
Abstract
The unique environment of the Qinghai-Tibetan Plateau provides a great opportunity to study how primate intestinal microorganisms adapt to ecosystems. The 16S rRNA gene amplicon and metagenome analysis were conducted to investigate the correlation between gut microbiota in primates and other sympatric animal species living between 3600 and 4500 m asl. Results showed that within the same geographical environment, Macaca mulatta and Rhinopithecus bieti exhibited a gut microbiome composition similar to that of Tibetan people, influenced by genetic evolution of host, while significantly differing from other distantly related animals. The gut microbiota of plateau species has developed similar strategies to facilitate their hosts’ adaptation to specific environments, including broadening its dietary niche and enhancing energy absorption. These findings will enhance our comprehension of the significance of primate gut microbiota in adapting to specific habitats.
Similar content being viewed by others


Introduction
The Qinghai-Tibetan Plateau, the highest plateau in the world, is also known as “the roof of the world” or the “water tower of Asia”1. High-altitude environments pose a unique challenge to the physiology of indigenous Tibetan people and non-human primates (NHPs), who are exposed to extreme cold and low oxygen2. In cold environments, homothermal animal require additional energy to sustain their core body temperature. For instance, when rodents live at 4 °C, their daily food intake can double, suggesting that 60% of their total energy expenditure is utilized for thermogenesis3. The low-oxygen environment of the plateau prompts an increase in the body’s basic metabolic rate, leading to elevated energy expenditure as a means of adapting to the thin oxygen atmosphere4. The relatively homogeneous vegetation types in the temperate coniferous forests of the Tibetan Plateau exacerbates the scarcity of primate food resources and poses challenges for their energy acquisition5,6. However, primates can be well-adapted to this harsh environment by genetic adaptation7.
The gut microbiota is an essential component of acclimatization to extreme environments2,8. Evidence has shown that the gut microbiota is critical to the well-being of hosts9; as an internalized “microbial organ”10, trillions of microbes colonize a host’s gut tract, forming a complex microecosystem11. Gut microbial composition has myriad effects on nutrient intake, energy metabolism, immune function, physiological regulation, pathogen resistance, toxin transformation, behavior, and even adaptation of their hosts12,13. For instance, the gut microbiota of higher-altitude mammals displays a higher ratio of Firmicutes to Bacteroidetes (F/B ratio) than those of lower-altitude mammals2,6. The main functions of Firmicutes and Bacteroidetes are to help the host digest and absorb nutrients and to degrade carbohydrates and proteins, respectively6,14. In addition, Prevotella may play a crucial role in facilitating energy supply and adaptation to hypoxic conditions. The relative abundance of Prevotella in Tibetan populations has been consistently observed to be higher compared to lowland populations8.
The gut microbiota also influences on niche differentiation within host feeding strategies15. Niche differentiation is the basis for maintaining species diversity because resource partitioning promotes sympatric coexistence16, particularly in environments characterized by limited food resources such as the Tibetan Plateau. The Gut microbiota of sympatric animals can be transmitted through direct or indirect contact; however, variation in species-specific activity rhythms and dietary preferences contribute to the diversification of gut microbial communities in these animals16,17. The gut microbiota serves as a valuable tool for gaining novel insights into the interrelationships among diverse species, particularly regarding the mechanisms underlying the coexistence of similar niches15,18.
There have been numerous studies conducted on the gut microbiota of primates inhabiting the Qinghai-Tibetan Plateau, however, most of these studies have focused on a single species of primates, and these study sites did not reach the maximum altitude range for the species2,6. At present, there are scarce comparative studies regarding the gut microbiota of diverse primates within the Tibetan Plateau and between primates and other co-occurring animals in the same area.
As one of the world’s highest-living primates, the black-and-white snub-nosed monkey (Rhinopithecus bieti) inhabits an altitudinal range of 3600–4500 m19. Within their home range, near the natural northernmost population (Zhina Group) there is also a traditional Tibetan village (altitude 4000 m) and a population of rhesus macaque (Macaca mulatta) as well. This area represents the highest distribution point for both R. bieti and M. mulatta and approaches the upper limit for human survival19,20,21. This unique setting offers favorable conditions for studying the composition and interrelationships of gut microbes in different species under extreme altitude conditions. In this study, we have chosen Tibetan people, M. mulatta, and R. bieti as representatives of primates; yaks (Bos mutus) and goat (Capra hircus) as representatives of domestic animals; tufted deer (Elaphodus cephalophus) and Chinese forest musk deer (Moschus berezovskii) as representatives of wild herbivores; and the ubiquitous white eared-pheasant (Crossoptilon crossoptilon) as a representative of the Galliformes. The fact that R. bieti is monogastric, like humans and M. mulatta, is worth noting. However, the R. bieti possesses a specialized S-shaped and partitioned stomach22,23. This adaptation enables them to eat food containing high levels of structural polysaccharides, i.e., cellulose and related compounds24. On the other hand, B. mutus, C. hircus, E. cephalophus and M. berezovskii are all ruminants characterized by possessing a complex stomach structure17. The digestive system of C. crossoptilon, by contrast, is typical of herbivorous birds and comprises the esophagus, crop, proventriculus, gizzard, intestines, and cloaca25 (Fig. 1).

①–④: the gastrointestinal morphology of animals. A–I is illustrations of their dietary composition. A Bush fruits; B lichens; C mushrooms; D invertebrate, E Bush flowers and young leaves; F grass; G Bush fruits and mature leaves; H bark; I highland barley.
We aimed to investigate (1) the factors influencing the gut microbiome composition of coexisting primates and other animals at extreme altitude in the Hengduan Mountains on the eastern margin of the Qinghai-Tibetan Plateau, (2) the contribution of the gut microbiome to primate adaptation to cold, alpine environments and to their ecological niche differentiation. We hypothesized that: (1) The composition of gut microbiota is primarily determined by host phylogeny and dietary niche; (2) The gut microbiota of plateau species have developed similar strategies to facilitate their hosts’ adaptation to specific environments, including broadening its dietary niche and enhancing energy absorption.
Results
Gut microbiome structure
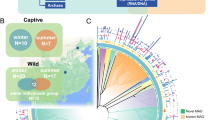
In total, we collected 51 fecal samples for 8 sympatric species (Supplementary Table 1). Each species harbored a structurally unique gut microbiome (Fig. 2). Non-MetricMulti-Dimensional Scaling (NMDS; Fig. 2A binary jaccard and Fig. 2B bray curtis distances) of 16S rRNA gene amplicon data illustrates stronger clustering of 8 species fecal samples by host phylogenetic clade (binary jaccard: PERMANOVA R2 = 0.271, P < 0.001; bray curtis: PERMANOVA: R2 = 0.486, P < 0.001) than diet (vegetarian vs omnivory, binary jaccard: PERMANOVA R2 = 0.053, P < 0.001; bray curtis: PERMANOVA: R2 = 0.101, P < 0.001). Notably, there was higher similarity of microbiome communities among more closely related host species. The Gut microbiota of Primates, Cetartiodactyla, and Galliformes each form a distinct region, characterized by intra-group convergence and inter-group variations (Fig. 2A, B). The cluster analysis of the gut microbiota in host organisms aligns with their respective phylogenies (Fig. 2C). Our analysis revealed that the phylogenetic relationships of hosts were a vital factor influencing the composition of animal gut microbiota at the OTU level (Fig. 2).

