
Abstract
Invasive lung myofibroblasts are the main cause of tissue remodeling in idiopathic pulmonary fibrosis (IPF). A key mechanism contributing to this important feature is aberrant crosstalk between the abnormal/injured lung epithelium and pulmonary fibroblasts. Here, we demonstrate that lungs from patients with IPF and from mice with bleomycin (BLM)-induced pulmonary fibrosis (PF) are characterized by the induction of human epididymis protein 4 (HE4) overexpression in epithelial cells. HE4 knockdown primarily in epithelial cells attenuates BLM-induced PF in mice, whereas the administration of recombinant mouse HE4 exacerbates fibrosis after BLM stimulation. Mechanistic analysis shows that HE4 and annexin II (ANXA2) specific binding enhances the profibrotic phenotype in epithelial cells, and directly promotes lung fibroblast activation, leading to aberrant epithelial-fibroblast crosstalk and the persistent myofibroblast phenotype. The HE4 and ANXA2 binding site is located after the 30th amino acid at the N terminus of the HE4 molecule. Finally, intratracheal administration of HE4 shRNA lentivirus protects mice against BLM-induced PF. These data suggest that HE4 can serve as a potential therapeutic target in the treatment of IPF.
Similar content being viewed by others


Introduction
Idiopathic pulmonary fibrosis (IPF), a progressive fibrotic lung disease characterized by the histological appearance of usual interstitial pneumonia (UIP) and worsening respiratory symptoms, has high mortality and few treatment options1,2. Although the exact pathophysiological mechanisms underlying IPF remain unknown, a complex interplay of diverse cell types is involved. Sustained or repetitive lung epithelial injury is regarded as the key causative event for the disease. The injured epithelium produces profibrotic factors that contribute to subsequent fibroblast activation and myofibroblast differentiation. A persistent myofibroblast phenotype leads to excessive deposition of extracellular matrix (ECM)1,2,3,4. Thus, dysregulated epithelial cells triggering activation of fibroblasts and myofibroblasts via multiple mechanisms are central to IPF progression.
Human epididymis protein 4 (HE4), encoded by the WAP four disulfide core domain protein 2 (WFDC2) gene, was originally identified in the epithelium of the distal epididymis; it was also expressed in the normal glandular epithelium of the female generative tract and breast, in respiratory epithelium, and in epithelial ovarian cancer5,6. Thus far, little is known regarding the biological role of HE4, although previous studies revealed that HE4 is involved in fibrotic diseases, such as kidney fibrosis and cardiac interstitial fibrosis7,8. Additionally, considering that HE4 is a secreted glycoprotein, multiple studies have shown that it can serve as a novel biomarker of severity in multiple types of interstitial lung disease (ILD), including rheumatoid arthritis-associated ILD, systemic sclerosis-related ILD, idiopathic inflammatory myopathy-related ILD, and IPF9,10,11,12,13,14. These observations illustrate the relationship between HE4 expression levels and pulmonary fibrosis (PF). However, the underlying mechanism remains unclear. It is unknown whether HE4 acts alone, or by interacting with a receptor on the cell membrane, to affect the biological behavior of critical cells in IPF.
Here, we explored whether HE4 expression is associated with the clinical features of IPF severity and whether increased numbers of HE4+ epithelial cells in lung tissues are a robust feature of PF. We then interrogated the role of HE4 in various cell types in the context of PF. We found that HE4 mediated phenotypic changes in lung epithelial cells and fibroblasts, two key target cells for PF, leading to abnormal epithelial-fibroblast interaction. Moreover, we found that annexin II (ANXA2), a specific binding partner of HE4, was expressed in lung epithelial cells and fibroblasts; the interaction between HE4 and ANXA2 promoted the aberrant crosstalk described above. Finally, we developed a therapeutic strategy to inhibit HE4 activity in lung epithelium through HE4 shRNA-loaded lentivirus, offering a potential therapeutic approach for patients with IPF.
Results
HE4 is upregulated in PF and mainly colocalizes in epithelial cells
We first explored whether HE4 expression is aberrant in IPF. We analyzed HE4 expression in a previously published large lung dataset GSE47460 of patients with IPF and controls15. Dataset GSE47460 contained two platforms (Gene chips): GPL14550 and GPL6480. We found that HE4 was significantly upregulated in patients with IPF from both GPL14550 and GPL6480 (Fig. 1A and Supplementary Fig. S1). Next, we constructed linear regression models and assessed correlation of HE4 RNA expression with lung function. Intriguingly, we found a strong negative correlation between HE4 RNA expression and lung function parameters (e.g., FVC and DLCO) in patients with IPF (Fig. 1B and Supplementary Table 1). To investigate HE4 at the protein level, we performed enzyme-linked immunosorbent assays (ELISAs) using plasma and bronchoalveolar lavage fluid (BALF), and IHC staining using lung tissues from patients with IPF; we found that HE4 levels were significantly higher in patients with IPF than in Controls (Fig. 1C, D, Supplementary Fig. S2 and Supplementary Tables 2–3). These results suggested that HE4 expression is upregulated in IPF and associated with disease severity.

A HE4 gene expression was reanalyzed in patients with IPF (n = 122) and controls (n = 91) from subset GPL14550 in dataset GSE47460, with differential expression FDR-adjusted p-value. B Linear relationship and association between lung function, such as FVC % (n = 119) and DLCO % (n = 110) predicted, and log of HE4 gene expression in patients with IPF from subset GPL14550 in dataset GSE47460. C Plasma HE4 concentrations in control (n = 47) and patients with IPF (n = 30). D Representative images of HE4 immunostaining in lung sections of control and patient with IPF (n = 6). IgG staining was used as a control. Scale bar = 10 μm. E qRT-PCR analysis of HE4 mRNA expression in lung tissues of mice with BLM stimulation (n = 6). F Western blot analysis of HE4 and fibronectin protein expression in lung homogenates of BLM-challenged mice (n = 6). G-H HE4 concentrations in BALF and plasma from mice with BLM induction (n = 6). I Representative results of HE4 immunostaining in lung sections of mice 21 d after saline or BLM treatment (n = 6). IgG staining was used as a control. Scale bar = 10 μm. J Representative results for multiplex immunofluorescence of HE4 (red), SP-C (green), and vimentin (white) in lung sections of mice. The nuclei were stained blue by DAPI. Red arrows: HE4+SP-C+ cells. Scale bar = 20 μm. ns, not significant. *p < 0.05, **p < 0.01. Data are represented as means ± SEM. The group size ‘n’ refers to biologically independent samples/animals.
To test the above assumption, we estimated HE4 expression in a mouse model of bleomycin (BLM)-induced PF. Notably, BLM caused a time-dependent increase in HE4 mRNA expression in the mouse lung (Fig. 1E). We also observed overexpression of HE4 protein in fibrotic lungs at 21 days after BLM-mediated induction of PF, along with increased expression of fibronectin (Fig. 1F). Furthermore, BLM upregulated HE4 levels in the BALF and plasma from mice with PF; HE4 concentrations were higher in the BLM 21 days group than in the BLM 7 days and saline groups (Fig. 1G, H). These findings indicate that lungs from mice with BLM-induced PF are characterized by increased HE4 expression.
