
Abstract
The differentiation of Th17 and iTreg is tightly associated with fatty acid metabolism. TGFβ1-induced iTreg differentiation from Th0 relies on fatty acid oxidation (FAO), whereas IL-6 with TGFβ1 shifts metabolism to Th17-preferred fatty acid synthesis (FAS). However, how IL-6 reprograms fatty acid metabolism remains unclear. Here, we unveiled that TGFβ1-activated JNK is recruited to the Klhl25 promoter by NF-YA. JNK then phosphorylates histone H3 at Ser10 to activate Klhl25 transcription, leading to the ubiquitination-dependent degradation of ATP-citrate lyase (ACLY) and the switch from FAS to FAO, which supports iTreg generation. Whereas, upon IL-6 signaling, NF-YA is phosphorylated by ERK, losing its DNA binding ability, which shuts off TGFβ1-JNK-mediated Klhl25 transcription and ACLY ubiquitination, thereby increasing FAS and supporting Th17 differentiation. This study demonstrated that KLHL25-ACLY module functions as a switch in response to TGFβ1 and IL-6 signals, playing a decisive role in the fate determination of iTreg/Th17 differentiation.

Similar content being viewed by others


Introduction
CD4+ T cells consist of multiple subpopulations, including Th1, Th2, Th17 and Treg, all of which play critical roles in regulating immune responses and inflammatory diseases1,2. Among them, the anti-inflammatory regulatory T cells (Treg) and pro-inflammatory T helper 17 cells (Th17) constitute a balance that is crucial for immune homeostasis3,4. A decreased iTreg/Th17 ratio, resulting from impaired iTreg generation or excessive Th17 differentiation, is often associated with the autoimmune disorders such as inflammatory bowel disease (IBD), psoriasis, rheumatic arthritis, etc5,6. Interestingly, both iTreg and Th17 are differentiated from activated T cells (Th0). TGFβ1 signaling determines the differentiation of Th0 into iTreg, which relies on the expression of the core transcription factor Foxp37; whereas, along with TGFβ1, additional IL-6 signaling induces the expression of transcription factor RORγt and cytokines IL-17, leading to the differentiation of Th0 into Th178. Thus, the presence or absence of IL-6 signaling is a decisive factor in determining whether TGFβ1-responsive Th0 differentiating into Th17 or iTreg. However, the molecular mechanism by which IL-6 signaling counteracts pre-existing TGFβ1 signaling remains poorly understood.
Recent studies have shown that the differentiation fate of iTreg and Th17 is tightly associated with fatty acid metabolism9. Th17 differentiation relies on aerobic glycolysis and fatty acid synthesis (FAS), a primary metabolic mode adopted by almost all rapidly proliferating cells, including Th0; In contrast, iTreg differentiation requires the establishment of fatty acid oxidation (FAO)10,11. Impairment of FAO significantly inhibits Th0 differentiating into iTreg, while conversely, inhibition of FAS not only restrains Th17 differentiation but also promotes the transition of Th0 into iTreg12,13. FAS is controlled by three rate-limiting enzymes in cytoplasm, including ATP-citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN). ACLY catalyzes mitochondrial-derived citrate to produce acetyl-CoA, which is then utilized by ACC for the formation of malonyl-CoA. FASN uses both malonyl-CoA and acetyl-CoA as substrates to continuously synthesize long-chain fatty acids. Notably, malonyl-CoA functions as the physiological inhibitor of carnitine palmitoyltransferase 1 (CPT1), the rate-limiting enzyme in FAO pathway. Thus, when FAS is vigorous, FAO is naturally inhibited by the adequate malonyl-CoA. On the contrary, a reduction of FAS, caused by decreased ACLY or ACC activity, relieves this inhibition of CPT1, allowing FAO to proceed. During the activation of naïve CD4+ T cells into Th0 upon the ligation of TCR/CD28 co-stimulation, the expression of ACLY, ACC and FASN is upregulated through mTOR-SREBP1 signaling14. Interestingly, our previous study revealed that, among the three rate-limiting enzymes of the FAS pathway, ACLY exhibited the most significant decrease in iTreg15. In response to TGFβ1, ACLY is degraded via ubiquitin ligase CUL3-KLHL25-mediated ubiquitination, leading to the reduction of downstream product malonyl-CoA. This results in the inhibition of FAS and the promotion of FAO, thus favoring iTreg differentiation15. Although the role of IL-6 signaling in the differentiation of Th0 into Th17 has been well established, how TGFβ1-induce FAO is converted by IL-6 signaling to control the fate of cell differentiation has not yet been explored.
Given that Th17 differentiation relies on FAS, which requires relatively high expression and activity of ACLY, the reversal of TGFβ1-induced ACLY degradation is thought to be crucial for responding to IL-6 signaling and is essential for Th17 differentiation. In the present study, we showed that IL-6-activated ERK restrains the binding of JNK/NF-YA to the promoter of Klhl25, thereby suppressing TGFβ1-triggered ubiquitination and degradation of ACLY. This ultimately enhances FAS and thus promotes Th17 differentiation. Hence, the KLHL25-ACLY module function as a switch in response to TGFβ1 and IL-6 signaling, playing a decisive role in determining the differentiation fate of iTreg versus Th17.
Results
Reversal of TGFβ1-induced ubiquitination-dependent ACLY degradation is crucial for Th17 differentiation upon IL-6 addition
Our previous study unveiled that TGFβ1 signaling triggers ubiquitination-dependent ACLY degradation, leading to the establishment of FAO and favoring iTreg differentiation15. Given that along with TGFβ1, additional IL-6 signaling induces the differentiation of Th0 into FAS-preferred Th17, we speculated that IL-6 may modify fatty acid metabolism to stabilize and/or enhance FAS, thereby promoting Th17 differentiation. To test this, modes of iTreg and Th17 differentiation in vitro were set up (Fig. S1a) and [U-13C]-labeled glucose or palmitate tracing experiments were performed to assess de novo FAS and FAO, respectively. Intriguingly, we observed a significant reduction in de novo FAS and an enhancement of FAO during the differentiation of Th0 into iTreg (Figs. 1a, b, S1b, c); whereas, these alterations were not seen in Th17 (Figs. 1a, b, S1b, c), confirming that FAS is required for Th17 differentiation. Specifically, compared to the high expression of ACLY, ACC, and FASN, the key enzymes in de novo FAS pathway, in Th0, ACLY expression and activity were significantly reduced upon exposure to TGFβ1 alone for iTreg differentiation. However, when IL-6 was added along with TGFβ1, we observed significantly elevated levels of ACLY expression and activity (Fig. 1c, d, S2a–d). Next, to verify the key role of ACLY in IL-6-induced Th17 differentiation, ACLY activity was reduced by inhibitor SB204990, or Acly expression was knocked down by targeting siRNAs. While iTreg differentiation was enhanced as a control, IL-6-induced Th17 differentiation was significantly impaired by these pharmacological and genetic interventions, which however, was recovered by the supply of malonyl-CoA, the downstream product of ACLY in FAS pathway (Figs. 1e, S3a, b). Conversely, overexpression of ACLY reduced iTreg differentiation while increased IL-6-induced Th17 differentiation. This enhancement was abolished upon treatment with an ACC inhibitor (Figs. 1f, S3c). These results revealed that high-level and -activity of ACLY are indispensable for inducing Th17 differentiation.

Analyses of FAS and FAO in iTreg and Th17. Naïve CD4+ T cells were cultured under Th0, iTreg or Th17 polarization conditions for 48 h in the presence of [U-13C] glucose (11 mM) (a) or [U-13C] palmitate (100 μM) (b). Cells were collected and subjected to metabolic flux analysis for FAS (a) or FAO (b) by UHPLC-HRMS analysis. c,d Assay of the protein and enzymatic activity of ACLY in iTreg and Th17. Cells were cultured as described in the legend to (a) for 72 h and subjected to analyses for ACLY protein level by WB (c) and ACLY enzymatic activity by enzymatic activity assay kit (d). e Knockdown of Acly reduces Th17 differentiation. Naïve CD4+ T cells isolated from mice were transfected with negative control (NC) or Acly targeting siRNAs and were cultured as described in the legend to (a) for 72 h. Malonyl-CoA (50 μM) was added (or not) to the culture at the onset. Cells were stained for CD4/IL-17A to identify Th17 prior to flow cytometry (FCM) analysis (left) and quantification (right). f Overexpression of ACLY enhances Th17 differentiation. Cells transfected with pMIG-Acly overexpression virus were polarized as described in the legend to (e) and were stained for CD4+IL-17A+Th17 analysis by FAM (left) and quantification (right). For (a–f), data are representative as mean ± SD (n = 4 (a) or 3 (b–f) biologically independent samples) with p values determined by one-way ANOVA test. For WB in (c, e, f), one representative experiment out of three is represented. The values indicate mean intensities based on three biological replicas.
To explore the mechanism by which ACLY is regulated during Th17 differentiation, the expression and degradation of ACLY were examined. The transcription of Acly remained unchanged upon the treatment of TGFβ1, or TGFβ1 along with IL-6 (Fig. 2a). We then employed cycloheximide (CHX), a well-documented ribosome inhibitor commonly used to block protein synthesis, during either Th17 or iTreg differentiation to assess ACLY protein level. The time dynamic analysis showed that IL-6 stimulation led to a recovery of TGFβ1-triggered ACLY reduction, even in the presence of CHX (Fig. 2b), suggesting that IL-6 inhibits the degradation of ACLY. As expected, this inhibitory effect on ACLY degradation was attributed to the reversal of TGFβ1-triggered ACLY ubiquitination (Figs. 2c, S4a). These data indicated that IL-6 reverses TGFβ1-induced ubiquitination-dependent ACLY degradation, thereby stabilizing ACLY protein levels and promoting FAS, which ultimately facilitates Th17 differentiation.

