣ) exhibits a clear dependence on the pump energy, which is tuned to the A or B exciton resonance. Here, we have selected to excite only A and B excitons, but not the C-exciton. If one were to excite C excitons near the band nesting region, both excitons and free electrons would be generated. The generated C excitons undergo intervalley exciton-exciton scattering, leading to thermalization at the K valley. Free electrons interact with phonons, releasing energy as heat and subsequently forming bound electron–hole pairs48. After the excitation of carriers, the decay dynamics were probed using two energies, 0.62 and 0.31 eV, generated by a home-built difference frequency generation (DFG) setup59. The low-energy 0.31 eV probe, with a narrow line width of 0.31 ± 0.0085 eV, exactly matches the 1sB to 3pB transition48 and is sufficient to make the transition from 1s to 3p, allowing us to investigate intra-excitonic transitions. In the second case, we selected a probe energy of 0.62 eV, which is higher than the excitonic binding energy. This probe energy enables a broad range of transitions from any exciton level to the conduction band (CB). Transient measurements were performed at the same spatial location for both pump wavelengths, and additional measurements at nearby points confirmed that the decay dynamics remained consistent within experimental error. The transient signals are presented as the variation of ΔT/T as a function of delay time. Here, ΔT denotes the pump-induced change in probe transmission, defined as the difference between the transmission with and without the pump. T represents the probe transmission in the absence of the pump. The spectra exhibit negative ΔT/T dynamics, indicating pump-induced absorption. A schematic representation of the overall energy landscape and the decay channels of excited carriers in V-MoS2 is shown in Fig. 1.

a Pumping at A (1.85 eV, red) or B (2 eV, green) resonances and subsequently probing with 0.31 or 0.62 eV. The low-energy probe (0.31 eV) resolves intra-excitonic levels; the corresponding band splitting is illustrated. The grey arrow in a shows the exciton binding energy (Eb = 0.4–0.5 eV). The conduction band (CB) is shaded in grey. b The relaxation pathways at both probe energies. The excitons dissociate and form the hot carriers above the Mott density due to bandgap renormalization (BGR). Orange dashed lines indicate the Vanadium-induced localized states near the conduction and valence bands. Curved arrows represent exciton relaxation into the defect levels, while the grey curved arrow indicates radiative recombination of electrons into the valence band. For clarity, the CB splitting is exaggerated; in reality, the valence band splitting is an order of magnitude larger. Rydberg-like excitonic levels are shown only in the conduction band for schematic simplicity.
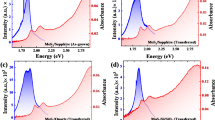
Monolayers of MoS2 and lightly doped V-MoS2 (1.8 atomic% V)44, were synthesized using the CVD method, as discussed in the Experimental methods and in our previous studies41. The doping percentage was confirmed using both the XPS and STEM measurements44. A comparison of the Raman spectra of MoS2 and V-MoS2 is presented in Fig. S2a. Monolayer MoS2 exhibits two Raman modes, E2g (in-plane vibration) and A1g (out-of-plane vibration), at 385 and 404 cm−1, respectively. The difference between these two modes is ~19 cm−1, indicating the formation of monolayers60,61. The standard deviation for the A1g mode is 0.3 cm−1, and for the E2g mode is 0.1 cm−1. In addition to A1g and E2g modes, a new set of broad peaks appears in the case of V-MoS2, between 100 and 260 cm−1, termed the longitudinal acoustic (LA) mode. The LA mode can be used to estimate the average distance (LD) between defect (here V) sites in MoS2 and hence to calculate the percentage of doping, as reported in our previous study44. The photoluminescence (PL) spectra of MoS2 and V-MoS2, which show a red shift for V-MoS2, are shown in Fig. S2b. The observed reduction in A exciton PL intensity for the Vanadium-doped MoS2 sample corresponds to the introduction of more localized defects due to V-doping41. The excitonic emission at room temperature in monolayer TMDs is dominated by the A exciton due to the rapid relaxation from B to A, and it is highly sensitive to the defects62. As the crystal becomes more defective, the IA/IB ratio decreases due to the enhanced B exciton emission following V doping42,63. Figure 2a shows the optical image of a large area of V-MoS2 grown on a Si/SiO2 substrate. The crystal structure indicates a 1H phase of MoS2 with a perfect hexagonal lattice structure found by systematic investigation of the crystal structure of MoS2 via HAADF-STEM image20. The STEM image confirms that in V-MoS2, the Mo sites are substituted by V atoms through substitutional doping. These defects are not localized at any specific region of the sample and are randomly distributed.

a Optical image of a large area of V-MoS2 on a Si/SiO2 substrate. b UV–visible absorption spectra of V-MoS2 (red) and pristine MoS2 (black curve), showing spectral broadening in the V-MoS2 sample.
The A and B excitons in the MoS2 will have a binding energy equal to the difference between the electronic band gap energy (Eg) and the optical energy of the excitons. This energy ranges from 0.4 to 0.54 eV, as reported by Hyunyong et al. 48 and is represented in Fig. 1a as Eb. Beyond the K-point, a band nesting region exists where the slope of the conduction band and valence band match, leading to the formation of C excitons64,65. The excitonic energy levels were determined with a UV–Vis spectrophotometer (Jasco-670). The steady-state absorption spectra for both monolayer MoS2 and V-MoS2 are shown in Fig. 2b. exhibit distinct peaks at 670 nm (1.85 eV), 620 nm (2 eV), and 450 nm (2.7 eV), corresponding to the A, B, and C excitons, respectively. A secondary peak is observed at the higher energy region of the absorption spectrum (~3.1 eV), which is the D excitonic state at the band nesting region away from the K point66. In V-MoS2, we observed noticeable peak broadening for all three excitonic peaks, which is attributed to the high density of defect states near the conduction and valence bands due to V doping. V-doping introduces new localized energy levels that modify the electronic structure, creating new defect states. The doping process can also introduce additional structural disorder, leading to a more defective crystal lattice. Such disorder not only broadens the excitonic resonances but can also increase the overall absorption baseline due to defect-induced scattering and tail states.
