
Abstract
Aha1 is one of the well-known co-chaperones of Hsp90. However, the action mode of Aha1 has not been fully elucidated yet, and the binding mode of Aha1’s C-terminal domain (Aha1-CTD) to Hsp90 is still under discussion. Meanwhile, since both Hsp90 and Aha1 contribute to tumorigenesis through controlling the homeostasis of onco-proteins, Hsp90-Aha1 system might serve as a target for anti-tumor drug development. A few of active compounds towards Hsp90-Aha1 system have been reported during the past years, but no compound binding pocket in Aha1 was pictured yet. Here in this manuscript, by using the discovered dual-modulator Benzbromarone as the probe, the pocket in Aha1 responsible for compound recognition is defined. Interestingly, as shown by the cryo-EM structures of Hsp90:Aha1 system, it is the same pocket that is involved in the in vitro interaction between Aha1-CTD and Hsp90-MD. Besides, Benzbromarone’s binding to Hsp90-NTD also exhibits unique structural features. Not surprisingly, due to the interference with the Hsp90 machinery, Benzbromarone could down-regulate the ATPase activity of the chaperone. Finally, according to the cellular-based experimental data, Benzbromarone has been shown to exhibit cytotoxicity against multiple cancer cell types, at least in part, through its modulation of the Hsp90 system.
Similar content being viewed by others


Introduction
Molecular chaperones are a category of housekeeping proteins that assist in the correct folding of proteins and prevent them from misfolding and/or aggregation. Heat shock protein 90 (Hsp90) is one of the most important molecular chaperones in living systems except archaeobacteria. And it plays crucial roles in various physiological processes through helping with protein folding, assembling and ligand binding etc. Proteins that undergo structural activation with the assistance of Hsp90 are collectively referred to as “client proteins” of the chaperones1,2,3,4,5. Up to date, approximately 400 proteins including kinases, transcription factors and hormone receptors etc. have been identified as client proteins of Hsp901,2,6. As it has been known, quite a number of Hsp90’s client proteins such as Src, AKT, CDK2 and CDK4 etc. play important roles in the development of tumors. Mainly due to its modulation of the cellular homeostasis of the onco-proteins, Hsp90 system has become a target for anti-tumor drug development7,8,9,10,11,12,13.
Hsp90 exists as a homodimer in cells. Each Hsp90 protomer consists of three highly conserved structural domains which include the N-terminal ATP binding and hydrolysis domain (NTD), the middle domain (MD) for client protein recognition, and the C-terminal domain (CTD) for Hsp90’s dimerization in the resting state2,3,4,5. Due to the fundamental role of Hsp90 in cellular homeostasis of client proteins, the chaperoning cycle related to Hsp90 is tightly modulated by multiple factors. The binding and hydrolysis of ATP can induce a series of conformational changes in the chaperone, which leads to the activation or assembly of its client proteins1,2,3,4,5. In the absence of ATP, Hsp90 primarily forms homodimer through its CTD and adopts an open V-shape conformation. With the binding of ATP, the remaining structural domains of Hsp90 (NTD and MD) are oriented towards their corresponding partner domains in the protomers, and the closed conformation with Hsp90’s N-terminal domain dimerized is finally formed. Then, upon the hydrolysis of ATP and the release of ADP and free phosphate (Pi), Hsp90 returns to its initial V-shape conformation. Apart from ATP and ADP, the chaperoning cycle of human Hsp90 is also regulated by over 20 co-chaperones1,14,15,16,17,18,19,20,21,22. Cdc37, p23, and Aha1 etc. are all well-known co-chaperones for Hsp90. While different from the other co-chaperones including Cdc37 and p23 etc., Aha1 is the only co-chaperone that could significantly stimulate the ATPase activity of Hsp9019,21,23,24.
Aha1 is composed of two structured domains (the N-terminal domain and the C-terminal domain) which are linked together by a long unstructured linker region25,26. Currently, the physiological function of Aha1 mediated by its interaction with Hsp90 has not been fully understood yet, but according to the published literatures, a multi-step molecular mechanism is indicated for the binding of Aha1 to Hsp90 which is in couple with the activation of Hsp90’s ATPase activity24,26,27,28. Initially, Aha1 is recruited to Hsp90 through the interaction between its N-terminal domain (Aha1-NTD) and the middle domain of the chaperone (Hsp90-MD). It is worth noting that the conserved R73KxK76 motif in Aha1-NTD is demonstrated to interact with the catalytic loop residing in Hsp90-MD (Supplementary Fig. S1)26,29. As it has been shown, the spatial rearrangement of Hsp90’s catalytic loop and its positioning in proximity of the nucleotide are required for the full acceleration of the ATPase activity of the chaperone1,2,30. After the initial binding driven by Aha1’s N-terminal domain, the C-terminal domain of the protein (Aha1-CTD) has been proposed to interact with the N-terminal domain of Hsp90 (Hsp90-NTD) upon the presence of nucleotide and thus favor Hsp90-NTD’s dimerization. Specifically, [1H, 15N] HSQC titration data demonstrated that the AMPPNP-bound form of Hsp90-NTD showed specific binding of Aha1-CTD24. Besides, accumulated evidence has also revealed that Aha1-CTD can interact with Hsp90-MD under certain conditions27,31. For example, upon the phosphorylation of Tyr313 in Hsp90-MD, significant chemical shift perturbations were observed for a large set of Ile signals from Hsp90-MD in response to the addition of Aha1-CTD31. Overall, it seems like that Aha1-CTD binds to multiple Hsp90 domains in a dynamic fashion. Although the binding modes of Aha1’s two structured domains to Hsp90 might have not been fully pictured yet, both of them are required for the fully activation of Hsp90’s ATPase activity. As it has been demonstrated, full-length Aha1 could stimulate the ATPase rate of Hsp90 15- to 30-fold19,23,24,32,33. In the meanwhile, Aha1-NTD alone could only enhance the ATPase activity of Hsp90 by 1.5- to 3-fold24,33. Taking all aforementioned findings into account, chemical compounds binding to either Aha1-NTD or Aha1-CTD might be capable of interfering with Hsp90-Aha1 interaction and modulating the functional display of the chaperone.
In a preliminary study aiming to discover chemical inhibitors against Hsp90, we found that Benzbromarone, which is an approved uricosuric drug by targeting urate anion transporter 1 (URAT1), could bind to both Aha1-CTD and Hsp90-NTD with the corresponding affinities at hundreds of nanomolar and low micromolar range, respectively. After the confirmation of Benzbromarone’s binding to Aha1-CTD and Hsp90-NTD, biophysical studies were carried out to reveal the binding modes between the compound and the proteins. According to the NMR and X-ray crystallography data, Benzbromarone interacts with Hsp90-NTD by binding to the ATP recognition pocket in the chaperone, while it forms complex with Aha1-CTD by binding to the hydrophobic cavity composed of α3 (α-helix 3), β3-β4 loop and α2-β2 loop. Besides, since Aha1-CTD is involved in the interaction between Aha1 and Hsp90, we then performed NMR-based competitive binding assay to test whether Benzbromarone’s presence is capable of interfering with Hsp90-Aha1 interaction. The obtained data verified that Benzbromarone could inhibit the binding of Aha1-CTD to Hsp90-NTD in the presence of ATP analogue AMPPNP. In the meanwhile, cellular-based experiments demonstrated that Benzbromarone exhibits cytotoxicity against multiple cancer cell lines, at least in part through its modulation of the Hsp90 system.
Results
Benzbromarone binds to both Hsp90-NTD and Aha1-CTD
In our previous work, we occasionally found that Benzbromarone could potentially bind to both Hsp90-NTD and Aha1-CTD (Fig. 1A). To confirm this finding, NMR techniques and ITC approach were applied to study the interactions between Benzbromarone and Hsp90-NTD/Aha1-CTD. All the obtained data demonstrates that Benzbromarone could specifically interact with Hsp90-NTD and Aha1-CTD (Fig. 1B–D and Supplementary Fig. S2). Upon the addition of either Hsp90-NTD or Aha1-CTD, significant line-broadening was observed for the NMR resonances of Benzbromarone, which indicating a specific interaction between Benzbromarone and Hsp90-NTD/Aha1-CTD. Besides, the data derived from [1H, 15N] HSQC titration experiments and ITC further confirms the aforementioned conclusion. And the binding affinities for Benzbromarone to Hsp90-NTD and Aha1-CTD were determined to be 15.10 ± 3.86 µM (by NMR method) and 0.84 ± 0.35 µM (by ITC approach), respectively.

A Chemical structure of Benzbromarone. B Ligand-observed CPMG spectra indicate that Benzbromarone could specifically bind to Hsp90-NTD and Aha1-CTD. The CPMG spectra are colored in red for Benzbromarone alone, and colored in cyan for Benzbromarone with the presence of either Hsp90-NTD or Aha1-CTD. C Isothermal titration calorimetry approach was applied to determine the thermodynamic parameters for the binding of Benzbromarone to Aha1-CTD. The Kd value for Aha1-CTD:Benzbromarone system was calculated to be 0.84 ± 0.35 µM. D The dissociation constant for the binding of Benzbromarone to Hsp90-NTD was determined by the global fitting analysis of normalized resonance intensity changes (1-I/I0) revealed by [1H, 15N] HSQC titration experiments. The Kd value for Hsp90-NTD:Benzbromarone system was calculated to be 15.10 ± 3.86 µM.