a Detection of Acly mRNA level in iTreg and Th17. Naive CD4+ T cells were cultured under iTreg or Th17 polarization conditions for 4 days. Total RNA and cDNA were obtained at indicated times for Acly mRNA level analysis. b IL-6 inhibits ACLY degradation. Naïve CD4+ T cells were cultured as described in the legend to (a) for 48 h and treated with CHX (10 μg/ml) as indicated prior to WB analysis (left). Quantification of ACLY levels (right). c IL-6 inhibits ACLY ubiquitination. Cells polarized as described in the legend to (a) for 48 h were treated with MG132 (10 μM) for 6 h and cell extracts were immunoprecipitated with antibodies against ACLY. IgG serves as a negative control. d IL-6 reduces the interaction of ACLY with CUL3-KLHL25. Cells polarized as described in the legend to (c) and cell extracts were immunoprecipitated with antibodies against ACLY. IgG serves as a negative control. e, f Impact of IL-6 on the transcription and expression of Cul3 and Klhl25. Cells were polarized as described in the legend to (a) for indicated time. The mRNA level of Klhl25 (up) and Cul3 (down) were analyzed by qPCR (e). The protein level of CUL3 and KLHL25 were determined by WB (f). g KLHL25 overexpression abolishes the inhibitory effect of IL-6 on the interaction of ACLY with CUL3. Naïve CD4+ T cells transfected with pMIG-Klhl25 were polarized as described in the legend to (c). Cell extracts were immunoprecipitated with antibodies against ACLY before WB analysis. IgG serves as a negative control. h KLHL25 overexpression abolishes the inhibitory effect of IL-6 on ACLY ubiquitination. Cells prepared as described in the legend to (g) were lysed and immunoprecipitated with antibodies against ACLY before WB analysis. IgG serves as a negative control. Data are representative as mean ± SD (n = 3 (a, e) or 4 (b–d, f–h) biologically independent samples) with p values determined by two-way ANOVA test (a, b, e, f) or one-way ANOVA test (c, d, g, h). For WB in (b–d, f–h), one representative experiment out of three is represented. The values indicate mean intensities based on three biological replicas.
IL-6-elicited suppression of TGFβ1-induced Klhl25 transcription accounts for the abolishment of ACLY ubiquitination
Our previous study showed that ubiquitin ligase CUL3-KLHL25 mediates TGFβ1-induced ACLY ubiquitination, facilitating the shift in fatty acid metabolism from FAS, utilized by Th0, into iTreg-preferred FAO15. CUL3-KLHL25 complex belongs to Cullin-RING ubiquitin ligase family, the largest class of ubiquitin ligases. In brief, CUL3 is the core scaffolding protein holding the entire complex together, while KLHL25 serves as an adaptor for substrate recognition16,17. So, the interaction of ACLY with CUL3-KLHL25 upon treatment of TGFβ1 alone or along with IL-6 was examined. The result showed that addition of IL-6 attenuated the binding of ACLY with CUL3-KLHL25, which was enhanced by TGFβ1 (Figs. 2d, S4b), and correspondingly altered ACLY ubiquitination (Fig. 2c). Then, the kinetics of the mRNA and protein levels of both Cul3 and Klhl25 were analyzed. The result showed that the mRNA and protein levels of Klhl25 were elevated upon TGFβ1 stimulation, which were abolished by the addition of IL-6; whereas, those of CUL3 were scarcely changed (Fig. 2e, f). To corroborate that transcription suppression of Klhl25 accounts for the effect of IL-6 on Th17 differentiation induction, we conducted retroviral infection-mediated overexpression of KLHL25, followed by exposing the cells to TGFβ1 and IL-6. The results from Co-IP assays showed that when overexpression of KLHL25 increased the interaction of CUL3 with ACLY in iTreg (Fig. S4c, d), the impairment of CUL3 binding with ACLY as well as the decrease in ubiquitination of ACLY both induced by IL-6 treatment were also reversed by overexpression of KLHL25 (Figs. 2g, h, S4e, f). In parallel, the enhancement of FAS and the reduction of FAO were also reversed in Th17 (Figs. 3a–c, S5a, b). Ultimately, the differentiation of Th17 was reduced upon the overexpression of KLHL25. Intriguingly, this decrease was recovered by supply with malonyl-CoA, an important intermediate in FAS pathway (Fig. 3d). Taken together, the combined data suggested that IL-6 suppresses ACLY ubiquitination by mitigating TGFβ1-mediated transcription of Klhl25, thereby sustains FAS to favor Th17 differentiation.

a,b KLHL25 overexpression reduces FAS and enhances FAO. Naïve CD4+ T cells transfected with pMIG-Klhl25 overexpression virus were cultured under iTreg or Th17 polarization conditions in the presence of [U-13C] glucose (11 mM) (a) or [U-13C] palmitate (100 μM) (b) for 48 h. Cells were collected and subjected to metabolic flux analyses for FAS and FAO by ultra-high performance liquid chromatography-high resolution mass spectrometry (UHPLC-HRMS) analysis. c KLHL25 overexpression reverses the inhibitory role of IL-6 on FAO. Naïve CD4+ T cells transfected with pMIG-Klhl25 overexpression virus were cultured under iTreg of Th17 polarization conditions for 48 h. For oxygen consumption rate (OCR) detection, cells were transferred to XF Base Medium containing palmitate and L-carnitine. Diagram (left) illustrating the OCR at various conditions and associated quantifications (right) are shown. d Overexpression of KLHL25 inhibits Th17 differentiation. Cells cultured as described in the legend to (c) were treated with (or without) malonyl-CoA (MCoA, 50 μM) for 72 h and then stained for CD4+IL-17A+Th17 analysis by FAM (left) and quantification (right). For (a–d), data are representative as mean ± SD (n = 4 (a) or 3 (b–d) biologically independent samples) with p values determined by one-way ANOVA test. For WB in (d), one representative experiment out of three is represented.
TGFβ1 promotes the transcription activation of Klhl25 through NF-YA-mediated binding of JNK to the Klhl25 promoter
To decipher how IL-6 suppresses TGFβ1-induced Klhl25 transcription to maintain the stability of ACLY protein, it is essential to first investigate the molecular mechanism underlying TGFβ1-induced Klhl25 transcription, which was not addressed in our previous study. The well-known effective signal mediators downstream of TGFβ1 receptor, such as Smad3, ERK, JNK, and p3818,19, were examined. The result showed that the TGFβ1-induced increase in both mRNA and protein levels of Klhl25 was abolished by inhibition of JNK using an inhibitor that targeting its phosphorylation; In contrast, inhibitors targeting the phosphorylation of Smad3, ERK or p38 had no effect (Fig. 4a, b). Accordingly, the effects of TGFβ1 on both the binding of CUL3 with ACLY and the ubiquitination-dependent degradation of ACLY were diminished upon JNK inhibition (Figs. 4c–e, S6a, b). Furthermore, these changes triggered by TGFβ1 alone and/or in conjunction with JNKi were largely enhanced upon KLHL25 overexpression (Fig. S6a–d). Moreover, JNK inhibition suppressed TGFβ1-induced FAO and iTreg differentiation, which were enhanced when KLHL25 was overexpressed (Fig. 4f, g). Likewise, knockdown of Acly using targeted siRNA could also reverse the effect of the JNK inhibitor on iTreg differentiation (Fig. 4h). Taken together, the combined data indicated that TGFβ1 promotes Klhl25 transcription, which is dependent on JNK activation; the expression of Klhl25 leads to ACLY degradation, resulting in enhanced FAO and iTreg differentiation.

a, b JNK inhibition reduces the transcription and protein level of Klhl25. Naïve CD4+ T cells cultured under iTreg polarization condition were treated with specific inhibitors against Smad3, ERK, JNK, and p38 at the onset for 48 h. a Klhl25 mRNA level was assessed by qPCR. b KLHL25 protein level was analyzed by WB. c,d JNK inhibition attenuates the binding of CUL3-KLHL25 with ACLY and ACLY ubiquitination. Naïve CD4+ T cells transfected with pMIG-Klhl25 overexpression virus were cultured under iTreg polarization condition in the presence of JNK inhibitor (10 μM) for 48 h. Cells were treated with MG132 (10 μM) for 6 h before immunoprecipitated with antibodies against ACLY, and then the binding of ACLY with CUL3-KLHL25 was assessed by WB (c), the ubiquitination of ACLY was determined by WB (d). IgG serves as a negative control. e JNK inhibition suppresses TGFβ1-induced ACLY degradation. Naïve CD4+ T cells were transfected and cultured as described in the legend to (c, d) before the analysis of ACLY protein level by WB. (f,g) Impact of KLHL25 overexpression on CPT1 activity and iTreg differentiation upon JNK inhibition. Cells were transfected and cultured as described in the legend to (c, d) before analysis for CPT1 enzymatic activity assay by kit (f). Cells transfected and cultured as described in the legend to (c, d) for 72 h were stained for CD4+ Foxp3+ iTreg analyses by flow cytometry (FCM) (left) and quantified (right) (g). h Impact of Acly knockdown on iTreg differentiation upon JNK inhibition. Naïve CD4+ T cells transfected with Acly targeting siRNAs were cultured as described in the legend to (g) and then stained for CD4+ Foxp3+ iTreg analyses by FCM (left) and quantified (right). For (a–h), data are representative as mean ± SD (n = 3 biologically independent samples) with p values determined by one-way ANOVA test. For WB in (b–h), one representative experiment out of three is represented. The values indicate mean intensities based on three biological replicas.
Since JNK directly binds to gene promoter regions and phosphorylates H3S10 to promote gene transcription20, a dual luciferase reporter analysis was conducted. The data showed that TGFβ1 treatment increased the transcriptional activity of Klhl25 promoter; however, this effect was abolished by JNK inhibition (Fig. 5a). Importantly, the chromatin immunoprecipitation (ChIP) experiments showed that JNK bound to the Klhl25 promoter upon TGFβ1 treatment, leading to enhanced H3S10 phosphorylation. However, both JNK binding and H3S10 phosphorylation were markedly attenuated following the administration of a JNK inhibitor (Fig. 5b, c).

a JNK inhibition reduces the promoter activity of Klhl25. HEK293T cells were transfected with pGL 4.0-Klhl25 and cultured in the presence of JNK inhibitor and (or) TGFβ1 before dual luciferase reporter assay. b JNK associates with Klhl25 promoter. Naïve CD4+ T cells were cultured under iTreg or Th17 polarization conditions in the presence (or absence) of JNK inhibitor for 48 h and subsequently subjected to ChIP analysis for JNK binding on Klhl25. IgG serves as a negative control. c JNK inhibition reduces the H3S10 phosphorylation on Klhl25 promoter. Cells polarized as described in the legend to (b) and subjected to ChIP analysis for H3S10 phosphorylation on Klhl25 promoter. IgG serves as a negative control. d Inhibition of JNK reduces the interaction of JNK with NF-YA. Cells were cultured as described in the legend to (b). Cell extract was immunoprecipitated with antibodies against JNK (left) and NF-YA (right) followed by WB measurement. IgG serves as a negative control. e NF-YA and JNK co-localizes on Klhl25 promoter. Cells were cultured as described in the legend to (b) and subjected to re-ChIP analysis for the co-localization of NF-YA and JNK on Klhl25 promoter. IgG serves as a negative control. f Inhibition of JNK scarcely changes the binding of NF-YA to Klhl25 promoter. Cells were cultured as described in the legend to (b) and subjected to ChIP analysis for NF-YA association with Klhl25 promoter. IgG serves as a negative control. g Knockdown of Nfya attenuates the association of JNK with Klhl25 promoter. Naive CD4+ T cells were electrotransfected with siRNAs against Nfya and cultured as described in the legend to (b). Cells were then subjected to ChIP analysis for JNK binding on Klhl25. IgG serves as a negative control. h Knockdown of Nfya reduces Klhl25 transcription. Cells were cultured as described in the legend to (g) and Klhl25 mRNA level was detected by qPCR. For (a–h), data are representative as mean ± SD (n = 3 biologically independent samples) with p values determined by one-way ANOVA test (a, d, h) or two-way ANOVA test (b, c, e–g). For WB in (d, h), one representative experiment out of three is represented. The values indicate mean intensities based on three biological replicas.