Depicted are measures of beta diversity by Non-MetricMulti-Dimensional Scaling (NMDS), including Binary Jaccard distances graphed (A) and Bray Curtis distances graphed (B). C Comparison between gut microbiota tree and host phylogeny relationships, based on Unweighted Pair-group Method with Arithmetic Mean and National Center for Biotechnology Information Taxonomy database. Vegetarian are shaded in the phylogenetic tree.
The pielou evenness index of microbiota in the Cetartiodactyla were significantly higher than those in the Primates (t-test, t = −7.849, df = 43, P < 0.001) and Galliformes (t-test, t = 9.119, df = 28, P < 0.001) (Supplementary Fig. 1). The predominant bacterial phylum isolated from the 8 species were Firmicutes and Bacteroidota (Supplementary Fig. 2). Firmicutes were most abundant in E. cephalophus (68.79%), intermediately abundant in M. berezovskii (57.64%), B. mutus (54.04%) and R. bieti (52.59%), and least abundant in C. crossoptilon (20.14%). In contrast, Bacteroidota were most abundant in C. hircus (42.54%) and Tibetan people (39.14%), intermediately abundant in B. mutus (36.44%), and least abundant C. crossoptilon (3.93%). C. crossoptilon had a significantly higher F/B ratio than other species (t-test, t = 5.176, df = 6, P < 0.01), and wildlife exhibited a higher F/B ratio compared to Tibetan people and domesticated animals (Kruskal-Wallis, z = −2.236, P < 0.05) (Supplementary Table 2). M. mulatta (11.94%) and R. bieti (8.18%) had the greatest proportion of Spirochaetota, and the relative abundance of Proteobacteria in M. mulatta (16.73%) was second only to that of C. crossoptilon (Supplementary Fig. 2).
At the family level (Fig. 3), Prevotellaceae were most abundant in Tibetan people (33.46%) and C. hircus (21.58%), and were similar in R. bieti (14.86%), B. mutus (13.98%) and M. mulatta (11.64%). Oscillospiraceae were present in large proportions in all Cetartiodactyla species (16.32% ± SD 3.1%), and least abundant in C. crossoptilon (0.19%). The relative abundance of Lachnospiraceae in the gut tract of mammals (9.42% ± SD 1.95%) was also higher than that of C. crossoptilon (0.78%).

Relative abundance of gut bacterial taxa in different animals on the family level based on 16S rRNA gene amplicon analysis.
In total, only 11 (0.04%, n = 27615) OTUs were shared by our 8 sympatric species (Fig. 4A). However, there were 35 (0.13%) OTUs shared between Cetartiodactyla and three primates, and 56 (0.20%) OTUs were shared among the three primates (Fig. 4B, C). Notably, the prevalence of OTUs shared between sympatric heterospecifics often indicates the horizontal transmission of gut microbes26. These horizontally transmitted bacterial taxa spanned 6 phyla and 8 genera, including notable genera such as Bacteroides, Prevotella-9, Faecalibacterium, Subdoligranulum, Treponema (Supplementary Table 3).

A 11 OTUs were shared by 8 sympatric species. B 35 OTUs were shared among the Cetartiodactyla and three primates. C 56 OTUs were shared among the three primates.
Interspecific differences in microbiomes
The LEfSe results showed that the 8 sympatric species harbored 69 significantly different taxa (Fig. 5, LDA > 4, P < 0.05). Lachnospiracea Muribaculaceae, Prevotella-7, and Christensenellaceae were markedly more abundant in the R. bieti gut microbiota. Succinivibrio and Treponema were the most abundant taxa in the M. mulatta gut microbiota. Tibetan people had high relative abundance of the genera Prevotella-9, Faecalibacterium, and Phascolarctobacterium. On the other hand, ruminants had a higher relative abundance of Oscillospiraceae and Rikenellaceae, and the genera Akkermansia, Prevotella, and Prevotellaceae. The Gut microbiota of C. crossoptilon were the most specialized, and with a different flora mainly composed of Escherichia, Shigella, Streptococcus, and Rickettsiella.

A taxonomic representation of statistically and biologically consistent differences between different geographical populations. Differences are represented by the color of the most abundant class. Each circle’s diameter is proportional to the taxon’s abundance and represent phylum, class, order, and family. B Histogram of the LDA scores computed for features differentially abundant among the sympatric species. LEfSe scores can be interpreted as the degree of consistent difference in relative abundance between features in the two classes of analyzed microbial communities. The histogram thus identifies which clades among all those detected as statistically and biologically differential explain the greatest differences between communities.
In addition to bacteria, some Archaea were also detectable using 16S rRNA gene sequencing. The majority of detected Archaeal OTUs in this study could be taxonomically assigned as methanogenic Archaea (methanogens), which are a phylogenetically diverse group of microorganisms. The relative abundance of Methanobacteria within archaea was higher in domestic animals compared to wild animals (t-test, t = −8.805, df = 37, P < 0.001) and humans (t-test, t = −3.671, df = 13, P < 0.01) (Fig. 6).

The boxes represent 25–75th percentiles, the lines in the box indicate the median and whiskers extend to the maximum and minimum values within 1.5× the interquartile range. Significant differences were marked as “**” (P < 0.01), “***” (P < 0.001) by t-test.
Gut microbiome function in primates
The Zhina group of R. bieti in the area occupied home range of 22.15 km² from March 2023 to December 2023 (ranging from 3600 to 4500 m above sea level), with M. mulatta occurring. These NHPs in the study area shared space and similar activity patterns with local Tibetan people (Fig. 7). Both species are diurnal, with activity concentrated between 8:00 and 9:00 and between 13:00 and 16:00 (Supplementary Fig. 3).

Map of sampling sites for 8 sympatric species in Markam, Tibet, China.