Characterization of the interactions between Benzbromarone and Hsp90-NTD/Aha1-CTD
To map the binding site of Benzbromarone on Hsp90-NTD, we conducted [1H, 15N] HSQC NMR titration experiments. Upon the addition of Benzbromarone, the NMR resonances for a number of residues in Hsp90-NTD, which include I26, R46, S53, K58, I78, D86, G95, G97, D102, G132, F134, G135, V136, F138, S140, L188, Q194, E199, H210 and E223, fully attenuated (Fig. 2A–C). Meanwhile, the NMR resonances for amino acid residues including Q28, L32, K41, L45, I49, S50, Y61, H77, V92, D93, T94, I96, A117, M119, G125, W162 and K185 in Hsp90-NTD underwent significant chemical shift perturbations in response to the presence of the compound (Fig. 2A, B). Overall, most of the significantly perturbed residues localize to the ATP/ADP binding pocket of Hsp90-NTD which is mainly composed of the “lid” segment spanning A111 to G135 and its preceding loop-helix-loop region D93-I110, part of the α-helical structure starting from K41 to E62, and a few of residues from two β-strands ranging around T152 and T184. Therefore, the [1H, 15N] HSQC data suggests that Benzbromarone could interact with Hsp90-NTD through occupying its ATP binding and hydrolysis pocket. It should be noted that conformational exchange and line broadening already occur in the ATP-binding pocket and its adjacent “lid” segment of Hsp90 in the apo state. Therefore, the observed signal attenuations in response to the presence of Benzbromarone for residues located in this specific region may either indicate a direct contact with the compound or reflect coupling to the modification of the pre-existing conformational exchange. In line with the NMR data, the model structure for Hsp90-NTD complexed with Benzbromarone deriving from molecular docking indicates that Benzbromarone primarily binds to the hydrophobic pocket consisting of N51, S52, A55, M98, L103, L107, T109, I110, V136, Y139, V150 and V186 in Hsp90-NTD (Fig. 2D). Notably, the π-π stacking interactions between the benzofuran moiety of the compound and the aromatic rings of F138 and W162 in Hsp90-NTD contribute significantly to the complex formation. Additionally, hydrogen bonding interactions between Benzbromarone and T184, as well as those formed through water molecules with D93 and G97, are also crucial for the binding of the compound to Hsp90’s N-terminal domain. Interestingly, although the scaffold of the benzofuran moiety in Benzbromarone has a high similarity to that of the adenine group in AMPPNP (a nonhydrolyzable analogue of ATP), the positions of them in Hsp90-NTD are totally different to each other (Fig. 2D). This might be caused by the significant polarity difference between the 3,5-dibrom-4-hydroxyphenyl moiety of Benzbromarone and the phosphonoamidodiphosphoric group in AMPPNP.

A The binding sites of Benzbromarone in Hsp90-NTD were characterized by carrying out [1H, 15N] HSQC NMR titration experiments. Superposition of [1H, 15N] HSQC spectra of Hsp90-NTD without (red) and with the presence of Benzbromarone (black, molar ratio of 1:1 for Hsp90-NTD to Benzbromarone) reveals spectral changes upon Benzbromarone’s binding. The residues in Hsp90-NTD that might be involved in the recognition of Benzbromarone were identified by performing chemical shift perturbation (CSP) analysis (B) and NMR signal intensity change analysis C. The mean and mean + S.D. CSP values for amino acid residues in Hsp90-NTD are represented by dashed line and solid line, respectively. The mean relative intensity change value for amino acid residues in Hsp90-NTD is represented by dashed line. Residues with chemical shift perturbations greater than mean + S.D. are labeled. Proline residues and those residues with fully attenuated resonances upon the addition of Benzbromarone are denoted by green dots and red dots, respectively. D Expanded view of the possible binding mode for Benzbromarone to Hsp90-NTD, which was revealed by molecular docking. Detailed interactions between Benzbromarone and its surrounding residues in Hsp90-NTD are shown, and intermolecular hydrogen bond details are highlighted. The PDB code for Hsp90-NTD:AMPPNP complex structure is 3T1K. E The binding sites of Benzbromarone in Aha1-CTD were characterized by [1H, 15N] HSQC NMR titration experiments. Superposition of [1H, 15N] HSQC spectra of Aha1-CTD without (red) and with the presence of Benzbromarone (black, molar ratio of 1:2 for Aha1-CTD to Benzbromarone) reveals spectral changes upon Benzbromarone’s binding. F The residues in Aha1-CTD that might be involved in the recognition of Benzbromarone were identified by performing chemical shift perturbation (CSP) analysis. The mean and mean + S.D. CSP values are represented by dashed line and solid line, respectively. Residues with chemical shift perturbations greater than mean + S.D. are labeled. Proline residues and those with significantly attenuated resonances upon Benzbromarone’s addition are denoted by green dots and red dots, respectively. G The crystal structure of Aha1-CTD complexed with Benzbromarone (PDB code: 8Z3J) reveals the binding mode of the compound to the protein. The expanded view of Benzbromarone’s binding pose to Aha1-CTD is depicted in the left panel, illustrating detailed interactions between Benzbromarone and its surrounding residues within Aha1-CTD, with intermolecular hydrogen bonds highlighted. For comparison, the right panel shows a superposition of the crystal structure of Aha1-CTD complexed with Benzbromarone (PDB code: 8Z3J, in gray) and the solution NMR structure of Aha1-CTD in its apo state (PDB code: 1X53, in light green). H The ribbon and the surface presentation for the crystal structure of Aha1-CTD in free state (PDB code: 8Z3H) are shown in the left and the right panel, respectively. I The ribbon and the surface presentation for the crystal structure of Aha1-CTD complexed with Benzbromarone (PDB code: 8Z3J) are shown in the left and the right panel, respectively.
After the characterization of Benzbromarone’s interaction with Hsp90-NTD, [1H, 15N] HSQC NMR titration experiments and X-ray crystallography were jointly used to determine the binding mode of the compound to Aha1-CTD (Fig. 2E–I and Supplementary Table S1). With the addition of Benzbromarone, significant chemical shift changes were observed for amino acid residues including F226, T227, F235, H237, A240, V253, N256, S258, R316, W319, Y322, G326, T330, and Y333 in Aha1-CTD (Fig. 2E, F). Meanwhile, the NMR resonances for a few of residues such as M252, V257, K277, S278, G318, and Q320 almost fully attenuated in response to the exposure of Benzbromarone (Fig. 2E, F). Most of the aforementioned residues localize to α3 (α-helix 3), β3-β4 loop, and α2-β2 loop regions of Aha1-CTD. As known, the interaction between protein and ligand could lead to the local environment changes and thus the chemical shift perturbations of the amino acid residues either being involved in the direct contacts with ligand or undergoing conformational changes to facilitate ligand binding34,35,36,37. Therefore, to definitively identify the specific residues responsible for the binding of Benzbromarone to the Aha1-CTD, the high-resolution crystal structure of the Aha1-CTD in complex with Benzbromarone was determined (Fig. 2G). This structure aligns with the findings from NMR analysis, revealing that a hydrophobic cavity formed by residues F235, T236, V253, N256, V257, W274, F276, R314, Q317, W319, Y322, and Y323, which are predominantly located in the α3 (α-helix 3), β3-β4 loop, and α2-β2 loop regions of Aha1-CTD, is crucial for Benzbromarone recognition (Fig. 2G–I). Compared with the crystal structure of Aha1-CTD in free state, Q320, R321, and Y322 in α3 of the protein shift outside a bit for a better accommodation of Benzbromarone’s benzofuran moiety (Figs. 2H, I and Supplementary Fig. S3). Consistently, as shown by the pocket volume analysis data, the size of the pocket to accommodate Benzbromarone increases from 188 Å3 to 205 Å3 when the compound is present. Meanwhile, as illustrated in Fig. 2G, the van der Waals contacts between Benzbromarone and those amino acids in the hydrophobic pocket of Aha1-CTD serve as a driving force for the binding of the compound to the protein.
Benzbromarone interferes with the Hsp90-NTD:Aha1-CTD interaction and modulates the ATPase activity of Hsp90 system
After the elucidation of the binding modes between Benzbromarone and Hsp90-NTD/ Aha1-CTD, [1H, 15N] HSQC-based competitive binding experiments were performed to test whether Benzbromarone could interfere with the weak interaction between Hsp90-NTD and Aha1-CTD in the presence of ATP analogue AMPPNP. A series of [1H, 15N] HSQC spectra for 15N-labeled Aha1-CTD alone, 15N-labeled Aha1-CTD:unlabeled Hsp90-NTD (with AMPPNP) mixture sample, 15N-labeled Aha1-CTD:Benzbromarone mixture sample, and 15N-labeled Aha1-CTD:unlabeled Hsp90-NTD (with AMPPNP):Benzbromarone mixture sample were recorded. As shown in Fig. 3A and Supplementary Fig. S4, upon the presence of both Hsp90-NTD and Benzbromarone, the chemical shift perturbation patterns for representative residues of F226, H237, S258, I270, E313, and T330 in Aha1-CTD are very similar to the observed resonance perturbation patterns of them with the addition of only Benzbromarone. Therefore, the [1H, 15N] HSQC-based competitive binding data suggest that the presence of Benzbromarone disfavors the weak interaction between Hsp90-NTD and Aha1-CTD. This conclusion is further supported by the [1H, 15N] HSQC-based competitive binding experiments conducted by using 15N-labeled Hsp90-NTD with AMPPNP, 15N-labeled Hsp90-NTD with AMPPNP:unlabeled Aha1-CTD mixture sample, and 15N-labeled Hsp90-NTD with AMPPNP:unlabeled Aha1-CTD:Benzbromarone mixture sample (Supplementary Fig. S5). It appears that the induced chemical shift perturbations for representative residues of 15N-labeled Hsp90-NTD in response to the addition of unlabeled Aha1-CTD were abolished by the presence of Benzbromarone (Supplementary Fig. S5C, S5D).