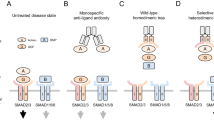
The site specificity of JNK binding to gene promoter relies on the transcription factor NF-Y, a trimeric complex including NF-YA, NF-YB and NF-YC20. NF-YA is the dominant DNA binding subunit within the NF-Y complex21. Consequently, Co-IP and Re-ChIP experiments were conducted, showing that upon TGFβ1 treatment, NF-YA and JNK bound to each other in a manner dependent on JNK activation; also, they co-localized at the promoter of Klhl25 (Fig. 5d, e). When JNK phosphorylation was inhibited, both the interaction and co-localization at Klhl25 promoter were declined (Fig. 5d, e); however, the binding of NF-YA’ s own to Klhl25 promoter was unchanged (Fig. 5f). Notably, knockdown of Nfya with targeted siRNAs significantly disrupted the binding of JNK to the Klhl25 promoter and attenuated TGFβ1-induced transcription of Klhl25 (Fig. 5g, h), indicating that NF-YA is indispensable for the effect of JNK on activating Klhl25 transcription. These data suggested that TGFβ1 activates JNK, which is then recruited by NF-YA to the Klhl25 promoter, thereby promoting transcription activation of the target gene.
IL-6-induced ERK activation abolishes NF-YA binding to the Klhl25 promoter
To investigate whether IL-6 reprograms fatty acid metabolism to promote Th17 differentiation by reversing JNK-mediated transcription activation of Klhl25, we conducted a dual luciferase reporter assay. The results showed that the addition of IL-6 dampened TGFβ1-triggered transcriptional activation of the Klhl25 promoter (Fig. 6a). Given the critical role of JNK in TGFβ1-induced Klhl25 transcription, the effect of IL-6 on JNK expression and phosphorylation was analyzed. IL-6 did not influence JNK expression or phosphorylation in the presence of TGFβ1 (Fig. S7a, b). Furthermore, the nuclear localization of JNK remained unchanged following IL-6 treatment (Fig. S7c). However, IL-6 treatment significantly inhibited the binding of JNK to the Klhl25 promoter (Fig. 6b). Additionally, phosphorylation of H3S10 was reduced (Fig. 6c). Notably, IL-6 treatment also diminished the binding of NF-YA to the Klhl25 promoter induced by TGFβ1 (Fig. 6d). Furthermore, the co-localization of JNK and NF-YA at the Klhl25 promoter triggered by TGFβ1 were reduced by IL-6 treatment, although the interaction between JNK and NF-YA remained unchanged (Fig. S8a, b). These data suggested that the loss of DNA binding activity of NF-YA upon IL-6 incubation is responsible for the decreased transcription of Klhl25.

a IL-6 inhibits the transcriptional activity of Klhl25 promoter. HEK293T cells were transfected with pGL 4.0-Klhl25 and treated with IL-6 and (or) TGFβ1 before dual luciferase reporter assay. b IL-6 attenuates JNK association with Klhl25 promoter. Naïve CD4+ T cells were cultured under iTreg or Th17 polarization conditions for 48 h and subsequently subjected to ChIP analysis for JNK binding on Klhl25. IgG serves as a negative control. c IL-6 reduces the H3S10 phosphorylation on Klhl25 promoter. Cells were cultured as described in the legend to (b) and subsequently subjected to ChIP analysis for H3S10 phosphorylation on Klhl25 promoter. IgG serves as a negative control. d IL-6 attenuates the association of NF-YA with Klhl25 promoter. Cells cultured as described in the legend to (b) were subjected to ChIP analysis for the binding of NF-YA on Klhl25 promoter. IgG serves as a negative control. e,f IL-6 enhances the binding of ERK with NF-YA and the phosphorylation of NF-YA. Cells cultured as described in the legend to (b) were treated with (or without) ERK inhibitor (15 μM). Cell extracts were immunoprecipitated with antibodies against NF-YA (e) or ERK (f) for WB analysis using indicated antibodies. IgG serves as a negative control. g ERK inhibition abolishes the inhibitory effect of IL-6 on the association of NF-YA on Klhl25 promoter. Cells were cultured as described in the legend to (f) and subsequently subjected to ChIP analysis for NF-YA binding on Klhl25 promoter. IgG serves as a negative control. h IL-6-triggered-ERK inhibits the binding of NF-YA to Klhl25 promoter in vitro. Cells cultured as described in the legend to (f) were lysed and nuclear extracts were harvested. Subsequently, the nuclear extracts were incubated with 100 fmol Cy5-labeled DNA oligos (WT, or with a mutation of NF-YA binding site) from Klhl25 promoter for 10 min on ice (total volume is 10 µL). Also, the non-labeled DNA oligos from Klhl25 promoter (10 pmol), called cold probes, was used. Retardation of protein/DNA complex was analyzed by EMSA. For (a–h), data are representative as mean ± SD (n = 3 biologically independent samples) with p values determined by one-way ANOVA test (a, e, f) or two-way ANOVA test (b–d, g, h). For WB in (e, f) and EMSA (h), one representative experiment out of three is represented. The values indicate mean intensities based on three biological replicas.
To further uncover the mechanism of IL-6 inhibiting NF-YA binding to Klhl25 promoter, the signaling pathways downstream of IL-6 receptor, mainly including JAK-STAT3 and ERK, were considered22,23. Given that NF-YA is a substrate of activated ERK, which phosphorylates NF-YA and reduces its DNA-binding activity24, the ERK inhibitor was applied in the presence of IL-6. The result showed that the addition of IL-6 promoted the interaction of ERK with NF-YA, and enhanced NF-YA phosphorylation, which were compromised in response to ERK inhibition (Fig. 6e, f). As a control, ERK inhibition couldn’t reverse these changes in the absence of IL-6 (Fig. S8c). Moreover, the ChIP result showed that the inhibition of IL-6 on the binding of NF-YA to Klhl25 promoter was reversed by ERK inhibitor treatment (Fig. 6g), likewise, the co-localization of JNK with NF-YA at the Klhl25 promoter was also recovered upon ERK inhibition (Fig. S8d). Importantly, the effect of IL-6 signaling-activated ERK on the binding of NF-YA/JNK to Klhl25 promoter was confirmed by using human peripheral blood T cells (Fig. S8e). Furthermore, an EMSA was performed using a probe derived from Klhl25 promoter, containing NF-YA binding site (located at −968 to −939). The results showed that the NF-YA from the nuclear extract of the cells treated with TGFβ1 was able to bind to the Klhl25 promoter-derived probe, which was significantly inhibited by IL-6 treatment; However, when ERK was inhibited, the IL-6-induced inhibitory effect was significantly reversed (Fig. 6h). Moreover, the binding of NF-YA to the probe was notably reduced upon the addition of a cold probe or due to the NF-YA binding-site mutation, confirming the specificity of NF-YA binding (Fig. 6h). These data suggested that IL-6 activates ERK, which subsequently phosphorylates NF-YA, thereby impairing its DNA-binding ability.
In addition, ERK inhibition abolished IL-6-induced reduction in Klhl25 promoter transcriptional activity, as well as the decrease in both mRNA and protein levels of Klhl25 (Fig. 7a–c). As a control, the expression of Klhl25 did not change upon STAT3 inhibition, and ERK inhibition did not suppress Klhl25 transcription in the absence of IL-6 (Figs. 7b, c, S8f), indicating the specificity of ERK in regulation Klhl25 transcription in response to IL-6. Importantly, the effects of IL-6 signaling-activated ERK on the Klhl25 expression, the interaction of ACLY with CUL3-KLHL25, as well as the ubiquitination and degradation of ACLY were also confirmed by using human T cells (Figs. 7d, S9a–e). To further signify the ERK/NF-YA circuit in controlling ACLY protein stability and Th17 differentiation, Nfya knockdown was performed in the presence of ERK inhibition. The results showed that IL-6-induced reduction in the mRNA and protein levels of Klhl25, ACLY ubiquitination, FAO metabolism, and the increase in Th17 differentiation were all reversed upon ERK inhibition. However, the effects of ERK inhibition were significantly attenuated following Nfya knockdown (Figs. 7e–i, S9f). Similarly, when Klhl25 was knocked down, the effects of ERK inhibition on the binding of ACLY with CUL3, ubiquitination-dependent ACLY degradation as well as Th17 differentiation were all abolished (Figs. 7j, S10a–e). Importantly, the effects of Nfya and Klhl25 knockdown on IL-6-induced Th17 differentiation were also confirmed by using human T cells (Fig. S9g). On the other hand, when ACLY was overexpressed, the reduction of Th17 differentiation triggered by ERK inhibition was significantly recovered (Fig. S10f). Taken together, these data suggested that NF-YA, regulated by TGFβ1-JNK or IL-6-ERK signaling, acts as an executor to activate or inactivate Klhl25 transcription, thereby determining the differentiation fate of iTreg or Th17.