When we pump and probe the monolayers with energy resonant with A and B excitonic transitions, both in the visible region, the depleted population from the valence band gives the ground state bleach signal due to increased transmission of probe27,67. In our visible pump mid-IR probe experiments, the differential transmission is positive for defect level bleach51,55, and negative for pump-induced excited-state absorption, as shown in Fig. 3a. This manuscript reports negative transient signals attributed to intraband transitions and their subsequent decay through defect levels. Transient spectra were recorded for different excitation carrier densities in both MoS2 and V-MoS2 by tuning the pump energies to the A and B excitons. Following resonant excitation by the pump, the probe monitors the change in absorption of excited carriers until the excitonic population reaches equilibrium. The relaxation process is generally mediated by defect states located between the CB and VB, and these states exist in several forms. In monolayer MoS2, carrier dynamics are governed by intrinsic mid-gap defect states that primarily originate from sulfur vacancies (single, double, and paired)68,69. These native defect states, such as single sulfur vacancies (Vs) and double sulfur vacancies (Vs2), introduce deep-level defect states ~0.5–0.6 eV below the conduction band minimum (CBM) and shallow defect states closer to the CBM68,69,70. Neighbouring sulfur vacancies (VsVs) form hybridized defect states due to the interaction between adjacent vacancies. These intrinsic mid-gap states act as efficient non-radiative recombination centres and strongly influence the carrier dynamics. In contrast, the V doping introduces additional energy levels near the CBM and VBM as discussed in the previous section. These new trap states, distinct from the intrinsic sulfur-related defects, play a central role in the modified carrier dynamics investigated in the work, and the extracted decay constants therefore reflect contributions from both types of trap states.

a Transient spectra at 0.31 eV and b 0.62 eV probe energies. The negative ΔT/T indicates pump-induced absorption. c Change in peak ΔT/T suggests an increase in carrier density as excitation fluence increases. d Decay times show that carrier relaxation remains independent of the excitation carrier density induced by the pump. e Percentage of the amplitude for different relaxation components, where A1 corresponds to fast decay, and A2 corresponds to slower decay. f Comparison of τr values for probe energies of 0.31 eV (black curve) and 0.62 eV (blue curve) for V-MoS2 under 1.85 eV pump excitation.
If one uses a mid-IR or NIR probe energy that can probe the transition of the electrons from the mid-gap defect states to the conduction band, the sign of the transient signal will be positive since the pump iodizes the defect level51,55. Also, the expected decay time from the defect states will be in hundreds of picoseconds67,71,72. Recently, Wang et al. have also shown that high-energy NIR pulses can probe transitions within the conduction band49. This confirms that in this study, the IR beams are not probing the transition from sulfur defects to the conduction band. Once the pump creates a population in the excitonic levels, and if the excited carrier density is near the Mott threshold, after a few femtoseconds, bandgap renormalization (BGR) can occur67,73,74,75. Due to screening effects, the probe detects the free carrier transitions within the conduction band. With doping, the rate of energy transfer to mid-gap defect states changes, altering the overall dynamics of free electron relaxation and recombination. To capture these effects, we systematically varied the pump fluence across a broad range and compared the transient defect-state dynamics in pristine MoS2 and V-MoS2.
Resonance pumping at the A exciton (E A = 1.85 eV) in V-MoS2
The pump-induced carrier density-dependent transient absorption spectra for V-MoS2 are shown in Fig. 3a for a pump energy of 1.85 eV with a probe energy of 0.31 eV, and in Fig. 3b for the same pump energy with a probe energy of 0.62 eV. The increases in peak ΔT/T with carrier density for both probe energies, as shown in Fig. 3c, indicate the increase in the number of excited carriers with pump fluence without any significant band filling effect76. After reaching the peak ΔT/T, relaxation back to equilibrium occurs at two different time scales. These timescales are determined by fitting the decay curve with a bi-exponential decay function convoluted with an error function, as described by Eqs. (1) and (2) below77,78,79.
From these equations, the decay time constants (τ1, τ2), the amplitudes of decay (A1, A2), the population rise time (τr), and the instrument response function (IRF) σ are obtained from fitting. Here, erf denotes the error function, and C and D are the baseline offsets. The terms t and t0 represent, respectively, the delay time and the time-zero offset associated with the instrument response and pulse width. A detailed curve fitting for one dataset with both equations is presented in Fig. S3a, b in the Supplementary Material. Both the above equations give similar values of τ1 and τ2. The calculated IRF from fitting, which represents the minimum timescale the transient absorption setup can resolve, is ~82 fs in our case. The pump pulse width near the sample was measured to be ~89 fs from the autocorrelation trace, as shown in Fig. S4a. The cross-correlation between the 1.85 eV pump and 0.31 eV probe pulses yields a FWHM of 114 fs (Fig. S4b). A comparison of the cross-correlation and transient data confirms that the experimental signals are not limited by the temporal resolution of the system (Fig. S4c). In the transient spectra, the time to reach the peak ΔT/T value is denoted by the time (τr), which is the characteristic time over which the excited state population builds up after the pump pulse excites the carriers. It is a measure of how fast the system attains the maximum transient population. The equilibrium recovery time constants τ1 and τ2 are associated with the fast and slow processes of carrier capture by defect states in the system48,53.
Haining Wang and coworkers proposed the following relaxation mechanisms in monolayer MoS2, where the two time scales are attributed to differences in the carrier capture rates of various defect levels53, and the change in this decay time with fluence can be attributed to the saturation of defect states73. In semiconductors with numerous defect levels exhibiting different carrier capture rates, saturation of all defect states does not occur. It would not necessarily cause a sudden shift in the transient behaviour, as other defect states remain active. In this scenario, the pump fluence would have little to no effect on the decay time constants, which is precisely what we observe in V-MoS2 at high pump fluence. The transient spectra of V-MoS2 over the full range of pump excitation carrier densities are presented in Fig. S5c. Both the fast and slow decay times decrease with increasing pump fluence and eventually plateau at high carrier densities (Fig. S5a). This reduction in decay constants at higher densities suggests the dominance of many-body-assisted carrier trapping into defect levels. Further, the peak ΔT/T increases monotonically with fluence as shown in Fig. S5b. The obtained time constants at high carrier densities are plotted in Fig. 3d for different pump fluences, confirming that they remain independent of the excitation carrier density. So, the fast and slow time scales can be attributed to the different carrier capture rates by different types of defect levels. As the carrier density increases, many-body interactions promote exciton dissociation to form free carriers80, which also results in the redshift of absorption spectra81. Once the carriers become trapped in a defect state, exciton formation reoccurs due to the reduced screening effect73,82. The fast decay channel (τ1) is dominated by ultrafast capture of free electrons into the shallow sulfur defects, located just below the CBM83, as shown in Fig. 1b. This is consistent with previously reported fast carrier capture times71,73. The slower decay component (τ2) arises from excitons trapping at deeper sulfur defect states, a process mediated by phonon-assisted capture and multiphonon emission, which typically occurs on a few-picosecond timescale. The amplitudes A1 and A2 of the slow and fast decay components represent the relative contributions of excitons participating in each process to the overall signal. In Fig. 3e, the percentage of amplitude for both time scales is plotted. The A1 component dominates the decay process, indicating that a larger fraction of carriers undergo fast decay. Notably, the amplitude percentages remain constant across different excitation carrier densities. This suggests that, despite an increase in the total number of carriers with higher fluence, the proportion of carriers decaying through different channels remains unchanged for this system.