A The results for [1H, 15N] HSQC-based competitive binding experiments are shown. Zoomed view of the superimposed [1H, 15N] HSQC spectra for 15N-Aha1-CTD alone (red), 15N-Aha1-CTD with the presence of Hsp90-NTD (with AMPPNP, blue, molar ratio of 1:5 for 15N-Aha1-CTD to Hsp90-NTD), 15N-Aha1-CTD with the presence of Hsp90-NTD and Benzbromarone (with AMPPNP, green, molar ratio of 1:5:8 for 15N-Aha1-CTD to Hsp90-NTD to Benzbromarone), and 15N-Aha1-CTD with the presence of Benzbromarone (purple, molar ratio of 1:8 for 15N-Aha1-CTD to Benzbromarone). The directional changes in chemical shift perturbations induced by Hsp90-NTD or Benzbromarone binding are indicated by black arrow and red arrow, respectively. B The determined ATPase activity data of Hsp90β (solid circle) and Hsp90β:Aha1(solid triangle) upon the treatment of Benzbromarone are shown. The ATPase rates obtained from the NADH-coupled ATPase experiments were plotted, along with the mean ± SD derived from three times of independent repeats (n = 3; each repeat contains one experimental data extracted from technical triplicates). Statistical significance was determined using unpaired t-test (***p < 0.001, ****p < 0.0001). C The ATP hydrolysis reaction catalyzed by Hsp90β was monitored by collecting 1D 31P NMR spectra. Overlay of 1D 31P spectra of the Hsp90β:ATP (4 μM:1 mM) reaction system without (red) and with the addition of Benzbromarone (400 µM, green) acquired at the time points of 2 h and 4 h after the initiation of reaction, are shown. D The ATP hydrolysis reaction catalyzed by Hsp90β and Aha1 was monitored by collecting 1D 31P spectra. Overlay of 1D 31P spectra of the Hsp90β:Aha1:ATP (4 μM:20 μM:1 mM) reaction system without (red) and with the addition of Benzbromarone (400 µM, green) are presented. The spectra were acquired at the time points of 20 min and 1 h after the initiation of reaction.
As demonstrated, Benzbromarone could bind to both Hsp90-NTD and Aha1-CTD, which may consequentially disrupt the interaction between two protein domains. However, given that the interaction between Aha1 and Hsp90 is primarily driven by the binding of Aha1-NTD (Aha1 N-terminal domain) to Hsp90-MD (Hsp90 middle domain), it is essential to evaluate the potential modulatory effect of the compound on Aha1’s Hsp90 activation activity. Therefore, the in vitro NADH-coupled ATPase assay and the ATP hydrolysis assay monitored by 1D 31P NMR spectra were carried out. For the in vitro NADH-coupled ATPase assay, the ATPase activity of full-length Hsp90β was measured by tracking the decrease of the absorbance at 340 nm for NADH in the reaction system containing full-length Hsp90β, ATP without or with Benzbromarone. For the 31P NMR approach, 1D 31P NMR spectra were acquired to detect the level changes of ATP, ADP, and free phosphate Pi in the reaction system containing full-length Hsp90β, ATP without or with Benzbromarone/NVP-AUY922 and in the reaction system containing full-length Hsp90β, ATP, Aha1 without or with Benzbromarone/NVP-AUY922. Compared to the reaction system without the addition of Benzbromarone/NVP-AUY922, the addition of either one of two compounds led to a decrease in the turnover rate or the ADP and Pi concentrations in the reaction system, which indicating that Benzbromarone and NVP-AUY922 could inhibit the ATPase activity of Hsp90β in its free state (Fig. 3B, C and Supplementary Fig. S6). Similarly, Benzbromarone was also found to inhibit the activation effect of full-length Aha1 on the activity of Hsp90β (Fig. 3B, D).
Benzbromarone interacts with Hsp90 and Aha1 in cellular context and causes cytotoxicity against multiple human cancer cell lines
Hsp90 facilitates the structural folding and activation of about 400 client proteins, which include onco-proteins such as AKT, cyclin-dependent kinases, and v-Src etc. Through regulating the cellular homeostasis of these onco-proteins, Hsp90 contributes to the pathological process of multiple types of tumors and serves as a target for anti-tumor drug development7,8,9,10,11,12,13. Meanwhile, Aha1’s contribution in cell proliferation and survival has not been fully elucidated yet. However, published data suggests that the overexpression of Aha1 contributes to the metastatic advantage of colorectal cancer and promotes the growth and metastasis of osteosarcoma38,39. Therefore, active compounds interfering with Hsp90-Aha1 system might exhibit therapeutic effects against cancer. Here in this study, cellular-based assays were carried out to evaluate the biological effects of Benzbromarone by mainly using colorectal cancer and osteosarcoma cell lines as model systems.
As aforementioned, Hsp90 system plays an important role in the pathogenesis of multiple types of cancers such as breast cancer, prostate cancer, colorectal cancer, and osteosarcoma etc40,41,42,43,44. Here in our study, the potent Hsp90-NTD inhibitor NVP-AUY922 was utilized to experimentally verify the dependency of selected cancer cell lines including human prostate cancer cells (LNCaP), human osteosarcoma cells (MNNG/HOS Cl #5 and MG-63), and human colorectal cancer cells (LS174T, HCT116 and HT-29) to Hsp90 system (Supplementary Fig. S7). Based on the data obtained, NVP-AUY922 demonstrates strong pharmacological potency against the aforementioned cancer cell lines (Supplementary Fig. S7). Then, cellular thermal shift assay was conducted to confirm the interaction between Benzbromarone and Hsp90/Aha1 in the cellular context. In this assay, cell extracts (MNNG/HOS Cl #5 and LS174T) were pre-incubated with Benzbromarone in an ice bath for 1 h. After pre-incubation, the mixtures were divided into aliquots and subjected to incubation at different temperatures ranging from 49 °C to 67 °C for 5 min. The supernatants after centrifugation were then analyzed by immunoblotting analysis. Compared with the DMSO treatment, positive shifting of the thermal stability for both Hsp90 and Aha1 in MNNG/HOS Cl #5 and LS174T cells was observed upon pre-incubation with Benzbromarone (Fig. 4), indicating that Benzbromarone could interact with Hsp90 and Aha1 in tested cancer cells. In the following studies, cell viability assay and colony formation assay were conducted to assess whether the application of Benzbromarone would cause cytotoxicity in human prostate cancer cells (LNCaP), human osteosarcoma cells (MNNG/HOS Cl #5 and MG-63), and human colorectal cancer cells (LS174T, HCT116 and HT-29). We then observed that the growth and proliferation of all tested six cancer cell lines was inhibited in a dose-dependent manner upon the application of Benzbromarone, and the corresponding IC50 values were determined to be at tens of micromolar level (Fig. 4). Meanwhile, the results of the colony formation experiments demonstrated that the administration of Benzbromarone could significantly suppress the colony formation capacities of six tested cancer cell lines (Fig. 4).

A Cellular thermal shift assay was applied to detect the binding of Benzbromarone to Hsp90 and Aha1 in cellular context. Upon the treatment of Benzbromarone, the cellular thermal stabilities of Hsp90 and Aha1 were up-regulated, and the positive shifting for the temperature-dependent melting of both Hsp90 and Aha1 was observed. The cellular thermal shift experiments were all repeated for three times. Raw blot data from multiple runs (n = 3) were processed by ImageJ and graphs were plotted with GraphPad Prism 8. The quantitative analysis plots for Hsp90 and Aha1 are shown in the middle panel and the right panel, respectively. B Cell viability of LNCaP, MNNG/HOS Cl #5, MG-63, LS174T, HCT116 and HT-29 cells were assessed after their exposure to vehicle and different concentrations of Benzbromarone for 72 h. Data were analyzed by GraphPad Prism 8 and is presented as means ± standard deviation (n = 3). The IC50 values of Benzbromarone against LNCaP, MNNG/HOS Cl #5, MG-63, LS174T, HCT116 and HT-29 cells were determined to be 44.05 ± 2.93 µM, 45.54 ± 3.47 µM, 69.15 ± 3.59 µM, 76.28 ± 6.79 µM, 55.93 ± 1.82 µM and 59.62 ± 3.73 µM, respectively. (C) Colony formation experiments were performed by using LNCaP, MNNG/HOS Cl #5, MG-63, LS174T, HCT116 and HT-29 cells treated with different concentrations of Benzbromarone (30 µM for LNCaP, LS174T, HCT116 and HT-29 cells, 20 µM for MNNG/HOS Cl #5 cells, 25 µM for MG-63 cells), and the quantitative results are presented in lower panel (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, t-test).