a IL-6-triggered-ERK reduces the transcriptional activity of Klhl25 promoter. HEK293T cells were transfected with pGL 4.0-Klhl25 and treated with IL-6 and (or) TGFβ1 in the presence (or absence) of ERK inhibitor (15 μM) before dual luciferase reporter assay. b,c ERK but not STAT3 inhibition rescues Klhl25 mRNA and protein level. Naïve CD4+ T cells were cultured under iTreg or Th17 polarization conditions for 48 h in the presence (or absence) of ERK inhibitor and STAT3 inhibitor (10 μM). The mRNA level of Klhl25 was assayed by qPCR (b) and the protein level was detected by WB (c). d IL-6-triggered-ERK inhibits the association of ACLY with CUL3-KLHL25 during human Th17 differentiation. Naive CD4+ T cells isolated from human peripheral blood were cultured under iTreg or Th17 polarization conditions for 48 h in the presence (or absence) of ERK inhibitor. Cells were immunoprecipitated with antibodies against ACLY for WB analysis. IgG serves as a negative control. e,f Knockdown of Nfya abolishes the effect of ERK inhibition on the transcription and expression of Klhl25. Naive CD4+ T cells transfected with Nfya targeting siRNAs were cultured under iTreg or Th17 polarization conditions in the presence (or absence) of ERK inhibitor and subsequently subjected to qPCR (e) or WB (f) analysis. g Knockdown of Nfya abolishes the effect of ERK inhibition on ACLY ubiquitination. Cells transfected and cultured as described in the legend to (e, f) were treated with MG132 before immunoprecipitated with antibodies against ACLY (g) or Ub (h) for WB analysis. IgG serves as a negative control. h,i Knockdown of Nfya abolishes the effect of ERK inhibition on FAO and Th17 differentiation. h Cells were cultured as described in the legend to (e, f). For oxygen consumption rate (OCR) detection, cells were transferred to XF Base Medium containing palmitate and L-carnitine. Diagram (up) illustrating the OCR at various conditions and associated quantifications (down) are shown. i Cells cultured as described in the legend to (e, f) for 72 h were analyzed by flow cytometry (FCM) for the differentiation of CD4+ IL-17A+Th17 (left) and quantified (right). j Knockdown of Klhl25 abolishes the effect of ERK inhibition on Th17 differentiation. Naive CD4+ T cells transfected with Klhl25 targeting siRNAs were cultured under iTreg or Th17 polarization conditions in the presence (or absence) of ERK inhibitor and subsequently analyzed by FCM for the differentiation of CD4+ IL-17A+Th17 (left) and quantified (right). For (a–j), data are representative as mean ± SD (n = 3 biologically independent samples) with p values determined by one-way ANOVA test. For WB in (c, d, f, g), one representative experiment out of three is represented. The values indicate mean intensities based on three biological replicas.
Due to the limitations of the in vitro differentiation system, where only a small portion of cells become polarized, pure iTreg and Th17 were obtained from Foxp3YFP and IL-17AGFP mice to further confirm the crucial role of NF-YA in the transcriptional regulation of Klhl25 in iTreg and Th17. The data showed that iTreg expressed higher levels of KLHL25, displayed more interaction between CUL3 and ACLY, and exhibited more ACLY ubiquitination compared to Th0 and Th17 (Fig. S11a–e). Furthermore, the co-binding of NF-YA/JNK to the Klhl25 promoter and the H3S10 phosphorylation of this region were enhanced in iTreg, while these effects were correspondingly reversed in Th17 (Fig. S11f, g). In addition, we found that the phosphorylation of NF-YA was increased in Th17 compared to Th0 or iTreg (Fig. S11h), suggesting that the activation of the IL-6 signaling pathway during Th17 differentiation promotes the phosphorylation of NF-YA and inhibits its DNA-binding activity. These data strongly support the important role of NF-YA in regulating Klhl25 transcription in iTreg and Th17.
KLHL25 may play an important role in autoimmune diseases by regulating Th17 differentiation
IBD, as a common digestive system disease, poses a serious threat to public health, and modulation of Th17 function holds promise for the therapeutic treatment25,26. We utilized a classic mouse IBD model by the adoptively transferring of naive CD4+ T cells into Rag1-/- recipient mice27, to perform a functional assay of KLHL25 in IBD pathogenesis. Naive CD4+ T cells were isolated and infected with a retrovirus overexpressing KLHL25 prior to their polarization into iTreg, Th17, or Th17 (KLHL25OE) (Fig. 8a). Compared to the control, Th17 differentiation was reduced by KLHL25 overexpression (Fig. S12a). Mice injected with naive CD4+ T cells alone developed severe colitis, manifested by body weight loss (Fig. 8b), an increase in disease activity index (DAI) score (Fig. 8c), colon length shortening (Fig. 8d), inflammatory cell infiltration, mucosal edema and injury, and an increase in crypt damage (Fig. 8e), indicating successful model establishment. As expected, mice injected with naive CD4+ T cells along with iTreg showed more Treg and fewer Th17 in the colon lamina propria (cLP) and mesenteric lymph nodes (MLNs) (Figs. 8f, g, S12b, c), and significantly alleviated pathological alterations associated with colitis (Fig. 8b–e). In contrast, mice injected with naive CD4+ T cells along with Th17 showed fewer Treg and more Th17 in both the cLP and MLNs, accompanied by more severe symptoms (Figs. 8b–g, S12b, c). Notably, when injected with naive CD4+ T cells along with Th17 (KLHL25OE), mice showed fewer Th17 and more Treg in both the cLP and MLNs, resulting in further alleviation of colitis compared to those injected with Th17 (Figs. 8b–g, S12b, c). Using the same T-cell adoptive transfer IBD model, we found that injection of Th17, treated with ACLY inhibitor SB204990 during its differentiation, also effectively relieved T cell transfer-induced colitis in mice, compared to that injected with normal differentiated Th17 (Fig. S13a–f). Moreover, oral administration of the ACLY inhibitor to mice resulted in a reduction of Th17, an increase of Treg population in colonic lamina propria (cLP), and alleviation of dextran sodium sulfate (DSS)-induced colitis, another classic mouse IBD model (Fig. S14a-f). Together, these in vivo data confirm the important role of KLHL25-mediated Th17 and iTreg differentiation in colitis alleviation. Additionally, we employed an imiquimod (IMQ)-induced murine model of psoriasis, another autoimmune disease closely associated with the aberrant differentiation of Th1728. The results showed that, similar to the positive control dexamethasone, SB204990 administration significantly ameliorated the pathological alterations of psoriasis-like dermatitis compared to the IMQ-alone group, as indicated by the skin pathological changes, scores of skin erythema, thickness, scaling, psoriasis area and severity index (PASI) score, and spleen size (Fig. S15a–f). Moreover, fewer Th17 and more Treg were observed in the spleens of mice treated with SB204990 (Fig. S15g, h). Taken together, these data revealed that the “KLHL25-ACLY” module may be an important target for the treatment of various autoimmune diseases.

a Experimental design. b–g Systematic evaluation of colitis in mice adoptively transferred with different cells. Body weight (b) and disease activity index (DAI) score (c) based on body weight loss, stool consistency, and blood in the stool were recorded weekly. 7 weeks later, entire colons from mice treated in different ways were removed for length assessment (d) and hematoxylin and eosin (H and E) staining, which is used for revealing histopathological changes (e). Scale bar, 200 mm. One representative experiment out of four biological replicas is represented. Treg (f) and Th17 (g) frequency in the lamina propria of colon (cLP) were analyzed by flow cytometry. For (b–g), data are representative of three independent experiments. Graphs represent the mean ± SD (n = 4 mice) with p values determined by two-way ANOVA test (b, c) or one-way ANOVA test (d, f, g).
Discussion
The differentiation fate of Th0 cells into the inflammation-suppressing iTreg or the inflammation-promoting Th17 is primarily determined by the specific cytokine environment8. Recent evidence also suggested that the differentiation of iTreg and Th17 is tightly associated with fatty acid metabolism12,13. However, how fatty acid metabolism is programmed or reprogrammed by different cytokine signaling, thereby impacting cell differentiation, remains poorly understood. While our previous study uncovered that TGFβ1 promotes the differentiation of FAO-preferred iTreg, relying on CUL3-KLHL25-mediated ubiquitination-dependent degradation of ACLY15, the present study further identified that KLHL25-ACLY module functions as a switch in responses to TGFβ1 and IL-6 signaling, dictating the type of fatty acid metabolism and playing a decisive role in the fate determination of iTreg/Th17 differentiation.
IL-6 is a classical proinflammatory cytokine that is rapidly produced during infection and tissue injury, contributing to host defense by stimulating acute phase responses, hematopoietic and immune responses22,29. Promotion of Th17 differentiation is one of the major pro-inflammatory functions of IL-622. Regarding the underlying mechanism, it is well established that IL-6 promotes the expression of the Th17 lineage-specific transcription factor Rorc by activating STAT3, STAT3 then enhances the transcription and expression of Hif1α, and subsequently, STAT3/HIF1α complex promotes the transcriptional activation of Rorc. Ultimately, STAT3/HIF1α/RORγt form a transcription factor complex with Smad2/3-Trim33, leading to the upregulation of the proinflammatory cytokine IL-1730,31. Interestingly, IL-6-induced HIF1α also binds to the iTreg lineage-specific transcription factor Foxp3, mediating its ubiquitination-dependent degradation and inhibiting iTreg differentiation30. Our previous study documented that the level of ACLY significantly declines during iTreg differentiation upon TGFβ1 stimulation15. This reduction is due to CUL3-KLHL25-mediated ACLY ubiquitination15. Here, we unveiled that in response to IL-6 signaling, CUL3-KLHL25-mediated ubiquitination-dependent degradation of ACLY is suppressed, leading to a shift in fatty acid metabolism from FAO to FAS, thereby promoting the differentiation of Th0 into Th17. Evidently, rewriting the protein ubiquitination profile constitutes a layer of control that IL-6 signaling employes to direct Th17 differentiation.
Importantly, the present study elucidated that the transcription regulation of Klhl25 is crucial for the stability of ACLY, thereby controlling the fate of Th0 differentiation in response to different cytokine contexts. KLHL25 is a newly discovered scaffold protein, and by forming a complex with the ubiquitin ligase CUL3, KLHL25 is responsible for recognizing and binding substrates, thereby mediating their ubiquitination-dependent degradation16. This study demonstrated that the regulation of ACLY stability during iTreg/Th17 differentiation depends on the presence or absence of the ubiquitin ligase adaptor protein KLHL25 under distinct cytokine contexts. Notably, recent studies have revealed a significant correlation between KLHL25 expression and the survival of patients with childhood asthma, tick-borne encephalitis, and non-small-cell lung cancer32,33. Mutations in KLHL25 are also likely involved in the development of thyroid cancer34. Given that immune balance, mediated by iTreg and Th17, plays a critical role in the development of asthma, viral infection, and tumors35, the present study may provide new insights into the role of KLHL25 in these pathological processes, likely related to the imbalance of the differentiation of Th17 and iTreg.