We obtained a total of 1,241,233,976 clean reads remained from 16 primates after metagenomic sequencing and quality control. After optimizing the read assembly, 5,958,394 contigs were obtained, and 5,452,782 microbiota non-redundant gene data were obtained after gene prediction and de-redundancy. Analysis of Bray Curtis distances indicated an effect of species on pathway composition (PERMANOVA: R2 = 0.704, P < 0.001; Fig. 8A). A Kruskal-Wallis test confirmed that the three primates exhibited notable disparities in the relative abundance of KEGG level 3 categories (Fig. 8B). Among these pathways, Tibetan people showed significantly higher enrichment in pathways such as “Butanoate metabolism”, “beta-Alanine metabolism”, “Starch and sucrose metabolism”, “Thiamine metabolism”, “Folate biosynthesis”, “Carbapenem biosynthesis”. However, NHPs showed significantly higher enrichment in pathways such as “Carbon fixation pathways in prokaryotes”, “DNA replication”, “Nucleotide excision repair”, “Mismatch repair”, “Homologous recombination”. Then, we note that some modules are enriched in three primates, such as: Gluconeogenesis (M00003), Acetyl-CoA pathway (M00422), Methionine biosynthesis (M00017, M00338) and Phosphate acetyltransferase-acetate kinase pathway (M00579) (Fig. 9).

A analysis of Bray Curtis distances indicated an effect of species on pathway composition; B a comparison of the microbiome gene functions among primates in the KEGG 3 classification was conducted; C the functional annotations of primates based on the CAZy database, at family level.

A The gluconeogenesis module (M00003); B the acetyl-CoA pathway module (M00422); C the methionine biosynthesis module (M00017, M00338); D the phosphate acetyltransferase-acetate kinase pathway module (M00579). Different colors signify diverse species. The boxes represent 25–75th percentiles, the lines in the box indicate the median and whiskers extend to the maximum and minimum values within 1.5× the interquartile range.
According to the functional annotation of the CAZy database, at family level, two NHPs are more alike (Fig. 8D). They exhibited enrichment in acetyl xylan esterase (EC 3.1.1.72), pectin methylesterase (EC 3.1.1.11), endoglucanase (EC 3.2.1.4), beta-glucosidase (EC 3.2.1.21), lysozyme (EC 3.2.1.17), cellobiose phosphorylase (EC 2.4.1.20), diacetylchitobiose deacetylase (EC 3.5.1.-). On the other hand, the relative abundance of glucoamylase (EC 3.2.1.3) and beta-galactosidase (EC 3.2.1.23) in humans is higher than that of the two NHPs.
Discussion
By comparing gut microbiota across 8 sympatric species, we show that hosts with similar habitat harbor strikingly different gut microbiomes. The impact of host phylogeny and physiology on the gut microbiota of distantly related species was significantly more pronounced than that of the host’s dietary niche. The gut microbiomes of closely related species, such as various animals within the ruminantia, demonstrated notable resemblances. Traditional Tibetan people and NHPs showed parallel gut microbiome adaptations to analogous ecological conditions. This might be the result of long-term co-evolution between the host and environment and suggests that the composition of gut microbiota in sympatric species is influenced by both host phylogeny and physiology as well as dietary niches15,17,18. Interestingly, R. bieti are foregut fermenters with a multi-compartmental, sac-like stomach that is divided into fundus I and fundus II from the beginning of lactating larva22,23. This unique structure resembles that of ruminant stomachs, enabling them to efficiently digest high-fiber food22,23. However, our findings demonstrate that the gut microflora of R. bieti exhibits a higher degree of similarity to that of humans and M. mulatta, while displaying distinct dissimilarities from ruminants. This represents an additional indication that evolutionary trends in host phylogeny exert a greater influence than dietary niche and physiology on the structuring of gut microbiomes27.
Shifts in dietary substrates provided to gut microbes as a result of host diet changes could result in the same processes, explaining the dual impact of host phylogeny and dietary niche that we observed15,28. The differentiation of ecological niches is crucial for the maintenance of species diversity, as it facilitates resource partitioning and enables sympatric coexistence, particularly in environments characterized by limited food availability, such as the Qinghai-Tibetan Plateau16. Dietary partitioning represents a mechanism for sympatric species to avoid competition, and is typically demonstrated by documenting diets with minimal overlap and describing the morphological traits that facilitate dietary specialization29. In our study, higher relative abundance of Rikenellaceae and Akkermansia were observed in ruminants than in Primates and Galliformes. Rikenellaceae exhibited a positive correlation with food intake and body weight, while also facilitating lipid metabolism30. Increased Akkermansia relative abundance indicates a response of this gut microbe to high fiber and low protein diets31. The arginine and fatty acid anabolic pathways encoded by Akkermansia can significantly enhance the energy and nitrogen utilization efficiency of ruminants, thereby aiding their survival during periods of severe nutritional stress in cold environments31. The abundance of evidence indicates that ruminants possess a highly efficient gut microbial phenotype.
The plant species present in R. bieti habitat of our study are chiefly Abies squamata, Picea likiangensis, and Larix gmelinii, accompanied by hard-leaved broadleaf forests and some other evergreen/deciduous shrubs32. The plant diversity within their habitat is relatively low. Lichen constitutes the main food source in the annual diet (74.8%), while the proportions of other food sources are significantly lower: buds and leaves account for 15.9%, flowers, fruits, and seeds constitute 2.5%, bark, roots, resin, grass, etc. amount to 4.9%, and invertebrates make up 2.0%33. The consumption of lichens gradually increases from south to north in the R. bieti diet, with the northern Xiaochangdu group (near our study population) relying on lichens as their primary food source during winter, accounting for 98.2%33. In contrast, the central Samage group shows a decrease in lichens consumption at 67%34, while the southern Jinsichang group only consumes approximately 5% throughout the year35. It is worth noting that lichens contain a higher proportion of hemicellulose compared to other plant parts, making them an unfavorable dietary choice for R. bieti36. The significant intake of lichens may serve as an adaptive strategy for R. bieti facing food limitations at high altitudes33. On the other hand, the M. mulatta in this study were exclusively confined to a single vegetated habitat, while their diet primarily consisted of bark, leaves, and the fruiting bodies of fungi. The gut microbiota plays a crucial role in facilitating NHPs adaptation to specific dietary requirements37. The metagenomic results concerning CAZymes demonstrated that NHPs possess a considerable ability to digest complex carbohydrates, such as cellulose, hemicellulose, and pectin. Acetylxylan esterases (AcXEs; EC 3.1.1.72) are components of microbial xylanolytic and cellulolytic systems, role of which is to remove acetyl groups esterifying d-Xyl p residues of xylan main chain at position 238. Endo-β-1,4-glucanase predominantly acts upon the non-crystalline regions of cellulose and decomposes the cellulose by randomly severing the beta-1,4-glycosidic bonds, liberating short-chain oligosaccharides of varying lengths, such as fructooligosaccharides and cellobiose39. Cellobiose phosphorylase (EC 2.4.1.20), a glycosyltransferase and hexosyltransferase, pertains to the transferase category of enzymes. Its principal function is to catalyze the reversible phosphorylation of cellobiose, which converts it into α-D-glucose 1-phosphate and D-glucose40. Diacetylchitobiose deacetylase (EC 3.5.1.-) exerts a crucial role in the chitin degradation pathway, which is prevalently abundant in the insect exoskeleton, thereby reflecting the insectivorous diet of NHPs41,42. Conversely, the results of CAZy database annotation showed that Tibetan People have a strong ability to digest starch (glucoamylase, EC 3.2.1.3)43 and lactose (beta-galactosidase, EC 3.2.1.23)44, which is in correspondence with their diet consisting of highland barley and butter8.