To further characterize the cell types that expressed the HE4 gene in fibrotic lungs, we reanalyzed and annotated 43,165 single cells (19,507 cells from 4 nonfibrotic control and 23,658 cells from 6 fibrotic lungs) from single-cell dataset GSE128033. The t-Distributed Stochastic Neighbor Embedding (tSNE) and projection plot showed that HE4 gene was mainly expressed by the clusters of epithelial cells (82.6%), including goblet cells, basal cells, ciliated cells, AT2 cells, and club cells. In addition, the results showed that HE4 gene expression was increased in AT2 and ciliated cells from IPF lungs compared with control lungs (Supplementary Fig. S3). Additionally, in lungs from patients with IPF and mice with BLM-induced PF, we observed considerably increased immunohistochemical staining of HE4; there were numerous HE4+ cells with epithelial morphology in fibrotic lungs (Supplementary Fig. S4, Fig. 1D, I). Upon investigation of these observations by multiplex immunofluorescence, we found a large increase in the proportion of HE4+SP-C+ cells after BLM stimulation. Moreover, in both saline and BLM groups, the proportion of HE4+SP-C+ cells were higher than the proportion of HE4+vimentin+ cells (Fig. 1J). Altogether, we suspected that HE4+ epithelial cells were a critical component of the fibrosis response.
HE4 is a profibrotic cytokine in the lung
To experimentally determine whether HE4 has a pro- or antifibrotic role in the lung, we examined the response to BLM-induced PF in HE4 knockdown mice. We designed three lentiviral shRNA constructs for HE4 silencing and selected shRNA3 for in vivo experiments because of its silencing efficiency. Additionally, we found that lentiviruses were predominantly located in epithelial cells and BLM-induced increase of HE4 protein expression was dramatically reduced in mice administrated with HE4 shRNA (Supplementary Figs. S5–S6). HE4 knockdown mice were significantly less susceptible to BLM and showed an increase in body weight compared with wild-type mice (Supplementary Fig. S7). Importantly, HE4 knockdown mice exhibited reduced PF. The extent and intensity of lung injury, and collagen accumulation, were significantly reduced in the lung tissues from HE4 knockdown mice (Fig. 2A and Supplementary Fig. S8). Consistent with these findings, the expression levels of fibrotic markers (collagen I, collagen III, α-SMA, and fibronectin) were significantly decreased in HE4 knockdown mice after BLM injury (Fig. 2A, B and Supplementary Fig. S9). Moreover, to rule out off-target effect, we administrated shRNA1 or shRNA2 lentivirus to BLM model mice. The results showed that shRNA1 or shRNA2 lentivirus administration significantly alleviated BLM-induced PF to varying degree. Moreover, the suppression of PF severity was correlated with knockdown efficiency (Supplementary Fig. S10). Next, we analyzed the inflammatory response in HE4 knockdown mice at 7 days after BLM stimulation using shRNA3 (Supplementary Fig. S11A). H&E staining showed no major differences in alveolar wall thickness or inflammatory cell infiltration between BLM + HE4 scr shRNA-treated and BLM + HE4 shRNA-treated mice (Supplementary Fig. S11B). Consistent with these results, HE4 silencing did not affect the expression levels of the inflammation-related genes Il-1β, Il-6, Mcp-1, Ifn-γ, and Cxcl10 (Supplementary Fig. S11C).

A Representative H&E and Masson’s trichrome staining of lung sections, and immunohistochemical analysis of α-SMA in lung sections from HE4 knockdown and WT mice 21 d after BLM treatment. Scale bar = 100 μm. B Western blot analysis of fibronectin, collagen I, and C expression of phosphorylated and total p65, JNK, ERK, and p38 in lung homogenates of mice. D Histological analysis of the severity of lung fibrosis in mice and immunohistochemical analysis of α-SMA and collagen III in lung sections. Representative images of the staining are shown. Scale bar = 100 μm. E Western blot analysis of fibronectin and collagen I in lung homogenates of mice. n = 6 biologically independent animals. ns, not significant. *p < 0.05, **p < 0.01. Data are represented as means ± SEM.
The results of recent studies have suggested that HE4 promotes cystic pulmonary fibrosis and renal fibrosis via MAPK and NF-κB signaling, respectively16,17. Thus, to investigate the impact of HE4 knockdown on downstream signaling pathways in PF, we evaluated the levels of critical signaling molecules in MAPK and NF-κB signaling pathways by western blotting using lung homogenates from mice stimulated with BLM for 21 days. The results showed that the p-ERK/ERK ratio was significantly decreased in the lungs of HE4 knockdown mice after BLM stimulation; However, HE4 knockdown did not cause significant changes in the ratio of p-JNK/JNK, p-p38/p38, or p-NF-κB p65/NF-κB p65 (Fig. 2C). Nevertheless, because of potential secondary changes in cell composition or cytokine expression in vivo, it is unclear whether the effects on the ERK signaling pathway are directly or indirectly driven by HE4. Overall, these results indicate that HE4 silencing attenuates BLM-induced PF and ECM deposition without affecting the inflammatory response to lung injury.
Because HE4 silencing ameliorated pathology in the mouse model of fibrosis, we examined whether HE4 augmentation could exacerbate PF. Accordingly, we treated experimental mice with various concentrations of His-tagged recombinant mouse (rm) HE4 (4 µg/kg, 40 µg/kg, 100 µg/kg) after BLM stimulation. Exogenous rmHE4 was delivered to the lungs via subcutaneous injection (Supplementary Fig. S12); this treatment significantly enhanced HE4 protein expression in mouse lung epithelial cells (Supplementary Figs. S12–S13). rmHE4 administration alone did not directly induce evident PF, compared with controls. However, the coadministration of rmHE4 (40 µg/kg or 100 µg/kg) induced further body weight loss and significantly enhanced BLM-induced collagen deposition and pathology score in mice, compared with BLM stimulation alone (Supplementary Fig. S14 and Fig. 2D). Consistent with these findings, there were significant increases in the expression levels of α-SMA, collagen III, fibronectin, and collagen I in the lungs of mice in either coadministration group (40 µg/kg or 100 µg/kg) (Fig. 2D, E). Taken together, these data provide evidence that HE4 augmentation exacerbates fibrosis in the BLM-induced model of PF.
HE4 induces a profibrotic phenotype in epithelial cells and activates lung fibroblasts
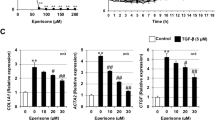
Many studies have shown that epithelial cell-secreted HE4 affects epithelial cell function through autocrine pathways17,18. To more fully elucidate the mechanism by which HE4 promotes PF, we used gain-of-function and loss-of function approaches in Beas-2B cells. We first detected the expression patterns of fibrosis genes in Beas-2B cells treated with various concentrations of recombinant human (rh) HE4 (0, 10 ng/ml, 20 ng/ml, 50 ng/ml) and/or TGFβ1. rhHE4 significantly increased the expression levels of fibrosis genes (Acta2, Col2a1, Fn1, vimentin, Ccn2, and Col1a2) in a concentration-dependent manner (from 10 to 50 ng/ml), as indicated by RT-qPCR (Supplementary Fig. S15). Western blotting showed that Beas-2B cells treated with 50 ng/ml rhHE4 had significantly increased expression levels of fibronectin, collagen I, α-SMA, and p-ERK. However, compared with Beas-2B cells that had been treated with 4 ng/ml TGFβ1, cells treated with 50 ng/ml rhHE4 alone exhibited significantly lower induction of collagen I expression (Fig. 3A). In contrast, the expression levels of fibrosis markers (fibronectin, collagen I, and α-SMA) were decreased in siRNA-HE4-transfected Beas-2B cells (Supplementary Fig. S16) compared with control (siRNA-NC-transfected) cells exposed to exogenous rhHE4 (Fig. 3B). These results indicated that rhHE4 induces the expression of fibrosis-related genes in epithelial cells in vitro by activating the ERK signaling.