The present study unveiled that transcription factor NF-Y is crucial for the transcription upregulation of Klhl25. NF-Y is a heterotrimeric complex consisting of three different subunits: NF-YA, NF-YB, and NFY-C. NF-YB and NF-YC form a stable heterodimer that interacts with the conserved segment of NF-YA. Mediated by NF-YA, NF-Y binds to the CCAAT DNA consensus sequence36. NF-Y regulates the expression of various cell type-specific genes that are activated by different physiological signaling pathways and are essential for normal tissue homeostasis, survival, and metabolic functions37. The roles of NF-Y in immune response have not been well understood. As a key factor for the expression of Tet1 and Tet2, NF-Y participates in the establishment of Treg-specific hypomethylation pattern and the stability of Foxp3 expression38. In the present study, NF-YA was revealed to interact with Klhl25 promoter and recruit JNK, leading to the transcriptional upregulation of Klhl25 in response to TGFβ1, subsequently promoting the differentiation of iTreg. However, the addition of IL-6 activates ERK, which then phosphorylates NF-YA, weakening its binding to DNA and resulting in the transcriptional suppression of Klhl25, thereby shifting Th0 differentiation toward Th17. Thus, our studies uncovered the pivotal role of NF-Y in fate decision of iTreg/Th17 differentiation, providing new insights into the role of NF-Y in immune response and relevant diseases.
Conventionally, TGFβ1 signaling activates the transcription factors Smad2/Smad3, which promote the transcriptional activation of Foxp3, and Foxp3 then plays a crucial role in the induction of iTreg differentiation39. On the other hand, classical signaling downstream of IL-6 mainly involves JAK/STAT3, which participates in RORγt expression and subsequently Th17 differentiation30. In addition, non-canonical MAPK (ERK/JNK/p38) signal pathways also affect iTreg/Th17 differentiation18; however, the underlying mechanisms are not well understood. The present study elucidated that TGFβ1-activated JNK interacts with the transcription factor NF-YA to promote the transcription activation of Klhl25, leading to ubiquitination-dependent ACLY degradation and ultimately inducing iTreg differentiation. In contrast, IL-6-induced activation of ERK results in the phosphorylation of NF-YA, reducing its DNA binding activity and thus reversing the JNK-activated Klhl25 transcription. Then the ubiquitination-dependent ACLY degradation is halted, and the fatty acid anabolism gets sustained. JNK and ERK are the classic members of the MAPK family involved in multiple biological processes, such as cell proliferation, differentiation, apoptosis, and tumorigenesis. Interestingly, they often play opposite roles in these processes. For instance, JNK promotes the activation of FOXO3 by phosphorylation at Ser574 in response to stress signals in tumor cells40, while ERK phosphorylates FOXO3 at Ser294, Ser344, and Ser425 to recruit the MDM2 E3 ubiquitin ligase, leading to FOXO3 ubiquitination and proteasomal degradation41. Additionally, BAD is a pro-apoptotic BCL2 family BH3-only protein that plays an essential role in promoting apoptosis and neuron development. JNK catalyzes the phosphorylation of the BAD at Ser128 to directly activate it, thereby promoting apoptosis42, whereas ERK-activated RSK phosphorylates BAD at Ser112, inactivating it and thereby inhibiting apoptosis43. The present study demonstrated that JNK and ERK differentially regulate Klhl25 transcription and fatty acid metabolism, which in turn plays a crucial role in the fate determination of Th0 differentiation into iTreg or Th17.
The imbalance between iTreg and Th17 is a key factor in a variety of autoimmune diseases, such as IBD and psoriasis3. Our study revealed that increasing the ubiquitination of ACLY through KLHL25 overexpression significantly inhibits Th17 differentiation and alleviates T cell adoptive transfer-induced IBD in mice. Meanwhile, administration of an ACLY inhibitor significantly reduced the frequency of Th17 and increased that of iTreg in mice, subsequently alleviating both IBD and psoriasis. Since the regulation of iTreg and Th17 in human is more complicated than in mice44,45, whether the growing list of ACLY inhibitors also exerts a protective effect in alleviating human IBD and other autoimmune diseases requires further investigation, despite our study demonstrating the regulatory role of ACLY inhibitors in human iTreg and Th17 differentiation in vitro.
Being used to treat immune diseases, ACLY inhibitor, hydroxycitrate (HCA), Radicicol (RAD) and SB204990 are able to reduce the production of pro-inflammatory cytokine prostaglandin E2 in macrophages, thereby alleviating the inflammatory response46. ACLY inhibition by HCA reduced the secretion of inflammatory cytokines IL-6 and IL-1β in TNFα-triggered human hepatocytes and in macrophages from subjects with Metabolic (Dysfunction)-Associated Steatohepatitis (MASH)47. ACLY-deficient CD4+ T cells exhibit an impaired capacity to induce intestinal inflammation in a transfer colitis model compared to wild-type T cells48. Furthermore, ACLY inhibitors have also been shown to lower blood glucose levels and triglyceride levels in the liver in diabetic animal models49. Notably, BemA, a small molecule that inhibits liver-specific ACLY, was approved by the U.S. Food and Drug Administration (FDA) in 2020 for the control and treatment of hypercholesterolemia49. Thus, our study, in conjunction with previous studies, clarified that ACLY inhibitors may possess important pharmacological potential. Moreover, developing targeted delivery strategies for ACLY inhibitors into CD4+ T cells or specific tissues is of great importance, as this approach could minimize the risk of toxicity and side effects in other tissues.
Our previous and present studies pinpointed KLHL25-ACLY module as a regulatory node that integrates TGFβ1 and IL-6 signaling with iTreg/Th17 differentiation, offering new insights into the modulation and maintenance of immune homeostasis. Moreover, ACLY inhibitors, which hold important pharmaceutical potentials46,49,50, are of considerable interest for immunotherapy targeting autoimmunity and unwanted inflammation, warranting further investigation.
Materials and Methods
Animals
C57BL/6 (B6) and BALB/c mice were purchased from Beijing HFK Bioscience Co., Ltd, China, and B6/Rag1−/− mice were purchased from Jiangsu Gempharmatech Co., Ltd, China. Foxp3YFP and Il17AGFP mice were provided by Bin Li (Shanghai Institute of Immunology, Shanghai Jiao Tong University School of Medicine, Shanghai, China) and Zhinan Yin (Guangdong Provincial Key Laboratory of Tumor Interventional Diagnosis and Treatment, Zhuhai Institute of Translational Medicine, Zhuhai People’s Hospital Affiliated with Jinan University, Jinan University, Zhuhai, China; The Biomedical Translational Research Institute, Faculty of Medical Science, Jinan University, Zhuhai, China), respectively. Mice were bred and cohoused 4-6 mice per cage, in a specific pathogen-free facility with a standard 12 h alternate light/dark cycle at an ambient temperature of 22 ± 2 °C and 30–70% humidity as described previously51 at the Animal Research Center of Northeast Normal University (Changchun, China). Health status of mice was determined via daily observation by technicians supported by veterinary care. Humane endpoint was established for monitoring mice, such that if there is a rapid weight loss reaching 10–15% of their body weight, the mice will be euthanized immediately to prevent them from suffering severe pain or distress. We have complied with all relevant ethical regulations for animal use. All experiments involving mice were conducted in accordance with the protocols for animal use, treatment, and euthanasia that were approved by the Animal Care Committee of Northeast Normal University (Authorization number: 202302035).
Induction of mouse iTreg and Th17 in vitro
Naïve CD4+ T cells were isolated from the spleens of 6- to 8-week-old mice using the MojoSort™ Mouse CD4+ Naïve T Cell Isolation Kit (480040, Biolegend) according to the manufacture’s instruction. For Th0 induction, 2–3 × 105 naïve CD4+ T cells were stimulated with Dynabeads® Mouse T-Activator CD3/CD28 (11453D, Gibco) at a cell:bead ratio of 1:1 and cultured in RPMI 1640 medium (R66504, Sigma-Aldrich) supplemented with 10% FBS (04-001-01, Biological Industries), penicillin-streptomycin (500 U, P06-07100, PAN biotech), β-mercaptoethanol (50 μM, M6250, Sigma-Aldrich), anti-IFNγ (10 μg/ml) and anti-IL-4 (10 μg/ml) as previous described with minor modifications15. To obtain iTreg, rhTGFβ1 (2 ng/ml, Peprotech) was simultaneously added along with the reagents used for inducing Th0 cells13,15. To obtain Th17, rhTGFβ1 (2 ng/ml, Peprotech) and IL-6 (20 ng/ml, Peprotech) were simultaneously added along with the reagents used for inducing Th0 cells as described previously52 Based on different experimental designs, cells were kept in medium containing the following regents. ACLY inhibitor: SB204990 (154566-12-8, Tocris Bioscience); ACC inhibitor: PF05175157 (TOCRIS, 5709); Proteasome inhibitor: MG132 (S2619, Selleck); ERK inhibitor: U0126 (tlrl-u0126, InvivoGen); JNK inhibitor: SP600125 (Selleck, S1460); p38 inhibitor: SB203580 (tlrl-sb20, InvivoGen); Smad3 inhibitor: SIS3 HCl (S7959, Selleck); STAT3 inhibitor: S31-201(S1155, Selleck).
To obtain pure iTreg or Th17, naïve CD4+ T cells were isolated from spleen of Foxp3YFP mice or Il17AGFP mice, followed by cultured under iTreg or Th17 polarization conditions, as shown above, for 3 days. The pure iTreg (YFP+) or Th17 (GFP+) were harvested by flow cytometry for subsequent experiments.
Induction of human iTreg and Th17 in vitro
Human peripheral blood samples were collected from the Jilin Blood Center (Changchun, China). All procedures involving human blood samples received approval from the local ethics committee (The ethical committee of the Northeast Normal University) (Authorization number: 202302035), and informed consent was obtained from all participants. After performing a Ficoll (Sigma Aldrich) gradient, naïve CD4+ T cells were isolated using the MojoSort™ Human CD4+ Naïve T Cell Isolation Kit (480042, BioLegend) in accordance with the manufacturer’s instructions53. For Th0 induction, 2–3 × 105 naïve CD4+ T cells were stimulated with Dynabeads® human T-Activator CD3/CD28 (11132D, Gibco) at a cell:bead ratio of 1:1 and cultured in RPMI 1640 medium (Sigma) supplemented with 10% FBS (Biological Industries), penicillin-streptomycin (500 U, PAN biotech), β-mercaptoethanol (50 μM, Sigma-Aldrich), anti-IFNγ (10 μg/ml) and anti-IL-4 (10 μg/ml) as previous described with minor modifications15. To obtain iTreg, rhTGFβ1 (2 ng/ml, Peprotech) was simultaneously added along with the reagents used for inducing Th0 cells as previous described with minor modifications15,53. To obtain Th17, rhTGFβ1 (2 ng/ml, Peprotech) and IL-6 (20 ng/ml, Peprotech) were simultaneously added along with the reagents used for inducing Th0 cells as previous described with minor modifications54.