Meanwhile, eight species exhibited a shared presence of 11 OTUs, with a notable occurrence of 47 OTUs among mammalian taxa. Notably, increased prevalence of OTUs shared between sympatric species relative to heterospecific species often indicates the horizontal transmission of gut microbes, and the phenomenon has been observed in sympatric chimpanzees and gorillas, as well as in sympatric Malagasy mammals28. The presence of symbiotic relationships enhances the microbial diversity within the gastrointestinal tract of animals45, and hosts acquire gut microbiota from other species to maintain a significantly greater species richness and biodiversity than in non-gut environments46,47. The acquisition of bacterium by animals may have occurred through cohabitation within their shared habitat, thereby enhancing their level of health6,17. For instance, Faecalibacterium, regarded as a next-generation probiotic or live biotherapeutic productis, is a bacterial genus with promising human health applications48. And the prevalence of Subdoligranulum is significantly associated with the diversity of the gut microbiota, serving as an indicator of the host’s metabolic well-being. Additionally, a high level of gut microbial diversity often enhances the host’s adaptive capacity by broadening the dietary niche of wild animals, as a diverse gut microbiota confers an extensive capability to handle the intricate compounds found in plant-based food sources49.Such as Bacteroides, Treponema and Prevotella-9, they aid the host in breaking down complex carbohydrates, produce SCFAs, and enhance digestion, nutrient absorption, metabolic regulation, and immune response50,51. In conclusion, the presence of symbiotic relationships enhances the microbial diversity within the gastrointestinal tract of animals45, and hosts acquire gut microbiota from other species to maintain a significantly greater species richness and biodiversity than in non-gut environments46,47.
Concurrently, low temperatures intensify the energy requirements of the animals. The austere environment of the Qinghai-Tibet Plateau poses a substantial challenge to the survival of animals, demanding that they possess effective means of energy acquisition31,52,53. In this study, we found that the gut microbiota of these 8 species predominantly consists of Firmicutes and Bacteroidetes at the phylum level, which aligns with findings from previous research54,55. Moreover, wild animals displayed higher F/B ratios in comparison to domestic animals and humans. Firmicutes possess a multitude of genes encoding enzymes involved in energy metabolism and exhibit the ability to synthesize diverse digestive enzymes, thereby facilitating nutrient digestion and absorption by their hosts56. In contrast, Bacteroidetes primarily specialize in the degradation of carbohydrates and proteins57. The higher F/B ratio in a microbial community thus should be positively associated with increased absorption of food energy, facilitating the maintenance of energy homeostasis and core body temperature in challenging environments6,14,58. At the family level, the bacteria with the highest relative abundance among mammals were Prevotellaceae, Oscillospiraceae, and Lachnospiraceae. The relative abundance of Prevotellaceae in Tibetan human populations is commonly higher than that in lowland populations and thus positively correlated with altitude8. Prevotellaceae is a saccharolytic anaerobe associated with the production of short-chain fatty acids (SCFAs), which can serve as a source of energy for the host59,60,61. In addition, SCFAs produced by the gut flora might influence blood pressure regulation by acting on SCFAs receptors, such as olfactory receptor 78 and G-protein couple receptor 418. On the other hand, it was reported that the Oscillospiraceae was more abundant in leaner individuals. A higher relative abundance of Oscillospiraceae might lead to decreased weight, which facilitates animal movement to forage more food resources in high-altitude mountains, thus allowing them to gain more energy to adapt to high- altitude mountains6. Additionally, previous research in Tibetan populations has demonstrated that the richness of Lachnospiraceae is higher than in groups living at lower altitudes8. In our research, we also observed a high relative abundance of Lachnospiraceae in mammals. These taxa are known to produce SCFAs, which serve as an important energy source and can also regulate the blood pressure of the host. This might help plateau animals maintain a healthier gut environment and adapt to life at higher altitudes46,62.
Meanwhile, in our metagenomic analysis, we observed pathways crucial for SCFAs synthesis, encompassing the M00422, M00579, and M00003 modules. The gut microbiota’s fermentation process yields SCFAs, primarily acetate, propionate, and butyrate, which are swiftly absorbed by the host’s gut epithelium, supplying energy and enhancing the host’s metabolic efficiency in food utilization37. Among these SCFAs, butyrate emerges as a preferred energy source; however, its production is largely dependent on the availability of intestinal carbohydrates37,60. In human gut microbiota, diverse fermentation strategies have evolved to harness additional energy. Pyruvate, a key intermediate, can be broken down and converted into succinate, lactic acid, or acetyl-CoA37. These compounds can subsequently be further metabolized by the host to generate acetate, propionate, and butyrate37,63. Herein, the modules related to SCFAs can generate the energy substrates acetyl-CoA and pyruvate, mediate the energy metabolism of the host and offer energy compensation to the host. This enhancement in energy metabolism has been instrumental in improving the host’s adaptation to the challenging environmental conditions of high-altitude plateaus, characterized by low temperatures and food scarcity37. In addition, we found that the enrichment rate of genes related to the DNA replication, nucleotide excision repair and mismatch repair pathways was significantly higher in NHPs. These pathways are enriched in animals at high altitudes, which have enhanced DNA repair capacity37.
In addition, our results show that Methanobacteria relative abundance in primates, wild ruminants, and C. crossoptilon is comparatively lower than that observed in domestic animals. Methanobacteria are predominantly found in the rumen of ruminant animals, accounting for their limited presence in non-ruminants8,31. Methane, a by-product of rumen fermentation by methanogenic archaea, leads to energy loss64. Low-methane production in the gut microbiota might help maintain highly efficient energy metabolism to survive in the harsh high-altitude environment65. The evidence thus suggests that wildlife might possess more efficient gastrointestinal functions compared to domestic animals, enabling them to minimize energy loss and adapt effectively to challenging natural environments.
In addition, the gut microbiota can play a pivotal role in the degradation of environmental chemicals and toxic pollutants, thereby mitigating their potential harm and enhancing host adaptability to the surrounding environment66. Our study demonstrates that the gut microbiota of M. mulatta exhibits functional enrichment in degrading hazardous substances such as atrazine and styrene. Atrazine is a commonly used herbicide that has been shown to be harmful to the health of animals67. Styrene and styrene polymers are released into the environment during the process of synthetic rubber and plastic production and incineration68. During our field investigation, we discovered a significant quantity of plastic bags negligently discarded. Styrene and styrene polymers are released into the environment during the process of synthetic rubber and plastic production and incineration68. M. mulatta frequently traverse and forage in close proximity to human settlements, thereby heightening the potential for exposure to hazardous substances. Functional enrichment of harmful substance degradation in the gut microbiota of M. mulatta can protect against the ingestion of such substances and enhance adaptability to worsening habitat pollution.