Benzbromarone suppresses the migration, invasion and epithelial-mesenchymal transitions of colorectal cancer and osteosarcoma cells
According to the published literatures, Hsp90 could enhance the migration and invasion capabilities of cancer cells through facilitating the maturation and activation of its client proteins, such as MMP-2 and Annexin II45,46,47. Besides, Aha1 has also been shown to promote the metastatic process of colorectal cancer and the metastasis of osteosarcoma38,39. Therefore, the active compound against Hsp90-Aha1 system, such as Benzbromarone could potentially interfere with the metastasis progress of cancer cells. Here in this study, wound healing assay, transwell migration assay, and invasion assay were performed to assess the modulation effects of Benzbromarone on the migration and invasion capacities of human cancer cells including HCT116 and MNNG/HOS Cl #5. Upon the exposure of 20 µM Benzbromarone, the wound healing rates of HCT116 and MNNG/HOS Cl #5 cells were significantly down-regulated in comparison with those of vehicle HCT116 and MNNG/HOS Cl #5 cell samples (Fig. 5A). Meanwhile, in line with the results of the wound healing assay, the migration and invasion capacities of HCT116 and MNNG/HOS Cl #5 cells were also notably suppressed in a dose-dependent manner to the treatment of Benzbromarone (Fig. 5B, C). Moreover, since the epithelial-mesenchymal transition (EMT) plays a critical role in tumor metastasis and progression, the expression levels of EMT-related protein markers such as N-Cadherin and E-Cadherin were then checked by immunoblotting analysis to further clarify the anti-metastasis effect of Benzbromarone towards cancer cells. As shown in Fig. 5, after treating the HCT116 cells with Benzbromarone at different concentrations (20 μM, 40 μM, 80 μM) for 48 h, the dose-dependent down-regulation tendency of N-Cadherin and significant up-regulation of E-Cadherin were observed, and the obtained immunoblotting analysis data indicates that Benzbromarone is capable of inhibiting the EMT process of tested cancer cells. Overall, the aforementioned experimental results demonstrate that Benzbromarone could down-regulate the migration and invasion process of HCT116 and MNNG/HOS Cl #5 cells.

A The wound healing rates of HCT116 and MNNG/HOS Cl #5 cells were down-regulated by the application of Benzbromarone. Wound healing experiments were conducted on HCT116 and MNNG/HOS Cl #5 cells treated with 20 µM Benzbromarone, and the quantitative results are presented in the lower panel (n = 3, *p < 0.05, **p < 0.01, ****p < 0.0001, t-test). B Transwell migration experiments were conducted on HCT116 and MNNG/HOS Cl #5 cells treated with Benzbromarone (10 µM and 30 µM for HCT116, 20 µM and 40 µM for MNNG/HOS Cl #5). The presence of Benzbromarone led to a significant decrease in the number of migrating cells, and the quantitative results are presented in the lower panel (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, t-test). C Transwell invasion experiments were conducted on HCT116 and MNNG/HOS Cl #5 cells treated with either 20 µM or 40 µM Benzbromarone. The presence of Benzbromarone led to a significant decrease in the number of invading cells, and the quantitative results are presented in the lower panel (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, t-test). D The application of Benzbromarone could modulate the cellular levels of EMT-related proteins, including N-cadherin and E-cadherin. After the treatment of HCT116 cells with 0, 20 µM, 40 µM, and 80 µM Benzbromarone for 48 h, the level of E-cadherin was significantly up-regulated. Statistical analysis was performed using Graphpad Prism 8 software (n = 3, *p < 0.05 compared to control, t-test).
Benzbromarone induces cell apoptosis and G2/M cell cycle arrest
In the following studies, flow cytometry technique was subjected to test whether Benzbromarone could induce apoptosis in colorectal cancer and osteosarcoma cells. In response to the treatment of Benzbromarone, the percentages of apoptotic MNNG/HOS Cl #5 and LS174T cells were significantly up-regulated in a dose-dependent manner (Fig. 6). Moreover, to further verify the flow cytometry analysis results, the cellular levels of cleaved PARP and cleaved caspase-3 in MNNG/HOS Cl #5 and LS174T cells without or with the administration of Benzbromarone were detected by using immunoblotting method. Not surprisingly, after treating the cancer cells with different concentrations of Benzbromarone (0, 20 µM, 40 µM and 60 µM Benzbromarone for MNNG/HOS Cl #5 cells; 0, 20 µM, 40 µM and 80 µM Benzbromarone for LS174T cells) for 48 hours, these two biomarkers for apoptosis were up-regulated in tested cancer cells (Fig. 6). The obtained data from both flow cytometry and immunoblotting analysis revealed that Benzbromarone could inhibit cell proliferation by inducing cell apoptosis. Furthermore, the cell cycle analysis by flow cytometry for MNNG/HOS Cl #5 and LS174T cells in response to the application of Benzbromarone indicates that the compound could enhance the cell population arrested at G2/M cell cycle phases (Fig. 6).

A Cell apoptosis was evaluated by flow cytometry analysis after the cells were treated with Benzbromarone for 48 h. MNNG/HOS Cl #5 cells were treated with 0, 20 µM or 40 µM Benzbromarone, and LS174T cells were treated with 0, 60 µM or 80 µM Benzbromarone. The cells were stained with PI and Annexin V-FITC and then analyzed using flow cytometry. The recorded data were analyzed by using FlowJo. The gating strategy for the flow cytometry (FACS) plots are shown in the first and third panels. The quantitative results derived from the FACS plots are presented in the upper-middle panels (n = 3, *p < 0.05, ***p < 0.001, t-test). B Immunoblotting analysis was used to detect the level changes of PARP, cleaved PARP, caspase-3, and cleaved caspase-3 after the treatment with Benzbromarone lasting for 48 h. MNNG/HOS Cl #5 cells were treated with 0, 20 µM, 40 µM, or 60 µM Benzbromarone, and LS174T cells were treated with 0, 20 µM, 40 µ,M or 80 µM Benzbromarone. Raw blot data for cleaved PARP from multiple runs (n = 3) were processed by ImageJ, and graphs were plotted with GraphPad Prism 8 (n = 3, **p < 0.01 compared to control, t-test). The unpaired two-tailed t-test comparing cleaved PARP levels between untreated and 60 µM Benzbromarone-treated MNNG/HOS Cl #5 cells showed a p-value of 0.094 (p = 0.094). C Benzbromarone induces G2/M cell cycle arrest in MNNG/HOS Cl #5 cells and LS174T cells. MNNG/HOS Cl #5 cells and LS174T cells were treated with 40 µM Benzbromarone and 60 µM Benzbromarone, respectively. The cells were stained with PI and analyzed by using flow cytometry. The recorded data were analyzed to extract cell cycle distribution information by using FlowJo. The quantitative results are presented in the right panel (n = 3).
Benzbromarone destabilizes Hsp90’s client proteins
Hsp90 is a key component of molecular chaperone system. It is involved in the quality control of hundreds of client proteins including kinases, transcription factors, and hormone receptors etc1,2,6. Due to its intimate relationship with the homeostasis control of multiple clients associated with cell proliferation, cell survival, and cell metastasis, Hsp90 plays a crucial role in the pathological process of malignant tumors7,8,9,48,49. Chemical modulation of the Hsp90 system’s chaperoning function by small molecules is expected to modify cell fate. As illustrated, Benzbromarone specifically binds to Hsp90-NTD and Aha1-CTD, which consequentially disrupting the interaction between these two proteins. Also, the compound was verified to interact with Hsp90 and Aha1 in cellular context. Therefore, the inhibition effects of Benzbromarone on the growth and metastasis of selected cancer cell lines might be at least partially attributed to its potentially interfering with the cellular function of Hsp90 system. To testify this inference, the cellular levels of representative clients of Hsp90 were detected in response to the exposure of Benzbromarone. After the application of the compound at indicated concentrations, the cellular levels of Hsp90’s client proteins including AKT, CDK2, and CDK4 were downregulated in LNCaP, MNNG/HOS Cl #5, MG-63, LS174T, HCT116 and HT-29 cells (Fig. 7). Besides, since Aha1 helps Hsp90 in the proper folding of steroid hormone receptors (SHRs)50, the level change of androgen receptor (AR) in prostate cancer cells (LNCaP) was also checked upon the exposure to Benzbromarone. According to the obtained data, the level of AR in LNCaP cells was also significantly down-regulated (Fig. 7). Notably, the immunoblotting data also demonstrate that Benzbromarone did not trigger up-regulation of additional heat shock response-associated proteins, such as Hsp70/Hsc70 and HSF1 (Fig. 7).

A, B Dose-dependent decreasing of the level for AR in LNCaP cells and dose-dependent decreasing of the levels for AKT, CDK2, and CDK4 in LNCaP, MNNG/HOS Cl #5, MG-63, LS174T, HCT116, and HT-29 cells were observed upon the application of increased amounts of Benzbromarone. Additionally, treatment with Benzbromarone did not significantly up-regulate the levels of Hsp70/Hsc70 and HSF1. Each experiment was repeated for three times, and the results for one of the experiments are shown here. Statistical analysis was performed using Graphpad Prism 8 software (*p < 0.05; **p < 0.01; ***p < 0.001 compared to control, t-test).