In V-MoS2, similar to the 0.31 eV probe, at the 0.62 eV probe, the fast and slow time constants remain unchanged (Fig. 3d). However, the slower time constant (τ2) is longer for the 0.31 eV probe than the 0.62 eV probe. The two-time constants for different densities are summarized in Table S1 for the 1.85 eV excitation. The longer τ2 for 0.31 eV arises because lower-energy probes can monitor the excited-state population over longer timescales than higher-energy probes, as the differences in energy exchange pathways result in distinct transfer rates. This behaviour is in agreement with previous reports48.
The characteristic rise time τr values are determined to study the time required to create the maximum exciton population. From Fig. 3f, the faster rise time at 0.31 eV probing compared to the 0.62 eV probing can be attributed to the low-energy probe accessing the intra-excitonic transition from 1SB to 3PB state, which is allowed by selection rules. Exciton formation itself is nearly instantaneous, occurring on a timescale of ~30 fs after pump excitation29. At high excitation densities, strong Coulomb screening leads to exciton dissociation into free carriers75, resulting in the formation of the electron–hole plasma. Complete dissociation typically occurs within a few hundred femtoseconds. For instance, Alexey Chernikov et al. reported that the carrier screening and bandgap renormalization begin within ~100 fs in monolayer WS275. The dissociation of excitons requires a slightly longer time to entirely disrupt the bound electron–hole pairs. Thus, some fraction of excitons can survive even after 100 fs before they completely dissociate into free carriers. The exciton can persist up to 0.3 ps after pump excitation in the WS2 sample75. In our experiment, the maximum absorption in pump-induced absorption occurs at 0.11 ± 0.002 ps for the 0.31 eV probe and 0.2 ± 0.001 ps for the 0.62 eV probe. The 0.31 eV probe pulse is resonant with discrete internal transitions of the exciton from the 1s to 3p states of the B exciton. Thus, during the time (τr) for a 0.31 eV probe, the pump excites carriers to the 1sA energy level of the A exciton, and a transition can occur to the lower 1sB level via phonon-mediated processes. The transition from A excitonic states to B excitonic states involves a change in the spin–orbit coupling configuration of the valence and conduction bands84. While a direct spin–flip transition is unlikely due to the spin conservation rule, phonon-mediated spin change can facilitate this process85,86. The rise time is independent of the pump fluence or carrier density, as shown in Fig. S6c, confirming that the rise time is attributed to exciton formation. The intra-excitonic transition can be attributed to the initial pump-induced signal. After the first few hundred femtoseconds (τr), the transient spectra are mainly from the free carrier absorption in the conduction band due to band renormalization at high densities. The black arrows in Fig. 1a illustrate this intra-excitonic transition. From the 1sB energy level, carriers release energy through non-radiative relaxation and become trapped in defect states, as shown by the decay channels in Fig. 1b. For further confirmation, we have performed visible pump-probe experiments in Monolayer MoS2 at low (1.3 × 1013 cm−2) and high (2 × 1014 cm−2) carrier densities, as shown in Fig. S10. The sample is excited with a pump energy of 2.17 eV, just above the two excitonic energy levels, resulting in a clean transient spectrum without pump scattering. The transient spectra are recorded using a white light probe that ranges from 580 nm (2.13 eV) to 720 nm (1.72 eV). A visible shift in excitonic bleach signals is observed at high carrier densities (Fig. S10d). However, there is no transient peak shift at 620 or 670 nm up to 0.5 ps. We are unable to reduce the pump to 620 nm (2 eV) due to scattering, but the time scales could be a little faster or remain around this range, where peaks do not shift. From all these observations using the mid-IR probe, we conclude that within the observed maximum τr values, the excitonic levels exist irrespective of the carrier density.
In contrast, the 0.62 eV probe energy is higher than the excitonic Rydberg-like energy levels. This energy allows carrier transition to higher energy levels in the conduction band, as shown by the blue arrow in Fig. 1a, b. Figure 3f shows that τr is longer for 0.62 eV (blue curve) than for 0.31 eV probe(black curve), confirming that the energy levels probed at these two wavelengths are different. The rise time (τr) for both probe energies remains independent of excitation carrier density (Table S1), indicating no significant saturation of the band-edge filling effect. The average Δτr value between 0.31 and 0.62 eV probe is ~0.09 ps. The 0.62 eV energy is higher than the exciton binding energy, which can allow the transition of stable excitons into free carrier states. This transition is sensitive to exciton delocalization and dissociation, as it requires overcoming the binding energy before the transition can occur. Additional processes, such as exciton–phonon interactions87,88 are necessary for excitons to dissociate into free carriers, leading to a longer rise time for the 0.62 eV probe. This is consistent with previous reports, which show that mid-IR beams probe transitions within excitonic levels, while NIR beams detect a convolution of multiple transition pathways within the conduction band48,53.
Resonance pumping at the B exciton energy (E B = 2 eV) in V-MoS2
When excited with 2 eV pump energy, the pump directly excites carriers into the B excitonic level. Figure 4a and b represents the transient spectra under different pump fluences with 2 eV pump energy and the corresponding probe energies. Similar to the case of the 1.85 eV pump, for the 2 eV excitation with a 0.31 eV probe, the fast and slow time scales remain almost independent of the excitation density (Fig. 4d). The maximum optical density at τr also increases linearly with pump fluence, as shown in Fig. 4c. The amplitude coefficients of relaxation also remain almost unchanged with increasing pump density (Fig. 4e), confirming that the fast and slow time scales correspond to relaxation through different types of defect states without saturation.

Carrier dynamics for different pump fluences at a pump energy of 2 eV. a Transient spectra for 0.31 eV and b 0.62 eV probe energies. c Peak ΔT/T as a function of carrier density. d Variation of fast and slow time scales with carrier density. e Percentage of the amplitude contribution from fast and slow decay channels. f Comparison of rise times for two different probe energies in V-MoS2.
Also, at 2 eV pump in resonance with B exciton and a 0.62 eV probe, the transient decay dynamics, indicating carrier capture by mid-gap defect states, exhibit a trend similar to that observed with A exciton resonance pumping. For the 1.85 eV pump energy and 0.62 eV probe energy, the fast relaxation time varies from 0.61 ± 0.01 to 0.63 ± 0.01 ps, as shown in Table S1, while for the 2 eV excitation, it ranges from 0.64 ± 0.01 to 0.67 ± 0.01 ps (Table S2). The slow relaxation time changes from 2.6 ± 0.1 to 2.9 ± 0.1 ps for the 1.85 eV pump and from 2.3 ± 0.1 to 2.9 ± 0.1 ps for the 2 eV pump (Tables S1 and S2). These decay time variations are too small to draw a definitive conclusion, especially considering the proximity of the two pumping energies.