A Western blot analysis of fibronectin, collagen I, α-SMA, phosphorylated and total ERK expression in Beas-2B cells with 4 ng/ml TGFβ1 and/or 50 ng/ml rhHE4. B Western blot analysis of fibronectin, collagen I, and α-SMA expression in Beas-2B cells transfected with NC siRNA or HE4 siRNA followed by PBS or 50 ng/ml rhHE4 stimulation. C The proliferation and D migratory ability of MRC-5 cells with different concentration of rhHE4, as detected by the CCK8 and wound healing assay, respectively. E Western blot analysis of fibronectin, collagen I, α-SMA, phosphorylated and total ERK expression in MRC-5 cells with different concentration of rhHE4. n = 3 biologically independent experiments. ns, not significant. *p < 0.05, **p < 0.01. Data are represented as means ± SEM.
In IPF pathogenesis, aberrant crosstalk between the abnormal pulmonary epithelium and lung fibroblasts is central to disease progression3,4,19. To determine whether secretory HE4 (sHE4) from epithelial cells affects fibroblast function, firstly, we overexpressed the HE4 gene in Beas-2B cells using an HE4 overexpression lentivirus. We found that introducing the HE4 expression vector (OE-HE4) led to increased HE4 protein secretion in the supernatant of Beas-2B cells (Supplementary Fig. S17). Our previous results showed that BALF HE4 levels were increased in patients with IPF and mice with BLM induction (Supplementary Fig. S2 and Fig. 1G). Altogether, these data indicated that epithelial cells derived HE4 is secreted to the extracellular medium. Next, we found that conditioned media from HE4-OE lentivirus-infected Beas-2B cells significantly increased the expression levels of fibrotic genes in MRC-5 cells (Supplementary Fig. S18). Then, we treated MRC-5 cells with rhHE4 in vitro to further explore the role of sHE4 in lung fibroblast activation. CCK8 assays revealed that various concentration of rhHE4 (10 ng/ml, 20 ng/ml) had pro-proliferative effects on MRC-5 cells at multiple time points (Fig. 3C). Wound healing assays showed that 10 ng/ml or 20 ng/ml rhHE4 increased the migratory capacity of lung fibroblasts (Fig. 3D). Intriguingly, compared with control cells, rhHE4-treated MRC-5 cells displayed substantially increased RNA expression levels of Acta2, Fn1, Col1a2, and vimentin in a concentration-dependent manner, as indicated by RT-qPCR. However, the effect of rhHE4 on fibroblast activation was weaker than the effect of 4 ng/ml TGFβ1 (Supplementary Fig. S19). Additionally, western blotting analysis showed that treatment with rhHE4 at 20 ng/ml significantly enhanced the expression levels of fibrotic markers, such as fibronectin, collagen I, and α-SMA, and activated ERK signaling (Fig. 3E). Therefore, we concluded that epithelial cell-secreted HE4 strongly promotes lung fibroblast activation in vitro by enhancing the level of phosphorylated ERK.
HE4 binding to annexin A2
To identify specific HE4-binding proteins, coimmunoprecipitation assays involving Beas-2B cells were performed. Proteins bound to HE4 were separated by electrophoresis and examined by Coomassie brilliant blue staining. The results showed that annexin A2 (ANXA2) had the highest Mascot score (Fig. 4A and Supplementary Fig. S20). Next, binding between HE4 and ANXA2 was verified through a series of in vitro and in vivo experiments. Whole cell lysates from Beas-2B cells were precipitated with anti-HE4 or anti-ANXA2 antibodies; the expressions patterns of ANXA2 and HE4 in Beas-2B cells were investigated. We found that the contents of cell lysates precipitated with either anti-HE4 or anti-ANXA2 antibodies could be detected by western blotting with anti-ANXA2 or anti-HE4 antibodies, respectively, suggesting that HE4 and ANXA2 formed a complex (Fig. 4B). To further explore the distribution of HE4 and ANXA2 in Beas-2B cells, membrane and cytoplasmic proteins in Beas-2B cells were isolated. The results showed the presence of HE4 and ANXA2 in the membrane and plasma and confirmed that they are binding partners (Fig. 4C). Similarly, we also isolated the membrane and cytoplasmic proteins in MRC-5 cells and found that the presence of HE4-ANXA2 complex in the membrane and cytoplasm (Fig. 4D). Additionally, coimmunostaining assays of HE4 and ANXA2 were performed using lung tissues from mice. We found that HE4 was colocalized with ANXA2 in epithelial cells (Fig. 4E). These results suggested that ANXA2 is the main protein that interacts with HE4 in epithelial cells.

A Coimmunoprecipitation assays in Beas-2B cells using an anti-HE4 antibody and Coomassie brilliant blue-stained. Lane 1, marker band. Lane 2, sample band. Lane 3, IgG band. Lane 4, marker band. The protein band E was the one with the highest Mascot score. The molecular weight of this protein is 36 kDa. B Immunoprecipitation (IP) of ANXA2/HE4 complex by anti-HE4 or anti-ANXA2 antibody and western blot analysis with anti-ANXA2 or anti-HE4 antibody in whole cell proteins from Beas-2B cells. Lanes 1 and 2, positive control; lane 3, negative control (Ig G); lane 4, IP by anti-HE4 or anti-ANXA2 antibody. C IP of membrane and cytoplasmic proteins by anti-HE4 antibody and western blot analysis with anti-ANXA2 antibody in Beas-2B cells. Lane 1, positive control; lane 2, negative control (IgG); lane 3, IP of cytoplasmic proteins by anti-HE4; lane 4, IP of membrane proteins by anti-HE4. D IP of membrane and cytoplasmic proteins by anti-HE4 antibody and western blot analysis with anti-ANXA2 antibody in MRC-5 cells. Lane 1, positive control; lane 2, negative control (IgG); lane 3, IP of cytoplasmic proteins by anti-HE4; lane 4, IP of membrane proteins by anti-HE4. E Representative results for coimmunostaining of HE4 (red) and ANXA2 (green) in the lung sections from mice. Scale bar = 50 μm. n = 6 biologically independent animals. F Schematic strategy for HE4 truncation 1 (del15) and HE4 truncation 2 (del30) construction. G The site of binding between HE4 and ANXA2, as detected by the GST pull-down assays.