Discussion
Due to the involvement in multiple biological activities through facilitating the proper folding and activation of client proteins such as kinases, transcription factors, and hormone receptors, etc., molecular chaperone Hsp90 has attracted extensive attention during the past several decades1,2,3,4,5,21,51,52. Specially, since it also serves as a target for anti-tumor drug development, quite a number of active compounds and candidate drugs have been developed towards Hsp90 system8,9,10,12,13,53,54,55. Most of the obtained compounds modulate the function of Hsp90 by binding to the ATP/ADP recognition pocket in Hsp90-NTD. The remaining active compounds targeting Hsp90 system function by either binding to the other two domains of the chaperone (Hsp90-MD and Hsp90-CTD) or interacting with the co-chaperones10,54,55,56,57,58,59. As the only co-chaperone capable of significantly enhancing Hsp90’s ATPase activity, a few of compounds have also been developed towards either Aha1 itself or the complex formed by Aha1 and Hsp9060,61. However, up to date, no high resolution structure has been reported for Aha1 complexed with active compound. Here in this study, the mechanisms of action for Benzbromarone, an active compound against both Hsp90 and Aha1, were explored at both molecular and cellular level. The data presented in this manuscript provides extra information for a further understanding of Aha1’s interaction with Hsp90 by using Benzbromarone as a chemical probe. Besides, the active compound itself and the solved high resolution complex structure shed a light on the development of chemical modulators with high potency and high specificity against Aha1.
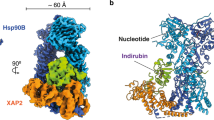
Benzbromarone, a benzofuran derivative, is an approved drug and clinically used to treat primary hyperuricemia and gouty arthritis etc. As reported, Benzbromarone’s anti-hyperuricemia effect could mainly be attributed to the increasing of urate excretion in the kidney’s proximal tubule through its inhibition on the urate transporter protein URAT162,63. Interestingly, we found that Benzbromarone could also bind to Hsp90-Aha1 system. In the following studies, one-dimensional CPMG NMR experiments, isothermal titration calorimetry (ITC), two-dimensional [1H, 15N] HSQC titration analysis, molecular docking and X-ray crystallography were jointly applied to verify and explore the interactions between Benzbromarone and Hsp90/Aha1 (Figs. 1 and 2). As illustrated by the obtained data, Benzbromarone could specifically bind to Hsp90-NTD and Aha1-CTD with the corresponding dissociation constants at 15.10 ± 3.86 µM and 0.84 ± 0.35 µM, respectively. Moreover, although Hsp90-NTD recognizes the compound by using its canonical ATP/ADP recognition cavity, the binding pose of Benzbromarone in Hsp90-NTD exhibits unique features. At least partially due to the huge difference in the polarity of the 3,5-dibrom-4-hydroxyphenyl group in Benzbromarone and the phosphonoamidodiphosphoric group in AMPPNP, Benzbromarone’s benzofuran moiety, which sharing a similar scaffold with AMPPNP’s adenine moiety, occupies an induced sub-pocket and forms π-π interactions with F138 and W162 in Hsp90-NTD (Fig. 2). While the 3,5-dibrom-4-hydroxyphenyl group of the compound is accommodated by the same sub-pocket for the recognition of AMPPNP’s adenine moiety (Fig. 2). For Aha1-CTD:Benzbromarone complex, the detailed interacting picture was revealed by the high resolution structure of the formed complex determined using X-ray crystallography (Fig. 2). Benzbromarone binds to the hydrophobic cavity composed of F235, T236, V253, N256, V257, W274, F276, R314, Q317, W319, Y322 and Y323 in Aha1-CTD, and these residues mainly distribute in α3 (α-helix 3), β3-β4 loop and α2-β2 loop regions of the protein. Interestingly, the dynamics behavior of the local regions responsible for the formation of the pocket and the recognition of Benzbromarone is featured with slow motions on a time scale of microseconds to milliseconds (Fig. 3D in reference 25), which indicating that the amino acid residues residing at these regions potentially undergo slow conformational exchange in solution64. The unique dynamics feature of the aforementioned regions suggest that they are potentially involved in protein-protein or protein-small molecule ligand interactions. Consistently, the cryo-EM structures of Hsp90:Aha1 complexes in different states demonstrate that besides interacting with Hsp90-NTD, Aha1-CTD could also bind to Hsp90-MD mainly through utilizing its residues crucial for Benzbromarone recognition (Supplementary Fig. S8)27. Therefore, a competitive binding for Benzbromarone and Hsp90-MD to Aha1’s C-terminal domain is expected. Meanwhile, Aha1-CTD has also been reported to interact with Hsp90-NTD in the presence of ATP analogue24. We thus performed NMR-based competitive binding experiments and ATP hydrolysis assay to verify whether Benzbromarone’s binding is capable of interfering with the interactions between Aha1 and Hsp90. Not surprisingly, upon the presence of the compound, the interaction between Aha1-CTD and Hsp90-NTD was inhibited (Fig. 3 and Supplementary Figs. S4 and S5). Consistently, it appears that the application of Benzbromarone could down-regulate the ATP hydrolysis rate of Hsp90-Aha1 system (Fig. 3D).
After the elucidation of the binding modes of Benzbromarone to Hsp90-NTD/Aha1-CTD, cellular-based experiments were carried out to test whether the compound could interact with Hsp90-Aha1 system in cellular context and thus modulate the corresponding down-stream cellular events. In line with the direct binding of Benzbromarone to Hsp90 and Aha1 indicated by the CESTA data (Fig. 4), Benzbromarone exhibited cytotoxicity against multiple tested cancer cell lines (Fig. 4). Meanwhile, it suppressed the migration and invasion capabilities as well as induced apoptosis and G2/M cell cycle arrest of colorectal cancer cells and osteosarcoma cells (Figs. 5 and 6). The following conducted immunoblotting analysis results demonstrated that the cellular levels of Hsp90’s representative client proteins including AKT, CDK2, CDK4 and AR were down-regulated upon the treatment of Benzbromarone (Fig. 7). Finally, to distinguish the cellular effects of the drug acting through its interaction with Hsp90 from those mediated by its binding to Aha1, we conducted drug combination experiments. We tested whether a synergistic effect against colorectal cancer could be achieved by combining the Hsp90-specific inhibitor NVP-AUY922 with Benzbromarone. The results indicate that no synergistic benefits were observed with the combination of these two drugs (Supplementary Fig. S9), suggesting that Benzbromarone primarily causes cytotoxicity through its binding to Hsp90. Overall, the cellular experiments revealed that Benzbromarone exhibits medium potency anti-tumor activity, at least partially through its interference with the Hsp90 system (Fig. 8).

Benzbromarone suppresses tumor cell growth and metastasis through dual mechanisms: competitively inhibiting Hsp90 ATPase activity and interfering with Hsp90-Aha1 interactions.
Methods
Protein sample preparation
The genes encoding full-length Aha1, Hsp90-NTD (Hsp909-236), and Aha1-CTD (Aha1204-338) were cloned into the pET28a vector, and the gene encoding full-length Hsp90β was cloned into the pET23a vector. Then, His-tagged Hsp90β, Aha1, Hsp90-NTD and Aha1-CTD were expressed in BL21 (DE3) cells. The cells were transfected with expression plasmid and cultured in Luria-Bertani medium at 37 °C. Once the OD value at 600 nm reached 0.6–0.8, protein expression was triggered by adding 0.3 mM IPTG (isopropyl β-D-thiogalactoside) to the medium. Following an 18-hour incubation at 16 °C, the cells were harvested by centrifugation, re-suspended in lysis buffer (for Hsp90β and Hsp90-NTD preparation: 300 mM NaCl, 100 mM Tris-HCl, 5% (v/v) glycerol, 5 mM β-mercaptoethanol (β-ME), pH 7.5; for Aha1 and Aha1-CTD preparation: 200 mM KCl, 20 mM Tris-HCl, 5 mM β-ME, 20 mM imidazole, pH 7.5) and then stored in −80 °C freezer for future use. To produce 100% 15N-labeled Hsp90-NTD and Aha1-CTD, cells were cultured in M9 media with 15N-labeled ammonium chloride as the only nitrogen source. Protein expression was triggered by adding 0.5 mM IPTG when the OD600 value reached 0.4–0.6. The cells were then incubated at 16 °C for 24 h before being harvested.