For excitation resonant with the B exciton and probing at mid-IR (0.31 eV) and near-IR (0.62 eV) wavelengths, the rise time exhibits a consistent difference, similar to the case of A-exciton pumping as shown in Fig. 4f. For the 0.31 eV probe, the rise time is 0.09 ± 0.001 ps (black curve), whereas for the 0.62 eV probe, it is ~0.20 ± 0.001 ps (blue curve) (Table 1). The time required to reach the peak ΔT/T is found to be independent of pump fluence for both cases. For the 0.31 eV probe energy, the τr value is consistently slightly higher for A exciton pumping than for B exciton pumping (Tables 1, S1, and S2). The difference in rise time is pronounced for the 0.31 eV probe but not for the 0.62 eV probe, as shown in Fig. S6a, b. As the pump energy increases, the rise time is expected to increase if we are probing free carriers67. However, in our experiment, the rise time decreases or remains constant when tuning the excitation energy from A to B exciton resonance, consistent with the A and B exciton pump-induced absorption reported by Wang et al. 67. The rise time at 0.31 eV reflects how rapidly the B-exciton ground state is populated before being absorbed into the 3pB level by a 0.31 eV probe. While probing at 0.31 eV, at 1.85 eV pump, the pump is off-resonant with the B-exciton, carriers must scatter into the 1sB state before the 1s to 3p transition can occur, resulting in a rise time of ~110 fs. As discussed earlier, the direct spin–flip transition is unlikely due to the spin conservation rule; phonon-mediated spin changes can facilitate this process. In contrast, with a 2 eV pump resonant with B-exciton, electrons are directly excited to the 1sB level, leading to faster intra-excitonic absorption, ~0.09 ± 0.001 ps. The additional rise time observed for the 1.85 eV pump can thus be attributed to the spin–flip mediated conversion from A exciton to B exciton, which can occur in tens of femtoseconds84. In literature, the rise times are about ∼400 fs, the typical time for exciton formation observed when pumping into continuum48, because of the transit time to reach the band edge. In the present case, however, dynamics are much faster because we directly access the exciton branch through resonant pumping. The more pronounced difference in rise time confirms that 0.31 eV probe captures the intraexcitonic transition from 1sB to 3pB at both excitation energies. In contrast, the 0.62 eV probe reflects a convolution of multiple processes involving all the excitonic levels; hence, the rise time remains unchanged for different pump energies. The corresponding absorption appears at a later time (~200 fs) compared to the 0.31 eV probe, as it requires overcoming the exciton binding energy.
Resonance excitation at the A exciton energy (E A = 1.85 eV) in MoS2
The carrier dynamics exhibit a distinct lifetime in MoS2, which has a lower defect density compared to V-MoS2. The increased number of defect states near the conduction and valence bands in V-MoS2 means that even after some defect states are filled, many remain available for electron occupation. A high defect density can affect the non-radiative recombination processes, where the energy from excited electrons dissipates as heat. As expected, relaxation times are longer in V-MoS2 due to the delayed trapping caused by localized Vanadium-induced energy levels. Transient decay dynamics across the full range of carrier densities, measured with a 1.85 eV pump and a 0.31 eV probe, for MoS2 are presented in Fig. S8a. At low carrier densities, the decay time constants are relatively long. With increasing carrier density, the decay time decreases at first due to carrier–carrier scattering and many-body interactions. After that, the decay time increases again with an increase in carrier density due to the saturation of defect states or a phonon bottleneck effect67,89. The peak ΔT/T increases as we increase the carrier density, as shown in Fig. S8b, c. To further study the decay dynamics in the MoS2 monolayer at different fluences, we have investigated the dynamics at different pump excitation energies and probe energies, similar to the V-MoS2 case. The transient spectra of MoS2 are shown in Fig. 5a, b, and the variation of time constants with fluence is shown in Fig. 5c.

a, b Transient spectra of the MoS2 monolayer for a 1.85 eV pump and probe energies of 0.62 and 0.31 eV. c Fitted values of the fast and slow relaxation timescales. d Variation in the percentage of the amplitude with excitation carrier density.
Even when the excitation energy is resonant with the exciton energy, the decay dynamics are governed by the free electrons generated under the strong screening effect at high carrier densities. In this regime, the phonon bottleneck effect arises as hot carriers transfer their energy to the lattice through phonons, while the phonons themselves are unable to dissipate this energy efficiently. This leads to an accumulation of hot phonons, which in turn reduces the effectiveness of phonon-mediated carrier energy loss. At high carrier densities, the reabsorption of longitudinal–optical (LO) phonons slows down the cooling process of excitons90. However, in V-MoS2, this process is suppressed due to the high density of V-induced defect states. These defects act as scattering centres for phonons91, which facilitates energy dissipation, enabling phonons to lose energy more efficiently92. The intrinsic cooling mechanism in V-MoS2 is, however, still slower because the localized states temporarily trap carriers, delaying their recombination. A comparison of the decay times between MoS2 and V-MoS2 highlights these significant variations in relaxation dynamics, as illustrated in Fig. 6a, b.

a Normalized transient response for a 1.85 eV pump and 0.62 eV probe, at a 1.3 × 1014 cm−2 pump excitation density in MoS2 and V-MoS2 samples showing distinct relaxation dynamics. b Transient spectra for a 1.85 eV pump and 0.31 eV probe highlighting significant differences in time constants between MoS2 and V-MoS2. c Rise time comparison for different probe energies, demonstrating a longer time to reach the peak ΔT/T for the 0.62 eV probe.
Since the pump excitation is resonant with the exciton energy, the τ1 component cannot be attributed to carrier thermalization. Carrier thermalization is generally fluence-independent or becomes faster at higher fluence93. In MoS2, the fast channel with time constant τ1 (with amplitude A1) is dominated by ultrafast capture of free electrons into shallow defect levels after bandgap renormalization, which is consistent with the previously reported fast defect capture process observed in MoS2 at high carrier densities71,94. The dissociated free electron can transfer its energy to other electrons and subsequently get captured at the defect sites. While the main contribution to the decay of the pump-induced signal comes from these processes, a minor component may originate from slower trapping into deep defect levels. At higher carrier densities, the phonon bottleneck becomes more prominent, and the saturation of trap states leads to slower overall decay dynamics. The overall processes in monolayer MoS2 are shown in Fig. S7, essentially without V-induced defect states as in Fig. 1. The decay coefficients A1 and A2, representing the probability of carriers being trapped, are plotted in Fig. 5d. These plots show that the fraction corresponding to fast decay increases with fluence, indicating that as more carriers are generated, a higher percentage undergoes fast trapping and recombination by Auger-mediated decay. The detailed decay constants for MoS2 at 1.85 eV pump are provided in Table S3.