To characterize the site of binding between HE4 and ANXA2, we constructed fusion proteins containing GST and His tags. We first examined whether the N-terminal region of HE4 is involved in the binding of HE4 to ANXA2, using two truncated forms of HE4. HE4 truncation 1 (del15) lacked the first 15 amino acids at the N terminus, and HE4 truncation 2 (del30) lacked an additional 15 amino acids at the N terminus (Fig. 4F). GST pull-down assays showed that mutants HE4-del15 and HE4-del30 clearly bound to ANXA2 (Fig. 4G), suggesting that the HE4 and ANXA2 binding site is located after the 30th amino acid at the N terminus of the HE4 molecule.
HE4-ANXA2 binding enhances the profibrotic phenotype in epithelial cells and promotes lung fibroblast activation
Next, we evaluated the regulatory effect of the HE4 and ANXA2 interaction on the profibrotic phenotype in epithelial cells. We transfected Beas-2B cells with siRNA-ANXA2, then confirmed the downregulations of ANXA2 RNA and protein expression levels in cells (Supplementary Fig. S21). RT-qPCR and western blotting showed that interference with ANXA2 attenuated the exogenous rhHE4-induced expression of fibrosis-related genes in Beas-2B cells (Supplementary Fig. S22 and Fig. 5A). Intriguingly, previous studies indicated that ANXA2 was also expressed on human lung fibroblasts20. Therefore, we investigated whether the binding of HE4 to ANXA2 regulated lung fibroblast activation. The results showed that supplementation with extrinsically activated rhHE4 protein induced increases in fibrotic markers, which could be suppressed by ANXA2 silencing (Supplementary Fig. S23 and Fig. 5B).

A Western blot analysis of fibronectin, collagen I and α-SMA expression in Beas-2B cells transfected with NC siRNA or ANXA2 siRNA followed by PBS or 50 ng/ml rhHE4 stimulation. n = 3 biologically independent experiments. B Western blot analysis of fibronectin, collagen I, and α-SMA expression in MRC-5 cells transfected with NC siRNA or ANXA2 siRNA followed by PBS or 20 ng/ml rhHE4 stimulation. n = 3 biologically independent experiments. C qRT-PCR and western blot analysis of ANXA2 expression in Beas-2B or MRC-5 cells with different concentration of rhHE4. n = 3 biologically independent experiments. D Western blot analysis of ANXA2 expression in lung homogenates of mice. n = 6 biologically independent animals. ns, not significant. *p < 0.05, **p < 0.01. Data are represented as means ± SEM.
Next, we performed in vitro and in vivo studies to determine whether HE4 regulated ANXA2 expression. We first examined the expression patterns of ANXA2 in Beas-2B and MRC-5 cells treated with various concentrations of rhHE4; we found that the upregulation of HE4 was not accompanied by significant changes in ANXA2 expression in either Beas-2B or MRC-5 cells (Fig. 5C). The results of animal experiments revealed that BLM stimulation led to increased ANXA2 expression in mouse lung tissue. However, ANXA2 expression was not significantly altered by HE4 knockdown (Fig. 5D). These data confirmed that increases or decreases in HE4 expression do not influence the expression of ANXA2.
Intratracheal administration of HE4 shRNA lentivirus protects mice against BLM-induced PF
Thus far, our data support a profibrotic role for HE4 in PF. Accordingly, we sought to translate the above discoveries into an anti-PF therapy. To determine the therapeutic effects of HE4 shRNA lentivirus administration in an in vivo model of PF, anesthetized C57BL/6 mice were intratracheally administered HE4 shRNA lentivirus at 7 days after BLM stimulation (Fig. 6A). The BLM-induced increase of HE4 protein expression was dramatically reduced in mouse lung tissue after administration of HE4 shRNA lentivirus (Supplementary Fig. S24). This treatment significantly alleviated BLM-induced PF, as determined by histopathological analysis and fibrosis scores (Fig. 6B). Consistent with these findings, as shown in Fig. 6B-C, lung tissues from mice administered HE4 shRNA lentivirus showed significant reductions in the expression levels of fibrotic markers (collagen I, collagen III, and fibronectin). Overall, these data suggest that targeting HE4 with HE4 shRNA-loaded lentivirus had therapeutic effects in a mouse model of established PF.

A Schematic for experimental design and time line of BLM-treated mice administered with either scrambled or HE4 shRNA-loaded lentivirus. B Representative H&E and Masson’s trichrome staining, and immunohistochemical analysis of collagen I and collagen III in lung sections of mice. Scale bar = 100 μm. C Western blot analysis of fibronectin and collagen I in lung homogenates of mice. n = 6 biologically independent animals. **p < 0.01. Data are represented as means ± SEM.
Discussion
IPF is a refractory disease, partially characterized by recurrent epithelial cell injury and the pathological accumulation of myofibroblasts that secrete ECM. More effective therapeutic options are needed; obstacle to new treatments is the limited understanding of molecular mechanisms underlying IPF pathogenesis. Our findings in the present study provide different insights concerning the regulation of pulmonary fibrogenesis and demonstrate a key role for lung epithelial cell-secreted HE4 in promoting PF. We provide evidence that HE4, a secreted glycoprotein, is associated with IPF severity while regulating phenotypic changes in lung epithelial cells and fibroblasts. Therefore, the targeted inactivation of HE4 could have potent anti-fibrotic effects in clinical settings.
We obtained the following evidence to concerning the profibrotic role of lung epithelial cell-derived HE4 in pulmonary fibrogenesis. Effective knockdown of HE4, primarily in epithelial cells, significantly attenuated BLM-induced PF without affecting the inflammatory response in mice. In the gain-of-function model, systemic administration of rmHE4 exacerbated BLM-induced PF. HE4 belongs to the WFDC family of proteins, which is a relatively unexplored family of low-molecular-weight proteins. Thus far, most published studies of HE4 indicates that this small secretory protein constitutes a crucial fibrosis-associated molecule across various organs. In kidney fibrosis, HE4 was shown to have a pro-fibrogenic effect7,17. HE4 silencing inhibited ECM deposition and alleviated fibrosis in mice with unilateral ureteral obstruction17. In dilated cardiomyopathy, HE4 induced cardiac interstitial fibrosis by activating cardiac fibroblasts8. Additionally, published experimental evidence suggested the role of HE4 in fibrotic related respiratory diseases. HE4 reportedly destroys normal ECM and promote fibrosis in the setting of cystic PF16; it participates in chronic obstructive pulmonary disease (COPD)-related airway remodeling21. Intriguingly, reanalysis of HE4 gene expression in dataset GSE47460 revealed a significant increase among patients with IPF relative to patients with COPD, suggesting that HE4 levels are closely related to the extent of tissue remodeling and fibrosis. However, no significant difference in HE4 gene expression was detected between control participants and patients with COPD in dataset GSE47460 (Supplementary Fig. S25), which contrasts with a previous study21. This discrepancy may be due to the fact that these studies did not classify COPD patients, particularly those with fibrotic interstitial lung abnormality (ILA). ILA is a common radiologic finding in COPD, with patients with ILA having a higher risk of PF, which can manifest as subpleural interstitial fibrosis or fibroblast foci22,23. Therefore, future studies should assess the clinical relevance of HE4 levels in COPD patients with fibrotic ILA. Additionally, a recent report provided the first description of WFDC2 deficiency as a cause of chronic destructive airway disease24, in contrast to ILDs9,10,11,12,13,14 which show elevated WFDC2 expression patterns. Dougherty et al. also highlighted the etiology of chronic destructive airway disease caused by the deficiency of secreted WFDC2, indicating that protein replacement therapy may be a potential treatment option24. This discrepancy may be explained that bronchiectasis in WFDC2 deficiency more closely mimics cystic fibrosis (all lobes) than IPF (lower lobes). Additionally, the pathological mechanisms underlying bronchiectasis and PF are different. The final common pathway in the pathological mechanisms underlying bronchiectasis results from a multifactorial combination of chronic or recurrent infection and an excessive inflammatory response, followed by abnormal lung tissues remodeling25. However, IPF is generally not regarded as an inflammatory disease. Most importantly, Dougherty et al. demonstrated the phenotypic variation between mouse WFDC2 deficiency and human WFDC2 deficiency24. Therefore, future studies should use humanized mouse model to further explore the function of WFDC2 in PF.