When needed, the cell pellets were lysed using a homogenized instrument (AH-Nano, ATS Engineering Inc., Brampton, ON, Canada), the insoluble materials were removed by centrifugation, and the supernatants were loaded onto the Ni-NTA column (GE Healthcare, Boston, MA, USA). The target proteins were eluted with elution buffer (for Hsp90β purification: 50 mM KCl, 30 mM Tris-HCl, 6 mM β-ME, 250 mM imidazole, pH 8.0; for Hsp90-NTD purification: 300 mM NaCl, 100 mM Tris-HCl, 5% (v/v) glycerol, 5 mM β-ME, 300 mM imidazole, pH 7.5; for Aha1 and Aha1-CTD purification: 200 mM KCl, 20 mM Tris-HCl, 5 mM β-ME, 300 mM imidazole, pH 7.5). The His6-tag of Aha1 was then removed by ulp1 protease, and the His6-tag of Aha1-CTD was removed by thrombin protease. Hsp90-NTD, Aha1 and Aha1-CTD were further purified by size-exclusion chromatography on a HiLoad 16/600 Superdex 200 column (GE Healthcare, Boston, MA, USA) equilibrated with size-exclusion chromatography buffer (for Hsp90-NTD purification: 75 mM NaCl, 20 mM Tris-HCl, 1 mM β-ME, pH 7.4; for Aha1 and Aha1-CTD purification: 150 mM KCl, 20 mM Tris-HCl, 0.5 mM ethylene diamine tetraacetic acid (EDTA), 5 mM β-ME, pH 7.5). Hsp90β was further purified by anion-exchange chromatography (GE Healthcare, Boston, MA, USA) using an elution buffer with KCl linearly increasing from 50 mM to 1000 mM. The fractions containing Hsp90β were subsequently concentrated using centrifugal filter tube (Millipore, Burlington, MA, USA) and purified by size-exclusion chromatography on a HiLoad 16/600 Superdex 200 column using the buffer containing 20 mM KCl, 30 mM Tris-HCl, and 1 mM DTT. Produced Hsp90β was then concentrated, supplemented with 10% (v/v) glycerol, flash-frozen in the liquid nitrogen and stored at −80 °C for future use. Purified proteins were analyzed by SDS/PAGE (10% or 15% gel), and the protein concentrations were determined by using the instrument of Nanodrop (Thermo Scientific, Waltham, Massachusetts).
Isothermal titration calorimetry measurements
ITC experiments were performed on a MicroCal ITC200 instrument (Malvern Panalytical, Malvern, UK) at 25 °C. Aha1-CTD and Benzbromarone were dissolved in the ITC buffer (150 mM KCl, 20 mM Tris-HCl, 2% DMSO, pH 7.5). Aha1-CTD (20 µM) was added to the cell of the instrument, titrated with 200 µM Benzbromarone contained in the instrument injection syringe. The ITC titrations were performed using an initial injection of 0.4 µL followed by 19 identical injections of 2 µL of Benzbromarone into the cell. The interval time between each injection was 160 s. Data were analyzed by using the Origin software package.
NMR spectroscopy
1D Carr-Purcell-Meiboom-Gill (CPMG) NMR experiments were performed on Bruker 600 MHz NMR spectrometers with a cryogenically cooled probe (Bruker, Billercia, MA, USA) at 25 °C. Samples containing 200 µM Benzbromarone, as well as 200 µM Benzbromarone in the presence of 5 µM Hsp90-NTD/Aha1-CTD, were dissolved in phosphate buffer (20 mM NaH2PO4, 20 mM Na2HPO4, 100 mM NaCl, pH 7.4, 2% DMSO). Data analysis was carried out using MestReNova software.
[1H, 15N] HSQC NMR experiments were performed on either Bruker 600 MHz or Bruker 800 MHz NMR spectrometer equipped with cryogenically cooled probe (Bruker, Billercia, MA, USA) at 25 °C. NMR samples of 100 µM 15N-labeled Hsp90-NTD and Benzbromarone at different concentrations (0 µM, 25 µM, 50 µM, 100 µM, 200 µM and 400 µM) were dissolved in the buffer containing 75 mM NaCl, 20 mM Tris-HCl, 1 mM β-ME, 2% DMSO and 10% D2O at pH 7.4. NMR samples of 100 µM 15N-labeled Aha1-CTD and Benzbromarone at different concentrations (0 µM, 50 µM, 100 µM, 200 µM and 400 µM) were dissolved in the buffer containing 150 mM KCl, 20 mM Tris-HCl, 0.5 mM EDTA, 5 mM β-ME 2% DMSO and 10% D2O at pH 7.5. As for the [1H, 15N] HSQC-based competitive binding experiments, NMR samples of 50 µM 15N-labeled Aha1-CTD without or with the addition of Hsp90-NTD (250 µM, with 2.5 mM AMPPNP and 5 mM MgCl2) or Benzbromarone (400 µM) or both of Hsp90 (250 µM, with 2.5 mM AMPPNP and 5 mM MgCl2) and Benzbromarone (400 µM) were dissolved in the buffer containing 150 mM KCl, 20 mM Tris-HCl, 0.5 mM EDTA, 5 mM β-ME, 2% DMSO and 10% D2O at pH 7.5. For another set of [1H, 15N] HSQC-based competitive binding experiments, NMR samples of 50 µM 15N-labeled Hsp90-NTD (with 0.5 mM AMPPNP and 5 mM MgCl2) without or with the addition of Aha1-CTD (250 µM), or Benzbromarone (400 µM) or both of Aha1-CTD (250 µM) and Benzbromarone (400 µM) were dissolved in the buffer containing 150 mM KCl, 20 mM Tris-HCl, 0.5 mM EDTA, 5 mM β-ME, 2% DMSO and 10% D2O at pH 7.5. The NMR spectra were processed by using NMRPipe65 and analyzed by using Sparky66,67. The chemical shift perturbation (CSP) values for 15N and 1H nuclei were calculated according to Eq. (1):
in which ΔδN represents the chemical shift change of the amide nitrogen and ΔδH represents the chemical shift change of the amide proton.
The Kd for Benzbromarone:Hsp90-NTD interacting system was derived from a global fitting to the intensity decay curves of selected residues (residues A24, M30, K41, L45, S50, I59, Y61, N83, I91, I96, A101, S129, 144, A145, T152, E158, G168, R173, M180, G181, Y197 and T214) according to Eq. (2) derived from the published literature68:
in which I represents the intensity value of selected residues along the titration, I0 represents the intensity value of selected residues in free state, P0 represents the concentration of Hsp90-NTD, Imax is a constant, Kd represents the dissociation constant and x represents the concentration of Benzbromarone along the titration. The theoretical basis for Eq. (2) is provided as follows.
The observed intensity of NMR signals upon the binding of Benzbromarone is the weighted average of the intensities in the free and bound states, that is,
here, xB and xF represent the fractions of Hsp90-NTD that are bound to Benzbromarone and free in solution, respectively. Given that,
we can express the fraction of the ligand as:
We also know that,
where [L]t and [P]t are the total concentrations of the ligand and protein, which are the sums of their free and bound forms. Therefore, Kd can be expressed as:
Then,
Where ΔIobs represents the change in observed intensity compared to the free state, and ΔImax is the maximum intensity change upon saturation (typically determined through the fitting procedure, as it is often not directly measurable experimentally). This equation enables the determination of Kd by fitting the measured residue intensity changes across different protein and ligand concentrations.
All of the source data related to the aforementioned chemical shift perturbation analysis are summarized in Supplementary Data 1.
ATP hydrolysis assay
The ATP hydrolysis process catalyzed by full-length Hsp90β was monitored through the recording of 1D 31P NMR spectra using a Bruker 500 MHz NMR spectrometer equipped with a cryogenically cooled probe (Bruker, Billercia, MA, USA). NMR samples of Hsp90β:ATP (4 μM:1 mM) reaction system without and with the addition of either 400 µM Benzbromarone or 400 µM NVP-AUY922 were transferred into NMR tubes and incubated in the NMR spectrometer at 37 °C. The 31P NMR spectra were acquired at intervals of 20 min for a total duration of 4 h. Additionally, NMR samples of Hsp90β:Aha1:ATP (4 μM:20 μM:1 mM) reaction system without or with the addition of either 400 µM Benzbromarone or 400 µM NVP-AUY922 were acquired at intervals of 20 min for a total duration of 1 h. The reaction buffer for all NMR samples contained 50 mM NaCl, 6 mM MgCl2, 25 mM HEPES, 2% DMSO, and 10% D2O at pH 7.2. Data analysis was carried out using MestReNova software.
NADH-coupled ATPase assay
The ATPase activity of full-length Hsp90β was measured by tracking the decrease in the absorbance at 340 nm for 90 min at an interval of 60 s using a regenerating enzyme-coupled assay. The reaction was carried out in the 384-well plate with a 50 μL volume and the reaction buffer contained 25 mM HEPES at pH 7.2, 50 mM NaCl, 1 mM phosphoenol pyruvate (Sigma-Aldrich), 0.6 mM NADH, 2 μL of pyruvate kinase/lactate dehydrogenase enzymes (Sigma-Aldrich), 6 mM MgCl2, 1 mM DTT, 500 μM ATP, and 2% DMSO. The measurement was initiated by the addition of 4 μM full-length Hsp90β alone or Hsp90β-Aha1 complex (0.5 μM:2.5 μM) at 37 °C on SpectraMax M5, and the final concentration of Benzbromarone was 400 μM. The raw data were analyzed as previously described in ref. 69. The ATPase rates calculated were expressed as per μM of Hsp90 (1/min). All assays were done in triplicate.