The rise time in MoS2 exhibits notable differences depending on the probe energy, similar to the behaviour observed in V-MoS2. The rise time is found to be independent of the excitation carrier density, indicating that the rise time reflects the exciton population buildup (Fig. S9c). As shown in Fig. 6c, the peak ΔT/T occurs at ~0.11 ± 0.001 ps for the 0.31 eV probe and around 0.2 ± 0.001 ps for the 0.62 eV probe. These values are similar to the rise time obtained for V-MoS2. This indicates that the formation mechanism of the exciton in both MoS2 and V-MoS2 remains the same.
Resonance excitation at the B exciton energy(E B = 2 eV) in MoS2
Similar to V-MoS2, when MoS2 is excited with a 2 eV pump, it creates a population directly at the B excitonic level. The transient decay dynamics measured at 2 eV excitation closely resemble those observed for 1.85 eV excitation, as shown in Fig. 7. The details of these figures are explained in the previous cases. Nearly the same decay constants (τ1 and τ2) are obtained with different pump excitation energies, and the rise time remains independent of pump fluence, as shown in Tables S3 and S4. The rise time values are 0.09 ± 0.001 ps for the 0.31 eV probe and 0.2 ± 0.001 ps for the 0.62 eV probe, as shown in Fig. S9a. The trend in rise time variation across different pump excitations is consistent for both samples (Table 1), MoS2 and V-MoS2, the rise time at 0.31 eV corresponds to the 1sB population buildup.

Pump excitation-dependent transient spectra of MoS2, with two-time-scale fitting using a double-exponential decay function for a pump energy of 2 eV and probe energies of a 0.62 eV and b 0.31 eV. c The variation of the fast and slow time constants. d Show the variation in the percentage of amplitude with excitation carrier density.
Discussion
The difference in the energy alignment between the two samples arises from Vanadium doping, which introduces additional relaxation channels into the energy landscape. The decay time constants extracted from the transient spectra reveal a strong dependence on defect densities. The overall carrier dynamics differ between MoS2 and V-MoS2 as the number of excited carriers increases with pump fluence. Figure 8 presents the detailed values of the fast and slow decay components for both samples. Notably, both fast and slow decay times in MoS2 are faster than those in V-MoS2. In MoS2, the time constants increase with pump-induced carrier concentration, as indicated by the green lines in Fig. 8. This behaviour is attributed to the lower density of defect states and the absence of localized states near the conduction band and valence band in MoS2, providing direct evidence of defect state saturation or the phonon bottleneck effect. In contrast, in V-MoS2, the decay time constants remain nearly invariant with increasing pump fluence for both excitation energies. In V-MoS2, in addition to intrinsic MoS2 defects, V-induced defect states serve as efficient carrier trapping centres, resulting in longer time constants for fast and slow decay processes. All these findings indicate that Vanadium doping in MoS2 introduces additional defect states close to the conduction band and valence band. For both samples, Δτr values remain independent of pump fluence when comparing carrier dynamics at 0.31 eV and 0.62 eV probe energies. The absolute τr values in MoS2 are the same as those in V-MoS2, as summarized in Table 1 or Tables S2 and S4. In both materials, τr is higher for the 0.62 eV probe than for the 0.31 eV probe for any pump energy, similar to both samples.

Pump excitation carrier density-dependent carrier capture times for MoS2 (green) and V-MoS2 (blue) samples at a 2 eV pump and 0.31 eV probe, b 1.85 eV pump and 0.31 eV probe, c 2 eV pump and 0.62 eV probe, and d 1.85 eV pump and 0.62 eV probe. In all the plots, the time constants of V-MoS2 remain constant, and those of MoS2 increase with carrier density.
In summary, we have investigated the role of Vanadium doping in modulating non-radiative electronic decay dynamics in monolayer MoS2 using mid-infra-red transient absorption spectroscopy. Our results provide the first direct experimental evidence that Vanadium doping introduces defect states that prevent defect saturation, fundamentally altering carrier relaxation pathways. Unlike pristine MoS2, where defect trapping dominates at higher excitation fluences, V-MoS2 exhibits fluence-independent decay times, indicating multiple relaxation channels with distinct capture rates. By probing intra-excitonic transitions at 0.31 eV and intraband transitions at 0.62 eV, we demonstrate that phonon-mediated processes play a key role in free electron relaxation and that Vanadium doping modifies electron trapping without introducing Auger heating effects. These findings establish mid-IR transient spectroscopy as a powerful tool for quantifying defect states and tuning lifetimes in 2D materials. Our results not only provide deeper insight into defect-assisted recombination in TMDCs but also offer a pathway to engineering electronic properties via controlled doping. Given the importance of hot carrier dynamics in optoelectronic and quantum devices, these findings could have significant implications for the design of next-generation 2D material-based technologies. Further theoretical modelling and ultrafast spectroscopic studies on different doping concentrations could provide additional insights into the tunability of defect states and their interaction with excited carriers.
Methods
Sample preparation
MoS2 and lightly doped V-MoS2 samples were grown on Si/SiO2 (300 nm) substrates using a custom-designed two-zone CVD system, as reported in our previous studies41,43. The growth process involved loading of MoO3 and V2O5 powders and a small amount of KCl in an alumina boat, with the Si/SiO2 (300 nm) substrate placed over the powder mixture. The growth temperature was set to 710 °C for metal decomposition, while the sulfurization temperature was maintained at 210 °C. Throughout the growth process, nitrogen gas (190 sccm) was continuously supplied as a carrier gas. MoS2 grown on a Si/SiO2 substrate was coated with polymethyl methacrylate (PMMA) via spin coating, followed by heating at 90 °C for 10 min. The substrate was then immersed in a 2 M KOH solution overnight, allowing the MoS2/PMMA film to detach. The floating films were thoroughly washed with water and subsequently transferred to a CaF2 substrate, followed by the removal of PMMA using acetone.