The aforementioned data collectively emphasize that host phylogeny is the vital influence on the gut microbiota in both human and NHPs residing at high altitudes. In the same geographical environment, M. mulatta and R. bieti exhibited a gut microbiota composition similar to that of Tibetans, influenced by genetic evolution of host, while significantly differing from other distantly related animals. Meanwhile, various animals possess distinct dietary niches, offering diverse substrates to the gut microbiome, which in turn gives rise to variations in the composition of gut microbiota. For instance, M. mulatta and R. bieti were more proficient in digesting complex carbohydrates, whereas the gut microbiota of Tibetan People was characterized by the capacity to digest starch and dairy products. This confirms our first hypothesis: The composition of gut microbiota was primarily determined by host phylogeny and dietary niche. On the other hand, shared OTUs expand the dietary ecological niche of animals, and the efficient enzymes produced by gut microbiota can assist the host in digesting food. Additionally, the SCFAs produced through gut microbiota fermentation can be rapidly absorbed by the intestinal epithelial cells of the host to supply energy to the host. This substantiates our second hypothesis: The gut microbiota of plateau species have developed similar strategies to facilitate their hosts’ adaptation to specific environments, including broadening its dietary niche and enhancing energy absorption. In addition, the gut microbiota of M. mulatta can possibly help them degrade specific harmful substances.
Methods
Ethics approval and consent to participate
Sample collection and experimental protocols were performed in accordance with the Institutional Review Board (IRB202303) and the Institutional Animal Care and Use Committee of the Central South University of Forestry and Technology, China under permit number LXY-2022010004, as well as Administration for Wild Animal Protection in Tibet Autonomous Region, China and adhered to the American Society of Primatologists Principles for the Ethical Treatment of Primates. All of the participants provided informed consent to participant in this study under the approval the Ethics Committee at the Central South University of Forestry and Technology (Approval number: 2012-018). All ethical regulations relevant to human research participants were followed. All samples included in this paper were taken in compliance with the laws of China, and no animal experimentation was involved.
Study site and animals
The study was conducted from March 2023 to June 2024 in Rhinopithecus bieti National Nature Reserve in Markang, Tibet, China (Altitude: 3600–4700 m, Average annual temperature: 2–8 °C, Annual precipitation:400–800 mm, N: 28°48′–29°40′, E: 98°20′–98°59′) (Fig. 7). There is a Tibetan village, Zhina in the study site, with a total of five families, raising goats, yaks, horses and other domestic animals, and living in a semi-agricultural and semi-nomadic mode. Although detailed information on local fauna is insufficient, the area is home to two species of NHPs, the R. bieti (Zhina group) and the M. mulatta. Other wild animals are mainly E. cephalophus, M. berezovskii and C. crossoptilon. We selected local Tibetan people and the two kinds of domestic animals they raise (B. mutus and C. hircus), and these five common wild animals for our study.
Field survey and sample collection
Prior the collection and analysis on fecal samples, we carried out direct tracking and camera trapping to collect data on rudimentary knowledge on distribution and ecological functions of sympatric animals in the area, particularly two NHPs, M. mulatta and R. bieti (Zhina group)69,70,71. We carried out direct tracking for 2–5 days (07:30–18:30) per week based on terrain accessibility and weather conditions to search for wild animals (particularly R. bieti) and their feces. The duration of the work amounted to 110 days, ranging from March 2023 to June 2024. If we found R. bieti or other animals, we recorded the position of the animals via GPS receivers every 30 mins whenever possible69,70,71. We also collected feces samples and determined the freshness and the species who may defecate via viewing feces appearances and local experience. We also deployed 15 camera traps at animal trails to monitor wild animals and their potential defecation behaviors72. Images of camera traps were retrieved weekly to identify sympatric animals and to assist location of potential feces.
When we managed to follow R. bieti, M. mulatta and domestic animals, we observed their defecation behaviors whenever possible. If we found fresh feces (less than 12 h after defecation), we collected 3–4 g fecal samples from each individual’s fecal patch non-invasively73. The stool samples were also obtained from five adult Tibetan human males who had not received any recent antibiotic treatment. After collection, the samples were stored at ambient temperature in sterile plastic tubes that were prefilled with 5 ml of DNA preservation solution (Singen Inc., Zhejiang, China). All samples were labeled with collection time and individual information and transported to the laboratory via dry ice, where samples were stored at −80 °C.
Home range and activity pattern of R. bieti and sympatric animals
We combined all recorded GPS coordinates to estimate home ranges of R. bieti via the modified Minimum Convex Polygon method (excluding villages, farmlands and alpine meadow/bare rock ≥4600 m which R. bieti never use)69,70,74,75. The daily activity rhythms of R. bieti and M. mulatta were also collected via camera trap76. We identified all photographed animals and chose typical species from different taxa (Cetartiodactyla and Galliformes) for feces collection in the field. Species taxonomic identification followed current checklists of mammals and birds in China77,78.
Amplicon sequencing, bioinformatics, and statistics
Total DNA was extracted from the samples using the TGuide S96 Magnetic Stool DNA Kit (Tiangen Biotech (Beijing) Co., Ltd.) according to manufacturer’s instructions. The hypervariable region V3-V4 of the bacterial 16S rRNA gene were amplified with primer pairs 338 F: 5′- ACTCCTACGGGAGGCAGCA-3′ and 806 R: 5′- GGACTACHVGGGTWTCTAAT-3′. polymerase chain reaction (PCR, 98 °C for 2 min, followed by 30 cycles of 98 °C for 30 s, 50 °C for 30 s, 72 °C for 60 s, and 72 °C for 5 min.) products were checked on agarose gel and purified through the Omega DNA purification kit (Omega Inc., Norcross, GA, USA). The purified PCR products were collected and the paired ends (2 × 250 bp) was performed on the Illumina Novaseq 6000 platform.
The V3–V4 region of the archaea 16S-rRNA gene was also amplified by PCR (95 °C for 5 min, followed by 35 cycles of 95 °C for 30 s, 50 °C for 30 s, 72 °C for 40 s, and 72 °C for 7 min) using the primers Arch349F (50-GYGCASCAGKCGMGAAW-30) and Arch806R (50-GGACTACVSGGGTATCTAAT-30). The PCR products were mixed with an equal volume of 2 × loading buffer and electrophoresed in a 1.8% agarose gel for detection. Samples with a band at approximately 450 bp were mixed in equidensity ratios followed by purification using a GeneJET Gel Extraction Kit (Thermo Fisher Scientific, Waltham, MA, United States). Sequencing libraries were validated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, United States) and quantified with a Qubit 2.0 Fluorometer (Thermo Fisher). Finally, paired-end sequencing was conducted on an Illumina NovaSeq 6000 platform (Illumina, Inc., San Diego, CA, United States) by the Biomarker Bioinformatics Technology Co., Ltd. (Beijing, China).