Molecular docking
The crystal structure of HSP90 N-terminal domain (Hsp90-NTD) in complex with pochoxime B (PDB code: 3INX) was used as the receptor model for molecular docking in the Schrödinger 2017 suite. The receptor model was preprocessed using the Protein Preparation Wizard70, which included adding hydrogen atoms and the missing side chains of residues, retaining the water molecules forming more than three hydrogen bonds, optimizing the orientations of polar hydrogens and the protonated states, and refining the overall structure using the OPLS3 force field71. The three-dimensional structure of Benzbromarone was generated and optimized using the LigPrep tool. The docking grid was centered on the centroid of Benzbromarone using the Receptor Grid Generation tool. The molecular docking was performed using the Ligand Docking tool72 in the standard precision mode, and the top-scoring conformation of docked Benzbromarone was finally selected for presentation and further analysis.
Protein pocket volume analysis
The crystal structures of Aha1-CTD in the apo and complex states (PDB codes: 8Z3H and 8Z3J) were used to analyze the pocket volume. The water molecules and/or ligand in the two crystal structures were firstly removed. The volume and geometric information of the largest pockets in both conformations of Aha1-CTD were then calculated using the PyVOL 1.7.6 software73 in largest mode. The results showed that the largest pockets in both states (apo state: 188 Å3, complex state: 205 Å3) were located at the same region (the Benzbromarone-binding pocket in the complex state). This indicates that Benzbromarone binding led to an increase in the pocket volume of this pocket.
Crystallization and data collection
Mixtures of Aha1-CTD and Benzbromarone were incubated in an ice bath for ~7–8 h before being used for crystallization. The final concentration of Aha1-CTD was 15 mg/mL, with a 1:2 molar ratio of Aha1-CTD to Benzbromarone. Crystals of Aha1-CTD complexed with Benzbromarone were obtained by using the sitting drop vapor diffusion method in a solution containing 100 mM MMT buffer and 20% (w/v) polyethylene glycol 1500 at pH 6.5 and 18 °C. Crystals of Aha1-CTD in apo state were obtained by using the sitting drop vapor diffusion method in a solution containing 100 mM MIB, pH 7.0, 25% polyethylene glycol 1500 at 18 °C. Crystals were cryoprotected with 25% ethylene glycol and flash frozen by liquid nitrogen. Reflection data were collected at Shanghai Synchrotron Radiation Facility Beamline 18U1 and Beamline 02U1. Data were processed by XDS or xia2 at the facility. PDB code 1X53 was used as a starting search model, and structures were finally refined by Refmac in CCP4 suite74. The structures have been deposited into PDB with accession codes of 8Z3H and 8Z3J.
Cellular thermal shift assay
The MNNG/HOS Cl #5 and LS174T cells were collected using a phosphate-buffered saline (PBS) solution that contained PMSF. Cells were then lysed directly by three freezing-thawing cycles in liquid nitrogen. Subsequently, the cell lysates were subjected to centrifugation at 20,000 × g for 20 min to eliminate cellular debris. Following centrifugation, the supernatants were combined and incubated in an ice bath with 200 μM Benzbromarone for 1 h. Additionally, supernatants containing 1% DMSO were used as the control. After the pre-incubation, the mixtures were divided into aliquots and incubated at various temperatures ranging from 49 °C to 67 °C for 5 min. Finally, the precipitated denatured proteins were removed through centrifugation, and the supernatants were analyzed using immunoblotting.
Cell viability and colony formation assays
Cell proliferation was evaluated using the sulforhodamine B (SRB) assay. Cells were seeded into 96-well plates (8000 cells per well for LNCaP and HCT116, 6000 cells per well for MNNG/HOS Cl #5 and MG-63, 12000 cells per well for LS174T, 10000 cells per well for HT-29) and allowed to adhere overnight. Subsequently, cells were treated with either 0.1% (v/v) DMSO or Benzbromarone/NVP-AUY922 at indicated concentrations. Following another 72-h incubation, cells were fixed with 10% (w/v) trichloroacetic acid for 1 h, washed with water, and stained with 4 mg/mL SRB (Sigma, St. Louis, MO, USA) for 30 min in the dark. Excess dye was removed using 1% (v/v) acetic acid. Protein-bound dye was dissolved in 10 mM Tris solution at pH 10.5 and measured at 510 nm using a SpectraMax M5 spectrophotometer after shaking. Data analysis was carried out using GraphPad Prism 8 software.
As for colony formation assay, cells were seeded in 6-well plates (500 cells per well for MNNG/HOS Cl #5, MG-63, LNCaP, HCT116, and HT-29, 1000 cells per well for LS174T) and incubated at 37 °C overnight. Single cells were allowed to grow and form colonies over 7–15 days until visible macroscopic colonies showed up. The cells were gently washed with PBS buffer, fixed with methanol, and stained with 0.1% crystal violet for 15 min. After rinsing and air drying, the colonies were imaged and counted. Data analysis was carried out using GraphPad Prism 8 software.
LNCaP, MNNG/HOS Cl #5 and MG-63 cells were purchased from the Cell Bank affiliated with the Shanghai Institute of Biochemistry and Cell Biology (SIBCB, Shanghai, China). LS174T and HCT116 cells, which were initially obtained from the American Type Culture Collection, were generously provided by Dr. Qiang Yu (Shanghai Institute of Materia Medica, Chinese Academy of Sciences). HT-29 cells, which were initially obtained from the Cell Bank affiliated with the Shanghai Institute of Biochemistry and Cell Biology (SIBCB, Shanghai, China), were generously provided by Dr. Hu Zhou (Shanghai Institute of Materia Medica, Chinese Academy of Sciences).
Wound healing assay, transwell migration and invasion assay
For wound healing assay, cells were seeded in 6-well plates and incubated at 37 °C overnight until reaching confluence. After making scratches by using sterile tips, the culture medium was changed from 10% FBS to FBS at lower concentrations (1% FBS for MNNG/HOS Cl #5 and 2% FBS for HCT116). After the treatment with either Benzbromarone or NVP-AUY922 at indicated concentrations, images were captured at different time points of 0 h, 12 h, 24 h (MNNG/HOS Cl #5) or 0 h, 24 h, 48 h (HCT116). The wound areas were analyzed using ImageJ software to assess cell migration capabilities.
Transwell migration and invasion experiments were performed using a 24-well transwell chamber (3464, Corning, NY, USA). Cells (1.5 × 105 cells per well for MNNG/HOS Cl #5 and 2.0 × 105 cells per well for HCT116) were seeded into the upper chambers in basal medium containing 0.2% BSA and Benzbromarone at indicated concentrations, and enriched complete medium containing 10% FBS was added into the lower chambers. The plates were then incubated at 37 °C for either 24 h (HCT116) or 48 h (MNNG/HOS Cl #5). After incubation, the chambers were washed with PBS, fixed with methanol, and stained with 0.1% crystal violet. Images were captured after removing non-migrated cells with cotton swabs. For invasion assay, the Matrigel matrix (356234, Corning, NY, USA) was diluted at a ratio of one to eight with cooled basal medium and added into the upper chambers in advance. The subsequent steps for the invasion assay were similar to that for the transwell migration assay.
Apoptosis assay and cell cycle distribution analysis by flow cytometry
Cells were seeded into 6-well plates (2 × 105 cells per well for MNNG/HOS Cl #5 and 8 × 105 cells per well for LS174T) and allowed to adhere overnight. Then cells were treated with 0.1% (v/v) DMSO or Benzbromarone at indicated concentrations for 48 h. For apoptosis assay, after incubation, the cells were collected, washed with cold PBS, and resuspended in 1× binding buffer at a concentration of 2.5 × 105 cells/100 µL. 100 µL of the cell suspension was added into a tube, mixed with 10 µL of PI and 5 µL of Annexin V-FITC (40302ES50, Yeasen, Shanghai, China). Then, the mixture was incubated for 15 min at room temperature in the dark, and the fluorescence signal was immediately assessed using a flow cytometer (BD LSRFortessa, BD Biosciences, San Jose, CA, USA). The acquired data was subsequently analyzed using FlowJo software.
In terms of cell cycle distribution analysis, after washing the collected cells with cold PBS buffer, cells were fixed with 70% ethanol at 4 °C overnight. The fixed cells were then centrifuged, washed with PBS buffer, and re-suspended in PI/RNase staining buffer (C1052, Beyotime, Shanghai, China). The fluorescence signal was assessed using a flow cytometer for 24 h, and the acquired data were subsequently analyzed using FlowJo software. The figure exemplifying the gating strategy for aforementioned cell cycle distribution analysis is defined as Supplementary Fig. S10 and included in the supporting information file.
Immunoblotting analysis
Following the treatment with either 0.1% (v/v) DMSO or Benzbromarone or NVP-AUY922 at indicated concentrations for 48 hours, MNNG/HOS Cl #5, MG-63, LNCaP, L174T, HCT116, and HT-29 cells were lysed using cell lysis buffer for western and IP analysis (Beyotime, Shanghai, China) containing 1 mM PMSF. The supernatant was obtained through centrifugation at 12,000 rpm for 30 min, and the protein concentration of the samples was determined using the BCA method prior to western blot analysis. Equal amounts of total protein were loaded onto a 10% polyacrylamide gel for SDS-PAGE electrophoresis to separate the proteins. The protein samples in the gel were then transferred onto a 0.22 µm polyvinylidene fluoride (PVDF) membrane. The PVDF membranes were pre-blocked with 5% non-fat milk in Tris-buffered saline with Tween 20 (TBST) buffer before being incubated overnight at 4 °C with primary antibodies (Hsp70, HSF1, AKT, Hsp90, Aha1, CDK2, CDK4, AR, E-Cadherin, N-Cadherin, PARP, caspase-3, α/β tubulin, Vinculin, and GAPDH). After subsequent incubation with either horseradish peroxidase (HRP)-conjugated anti-mouse or HRP-conjugated anti-rabbit secondary antibodies, the proteins on the membrane were visualized by adding a chemiluminescent HRP substrate. The uncropped images for the immunoblotting analysis data presented in Figs. 4A, 5D, 6B, 7 are included and shown in Supplementary Fig. S11.