Transient absorption spectroscopy
To measure the transient spectra of the samples, we used a home-built visible pump mid-IR probe setup. The fundamental beam consisted of an 800 nm pulse with a 75 fs pulse width and a 1 kHz repetition rate, generated from a Ti:Sapphire laser. The pump beams with photon energies of 1.85 and 2.5 eV were generated from a TOPAS optical parametric amplifier (OPA). The probe energies 0.31 and 0.62 eV were generated by home-built difference frequency generation (DFG) in an AgGaS2 crystal, achieved by spatial and temporal overlap of signal and Idler beams produced by a second tunable OPA. The resulting IR beams are allowed to pass through the low-pass filter (LP filter) to remove the signal and idler. To control the delay between the pump and probe, we used a motorized Aerotech stage. To measure the differential transmittance, the pump beam was mechanically chopped at 500 Hz and synchronized with a TTL trigger. The pump and probe beams are focused on the sample with two CaF2 lenses. The change in transmittance of the transmitted IR probe beam is recorded using a mercury–cadmium–telluride (MCT) detector connected with the boxcar integrator (SR 280). In a separate optical path, the ANDOR spectrometer confirmed the wavelength tuning of the generated mid-IR beams.
Data availability
The raw data supporting the findings of this study are available in the open-access data repository, ZENODO, under accession: https://zenodo.org/records/17747822.
References
Rodin, A., Trushin, M., Carvalho, A. & Castro Neto, A. H. Collective excitations in 2D materials. Nat. Rev. Phys. 2, 524–537 (2020).
Manzeli, S., Ovchinnikov, D., Pasquier, D., Yazyev, O. V. & Kis, A. 2D transition metal dichalcogenides. Nat. Rev. Mater. 2, 17033 (2017).
Tedstone, A. A., Lewis, D. J. & O’Brien, P. Synthesis, properties, and applications of transition metal-doped layered transition metal dichalcogenides. Chem. Mater. 28, 1965–1974 (2016).
Splendiani, A. et al. Emerging photoluminescence in monolayer MoS2. Nano Lett. 10, 1271–1275 (2010).
Tian, H. et al. Optoelectronic devices based on two-dimensional transition metal dichalcogenides. Nano Res. 9, 1543–1560 (2016).
Peng, B., Ang, P. K. & Loh, K. P. Two-dimensional dichalcogenides for light-harvesting applications. Nano Today 10, 128–137 (2015).
Khan, F., Julien, C. M. & Islam, S. S. Tuning the optoelectronic properties of reduced graphene Oxide MoS2 nanocomposite for room temperature photodetection application. FlatChem 43, 100586 (2024).
Thakar, K. & Lodha, S. Optoelectronic and photonic devices based on transition metal dichalcogenides. Mater. Res. Express 7, 14002 (2020).
Shabir, A. et al. Optimizing graphene content in scaffolds for evenly distributed crumpled MoS2 paper wads as anodes for high-performance Li-ion batteries. Nanotechnology 35, 375402 (2024).
Sun, H. et al. A review of transition metal dichalcogenides-based biosensors. Front. Bioeng. Biotechnol. 10, 941135 (2022).
Li, S. et al. p–n-type modulation of 2D transition metal dichalcogenides for electronic and optoelectronic devices. Nano Res. 15, 123–144 (2022).
Kwon, K. C., Baek, J. H., Hong, K., Kim, S. Y. & Jang, H. W. Memristive devices based on two-dimensional transition metal chalcogenides for neuromorphic computing. Nano-Micro Lett. 14, 58 (2022).
Babu, R. S. & Georgiadou, D. G. 2D transition metal dichalcogenides for energy-efficient two-terminal optoelectronic synaptic devices. Device 3, 100805 (2025).
Wang, Q. H., Kalantar-Zadeh, K., Kis, A., Coleman, J. N. & Strano, M. S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 7, 699–712 (2012).
Sun, J. et al. High-performance 2D WS2 photodetector enhanced by charge-transfer doping through NH3 annealing. Mater. Today Phys. 35, 101133 (2023).
Ma, X.-Q. et al. Flexible neuromorphic devices based on two-dimensional transition metal dichalcogenides. IEEE J. Flex. Electron. 3, 10–28 (2024).
Loh, L., Zhang, Z., Bosman, M. & Eda, G. Substitutional doping in 2D transition metal dichalcogenides. Nano Res 14, 1668–1681 (2021).
Zeng, P. et al. Thickness-dependent enhanced optoelectronic performance of surface charge transfer-doped ReS2 photodetectors. Nano Res 15, 3638–3646 (2022).
Choi, D., Jeon, J., Park, T.-E., Ju, B.-K. & Lee, K.-Y. Schottky barrier height engineering on MoS2 field-effect transistors using a polymer surface modifier on a contact electrode. Discov. Nano 18, 80 (2023).
Maity, D. et al. Interfacial engineering of degenerately doped V0.25Mo0.75S2 for improved contacts in MoS2 field effect transistors. Small Methods 9, 2401938–2401947 (2024).
Liu, B. et al. High-performance WSe2 field-effect transistors via controlled formation of in-plane heterojunctions. ACS Nano 10, 5153–5160 (2016).
Ko, R., Lee, D. H. & Yoo, H. Annealing and doping effects on transition metal dichalcogenides-based devices: a review. Coatings 13, 1364 (2023).
Ugeda, M. M. et al. Giant bandgap renormalization and excitonic effects in a monolayer transition metal dichalcogenide semiconductor. Nat. Mater. 13, 1091–1095 (2014).
Das, S., Gupta, G. & Majumdar, K. Layer degree of freedom for excitons in transition metal dichalcogenides. Phys. Rev. B 99, 165411 (2019).
Qiu, D. Y., da Jornada, F. H. & Louie, S. G. Optical spectrum of MoS2: many-body effects and diversity of exciton states. Phys. Rev. Lett. 111, 216805 (2013).
Zollner, K., Junior, P. E. F. & Fabian, J. Strain-tunable orbital, spin–orbit, and optical properties of monolayer transition-metal dichalcogenides. Phys. Rev. B 100, 195126 (2019).
Sim, S. et al. Exciton dynamics in atomically thin MoS2: interexcitonic interaction and broadening kinetics. Phys. Rev. B 88, 75434 (2013).
Marinov, K., Avsar, A., Watanabe, K., Taniguchi, T. & Kis, A. Resolving the spin splitting in the conduction band of monolayer MoS2. Nat. Commun. 8, 1938 (2017).
Trovatello, C. et al. The ultrafast onset of exciton formation in 2D semiconductors. Nat. Commun. 11, 5277 (2020).
Xu, Y. et al. Tightly bound and room-temperature-stable excitons in van der Waals degenerate—semiconductor Bi4O4SeCl2 with high charge-carrier density. Commun. Mater. 4, 69 (2023).
Liang, Q., Zhang, Q., Zhao, X., Liu, M. & Wee, A. T. S. Defect engineering of two-dimensional transition-metal dichalcogenides: applications, challenges, and opportunities. ACS Nano 15, 2165–2181 (2021).