The most notable finding in this report is that secretory HE4 contributes to PF pathogenesis by modulating phenotypic changes in lung epithelial cells and fibroblasts, two key target cells for PF. Recurrent and/or non-resolving injury to the lung epithelium is involved in initiating fibrosis. Although the initial cause of this injury in IPF remains unclear, it manifests as increased epithelial cell death and phenotypic changes in surviving epithelial cells26. Dysregulated lung epithelial cells with a profibrotic phenotype subsequently induce fibroblast activation and myofibroblast differentiation through aberrant epithelial-fibroblast crosstalk26,27. The present study demonstrated that HE4 was mainly expressed in lung epithelial cells from patients with IPF and BLM-stimulated mice, and epithelial cells derived HE4 was secreted to the extracellular medium. Moreover, secretory HE4 induced a profibrotic phenotype in epithelial cells; it also activated lung fibroblasts, suggesting that HE4 promotes aberrant epithelial-fibroblast crosstalk. Previously published literature concerning HE4 has suggested that it affects the phenotypes and functions of epithelial cells in various tissues. For example, renal tubular epithelial cell-secreted HE4 increased the expression of the ECM genes in renal tubular epithelial cells17; ovarian cancer epithelial cell-derived HE4 promoted malignant biological behavior in ovarian cancer epithelial cells18. Additionally, Zhan et al. reported that human bronchial epithelial cell-derived HE4 promoted fibroblast activation in COPD21. These previous studies revealed that secretory HE4 may function through autocrine and paracrine pathways, consistent with our findings. In the gain-of-function animal model, the lungs of mice treated with subcutaneous His-tagged rmHE4 showed increased His protein staining in the epithelium, fibroblastic foci, and fibrotic interstitium, indirectly validating these findings.
Next, we explored possible molecular mechanisms underlying the profibrotic effects of secretory HE4. We found that ANXA2 interacts with HE4; this protein binding interaction induces a profibrotic phenotype in epithelial cells and activates fibroblasts through aberrant epithelial-fibroblast crosstalk. Moreover, the presence of HE4-ANXA2 complex in the membranes of Beas-2B and MRC-5 cells indicated that HE4 secreted into the extracellular region could bind to the cell membranes of the epithelial cells themselves, as well as to those of adjacent fibroblasts. ANXA2 possesses both intracellular and extracellular roles. The interaction between ANXA2 and S100A4 can promote activation of matrix metalloproteinases, leading to ECM remodeling and neovascularization28,29,30. The results of previous studies suggested that ANXA2 is a specific target of BLM31 and plays a profibrotic role in PF20,31,32. Moreover, in lung tissues, ANXA2 was located in epithelial cells and fibroblasts20, indicating that secretory HE4 could bind to ANXA2 and mediate dual regulation of phenotypic changes in lung epithelial cells and fibroblasts. ANXA2 includes two forms: monomer and heterotetramer. The monomer is located to the cytoplasm, while the heterotetramer exits in the membrane33. In certain cases, the ANXA2 monomer can migrate to the cell membrane to support different physiological functions, such as exocytosis, endocytosis, and membrane transport34,35. Previous studies have reported that HE4 and ANXA2, which co-localize in the cell membrane and cytoplasm, can interact in various malignant tumor cell types36,37. Secreted HE4 bounded to the membrane protein ANXA2 of the ovarian cancer cells themselves. This would allow ANXA2 to potentially facilitate HE4 translocation into the nucleus, where it can function as a transcription factor to promote the expression of downstream signaling molecules37. Moreover, Wang et al. reported that HE4-ANXA2-MMP2 form a protein complex and promote the malignant of various cells migration36. During this process, ANXA2 serves as “a bridge”. Our results showed the presence of HE4-ANXA2 complex on the cytoplasm and membrane of Beas-2B and MRC-5 cells. Preliminary data in present study indicated that HE4 is primarily secreted by epithelial cells, rarely by fibroblasts. Thus, the presence of HE4 and ANXA2 in the cytoplasm of MRC-5 cells suggested that ANXA2 may help HE4 translocate into the cells. Additionally, we found that increases or decreases in HE4 did not affect the expression of ANXA2. Therefore, we speculate that epithelial cell-derived HE4 is secreted into the extracellular medium, where it binds to ANXA2 on epithelial cell and fibroblasts membranes. ANXA2 may then help HE4 enter the cells, where it can activate downstream signaling molecules and induce phenotypic/behavioral changes.
Finally, we note that HE4 knockdown inhibited the activation of ERK but not JNK, p38, or NF-κB p65 after BLM stimulation. Furthermore, exogenous HE4 protein supplementation activated ERK signaling in vitro. HE4 reportedly promotes the proliferation of ovarian cancer cells by regulating ERK1/2 phosphorylation38. Additionally, recent studies have indicated that ANXA2 activates ERK signaling and promotes malignant biological behavior in tumor cells39,40. Considering these previous studies together with our findings, we speculate that secretory HE4 binds to ANXA2 on the cell membrane, then ANXA2 may help HE4 migrate into the cells, likely activating downstream ERK signaling, and induce phenotypic changes in epithelial cells and fibroblasts, ultimately leading to PF progression. Additionally, HE4 has been reported to play a role in alveolar cell apoptosis41 and promote angiogenesis by activating STAT3 signaling in epithelial ovarian cancer42. Moreover, a previous study showed that OE-HE4 or recombinant HE4 led to the upregulation of several transcripts encoding for ECM proteins in ovarian cancer cells, including LAMB3, SERPINB2, LAMC2, and GREM143. Therefore, other mechanisms linking HE4 to PF cannot be ruled out and should be explored in future studies.
There were some limitations in this study. First, we used HE4 shRNA lentivirus for HE4 knockdown in the lungs of the mice with BLM-induced PF. Future studies using knockout mice or conditional knockout mice in epithelial cells are needed to further clarify the effects of HE4 in the lung. Second, to determine the therapeutic impacts of anti-HE4 agents, blockage of HE4 with a neutralizing antibody is needed. Finally, the exact mechanisms by which HE4 avoids altering the expression of ANXA2 require further analysis, although we confirmed that HE4 binds ANXA2 to promote PF.