Flow cytometry analysis
Cells were collected in PBS containing 1% FBS (v/v) and subsequently fixed and permeabilized using the Transcription Factor Buffer Set (562574, BD Biosciences) following the manufacturer’s instruction. For the analysis of mouse iTreg and Th17, selected protein markers were stained with FITC rat anti-mouse CD4 (553046), PE rat anti-mouse IL-17A (561020), and Alexa Fluor 647 rat anti-mouse Foxp3 (560401) monoclonal antibodies (BD Biosciences)15,55. For the analysis of human Th17, protein markers were stained with FITC rat anti-human CD4 (357406) and PE mouse anti-human IL-17A (560438) monoclonal antibodies (Biolegend)55. Flow cytometry analysis was carried out with BD FACS Canto II flow cytometer (BD Biosciences, San Jose, USA), and acquired data were analyzed using FlowJo, version 10, software (FlowJo) as described previously51. For gating, we used FSC (forward scatter) and SSC (side scatter) for gating to find viable, single cell events. Then, two parameter FITC and APC were examined for analysis of iTreg; and FITC and PE were examined for analysis of Th17.
Quantitative real-time PCR
Total RNA was isolated using the TRIzol reagent (15596-018, Invitrogen). Following this, cDNA was synthesized with the PrimeScript™ RT reagent Kit plus gDNA Eraser (RR047A, Takara) according to manufacturer’s instruction. Real-time PCR was performed on the QuantStudio 3 Real-Time PCR Instrument (Applied Biosystems) with a TB Green® Premix Ex Taq™ (Tli RNaseH Plus) regent (RR420A, Takara). The mRNA expression levels of the target genes were normalized to the expression levels of the β-actin gene. Data analysis was performed using the comparative cycling threshold method as previously described56. Quantitative PCR primers were as follows: Acly-F: AAGAAGGAGGGGAAGCTGAT, Acly-R: TCGCATGTCTGGGTTGTTTA; Klhl25-F: TCAGTCAGTGTTCACGAGACC, Klhl25-R: GCCTCAAAGTATCGGCTGGAG; Cul3-F: AGCCGGAAGGACACCAAGA, Cul3-R: GCTCCTCAAAACTAAGACCACTG; β-Actin-F: GGCTGTATTCCCCTCCATCG; β-Actin-R: GGCTGTATTCCCCTCCATCG.
siRNA transfection
All siRNA transfections were performed with mouse primary T cell nucleofection kit (VPA1006, Lonza) following the instructions using X-001 program as described previously57. Cells were transiently transfected with siRNA (siRNAs against ACLY, KLHL25, NF-YA were commercially synthesized, Shanghai GenePharma) at a final concentration of 20 nM. Then, cells were rested in RPMI-1640 (with 10%FBS) for 4–5 h followed by incubation under different conditions (iTreg or Th17) for a desired period of time. siRNA sequences were as follows: Acly siRNA-F: GCAAAGAACUCCUGUACAATT, Acly siRNA-R: UUGUACAGGAGUUCUUUGCTT; Klhl25 siRNA-F: CCGAGAUAUGGUGUCCAAATT, Klhl25 siRNA-R: UUUGGACACCAUAUCUCGGTT; Nfya siRNA-F: CCAGCAGAUCAUCAUCCAATT, Nfya siRNA-R: UUGGAUGAUGAUCUGCUGGTT; Negative control siRNA-F: UUCUCCGAACGUGUCACGUTT, Negative control siRNA-R: ACGUGACACGUUCGGAGAATT.
Plasmid construction
DNA sequences coding mouse Klhl25 gene (NCBI Accession No. NM_029652.1) and Acly gene (NCBI Accession No. NM_001199296.1) from mouse CD4+ T cells were amplified by PCR and subsequently cloned into pMSCV-IRES-GFP II (pMIG) vectors to construct recombinant pMIG-Klhl25, pMIG-Acly plasmids. The promoter DNA sequences of mouse Klhl25 gene (NCBI Accession Gene ID: 207952) was amplified by PCR and subsequently cloned into pGL 4.2 vectors to construct recombinant pGL 4.2-Klhl25 plasmid. The pMSCV-IRES-GFP II, pGL 4.2 and pCL-Eco vectors were purchased from addgene.
Retrovirus transduction of CD4+ T cells
Platinum-E cells, free of mycoplasma contamination, were cultured in 10-cm cell-culture dishes until they reached 70% confluence. Subsequently, the cells were transfected with pMIG-Klhl25 or pMIG-Acly plasmids. The supernatant was collected at 48, 72, and 96 h after transfection and was concentrated 100-fold in an ultracentrifuge. Aliquots were stored at −80 °C. Naïve CD4+ T cells were cultured under Th0-inducing condition as described above for 16–24 h and were spin-infected twice with retrovirus supernatants in the presence of 8 μg/mL polybrene (H8761, Solarbio) at 1000 × g, 90 min at 32 °C as described previously58. After 24 h of infection, supernatants were removed and cells were selectively cultured under either iTreg- or Th17-inducing condition for a desired period of time.
Assessment of ACLY enzyme activity
Cells were lysed using ultrasonic treatment in extracting buffer from the ACLY activity assay kit (ACLY-1-Y, Suzhou Comin). Supernatants were collected and used for the analysis of enzymatic activities following the manufacturer’s instructions as described previously15.
Immunoprecipitation and western blotting
Cells were lysed in cold western blot-immunoprecipitation (WB-IP) lysis buffer for 30 min and centrifuged (at 4 °C, 5 min at 14,000 × g) to remove cell debris. The supernatant was collected as whole cell lysate. ACLY, Ubiquitin (Ub), CUL3, NF-YA, ERK and JNK were immunoprecipitated from the whole cell lysates using anti-ACLY (ab40793, abcam), anti-Ubiquitin (ab7780, abcam), anti-CUL3 (sc-166110, Santa), anti-NF-YA (sc-17753, Santa), anti-ERK (4695S, CST) and anti-JNK (9252, CST) antibodies coupled to Protein A/G Magnetic Beads (B23202, Selleck) overnight at 4 °C. Nonspecific rabbit or mouse IgG antibody was used as a negative control. Immunoprecipitated proteins were mixed with 1×loading buffer and boiled at 100 °C for 10 min followed by western blotting as described previously56. For western blotting, protein samples were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). The membranes were blocked with 5% nonfat milk for 1 h at room temperature before being incubated with primary antibodies overnight at 4 °C. Following this, Mouse- or rabbit-conjugated HRP second antibodies were added to the membranes for 50 min at room temperature, followed by washed 3 times with 1×Tris-buffered saline with Tween 20 (TBST) and visualized using Chemiluminescent HRP Substrate (Millipore) on Chemiluminescence Image System (Tanon Science & Technology Co., Ltd). The intensity of the bands was quantified using ImageJ software. The antibodies for WB were anti-ACLY, anti-Ubiquitin, anti-CUL3, anti-NF-YA, anti-ERK, anti-JNK, anti-ERK-P (9101S, CST), anti-Smad3-P (9520S, CST), anti-JNK-P (4668, CST), anti-p38 (9212S, CST), anti-p38-p (9211S, CST), and anti-actin (HC201, TransGen).
Sequential ChIP assays
ChIP assays were performed as described previously with slight modifications56,59. Briefly, T cells were cross-linked with formaldehyde (final concentration, 1%) for 10 min and treated with glycine for 5 min at room temperature to terminate the cross-linking reaction. The cells were then washed with ice-cold PBS, and the nuclear pellet was extracted with Chromatin IP Kit (Cell signaling technology, US, #9003), following the manufacturer’s instructions. After sonication with Bioruptor (UCD-300, Diagenode), which was set to ‘high mode,’ samples were centrifuged at 9400 g(10 min, 4 °C). Supernatant was immunoprecipitated using anti-JNK (9252, CST), anti-H3S10-P (53348S, CST), or anti-NF-YA (sc-17753, Santa) antibodies overnight and with isotype IgG as a negative control. ChIP-Grade Protein G Magnetic Beads were added into the mixture and incubated for 2 h at 4 °C with rotation. After washing, DNA and proteins bound to beads were eluted in ChIP Elution Buffer. During reversing cross-links at 65 °C, proteins were digested with proteinase K for 2 h. The resulting DNAs were purified and eluted in distilled water followed by RT-qPCR analysis. The primer sequences were as follows: Klhl25 pro-F (−1100, −850): GCTCACCTGTGAGGGATTTT, Klhl25 pro-R (−1100, −850): GGATATGGGGGACTTTTGGT; Klhl25 pro-F (-850, -600): TTTGCTCTTTGTCTGCCCTTA, Klhl25 pro-R (−850, −600): GTAGCCCAACAATGGCTGTC; Klhl25 pro-F (−600, −350): TCAGTTTTGTACCGCTGAGAG, Klhl25 pro-R (−600, −350): TAGTTTCGATGCGAGGGAAT; Klhl25 pro-F (−350, −100): AACGGCTGTGATTCGGTAAC, Klhl25 pro-R (−350, −100): CCGTAATACAGTCGCGTGAG.
For sequential ChIP analysis, chromatin-bound material was treated with ChIP elution buffer containing 10 mM DTT for 30 min at 37 °C after the first immunoprecipitation. Subsequently, samples were adjusted in Re-ChIP buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton). The second immunoprecipitation was performed overnight at 4 °C, and DNA was purified as described above59.
13C-tracing assessment
13C-tracing assessments were performed as described previously with slight modifications60. In brief, cell cultures grown in media supplemented with either [U-13C] glucose (10 mM) or [U-13C] palmitate (100 μM) were homogenized using a combination of 10 mL formate and 800 mL chloroform. The mixture underwent four rounds of ultrasonication (1 min each) with 1 min cooling intervals in an ice-water bath, followed by equilibration at room temperature for 30 min. Subsequent centrifugation (3000 × g, 15 min) separated the phases. The chloroform layer was transferred to a clean glass vial and combined with 500 mL of 75% ethanol containing 0.5 M KOH, followed by incubation at 80 °C for 60 min. Hexane (600 mL) was then introduced, and the solution was vortexed vigorously for 1 min before phase separation during a 30 min rest period. After repeating centrifugation (3000 × g, 15 min), the hexane phase was evaporated to complete dryness. The dried material was derivatized by sequential addition of 10 mL 1-hydroxybenzotriazole (HoBt in DMSO), 20 mL cholamine (in DMSO with 200 mM TEA), and 10 mL HATU (in DMSO), with a 5 min room-temperature reaction. Prior to UHPLC-HRMS analysis, 60 mL acetonitrile was incorporated, and the samples were centrifuged (3000 × g, 15 min, 4 °C) to ensure clarity.