Yang, J., Kawai, H., Wong, C. P. Y. & Goh, K. E. J. Electrical doping effect of vacancies on monolayer MoS2. J. Phys. Chem. C 123, 2933–2939 (2019).
Rosa, B. L. T. et al. Investigation of spatially localized defects in synthetic WS2 monolayers. Phys. Rev. B 106, 115301 (2022).
Kumar, R. et al. Substitutional doping of 2D transition metal dichalcogenides for device applications: current status, challenges and prospects. Mater. Sci. Eng. R Rep. 163, 100946 (2025).
Suh, J. et al. Doping against the native propensity of MoS2: degenerate hole doping by cation substitution. Nano Lett. 14, 6976–6982 (2014).
Baithi, M. & Duong, D. L. Doped, Two-dimensional, semiconducting transition metal dichalcogenides in low-concentration regime. Crystals 14, 832 (2024).
Liu, S. et al. Light–matter interactions in high quality manganese-doped two-dimensional molybdenum diselenide. Sci. China Mater. 64, 2507–2518 (2021).
Oh, S., Lim, J. Y., Im, S. & Choi, H. J. Stability, efficiency, and mechanism of n-type doping by hydrogen adatoms in two-dimensional transition metal dichalcogenides. Phys. Rev. B 100, 85416 (2019).
Iqbal, M. W., Elahi, E., Amin, A., Hussain, G. & Aftab, S. Chemical doping of transition metal dichalcogenides (TMDCs) based field effect transistors: a review. Superlattices Microstruct. 137, 106350 (2020).
Sousa, F. B. et al. Effects of vanadium doping on the optical response and electronic structure of WS2 monolayers. Adv. Opt. Mater. 12, 2400235 (2024).
Sahoo, K. R. et al. Enhanced room-temperature spin–valley coupling in V-doped MoS2. Phys. Rev. Mater. 6, 85202–85211 (2022).
Zheng, B. et al. Vapor growth of V-doped MoS2 monolayers with enhanced B-exciton emission and broad spectral response. Front. Optoelectron. 16, 42 (2023).
Upadhyay, B., Sharma, R., Maity, D., Narayan, T. N. & Pal, S. K. Ultrafast carrier dynamics in vanadium-doped MoS2 alloys. Nanoscale 15, 16344–16353 (2023).
Maity, D. et al. Tuning the electronic structure of monolayer MoS2 towards metal like via vanadium doping. Phys. Rev. Mater. 8, 84002 (2024).
Sousa, F. B. et al. Giant valley Zeeman splitting in vanadium-doped WSe2 monolayers. Small 20, 2405434 (2024).
Hader, J., Neuhaus, J., Moloney, J. V. & Koch, S. W. On the importance of electron–electron and electron–phonon scatterings and energy renormalizations during carrier relaxation in monolayer transition-metal dichalcogenides. J. Phys. Condens. Matter 34, 285601 (2022).
Wen, X. et al. Ultrafast probes of electron–hole transitions between two atomic layers. Nat. Commun. 9, 1859 (2018).
Cha, S. et al. 1s-intraexcitonic dynamics in monolayer MoS2 probed by ultrafast mid-infrared spectroscopy. Nat. Commun. 7, 10768 (2016).
Wang, H., Zhang, C. & Rana, F. Surface recombination limited lifetimes of photoexcited carriers in few-layer transition metal dichalcogenide MoS2. Nano Lett. 15, 8204–8210 (2015).
Steinleitner, P. et al. Direct observation of ultrafast exciton formation in a monolayer of WSe2. Nano Lett. 17, 1455–1460 (2017).
Chi, Z. et al. Ultrafast energy dissipation via coupling with internal and external phonons in two-dimensional MoS2. ACS Nano 12, 8961–8969 (2018).
Chen, H. et al. Ultrafast formation of interlayer hot excitons in atomically thin MoS2/WS2 heterostructures. Nat. Commun. 7, 12512 (2016).
Wang, H., Zhang, C. & Rana, F. Ultrafast dynamics of defect-assisted electron–hole recombination in monolayer MoS2. Nano Lett. 15, 339–345 (2015).
Chi, Z., Chen, H., Chen, Z. & Chen, H. Unveiling defect-mediated carrier dynamics in few-layer MoS2 prepared by ion exchange method via ultrafast Vis–NIR–MIR spectroscopy. Chin. J. Chem. Phys. 33, 547–553 (2020).
Rajput, V. K., Maity, D., Deka, B. K., Narayanan, T. N. & Naraharisetty, S. R. G. Anisotropic nonradiative recombination of carriers in a few-layered MoS2 probed by mid-IR ultrafast spectroscopy. J. Phys. Chem. C 127, 13120–13129 (2023).
Mathela, S. et al. Understanding and controlling vanadium doping and sulfur vacancy behavior in two-dimensional semiconductors: toward predictive design. ACS Nano 19, 34192–34203 (2025).
Saigal, N. & Ghosh, S. Evidence for two distinct defect related luminescence features in monolayer MoS2. Appl. Phys. Lett. 109, 122105 (2016).
Sahoo, C., Sethupathy, M., Saad, N. A., Rao, D. N. & Naraharisetty, S. R. G. Ultrafast pump-probe signal detection using a data acquisition card. J. Instrum. 13, P10027 (2018).
Rajput, V. K., Jana, P., Ponnan, S., Taher, M. A. & Naraharisetty, S. R. G. Compact optical scheme for the generation of ultrafast mid-IR laser pulses. Pramana 96, 198 (2022).
Lee, C. et al. Anomalous lattice vibrations of single- and few-layer MoS2. ACS Nano 4, 2695–2700 (2010).
Yang, X. & Li, B. Monolayer MoS2 for nanoscale photonics. Nanophotonics 9, 1557–1577 (2020).
McCreary, K. M., Hanbicki, A. T., Sivaram, S. V. & Jonker, B. T. A- and B-exciton photoluminescence intensity ratio as a measure of sample quality for transition metal dichalcogenide monolayers. APL Mater. 6, 111106 (2018).
Liu, Y., Shen, T., Linghu, S., Zhu, R. & Gu, F. Electrostatic control of photoluminescence from A and B excitons in monolayer molybdenum disulfide. Nanoscale Adv. 4, 2484–2493 (2022).
Wang, L. et al. Slow cooling and efficient extraction of C-exciton hot carriers in MoS2 monolayer. Nat. Commun. 8, 13906 (2017).
Bieniek, M., Sadecka, K., Szulakowska, L. & Hawrylak, P. Theory of excitons in atomically thin semiconductors: tight-binding approach. Nanomaterials 12, 1582 (2022).
Tran, T.-X. et al. Augmented extraction efficiency of a hot D exciton in MoS2 via intervalley scattering. Nano Lett. 24, 11163–11169 (2024).