The qualified sequences with more than 97% similarity thresholds were allocated to one operational taxonomic unit (OTU) using USEARCH (version 10.0). Taxonomic annotation of the OTUs was performed based on the Naive Bayes classifier in QIIME2 (version 2020.6.0)79 using the SILVA database80 (release 138.1) with a confidence threshold of 70%. Alpha was performed to identify the complexity of species diversity of each sample utilizing QIIME2 software. Beta diversity calculations were analyzed by Non-Metric Multi-Dimensional Scaling to assess the diversity in samples for species complexity. To evaluate the distances of gut microbiota among various species, we conducted a hierarchical clustering analysis tree based on the Beta diversity distance algorithm. Specifically, the UPGMA (Unweighted Pair-group Method with Arithmetic Mean) algorithm was employed to construct the inter - sample tree structure, thereby reflecting the differences in the composition of various species. At the same time, we constructed the phylogenetic tree of the hosts by utilizing the Taxonomy feature on the NCBI, and then achieved visualization with iTol (https://itol.embl.de/). One-way analysis of variance was used to compare bacterial and archaea abundance and diversity. Linear discriminant analysis (LDA) coupled with effect size (LEfSe) was applied to evaluate the differentially abundant taxa. The online platform BMKCloud (https://www.biocloud.net) was used to analyze the sequencing data.
Metagenomic sequencing, bioinformatics, and statistics
To explore the differences and similarities in the functions of gut microbiota among three sympatric primates, we selected 16 samples from Tibetan people (n = 6), R. bieti (n = 5) and M. mulatta (n = 5) for metagenomic sequencing. Total DNA was extracted from the samples using the TGuide S96 Magnetic Stool DNA Kit (Tiangen Biotech (Beijing) Co., Ltd.) according to manufacturer’s instructions. DNA quality and quantity were determined using Nanodrop (ND-1000) spectrophotometry (Nanodrop Technologies, Wilmington, DE, USA) and agarose gel electrophoresis, respectively. DNA samples were stored at −20 °C until use. Shotgun sequencing was performed using an Illumina NovaSeq 6000, with at least 10 Gb per sample. Raw data were filtered using Trimmomatic v0.3681 to trim low-quality reads: 3’ tailing sequences were removed when the average quality over a 4 bp sliding window was less than 20 and reads less than 70 bp were discarded. The metagenome was assembled using MEGAHIT software (version 1.2.6) with the default parameters82. QUAST software (version 5.0.2) was used to evaluate the assembly results83. MetaGeneMark software (version 3.26) was used to identify the coding regions in the contigs84. Non-redundant gene sets were constructed using MMseqs2 software (version 12-113e3)85. The similarity threshold was set to 95%, and the coverage threshold was set to 90%.
The non-redundant gene sets were compared with the kyoto encyclopedia of genes and genomes (KEGG, https://www.genome.jp/kegg/) databases using the DIAMOND software (version 0.9.24)86. The threshold was set to an e < 1e-05. In the case of multiple alignment results (hits), the best alignment result was selected as the annotation of the sequence. The non-redundant gene sets were compared with the hidden Markov model of each family in carbohydrate-active enzymes database (CAZy, v10_date20211003) using the hmmer software (version 3.0, Default comparison parameters, default screening threshold “if alignment >80aa, use E < 1e-5, otherwise use E < 1e-3; covered fraction of HMM > 0.3”). Identified all the families that met the filtering threshold. In that way, we could locate the carbohydrate-active enzymes within the non-redundant gene and analyze the number of conservative carbohydrate-related functional domains they contained. Functional genes composition was subjected to analysis of similarity using the R vegan package, respectively. Wilcoxon rank-sum tests were used for nonparametric tests of the difference between groups of functional genes. p < 0.05 were considered statistically significant.
Statistics and reproducibility
In all experiments, biological replicates were employed, with each replicate representing an individual animal. We selected 51 samples from H. sapiens (n = 6), R. bieti (n = 10), M. mulatta (n = 8), E. cephalophus (n = 6), M. berezovskii (n = 5), B. mutus (n = 5), C. hircus (n = 5) and C. crossoptilon (n = 6) for the 16S rRNA gene amplicon study. For the metagenomic study, 6 H. sapiens, 5 R. bieti and 5 M. mulatta were included. The t-test and Wilcoxon rank-sum tests were used to analyze the differences between the two groups. The significance threshold was set at P < 0.05. *P < 0.05, **P < 0.01, ***P < 0.001. Statistical analysis was performed using IBM SPSS Statistics (version 23).
Data availability
The data presented in the study are deposited in the National Genomics Data Center (NGDC, https://ngdc.cncb.ac.cn/gsa/), accession numbers CRA015247 and CRA015267. Source values underlying the figures can be found in supplementary data.
References
Xu, S. et al. A genome-wide search for signals of high-altitude adaptation in Tibetans. Mol. Biol. Evol. 28, 1003–1011 (2011).
Jia, Z. et al. Impacts of the plateau environment on the gut microbiota and blood clinical indexes in Han and Tibetan individuals. mSystems 5, e00660–19 (2020).
Lal, N. K. et al. Xiphoid nucleus of the midline thalamus controls cold-induced food seeking. Nature 621, 138–145 (2023).
Dünnwald, T. et al. Body composition and body weight changes at different altitude levels: a systematic review and meta-analysis. Front. Physiol. 10, 430 (2019).
Xiang, Z. F., Huo, S., Xiao, W., Quan, R. C. & Grueter, C. C. Diet and feeding behavior of Rhinopithecus bieti at Xiaochangdu, Tibet: adaptations to a marginal environment. Am. J. Primatol. 69, 1141–1158 (2007).
Zhao, J. et al. Characterization of the gut microbiota in six geographical populations of Chinese rhesus macaques (Macaca mulatta), implying an adaptation to high-altitude environment. Microb. Ecol. 76, 565–577 (2018).
Li, L. & Zhao, X. Comparative analyses of fecal microbiota in Tibetan and Chinese Han living at low or high altitude by barcoded 454 pyrosequencing. Sci. Rep. 5, 14682 (2015).
Liu, K., Yang, J. & Yuan, H. Recent progress in research on the gut microbiota and highland adaptation on the Qinghai-Tibet Plateau. J. Evol. Biol. 34, 1514–1530 (2021).