The UHPLC-MS/MS analysis was conducted using an Agilent 1290 Infinity II UHPLC system linked to a 6470 A Triple Quadrupole mass spectrometer (Santa Clara, CA, USA). The samples were injected onto a Waters UPLC BEH C18 column (100 mm × 2.1 mm, 1.7 μm) with a flow rate of 0.3 ml/min. The mobile phase consisted of water (phase A) and acetonitrile (phase B), both containing 0.1% formate. A gradient elution was used for chromatographic separation with the following conditions: 0 min, 10% B; 4 min, 40% B; 8 min, 45% B; 11 min, 50% B; 14 min, 70% B; 15 min, 90% B; 15.5 min, 100% B; 18 min, 100% B; 18.1 min, 10% B; 20 min, 10% B. The column was maintained at 40 °C. Data were processed using Agilent MassHunter Workstation Software (version B.08.00), applying the default parameters and conducting manual checks to ensure the accuracy of qualitative and quantitative results for each compound. The peak areas of the target compounds were integrated and subsequently subjected to IsoCorrection analysis.
Extracellular flux analysis
For the FAO-associated oxygen consumption rate (OCR) assay by Seahorse XFp Analyzer (Agilent), Palmitate-BSA reagent (Agilent) and Mito Stress Test Kits (Agilent) were used following the manufacturers’ instructions with minor modifications61. In brief, cells were harvested and seeded onto poly-D-lysine-coated XFp plates (4 to 6 × 105 cells per well) by centrifugation in serum-free XF Base Medium (Agilent). For the palmitate oxidation assay, cells were incubated in XF Base Medium (Agilent) in a non-CO2 incubator for 15–20 min at 37° C. Then, XF Palmitate-BSA FAO Substrates (Agilent) consisting of palmitate-BSA (167 mM palmitate conjugated with 28 mM BSA) and 0.5 mM L-carnitine (Sigma Aldrich) were added to the medium to assess the OCR. Immediately following, an XF Cell Mito Stress Test was conducted using a Seahorse XFp Analyzer (Agilent), where various mitochondrial function modulators were added sequentially, including 1 mM oligomycin (Oligo) (Agilent), 1.5 mM FCCP (Agilent), and 100 nM rotenone plus 1 mM antimycin A (Rot/Ant A) (Agilent). Data were processed with Wave software version 2.3.0 (Agilent) and analyzed using appropriate statistical methods with GraphPad Prism version 6 (GraphPad Software).
Luciferase reporter gene assay
To assess the activity of the Klhl25 promoter, HEK293T cells were plated in 6-well plates at a density of 1 × 105 cells per well. The pGL 4.2 or pGL 4.2-Klhl25 promoter reporter plasmids were transiently transfected into the cells along with a Renilla control plasmid. After approximately 6 h, the transfected cells were treated with ERK or JNK inhibitors, in the presence of TGFβ1 and/or IL-6, for 12 h before being harvested. Cell lysates were then analyzed using the Dual-Luciferase® Reporter Assay System (E1910, Promega) as previously described56. The relative luciferase activity of the Klhl25 promoter was normalized to the luciferase activity of the Renilla control plasmid.
Electrophoretic mobility shift assay
The EMSA was performed as described previously with slight modifications56. In brief, nuclear extracts (1 μg) were mixed with 100 fmol of Cy5-labeled oligonucleotide probe containing the NF-YA binding site or a Cy5-labeled mutant oligonucleotide probe in a binding buffer (20 mM HEPES (pH 7.6), 1.5 mM MgCl2, 10 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 1 mg/ml BSA) along with 1 μg of poly(dI-dC), making a total volume of 10 μl. In some experiments, a non-labeled oligonucleotide probe containing the NF-YA binding site was added to the reaction mixture. After a 10-min incubation on ice, the entire reaction was loaded onto a 6% non-denaturing polyacrylamide-Tris-borate-EDTA (TBE) gel and electrophoresed at 100 V for 1.5 h at 4 °C. The sequence information of the probe was as follows: Cy5-Klhl25 pro: AGCTCATTTACATTTCCAATGCTATACCAA; CP-Klhl25 pro: TTGGTATAGCATTGGAAATGTAAATGAGCT; Cold probe-Klhl25 pro: AGCTCATTTACATTTCCAATGCTATACCAA; Cy5-Klhl25 pro (Nfya-mut): AGCTCATTTACATTTAACTGGCTATACCAA; CP-Klhl25 pro (Nfya-mut): TTGGTATAGCCAGTTAAATGTAAATGAGCT.
Mouse model of colitis
T-cell transfer colitis based on adoptive transfer of naïve CD4+ T cells into Rag1−/− mice is widely used for studying chronic intestinal inflammation62. To validate the role of “KLHL25-ACLY” axis in regulating the differentiation of iTreg and Th17, and thus impacting IBD, same-sex Rag1−/− mice aged 6 to 8 weeks were randomly divided into different groups, Ctrl, Th0, iTreg, Th17, Th17 (KLHL25OE) or Th17 (SB204990), with four mice each group. Sample size was based on previous studies62. As described previously, 5 × 105 freshly isolated naïve CD4+ T cells were administrated into 6- to 8-wk old Rag1−/− mice by intraperitoneal injection to induce colitis. We induced the differentiation of iTreg and Th17 in vitro with/without the infection of KLHL25 overexpression virus or the incubation of ACLY inhibitor SB204990, to obtained Th0, iTreg, Th17, Th17 (KLHL25OE) or Th17 (SB204990) cells. Cells from differently-treated groups had distinct percentages of Th17/iTreg, and 5 × 105 of these cells were then respectively injected into Rag1−/− mice along with naïve CD4+ T cells. Meanwhile, Rag1−/− mice of blank control group were administrated with PBS. Moreover, the function of “KLHL25-ACLY” axis in acute colitis was also assessed in dextran sodium sulfate (DSS)-induced colitis model. Same-sex C57BL/6J mice aged 6 to 8 weeks were randomly divided into 3 groups, Ctrl, 2% DSS, 2% DSS + SB204990, with four mice each group. DSS (0216011080, MP Biomedicals) dissolved in drinking water (2%, w/v) was given ad libitum to mice for 7 days. Mice supplied with regular drinking water were included as negative control. ACLY inhibitor, SB204990, (37.5 mg/kg/d) (154566-12-8, Tocris Bioscience) was administrated orally every two days. To minimize potential confounders, administer the medication at fixed times each time, and each group should follow a fixed order of administration.
Evaluation of colitis in mice
To assess the severity of colitis, disease activity index (DAI) was weekly (T-cell transfer colitis) or daily (DSS-induced colitis) recorded by scoring the body weight loss, stool consistency and blood in the stool as described in the literature62,63,64. 7 weeks later (T-cell transfer colitis) or 7 days later (DSS-induced colitis) mice were euthanatized to assess histopathological changes in colon tissues. The whole colon was collected, and its length was measured. After cleaning with saline and fixation in formalin, the colon tissues were embedded in paraffin. The tissue sections (3 μm) were stained with hematoxylin and eosin (H&E) and then analyzed blindly for intestinal inflammation, following the previously described method63,64. In addition, the mononuclear cells from colon lamina propria (cLP) and mesenteric lymph nodes (MLNs) were prepared as described previously65 for the analysis of Treg or Th17 by flow cytometry. The experimental process adheres to the principle of blinding; individuals involved in group allocation are prohibited from participating in the conduction of the experiment, outcome assessment, and data analysis.
Imiquimod (IMQ)-induced psoriasis mouse model
Same-sex BALB/c mice aged 6 to 8 weeks were randomly divided into 5 groups, Ctrl, IMQ, IMQ+Dex, IMQ + DMSO, IMQ + SB204990, with 4 mice each group. Sample size was based on previous studies66. Mice were depilated on the back skin 2 days prior to treatment and subsequently treated daily with either Aldara (containing 5% IMQ, purchased from MED*SHINE) or sham cream on the back skin for consecutive 7 days as described previously66. SB204990 or DMSO, which were dissolved in vaseline, were spread on the skin twice a day. Dexamethasone as the positive control. To minimize potential confounders, administer the medication at fixed times each time, and each group should follow a fixed order of administration. Disease severity was evaluated using a scoring system that considered scaling, erythema, and skin thickness, akin to the human Psoriasis Area and Severity Index. However, this assessment did not account for the area affected, as it is determined by the experimenter. A cumulative score was derived from the three parameters mentioned. Erythema and scaling were rated on a scale from 0 to 4, where 0 indicated no severity and 4 represented high severity. Skin thickness was scored based on the increase in thickness compared to day 1, with scores assigned as follows: 1 for 20–40%, 2 for 40–60%, 3 for 60–80% and 4 for >80%. The experimental process adhered to the principle of blinding; individuals involved in group allocation were prohibited from participating in the execution of the experiment, outcome assessment, and data analysis.
Statistics and reproducibility
All experiments were done at least three times independently confirming consistent results. The number used for each experiment is shown in figure legend. Animals and cells were randomized before treatments and no data was deliberately excluded from the analysis. Statistical analysis was performed using GraphPad Prism version 8 (GraphPad Software). Data are displayed as mean ± standard (SD). The p values were calculated from Student unpaired t test when comparing within two groups. One-way or two-way ANOVA were performed in the indicated figures when more than two groups were compared as described previously51. p < 0.05 were considered significant. Exact p values were indicated on graphs.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw data for blots and quantification were uploaded in Figshare (https://doi.org/10.6084/m9.figshare.28451552; https://figshare.com/s/3d3c1e4a5a0b42f6677d). Plasmids were uploaded in Addgene (ID:234866, 234871). All data generated or analyzed during this study are available from the lead contact on reasonable request.
References
Hippen, K. L., Aguilar, E. G., Rhee, S. Y., Bolivar-Wagers, S. & Blazar, B. R. Distinct regulatory and effector T cell metabolic demands during graft-versus-host disease. Trends Immunol. 41, 77–91 (2020).
Brown, E. M., Kenny, D. J. & Xavier, R. J. Gut microbiota regulation of T cells during inflammation and autoimmunity. Annu. Rev. Immunol. 37, 599–624 (2019).
Noack, M. & Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 13, 668–677 (2014).
Lee, G. R. The balance of th17 versus treg cells in autoimmunity. Int. J. Mol. Sci. 19, 1–14 (2018).