Wang, W. et al. Investigation of hot carrier cooling dynamics in monolayer MoS2. J. Phys. Chem. Lett. 12, 861–868 (2021).
Zhao, Y. et al. Electrical spectroscopy of defect states and their hybridization in monolayer MoS2. Nat. Commun. 14, 44 (2023).
Komsa, H.-P. & Krasheninnikov, A. V. Native defects in bulk and monolayer MoS2 from first principles. Phys. Rev. B 91, 125304 (2015).
Carozo, V. et al. Optical identification of sulfur vacancies: Bound excitons at the edges of monolayer tungsten disulfide. Sci. Adv. 3, e1602813 (2025).
Soni, A., Kushavah, D., Lu, L.-S., Chang, W.-H. & Pal, S. K. Ultrafast exciton trapping and exciton–exciton annihilation in large-area CVD-grown monolayer WS2. J. Phys. Chem. C 125, 23880–23888 (2021).
Das, S., Wang, Y., Dai, Y., Li, S. & Sun, Z. Ultrafast transient sub-bandgap absorption of monolayer MoS2. Light Sci. Appl. 10, 27 (2021).
Khatua, D., Singh, A., Gurung, S. & Jayabalan, J. Excitation density dependent carrier dynamics in a monolayer MoS2: exciton dissociation, formation and bottlenecking. Micro Nanostruct. 165, 207205 (2022).
Bera, S. K. et al. Atomlike interaction and optically tunable giant band-gap renormalization in large-area atomically thin MoS2. Phys. Rev. B 104, L201404 (2021).
Chernikov, A., Ruppert, C., Hill, H. M., Rigosi, A. F. & Heinz, T. F. Population inversion and giant bandgap renormalization in atomically thin WS2 layers. Nat. Photonics 9, 466–470 (2015).
Li, Y. et al. The Auger process in multilayer WSe2 crystals. Nanoscale 10, 17585–17592 (2018).
Tsai, H.-S. et al. Ultrafast exciton dynamics in scalable monolayer MoS2 synthesized by metal sulfurization. ACS Omega 5, 10725–10730 (2020).
Stavrias, N. et al. Competition between homogeneous and inhomogeneous broadening of orbital transitions in Si:Bi. Phys. Rev. B 96, 155204 (2017).
Gatamov, R., Baydin, A., Krzyzanowska, H. & Tolk, N. Fluence and wavelength dependent ultrafast differential transmission dynamics in graphene. Mater. Res. Express 7, 95601 (2020).
Pogna, E. A. A. et al. Photo-induced bandgap renormalization governs the ultrafast response of single-layer MoS2. ACS Nano 10, 1182–1188 (2016).
Sie, E. J., Frenzel, A. J., Lee, Y.-H., Kong, J. & Gedik, N. Intervalley biexcitons and many-body effects in monolayer MoS2. Phys. Rev. B 92, 125417 (2015).
Wang, Z. et al. Unveiling anomalous ultrafast carrier dynamics of strong spectral overlapping in few-layer MoS2. J. Phys. Chem. Lett. 16, 3287–3294 (2025).
Spindler, C., Galvani, T., Wirtz, L., Rey, G. & Siebentritt, S. Excitation-intensity dependence of shallow and deep-level photoluminescence transitions in semiconductors. J. Appl. Phys. 126, 175703 (2019).
Chi, Z., Wei, Z., Zhang, G., Chen, H. & Weng, Y.-X. Determining band splitting and spin-flip dynamics in monolayer MoS2. J. Phys. Chem. Lett. 14, 9640–9645 (2023).
Plechinger, G. et al. Trion fine structure and coupled spin–valley dynamics in monolayer tungsten disulfide. Nat. Commun. 7, 12715 (2016).
Wang, Z. et al. Intravalley spin–flip relaxation dynamics in single-layer WS2. Nano Lett. 18, 6882–6891 (2018).
Coveney, C. J. N., Haber, J. B., Alvertis, A. M., Neaton, J. B. & Filip, M. R. Rearrangement collision theory of phonon-driven exciton dissociation. Phys. Rev. B 110, 54307 (2024).
Perea-Causín, R., Brem, S. & Malic, E. Phonon-assisted exciton dissociation in transition metal dichalcogenides. Nanoscale 13, 1884–1892 (2021).
Urayama, J., Norris, T. B., Singh, J. & Bhattacharya, P. Observation of phonon bottleneck in quantum dot electronic relaxation. Phys. Rev. Lett. 86, 4930–4933 (2001).
Guillemoles, J.-F. The role of nonequilibrium LO phonons, Pauli exclusion, and intervalley pathways on the relaxation of hot carriers in InGaAs/InGaAsP multi-quantum-wells. Sci. Rep. 13, 5601 (2023).
Polanco, C. A. & Lindsay, L. Ab initio phonon point defect scattering and thermal transport in graphene. Phys. Rev. B 97, 14303 (2018).
Wang, Y., Dai, H., Liu, Z. & Liu, D. Phonon scattering in monolayer molybdenum disulfide under different defect concentrations based on temperature-dependent Raman spectra. J. Phys. Chem. C 127, 1109–1116 (2023).
Nie, Z. et al. Ultrafast carrier thermalization and cooling dynamics in few-layer MoS2. ACS Nano 8, 10931–10940 (2014).
Soni, A. et al. Substrate-induced modulation of transient optical response of large-area monolayer MoS2. Sci. Rep. 15, 7537 (2025).
Acknowledgements
The authors acknowledge the financial support from the UoH Institute of Eminence (IoE) grant RC1-20-011, DST-SERB (EMR/000516), and DST-Purse. Bedanta K. Deka acknowledges the PMRF fellowship for pursuing his Ph.D. Authors from TIFR acknowledge the financial support from TIFR, DAE via the project# RTI4007.
Author information
Authors and Affiliations
Contributions
Shilpa Mangalassery and Bedanta K. Deka carried out the pump-probe experiments. Shilpa Mangalassery carried out the UV–Visible measurements and analysed all the transient data. Dipak Maity, with the supervision of Thangarattu N. Narayanan, prepared the MoS2 samples and performed the PL, STEM, Raman characteristics, and Imaging. Sri Ram G. Naraharisetty designed and supervised the experiments. Sri Ram G. Naraharisetty and Shilpa Mangalassery wrote the manuscript. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mangalassery, S., Maity, D., Deka, B.K. et al. Mid-IR probing unveils vanadium doping-induced unsaturation of defect states in monolayer MoS2. npj 2D Mater Appl 10, 14 (2026). https://doi.org/10.1038/s41699-025-00652-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41699-025-00652-3