Clemente, J. C., Ursell, L. K., Parfrey, L. W. & Knight, R. The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 (2012).
Zhang, J. et al. A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. ISME J. 9, 1979–1990 (2015).
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K. & Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 (2012).
Gomez, A. et al. Temporal variation selects for diet-microbe co-metabolic traits in the gut of Gorilla spp. ISME J. 10, 514–526 (2016).
Sieber, M., Traulsen, A., Schulenburg, H. & Douglas, A. E. On the evolutionary origins of host-microbe associations. P. Natl. Acad. Sci. USA. 118, e2016487118 (2021).
Murphy, E. F. et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 59, 1635–1642 (2010).
Greene, L. K. et al. A role for gut microbiota in host niche differentiation. ISME J. 14, 1675–1687 (2020).
Schoener, T. W. Resource partitioning in ecological communities. Science 185, 27–39 (1974).
Fu, H. et al. Sympatric yaks and plateau pikas promote microbial diversity and similarity by the mutual utilization of gut microbiota. Microorganisms 9, 1890 (2021).
Kartzinel, T. R., Hsing, J. C., Musili, P. M., Brown, B. R. P. & Pringle, R. M. Covariation of diet and gut microbiome in African megafauna. Proc. Natl Acad. Sci. USA 116, 23588–23593 (2019).
Xiang, Z. F. et al. Distribution, Status and Conservation of the Black-and-White Snub-Nosed Monkey Rhinopithecus Bieti in Tibet. Oryx. 41, 525–531 (2007).
Cohen, J. E. & Small, C. Hypsographic demography: the distribution of human population by altitude. Proc. Natl Acad. Sci. USA95, 14009–14014 (1998).
Mittermeier, R. A., Rylands, A. B. & Wilson, D. E. Handbook of the mammals of the world:3. primates. J. Mammal. 95, 906–907 (2014).
Peng, Y. Z., Zhang, Y. P., Ye, Z. Z. & Liu, R. L. Study on the stomachs in three species of snub-nosed monkeys. Zoonoses Res. 4, 167–175 (1983).
Chen, J. J., Lu, T., Liu, J. S. & Huang, Z. R. Observations on the stomach of Rhinopithecus Roxellanae. Acta Theriol. Sin. 15, 176–180 (1995).
Xu, B. et al. Metagenomic analysis of the Rhinopithecus bieti fecal microbiome reveals a broad diversity of bacterial and glycoside hydrolase profiles related to lignocellulose degradation. BMC Genom. 16, 174 (2015).
Xiang, Z. F. et al. Morphological comparison of the digestive systems from 10 species of birds in 4 kinds of ecological groups. Sichuan J. Zool. 28, 3 (2009).
Moeller, A. H. et al. Sympatric chimpanzees and gorillas harbor convergent gut microbial communities. Genome Res. 23, 1715–1720 (2013).
Amato, K. R. et al. Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J. 13, 576–587 (2019).
Perofsky, A. C., Lewis, R. J. & Meyers, L. A. Terrestriality and bacterial transfer: a comparative study of gut microbiomes in sympatric Malagasy mammals. ISME J. 13, 50–63 (2019).
Kartzinel, T. R. et al. DNA metabarcoding illuminates dietary niche partitioning by African large herbivores. Proc. Natl. Acad. Sci. USA 112, 8019–8024 (2015).
Sanguinetti, E. et al. Microbiota signatures relating to reduced memory and exploratory behaviour in the offspring of overweight mothers in a murine model. Sci. Rep. 9, 12609 (2019).
Guo, N. et al. Seasonal dynamics of diet-gut microbiota interaction in adaptation of yaks to life at high altitude. NPJ Biofilms Microbiomes 7, 38 (2021).
Xiang, Z. F., Huo, S. & Xiao, W. Habitat selection of black-and-white snub-nosed monkeys (Rhinopithecus bieti) in Tibet: implications for species conservation. Am. J. Primatol. 73, 347–355 (2011).
Xiang, Z. F. et al. Diet and feeding behavior of Rhinopithecus bieti at Xiaochangdu, Tibet: adaptations to a marginal environment. Am. J. Primatol. 69, 1141–1158 (2007).
Huang, Z. P. et al. Black-and-white snub-nosed monkey (Rhinopithecus bieti) feeding behavior in a degraded forest fragment: clues to a stressed population. Primates 58, 517–524 (2017).
Yang, S. J. & Zhao, Q. K. Bamboo leaf-based diet of Rhinopithecus bieti at Lijiang, China. Folia Primatol. 72, 92–95 (2001).
Hou, R. et al. Seasonal variation in diet and nutrition of the northern-most population of Rhinopithecus roxellana. Am. J. Primatol. 80, e22755 (2018).
Zhao, J. et al. Metagenome and metabolome insights into the energy compensation and exogenous toxin degradation of gut microbiota in high-altitude rhesus macaques (Macaca mulatta). NPJ Biofilms Microbiomes 9, 20 (2023).
Pratima, B. In Progress in Biochemistry and Biotechnology, Microbial Xylanolytic Enzymes (ed Pratima, B.) 29–57 (Academic Press, 2022).
Rahman, M. S. et al. Endoglucanase (EG) activity assays. Methods Mol. Biol. 1796, 169–183 (2018).
Chomvong, K. et al. Overcoming inefficient cellobiose fermentation by cellobiose phosphorylase in the presence of xylose. Biotechnol. Biofuels 7, 85 (2014).
Jiang, Z. et al. Secretory expression fine-tuning and directed evolution of diacetylchitobiose deacetylase by bacillus subtilis. Appl. Environ. Microbiol. 85, e01076–19 (2019).
Biniek-Antosiak, K. et al. Structural, Thermodynamic and Enzymatic Characterization of N,N-Diacetylchitobiose Deacetylase from Pyrococcus chitonophagus. Int. J. Mol. Sci. 23, 15736 (2022).
Kelly, J. J. & Alpers, D. H. Properties of human intestinal glucoamylase. Biochim. Biophys. 315, 113–122 (1973).
Azra, S. & Qayyum H. In Foundations and Frontiers in Enzymology, Glycoside Hydrolases (ed Arun, G. & Kedar, S.) 323–347 (Academic Press, 2023).
Song, S. J. et al. Cohabiting family members share microbiota with one another and with their dogs. Elife 2, e00458 (2013).
Li, K. et al. Comparative analysis of gut microbiota of native Tibetan and Han populations living at different altitudes. PLoS ONE. 11, e0155863 (2016).
Seedorf, H. et al. Bacteria from diverse habitats colonize and compete in the mouse gut. Cell 159, 253–266 (2014).
Martín, R. et al. Faecalibacterium: a bacterial genus with promising human health applications. FEMS Microbiol. Rev. 47, fuad039 (2023).