Baker, K. F. & Isaacs, J. D. Novel therapies for immune-mediated inflammatory diseases: what can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann. Rheum. Dis. 77, 175–187 (2018).
Wu, R. et al. MicroRNA-210 overexpression promotes psoriasis-like inflammation by inducing Th1 and Th17 cell differentiation. J. Clin. Investig. 128, 2551–2568 (2018).
Richards, D. M. et al. Treg cell differentiation: from thymus to peripheral tissue. Regul. T Cells Health Dis. 136, 175–205 (2015).
Bettelli, E. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 (2006).
MacIver, N. J., Michalek, R. D. & Rathmell, J. C. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 31, 259–283 (2013).
Zeng, H. & Chi, H. Metabolic control of regulatory T cell development and function. Trends Immunol. 36, 3–12 (2015).
Wang, R. & Solt, L. A. Metabolism of murine TH17 cells: impact on cell fate and function. Eur. J. Immunol. 46, 807–816 (2016).
Berod, L. et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 20, 1327–1333 (2014).
Michalek, R. D. et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303 (2011).
Lochner, M., Berod, L. & Sparwasser, T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 36, 81–91 (2015).
Tian, M. et al. ACLY ubiquitination by CUL3-KLHL25 induces the reprogramming of fatty acid metabolism to facilitate iTreg differentiation. Elife 10, 1–27 (2021).
Zhang, C. et al. Cullin3-KLHL25 ubiquitin ligase targets ACLY for degradation to inhibit lipid synthesis and tumor progression. Genes Dev. 30, 1956–1970 (2016).
Genschik, P., Sumara, I. & Lechner, E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J. 32, 2307–2320 (2013).
Lu, L. et al. Role of SMAD and Non-SMAD signals in the development of Th17 and regulatory T cells. J. Immunol. 184, 4295–4306 (2010).
Zhang, Y. E. Non-smad signaling pathways of the TGF-β family. Cold Spring Harb. Perspect. Biol. 9, a022129 (2017).
Tiwari, V. K. et al. A chromatin-modifying function of JNK during stem cell differentiation. Nat. Genet. 44, 94–100 (2011).
Bungartz, G., Land, H., Scadden, D. T. & Emerson, S. G. NF-Y is necessary for hematopoietic stem cell proliferation and survival. Blood 119, 1380–1389 (2012).
Neurath, M. F. & Finotto, S. Cytokine & Growth Factor Reviews IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 22, 83–89 (2011).
Mihara, M., Hashizume, M., Yoshida, H., Suzuki, M. & Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 122, 143–159 (2012).
Aklilu, S. et al. Nfya-1 functions as a substrate of ERK-MAP kinase during Caenorhabditis elegans vulval development. Cells Dev. 169, 203757 (2022).
Zhang, Y. Z. & Li, Y. Y. Inflammatory bowel disease: pathogenesis. World J. Gastroenterol. 20, 91–99 (2014).
Ueno, A. et al. Th17 plasticity and its relevance to inflammatory bowel disease. J. Autoimmun. 87, 38–49 (2018).
Ostanin, D. V. et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am. J. Physiol. Liver Physiol. 296, G135–G146 (2009).
Bedoya, S. K., Lam, B., Lau, K. & Larkin, J. Th17 cells in immunity and autoimmunity. Clin. Dev. Immunol. 2013, 986789 (2013).
Tanaka, T., Narazaki, M. & Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 6, a016295 (2014).
Dang, E. V. et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell 146, 772–784 (2011).
Tanaka, S. et al. Trim33 mediates the proinflammatory function of Th17 cells. J. Exp. Med. 215, 1853–1868 (2018).
Giannos, P., Kechagias, K. S. & Gal, A. Identification of prognostic gene biomarkers in non-small cell lung cancer progression by integrated bioinformatics analysis. Biology10, 1–16 (2021).
Ignatieva, E. V., Yurchenko, A. A., Voevoda, M. I. & Yudin, N. S. Exome-wide search and functional annotation of genes associated in patients with severe tick-borne encephalitis in a Russian population. BMC Med. Genom.12, 1–23 (2019).
Chang, Y. S. et al. Detection of molecular alterations in taiwanese patients with medullary thyroid cancer using whole-exome sequencing. Endocr. Pathol. 29, 324–331 (2018).
Knochelmann, H. M. et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 15, 458–469 (2018).
Di Silvio, A., Imbriano, C. & Mantovani, R. Dissection of the NF-Y transcriptional activation potential. Nucleic Acids Res. 27, 2578–2584 (1999).
Maity, S. N. NF-Y (CBF) regulation in specific cell types and mouse models. Biochim. Biophys. Acta - Gene Regul. Mech. 1860, 598–603 (2017).
Yang, R. et al. Hydrogen Sulfide Promotes Tet1- and Tet2-Mediated Foxp3 Demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity 43, 251–263 (2015).
Schlenner, S. M., Weigmann, B., Ruan, Q., Chen, Y. & von Boehmer, H. Smad3 binding to the foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J. Exp. Med. 209, 1529–1535 (2012).
Tikhanovich, I. et al. Regulation of FOXO3 by phosphorylation and methylation in hepatitis C virus infection and alcohol exposure. Hepatology 59, 58–70 (2014).
Yang, J. Y. et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 10, 138–148 (2008).
Donovan, N., Becker, E. B. E., Konishi, Y. & Bonni, A. JNK phosphorylation and activation of bad couples the stress-activated signaling pathway to the cell death machinery. J. Biol. Chem. 277, 40944–40949 (2002).
Jin, K., Mao, X. O., Zhu, Y. & Greenberg, D. A. MEK and ERK protect-hypoxic cortical neurons via phosphorylation of Bad. J. Neurochem. 80, 119–125 (2002).
Shevach, E. M. & Thornton, A. M. tTregs, pTregs, and iTregs. Immunol. Rev. 259, 88–102 (2014).
Romagnani, S., Maggi, E., Liotta, F., Cosmi, L. & Annunziato, F. Properties and origin of human Th17 cells. Mol. Immunol. 47, 3–7 (2009).
Infantino, V., Iacobazzi, V., Palmieri, F. & Menga, A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Biophys. Res. Commun. 440, 105–111 (2013).
Convertini, P. et al. ACLY as a modulator of liver cell functions and its role in metabolic dysfunction-associated steatohepatitis. J. Transl. Med. 21, 1–19 (2023).
Schulz-Kuhnt, A. et al. ATP citrate lyase (ACLY)-dependent immunometabolism in mucosal T cells drives experimental colitis in vivo. Gut 73, 601–612 (2024).
Feng, X., Zhang, L., Xu, S. & Shen, A. ATP-citrate lyase (ACLY) in lipid metabolism and atherosclerosis: an updated review. Prog. Lipid Res. 77, 101006 (2020).
Khwairakpam, A. et al. ATP Citrate Lyase (ACLY): a promising target for cancer prevention and treatment. Curr. Drug Targets 16, 156–163 (2015).
Damasceno, L. E. A. et al. PKM2 promotes Th17 cell differentiation and autoimmune inflammation by fine-tuning STAT3 activation. J. Exp. Med. 217, e20190613 (2020).
Qu, Z. et al. PDLIM2 restricts Th1 and Th17 differentiation and prevents autoimmune disease. Cell Biosci. 2, 1–7 (2012).
Ghosh, S. et al. Regulatory T cell modulation by CBP/EP300 bromodomain inhibition. J. Biol. Chem. 291, 13014–13027 (2016).
Buchacher, T. et al. PIM kinases regulate early human Th17 cell differentiation. Cell Rep. 42, 113469 (2023).
Chung, Y. et al. Critical regulation of early Th17 cell differentiation by Interleukin-1 signaling. Immunity 30, 576–587 (2009).
Bannwarth, S. et al. Cell-specific regulation of TRBP1 promoter by NF-Y transcription factor in lymphocytes and astrocytes. J. Mol. Biol. 355, 898–910 (2006).
Mantei, A. et al. siRNA stabilization prolongs gene knockdown in primary T lymphocytes. Eur. J. Immunol. 38, 2616–2625 (2008).
Hochrein, S. M. et al. The glucose transporter GLUT3 controls T helper 17 cell responses through glycolytic-epigenetic reprogramming. Cell Metab. 34, 516–532.e11 (2022).
Zou, Y. et al. H4S47 O-GlcNAcylation regulates the activation of mammalian replication origins. Nat. Struct. Mol. Biol. 30, 800–811 (2023).
Johnson, M. O. et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 175, 1780–1795.e19 (2018).
Miguel, V., Ramos, R., García-Bermejo, L., Rodríguez-Puyol, D. & Lamas, S. The program of renal fibrogenesis is controlled by microRNAs regulating oxidative metabolism. Redox Biol. 40, 101851 (2021).
Furusawa, Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013).
Wirtz, S. et al. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 12, 1295–1309 (2017).
Wirtz, S., Neufert, C., Weigmann, B. & Neurath, M. F. Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2, 541–546 (2007).
Atarashi, K. et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science331, 337–341 (2011).
Xia, X. et al. GLS1-mediated glutaminolysis unbridled by MALT1 protease promotes psoriasis pathogenesis. J. Clin. Investig. 130, 5180–5196 (2020).
Acknowledgements
We thank Ping Wang (Tongji University Cancer Center, Shanghai Tenth People’s Hospital, School of Medicine, Tongji University, Shanghai, 200092, China) for critical comments. We also thank the following funding sources for their support: National Natural Science Foundation of China grant 32200729 (M.T.); National Natural Science Foundation of China grant 32070896 (M.W.); Department of Science and Technology of Jilin Province grant QT202304 (M.T.); Education Department of Jilin Province grant JJKH20231314KJ (M.T.); Education Department of Jilin Province grant JJKH20231312KJ (X.J.).
Author information
Authors and Affiliations
Contributions
Miaomiao Tian, Xueqing Ba and Min Wei designed research; Miaomiao Tian, Fengqi Hao, Xinyu Wang, Tianyi Chang, Shuang He, Huiyue Wang and Ying Jiang performed research; Miaomiao Tian, Fengqi Hao and Xin Jin analyzed data; Dan Li, Zhinan Yin provided reporter mice; Yang Wang, Jia Liu, Yunpeng Feng, Xueqing Ba and Min Wei supervised research; Miaomiao Tian, Fengqi Hao, Xueqing Ba and Min Wei wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Sutatip Pongcharoen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Toril Holien and Mengtan Xing.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tian, M., Hao, F., Jin, X. et al. KLHL25-ACLY module functions as a switch in the fate determination of the differentiation of iTreg/Th17. Commun Biol 8, 471 (2025). https://doi.org/10.1038/s42003-025-07917-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07917-z