In conclusion, we have demonstrated that lung epithelial cell-secreted HE4 plays a profibrotic role in BLM-induced PF and that HE4 knockdown primarily in epithelial cells can attenuate PF in response to noninfectious lung injury. We speculate that the underlying mechanisms of these processes involve inhibiting the profibrotic phenotype in epithelial cells and aberrant epithelial-fibroblast crosstalk, leading to limited expansion of myofibroblasts. HE4 exerts its function dependent on the cell type, and ANXA2 is an HE4-binding protein. HE4-ANXA2 binding regulates phenotypic changes in lung epithelial cells and fibroblasts via ERK signaling. This study provides molecular insights into the understanding of the mechanisms underlying IPF; in conjunction with our ongoing research, the present findings may lead to the establishment of anti-PF strategies targeting HE4.
Methods
Participants
Microarray datasets of lung samples from healthy controls and patients with IPF (GSE47460) were reanalyzed to determine HE4 RNA expression. Additionally, HE4 protein expression levels were detected in lung tissues, plasma, and BALF from patients with IPF by immunohistochemistry and ELISA, respectively. Human lung tissues, blood, and BALF samples were collected from Chinese participants at Zhongnan Hospital of Wuhan University. IPF was diagnosed in accordance with the ATS/ERS consensus criteria44. Human lung tissues, blood, and BALF samples were obtained under the auspices of the Medical Ethics Committee of Zhongnan Hospital of Wuhan University-approved protocol (Scientific Ethical Approval No. [2021027]). All of the participants gave informed consent prior to the inclusion of people in the study. All ethical regulations relevant to human research participants were followed.
Mice and BLM administration
All experimental procedures were approved by the Animal Care and Use Committee at Zhongnan Hospital of Wuhan University (ZN2021163). We have complied with all relevant ethical regulations for animal use. For lentivirus administration in the BLM model, 8-week-old male C57BL/6 J mice under pentobarbital anesthesia were intratracheally administrated with scrambled shRNA or HE4 shRNA-loaded lentivirus (GeneChem, Shanghai, P.R. China) at a dose of 2 × 107 transduction units per mouse. At this point, the identical volume of sterile saline was administered into the control or BLM counterparts. Three days later, the anesthetized mice received an intratracheal injection of 2.5 mg/kg BLM or an equivalent volume of sterile saline. To investigate the role of rmHE4 during the fibrogenesis, rmHE4 (Sino biological, Beijing, China) was subcutaneously injected (4 µg/kg, 40 µg/kg, 100 µg/kg mouse/each time) on day 1 to 20, after 1.5 mg/kg BLM induction. For therapy in the BLM model, lentivirus was injected into the anesthetized animals via intratracheal on the days 7 after BLM injection. The mice were euthanized on day 21 following BLM challenge to analyze PF. The target sequences of scrambled shRNA and HE4 shRNA are shown in Supplementary Table 4.
BALF collection
After mice had been euthanized, BALF was collected by inserting a tracheal cannula and flushing the airway with PBS; the fluid was maintained for 20 s, then gently aspirated. This process was repeated two more times. The collected lavage fluid was centrifuged at 1500 rpm for 7 min at 4 °C, then stored at −80 °C for further analysis.
Cell culture and treatments
Beas-2B human bronchial epithelial cells and MRC-5 cells were both purchased from iCell Bioscience (Shanghai, China). Transfection was carried out using liposomes with a vector transfection kit according to the instructions. For example, in a six-well plate, the siRNA-lipofectamine 2000 complex was prepared as follows: lipofectamine 2000 (Thermo Fisher, USA, Cat. No.: 11668019) was thoroughly mixed and diluted in serum-free Opti-MEM medium (Thermo Fisher, Cat. No.: 31985070), then incubated at room temperature for 5 min. Separately, 100 pmol of siRNA (RiboBio, Guangzhou, China) was diluted in 250 µl of serum-free Opti-MEM. After a 5 min incubation, the diluted siRNA was added to the diluted lipofectamine 2000, mixed well, and incubated at room temperature for 15 min to form the transfection complex. The complex was then added to each well containing serum-free DMEM medium with high levels of glucose. Transfected cells were cultured in a humidified atmosphere with 5% CO2 at 37 °C. After 6 h, the medium was replaced with fresh complete medium.
In Beas-2B cells, the treatments were as follows: (1) Beas-2B cells were treated with 4 ng/ml TGF-β1 and/or different concentrations of rhHE4 (Sino Biological, Beijing, China) (0, 10, 20, 50 ng/ml) for 48 h. (2) Beas-2B cells were infected with HE4 siRNA or siNC, then stimulated with PBS or 50 ng/ml rhHE4 for 48 h. (3) Beas-2B cells were infected with ANXA2 siRNA or siNC, then treated with PBS or 50 ng/ml rhHE4 for 48 h.
In MRC-5 cells, the treatments were as follows:(1) MRC-5 cells were treated with different concentrations of rhHE4 (0, 5, 10, 20 ng/ml) for 48 h. (2) MRC-5 cells were infected with ANXA2 siRNA or siNC, then stimulated with PBS or 20 ng/ml rhHE4 for 48 h.
Histology, immunohistochemistry and Immunofluorescence staining
Formalin-fixed and paraffin-embedded lung sections were stained with hematoxylin and eosin or Masson’s trichrome to evaluate the degree of PF45. Lung sections were deparaffinized, rehydrated, recovered, and then incubated with primary antibodies at 4 °C overnight. The following anti-HE4 antibodies were used: anti-HE4 antibody (Thermo Fisher, US, Cat. No.: PA5-72770) reacted with human lung tissues in immunohistochemistry. Anti-HE4 antibody (Thermo Fisher, US, Cat. No.: PA5-80227) and anti-HE4 antibody (Biorbyt, UK, Cat. No.: orb101171) reacted with mouse lung tissues in immunohistochemistry and immunofluorescence staining, respectively. Anti-α-SMA, anti-collagen-I, and anti-collagen-III antibodies (all from Abcam, Cambridge, UK) reacted with mouse lung tissues in immunohistochemistry. Anti-SP-C antibody (Abcam, Cambridge, UK) and anti-vimentin antibody (Abcam, Cambridge, UK) reacted with mouse lung tissues in immunofluorescence staining. Next, lung sections were incubated with HRP-polymer secondary antibodies for 1 h and then developed using DAB solution. In Immunofluorescence staining, HRP-conjugated or Alexa Fluor 488- or Alexa Fluor 594-conjugated antibodies (ZSGB-BIO, China) were used as secondary antibodies. The nucleus was labeled with DAPI (ZSGB-BIO, China).
Single-cell analysis
Downstream analysis of unique molecular identifier (UMI) count matrices was applied using Seurat v. 4.3.0. The cell-UMI matrix was filtered using the criteria of cells with more than 200 detected gene features and a mitochondrial percentage of less than 15%. The scDblFinder package was used for the identification and filter of empty droplets and doublets. For normalization, variance stabilization, data scaling, and highly variable gene selection, Seurat’s NormalizeData, ScaleData, and FindVariableFeatures functions were used. Mitochondrial gene content percentage was regressed out when scaling data. Harmony package v. 0.1.1 was employed for batch effect correction. Plots and assigned clusters were generated using Seurat’s standard pipeline. Cell populations were identified by examining gene markers in the associated transcriptomes. Publicly available data integration required batch effect correction due to different single-cell chemistries, and we aligned all samples using Harmony, which interactively clusters and corrects low-dimensional space.
qRT-PCR
Total RNAs isolation, reverse transcription, and qRT-PCR analysis were sequential performed in lung tissues and cells. Among them, total RNAs was extracted from whole mouse lungs. Gene relative expression was normalized to GAPDH or β-actin mRNA expression by using the 2−ΔΔCT method. The sequences of the specific primer sets are described in Supplementary Table 5.