Li, G. et al. Dietary shifts influenced by livestock grazing shape the gut microbiota composition and co-occurrence networks in a local rodent species. J. Anim. Ecol. 88, 302–314 (2019).
Zafar, H. & Saier, M. H. Jr Gut Bacteroides species in health and disease. Gut Microbes 13, 1–20 (2021).
Xue, M. Y. et al. Multi-omics reveals that the rumen microbiome and its metabolome together with the host metabolome contribute to individualized dairy cow performance. Microbiome 8, 64 (2020).
Ma, Y. F. et al. Population genomics analysis revealed origin and high-altitude adaptation of Tibetan pigs. Sci. Rep. 9, 11463 (2019).
Liu, X. et al. EPAS1 gain-of-function mutation contributes to high-altitude adaptation in Tibetan horses. Mol. Biol. Evol. 36, 2591–2603 (2019).
Tanes, C. et al. Role of dietary fiber in the recovery of the human gut microbiome and its metabolome. Cell Host Microbe 29, 394–407 (2021).
Wang, X. C. et al. Unique characteristics of gut microbiota in black snub-nosed monkeys (Rhinopithecus strykeri) reveal an enzymatic mechanism of adaptation to dietary vegetation. Zool. Res. 44, 357–360 (2023).
Kaakoush, N. O. Insights into the Role of Erysipelotrichaceae in the Human Host. Front. Cell Infect. Microbiol. 5, 84 (2015).
Jami, E., White, B. A. & Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE. 9, e85423 (2014).
Sun, G. et al. Comparative analyses of fecal microbiota in European mouflon (Ovis orientalis musimon) and blue sheep (Pseudois nayaur) living at low or high altitudes. Front. Microbiol. 10, 1735 (2019).
Shah, H. N. & Collins, D. M. Prevotella, a new genus to include Bacteroides melaninogenicus and related species formerly classified in the genus Bacteroides. Int. J. Syst. Bacteriol. 40, 205–208 (1990).
den Besten, G. et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 54, 2325–2340 (2013).
Biddle, A., Stewart, L., Blanchard, J. & Leschine, S. Untangling the genetic basis of fibrolytic specialization by lachnospiraceae and ruminococcaceae in diverse gut communities. Diversity 5, 627–640 (2013).
Suzuki, T. A. Links between natural variation in the microbiome and host fitness in wild mammals. Integr. Comp. Biol. 57, 756–769 (2017).
Oliphant, K. & Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome 7, 91 (2019).
Morgavi, D. P., Forano, E., Martin, C. & Newbold, C. J. Microbial ecosystem and methanogenesis in ruminants. Animal. 4, 1024–1036 (2010).
Zhang, Z. et al. Convergent evolution of rumen microbiomes in high-altitude mammals. Curr. Biol. 26, 1873–1879 (2016).
Chen, H. Y., Li, C. Q., Chen, S. Y. & Xiao, H. Metagenomic analysis reveals hidden links between gut microbes and habitat adaptation among cave and surface dwelling Sinocyclocheilus species. Zool. Res. 44, 793–807 (2023).
Eisler, B. R. Atrazine Hazards to Fish, Wildlife and Invertebrates: a Synoptic Review vol. 85 (U.S. Department of the Interior, Fish and Wildlife Service, 1989).
Kwon, B. G. & Moon, K. R. Physicochemical properties of styrene oligomers in the environment. Sci. Total Environ. 683, 216–220 (2019).
Chen, Y. X. et al. Preliminary study of the newly discovered primate species Rhinopithecus strykeri at Pianma, Yunnan, China using infrared camera traps. Int. J. Primatol. 36, 679–690 (2015).
Chen, Y. X. et al. Population and conservation status of a transboundary group of black snub-nosed monkeys (Rhinopithecus strykeri) between China and Myanmar. Zool. Res. 43, 523–527 (2022).
Xiang, Z. F., Xiao, W., Huo, S. & Li, M. Ranging pattern and population composition of Rhinopithecus bieti at Xiaochangdu, Tibet: Implications for conservation. Chin. Sci. Bull. 58, 2212–2219 (2013).
Xiao, Z. S. et al. Developing camera-trapping protocols for wildlife monitoring in Chinese forests. Biodivers. Sci. 22, 704–711 (2014).
Yao, H. et al. Endozoochorous seed dispersal by golden snub-nosed monkeys in a temperate forest. Integr. Zool. 16, 120–127 (2021).
Grueter, C. C., Li, D., Ren, B. & Wei, F. Choice of analytical method can have dramatic effects on primate home range estimates. Primates. 50, 81–84 (2009).
Harris, S. et al. Home-range analysis using radio-tracking data–a review of problems and techniques particularly as applied to the study of mammals. Mammal. Rev. 20, 97–123 (1990).
Chen, Y. X. et al. Activity rhythms of coexisting red serow and Chinese serow at Mt. Gaoligong as identified by camera traps. Animals 9, 1071 (2019).
Wei, F. W. et al. Catalogue of mammals in China. Cent. Integr. Data Anal. Wis. Sci. Cent. 41, 487–501 (2021).
Zheng, G. M. A. Checklist on the Classification and Distribution of the Birds of China 4th edn (ed Zheng, G. M.) (Science Press, 2023).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, D. et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Gurevich, A. et al. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Zhu, W., Lomsadze, A. & Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 38, e132 (2010).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Acknowledgements
Z.F.X. discloses support for the research of this work from the National Natural Science Foundation of China [32171487] and China Huaneng Group [HY2022-D8]. Q.Q.S. discloses support for the research of this work from the National Natural Science Foundation of China [32400413], the Natural Science Foundation of Hunan [2023JJ41038] and Excellent Youth Project of Hunan Education Department [22B0257]. We express our gratitude for the support received from all aforementioned projects. We would also like to extend our gratitude to Prof. Ming Li and Dr. Xiaochen Wang from the CAS Key Laboratory of Animal Ecology and Conservation Biology, Institute of Zoology, Chinese Academy of Sciences, for their substantial support in the preparation of this article.
Author information
Authors and Affiliations
Contributions
Z.F.X. designed the experiments. C.Z. performed the experiments. C.Z., Y.X.C., and L.Y. compiled the data and conducted the analyses. C.Z. and Y.L. collected samples and data. C.Z., Y.X.C., L.Y., and Q.Q.S. wrote the paper. Y.Y., Y.C., and C.G. provided help in analysis. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Samuel Aroney, and Süleyman Yıldırım for their contribution to the peer review of this work. Primary Handling Editors: Sabina Leanti La Rosa and Tobias Goris. [A peer review file is available.].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, C., Yu, Y., Yue, L. et al. Gut microbiota profiles of sympatric snub-nosed monkeys and macaques in Qinghai-Tibetan Plateau show influence of phylogeny over diet. Commun Biol 8, 95 (2025). https://doi.org/10.1038/s42003-025-07538-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07538-6