Immunoprecipitation and protein identification
Beas-2B cells were immunoprecipitated using an anti-HE4 antibody (Abcam, Cambridge, UK, Cat. No.: ab200828) and resuspended with protein A/G magnetic beads, according to the manufacturer’s instructions (Beaver, Suzhou, China). Eluents were separated by SDS-PAGE and stained with Coomassie brilliant blue. The anti-HE4 antibody was replaced with rabbit IgG (Beyotime, Jiangsu, China) as a negative control. Peptide and proteins were identified from the tandem mass spectrometry spectra using the MASCOT algorithm (Matrix Science, Boston, MA, USA).
Cytoplasmic and membrane proteins extraction
Cytoplasmic and membrane proteins were extracted using the Membrane and Cytosol Protein Extraction Kit (Beyotime), in accordance with the manufacturer’s instructions. Briefly, membrane protein extraction reagent A was added to the cells and incubated at 4 °C for 15 min. The mixture was then centrifugated at 700 g for 10 min at 4 °C. The supernatant was collected and centrifuged again at 14,000 × g for 30 min at 4 °C to pellet the membrane debris. The supernatant was stored as the cytoplasmic proteins. Membrane protein extraction reagent B was then added, vigorously vortexed for 5 s, and incubated at 4 °C for 10 min. This step was repeated two more times. The mixture was centrifugated, and the supernatant was stored as membrane protein fraction.
Coimmunoprecipitation and western blotting analysis
Precooled radioimmunoprecipitation assay buffer was added to Beas-2B cells, and incubated at 4 °C for 30 min. After the lysate had been centrifuged at 15,000 × g for 30 min at 4 °C, supernatant fractions were collected and kept as whole cell proteins. Whole cell proteins, cytoplasmic and membrane proteins were incubated with anti-HE4 antibody (Abcam, Cambridge, UK, Cat. No.: ab200828) or anti-ANXA2 antibody (Abcam, Cambridge, UK) for 3 h at 4 °C. Protein A/G magnetic beads were added, and the mixtures were incubated on a rocker platform overnight at 4 °C. The negative control contained only anti-HE4 or anti-ANXA2 antibodies without cell proteins.
Immunoprecipitates and proteins extracted from cells and tissues were subsequently subjected to SDS-PAGE and analyzed via western blotting using antibodies. For western blotting analysis, primary antibodies to the following proteins were used: GAPDH, fibronectin, collagen-I, collagen-III, p-JNK, JNK, p-p65 and p65 (all from Abcam, Cambridge, UK); ERK, p-ERK, p-p38 and p38 (all from CST, Danvers, USA); and HE4 (Thermo Fisher, US, Cat. No.: PA5-80227).
Construction of truncated forms of HE4
RT-PCR products derived from human complementary DNA and corresponding to full-length human HE4 and ANXA2 were cloned into the pGEX-4 T vectors. N-terminal truncated forms of HE4 with its first 15 or 30 amino acids deleted, del15 and del30, respectively, were used for glutathione S-transferase (GST) pull-down assays.
ELISA
HE4 protein concentrations in plasma and BALF were detected using the Mouse and the Human HE4 ELISA Kit (both from ELK Biotechnology, Wuhan, China) according to the manufacturers’ instructions.
Statistics and reproducibility
All data represent at least three independent experiments and are expressed as mean ± standard error of the mean (SEM). The group size “n” refers to biologically independent samples/animals/independent experiments, rather than technical replicates. Statistics including error bars were derived from a minimum of three biologically independent replicates. Normal distribution of the data was assessed using the Shapiro-Wilk test; p-values > 0.05 indicated a normally distributed. Group differences were evaluated using Student’s t-test for comparisons between two groups and one-way analysis of variance followed by Tukey’s post-hoc test for comparisons among ≥ 3 groups. Correlation analysis was performed by estimating the Pearson correlation coefficient. A p-value of < 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 9.0 software (San Diego, CA, USA).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that all data supporting the findings of this study are available in the main article, figures, tables, and supplementary materials, or are available from the authors upon request. The numerical source data behind the graphs and the corresponding statistical analysis are added as supplementary data. The original and uncropped gel images of the western blots are added as supplementary information. The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the iProX partner repository with the dataset identifier PXD058959.
References
Podolanczuk, A. J. et al. Idiopathic pulmonary fibrosis: state of the art for 2023. Eur. Respir. J. 61, 2200957 (2023).
Martinez, F. J. et al. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Prim. 3, 17074 (2017).
Moss, B. J., Ryter, S. W. & Rosas, I. O. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 17, 515–546 (2022).
Camelo, A., Dunmore, R., Sleeman, M. A. & Clarke, D. L. The epithelium in idiopathic pulmonary fibrosis: breaking the barrier. Front. Pharm. 4, 173 (2014).
Bouchard, D. et al. Proteins with whey-acidic-protein motifs and cancer. Lancet Oncol. 7, 167–174 (2006).
Galgano, M. T., Hampton, G. M. & Frierson, H. F. Jr. Comprehensive analysis of HE4 expression in normal and malignant human tissues. Mod. Pathol. 19, 847–853 (2006).
LeBleu, V. S. et al. Identification of human epididymis protein-4 as a fibroblast-derived mediator of fibrosis. Nat. Med. 19, 227–231 (2013).
Yamamoto, M. et al. HE4 predicts progressive fibrosis and cardiovascular events in patients with dilated cardiomyopathy. J. Am. Heart Assoc. 10. https://doi.org/10.1161/JAHA.120.021069. (2021)
Tian, M. et al. Elevated serum human epididymis protein 4 is associated with disease severity and worse survival in idiopathic pulmonary fibrosis: a cohort study. Ann. Transl. Med. 10, 992 (2022).
Raghu, G. et al. Idiopathic pulmonary fibrosis: prospective, case-controlled study of natural history and circulating biomarkers. Chest 154, 1359–1370 (2018).
Zhang, M. et al. Increased levels of HE4 (WFDC2) in systemic sclerosis: a novel biomarker reflecting interstitial lung disease severity? Ther. Adv. Chronic Dis. 11, 2040622320956420. https://doi.org/10.1177/2040622320956420. (2020)
Lin, T. et al. Human epididymis protein 4 as a new diagnostic biomarker for rheumatoid arthritis-associated interstitial lung disease. Clin. Exp. Rheumatol. 40, 2167–2174 (2022).
Liang, L. et al. Serum human epididymis protein 4 as a novel biomarker in identifying patients with interstitial lung disease in rheumatoid arthritis. Front. Med. 8, 755268 (2021).
Sun, F. et al. Human epididymis protein 4 as a clinical biomarker in identifying interstitial lung disease in patients with idiopathic inflammatory myopathies. Int. Immunopharmacol. 115, 109609 (2023).
Bauer, Y. et al. A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 52, 217–231 (2015).
Wang, J. et al. Human epididymis protein 4 (HE4) protects against cystic pulmonary fibrosis associated-inflammation through inhibition of NF-κB and MAPK singnaling. Genes Genomics 41, 1045–1053 (2019).
Zhang, L. et al. Hypoxia-induced HE4 in tubular epithelial cells promotes extracellular matrix accumulation and renal fibrosis via NF-kappa B. FASEB J. 34, 2554–2567 (2020).
Wang, S. et al. ZNF703 promotes tumor progression in ovarian cancer by interacting with HE4 and epigenetically regulating PEA15. J. Exp. Clin. Cancer Res. 39, 264 (2020).
Mei, Q., Liu, Z., Zuo, H., Yang, Z. & Qu, J. Idiopathic pulmonary fibrosis: an update on pathogenesis. Front. Pharm. 12, 797292 (2022).
Schuliga, M. et al. Annexin A2 contributes to lung injury and fibrosis by augmenting factor Xa fibrogenic activity. Am. J. Physiol. Lung Cell Mol. Physiol. 312, L772–L782 (2017).
Zhan, Y. et al. Human epididymis protein 4 aggravates airway inflammation and remodeling in chronic obstructive pulmonary disease. Respir. Res. 23, 120 (2022).
Beasley, M. B. Interstitial lung abnormalities. Surg. Pathol. Clin. 17, 215–225 (2024).
Miller, E. R. et al. Histopathology of interstitial lung abnormalities in the context of lung nodule resections. Am. J. Respir. Crit. Care Med. 197, 955–958 (2018).
Dougherty, G. W. et al. Recessively inherited deficiency of secreted WFDC2 (HE4) causes nasal polyposis and bronchiectasis. Am. J. Respir. Crit. Care Med. 210, 63–76 (2024).
Flume, P. A., Chalmers, J. D. & Olivier, K. N. Advances in bronchiectasis: endotyping, genetics, microbiome, and disease heterogeneity. Lancet 392, 880–890 (2018).
Blackwell, T. S. et al. Future directions in idiopathic pulmonary fibrosis research. An NHLBI workshop report. Am. J. Respir. Crit. Care Med. 189, 214–222 (2014).
Winters, N. I., Burman, A., Kropski, J. A. & Blackwell, T. S. Epithelial injury and dysfunction in the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Med. Sci. 357, 374–378 (2019).
Semov, A. et al. Metastasis-associated protein S100A4 induces angiogenesis through interaction with Annexin II and accelerated plasmin formation. J. Biol. Chem. 280, 20833–20841 (2005).
Hedhli, N. et al. The annexin A2/S100A10 system in health and disease: emerging paradigms. J. Biomed. Biotechnol. 2012, 406273 (2012).
Bjørnland, K. et al. S100A4 involvement in metastasis: deregulation of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in osteosarcoma cells transfected with an anti-S100A4 ribozyme. Cancer Res. 59, 4702–4708 (1999).
Wang, K. et al. Identification of ANXA2 (annexin A2) as a specific bleomycin target to induce pulmonary fibrosis by impeding TFEB-mediated autophagic flux. Autophagy 14, 269–282 (2018).
Lei, Y. et al. Cell-surface translocation of annexin A2 contributes to bleomycin-induced pulmonary fibrosis by mediating inflammatory response in mice. Clin. Sci. 133, 789–804 (2019).
Waisman, D. M. Annexin II tetramer: structure and function. Mol. Cell Biochem. 149-150, 301–322 (1995).
Gerke, V., Creutz, C. E. & Moss, S. E. Annexins: linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 6, 449–461 (2005).
Gerke, V. & Moss, S. E. Annexins: from structure to function. Physiol. Rev. 82, 331–371 (2002).
Wang, J. et al. Interaction of HE4 and ANXA2 exists in various malignant cells-HE4-ANXA2-MMP2 protein complex promotes cell migration. Cancer Cell Int. 19, 161 (2019).
Zhuang, H. et al. Human epididymis protein 4 in association with Annexin II promotes invasion and metastasis of ovarian cancer cells. Mol. Cancer 13, 243 (2014).
Lu, R. et al. Human epididymis protein 4 (HE4) plays a key role in ovarian cancer cell adhesion and motility. Biochem. Biophys. Res Commun. 419, 274–280 (2012).
Zhang, Y., Zhou, Z. H., Bugge, T. H. & Wahl, L. M. Urokinase-type plasminogen activator stimulation of monocyte matrix metalloproteinase-1 production is mediated by plasmin-dependent signaling through annexin A2 and inhibited by inactive plasmin. J. Immunol. 179, 3297–3304 (2007).
Ortiz-Zapater, E. et al. Tissue plasminogen activator induces pancreatic cancer cell proliferation by a non-catalytic mechanism that requires extracellular signal-regulated kinase 1/2 activation through epidermal growth factor receptor and annexin A2. Am. J. Pathol. 170, 1573–1584 (2007).
Zhang, T. et al. WFDC2 gene deletion in mouse led to severe dyspnea and type-I alveolar cell apoptosis. Biochem. Biophys. Res Commun. 522, 456–462 (2020).
James, N. E. et al. The biomarker HE4 (WFDC2) promotes a pro-angiogenic and immunosuppressive tumor microenvironment via regulation of STAT3 target genes. Sci. Rep. 10, 8558 (2020).
Ribeiro, J. R. et al. Human epididymis protein 4 promotes events associated with metastatic ovarian cancer via regulation of the extracelluar matrix. Front Oncol. 7, 332 (2018).
Raghu, G. et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 205, e18–e47 (2022).
Ashcroft, T., Simpson, J. M. & Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 41, 467–470 (1988).
Acknowledgements
This work was supported by funds from the National Natural Science Foundation of China (Grant Nos: 82070062 and 82370075), the Climbing Project for Medical Talent of Zhongnan Hospital, Wuhan University (Grant No. PDJH202205), the research found from Medical Sci-Tech innovation platform of Zhongnan Hospital of Wuhan University (Grant No. PTXM2022001), and Translational medicine and interdisciplinary research Joint Found of Zhongnan Hospital of Wuhan University (Grant No. ZNJC202218).
Author information
Authors and Affiliations
Contributions
Z.C. and Y.L. administered the project. Z.C., Y.L. and W.Z. designed the research. W.Z., M.Z. and X.H. performed the experiments and analyzed the data. W.Z., M.Z., X.H., H.G., W.S. and Q.H. collected human samples. W.Z. and M.Z. wrote the original manuscript. W.Z., M.Z. and X.H. contributed equally to this study and shared first authorship. All the authors reviewed and approved the submitted manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Jin-Min Nam and Laura Rodríguez Pérez.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zheng, W., Zou, M., Hu, X. et al. Human epididymis protein 4-annexin II binding promotes aberrant epithelial-fibroblast crosstalk in pulmonary fibrosis. Commun Biol 8, 93 (2025). https://doi.org/10.1038/s42003-025-07529-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07529-7
This article is cited by
-
Human epididymis secretory protein 4 in idiopathic inflammatory myopathy-associated interstitial lung disease
Clinical Rheumatology (2025)