The following antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA): anti-Hsp70 (#4872S), anti-HSF1 (#4356T), anti-AKT (#9272S), anti-CDK4 (#12790S), anti-AR (#5153S), anti-PARP (#9532S), anti-caspase-3 (#9662S), anti-GAPDH (#8884S), anti-α/β tubulin (#2148S), anti-Vinculin (#13901T) and anti-mouse (#7076S). The following antibodies were purchased from AbCam (Cambridge, SC, USA): anti-CDK2 (ab 32147), anti-E-cadherin (ab40772), and anti-N-cadherin (ab76011). Anti-Hsp90 (341320) was purchased from R&D Systems (Minneapolis, MN, USA). Anti-Aha1 (14725-1-AP) and Anti-α tubulin (66031-1-Ig) were purchased from Proteintech (Wuhan, China). Anti-rabbit (abs 20002) was purchased from Absin Bioscience Inc. (Shanghai, China).
Statistics and reproducibility
Data were analyzed using GraphPad Prism 8 software and are presented as means ± standard deviation. Unpaired two-tailed t-tests were used to assess the significance of NADH-coupled ATPase experiments at a 95% confidence level, while unpaired two-tailed t-tests were applied for the statistical analysis of all other experiments at a 95% confidence level. Significant values and the number of replicates are indicated in the respective figure legends. In addition, all source data (the individual data points) used in the statistical analysis are compiled in Supplementary Data 1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The crystal structures for Aha1-CTD in apo state and in Benzbromarone-bound state have been deposited into PDB with accession codes of 8Z3H and 8Z3J. All other data presented in the manuscript are available from the corresponding author upon reasonable request.
References
Biebl, M. M. & Buchner, J. Structure, function, and regulation of the Hsp90 machinery. Cold Spring Harb. Perspect. Biol. 11, a034017 (2019).
Schopf, F. H., Biebl, M. M. & Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 18, 345–360 (2017).
Pearl, L. H. & Prodromou, C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 75, 271–294 (2006).
Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 473, 2439–2452 (2016).
Prodromou, C. Regulatory mechanisms of Hsp90. Biochem. Mol. Biol. J. 3, 2 (2017).
Taipale, M. et al. Quantitative analysis of Hsp90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001 (2012).
Prodromou, C. Strategies for stalling malignancy: targeting cancer’s addiction to Hsp90. Curr. Top. Med. Chem. 9, 1352–1368 (2009).
Sidera, K. & Patsavoudi, E. HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat. Anti Cancer Drug Discov. 9, 1–20 (2014).
Tatokoro, M., Koga, F., Yoshida, S. & Kihara, K. Heat shock protein 90 targeting therapy: state of the art and future perspective. EXCLI J. 14, 48–58 (2015).
Garg, G., Khandelwal, A. & Blagg, B. S. Anticancer inhibitors of Hsp90 function: beyond the usual suspects. Adv. Cancer Res. 129, 51–88 (2016).
Koren, J. & Blagg, B. S. J. The right tool for the job: an overview of Hsp90 inhibitors. Hsf1 Mol. Chaperones Biol. Cancer 1243, 135–146 (2020).
Li, L., Chen, N. N., You, Q. D. & Xu, X. L. An updated patent review of anticancer Hsp90 inhibitors (2013-present). Expert Opin. Therapeutic Pat. 31, 67–80 (2021).
Liu, J. F., Shu, H. L., Xia, Q. X., You, Q. D. & Wang, L. Recent developments of HSP90 inhibitors: an updated patent review (2020-present). Expert Opin. Therapeutic Pat. 34, 1–15 (2024).
Vaughan, C. K. et al. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol. Cell 23, 697–707 (2006).
Ali, M. M. et al. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440, 1013–1017 (2006).
Siligardi, G. et al. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J. Biol. Chem. 277, 20151–20159 (2002).
Siligardi, G. et al. Co-chaperone regulation of conformational switching in the Hsp90 ATPase cycle. J. Biol. Chem. 279, 51989–51998 (2004).
Karagoz, G. E. et al. N-terminal domain of human Hsp90 triggers binding to the cochaperone p23. Proc. Natl. Acad. Sci. USA 108, 580–585 (2011).
Meyer, P. et al. Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 23, 511–519 (2004).
Roe, S. M. et al. The Mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 116, 87–98 (2004).
Zuehlke, A. & Johnson, J. L. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 93, 211–217 (2010).
Martinez-Yamout, M. A. et al. Localization of sites of interaction between p23 and Hsp90 in solution. J. Biol. Chem. 281, 14457–14464 (2006).
Panaretou, B. et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 10, 1307–1318 (2002).
Retzlaff, M. et al. Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol. Cell 37, 344–354 (2010).
Hu, H. et al. Aha1 exhibits distinctive dynamics behavior and chaperone-like activity. Molecules 26, 1943 (2021).
Biebl, M. M. & Buchner, J. p23 and Aha1: distinct functions promote client maturation. Subcell. Biochem. 101, 159–187 (2023).
Liu, Y. et al. Cryo-EM structures reveal a multistep mechanism of Hsp90 activation by co-chaperone Aha1. bioRxiv, 1-16 (2020).
Mercier, R. et al. The conserved NxNNWHW motif in Aha-type co-chaperones modulates the kinetics of Hsp90 ATPase stimulation. Nat. Commun. 10, 1273 (2019).
Horvat, N. K. et al. A mutation in the catalytic loop of Hsp90 specifically impairs ATPase stimulation by Aha1p, but not Hch1p. J. Mol. Biol. 426, 2379–2392 (2014).
Meyer, P. et al. Structural and functional analysis of the middle segment of Hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol. Cell 11, 647–658 (2003).
Xu, W. et al. Hsp90 middle domain phosphorylation initiates a complex conformational program to recruit the ATPase-stimulating cochaperone Aha1. Nat. Commun. 10, 2574 (2019).
Koulov, A. V. et al. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 21, 871–884 (2010).
Wolmarans, A., Lee, B., Spyracopoulos, L. & LaPointe, P. The mechanism of Hsp90 ATPase stimulation by Aha1. Sci. Rep. 6, 33179 (2016).
Walters, K. J. et al. Characterizing protein-protein complexes and oligomers by nuclear magnetic resonance spectroscopy. Methods Enzymol. 339, 238–258 (2001).
Walters, K. J., Lech, P. J., Goh, A. M., Wang, Q. H. & Howley, P. M. DNA-repair protein hHR23a alters its protein structure upon binding proteasomal subunit S5a. Proc. Natl. Acad. Sci. USA 100, 12694–12699 (2003).
Matsuo, H. et al. Identification by NMR spectroscopy of residues at contact surfaces in large, slowly exchanging macromolecular complexes. J. Am. Chem. Soc. 121, 9903–9904 (1999).
Zhang, N. X. et al. NMR-based model reveals the structural determinants of mammalian arylamine N-acetyltransferase substrate specificity. J. Mol. Biol. 363, 188–200 (2006).
Zheng, D. et al. AHA1 upregulates IDH1 and metabolic activity to promote growth and metastasis and predicts prognosis in osteosarcoma. Signal Transduct. Target. Ther. 6, 25 (2021).
Kim, D. et al. AHA1 regulates cell migration and invasion via the EMT pathway in colorectal adenocarcinomas. Sci. Rep. 11, 19946 (2021).
Klemke, L. et al. Hsp90-stabilized MIF supports tumor progression via macrophage recruitment and angiogenesis in colorectal cancer. Cell Death Dis. 12, 155 (2021).
Zhou, J. H. et al. Identification and biological evaluation of fused tetrahydroisoquinoline derivatives as Wnt/β-catenin signaling inhibitors to suppress colorectal cancer. Eur. J. Med. Chem. 276, 116664 (2024).
Mori, M. et al. Hsp90 inhibitor induces autophagy and apoptosis in osteosarcoma cells. Int. J. Oncol. 46, 47–54 (2015).
Eskew, J. D. et al. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 11, 468 (2011).
Kang, B. H. et al. Preclinical characterization of mitochondria-targeted small molecule Hsp90 inhibitors, Gamitrinibs, in advanced prostate cancer. Clin. Cancer Res. 16, 4779–4788 (2010).
McCready, J., Sims, J. D., Chan, D. & Jay, D. G. Secretion of extracellular hsp90α via exosomes increases cancer cell motility: a role for plasminogen activation. BMC Cancer 10, 294 (2010).
Baker-Williams, A. J. et al. Co-chaperones TIMP2 and AHA1 competitively regulate extracellular HSP90: client MMP2 activity and matrix proteolysis. Cell Rep. 28, 1894–1906.e6 (2019).
Hance, M. W. et al. Secreted Hsp90 is a novel regulator of the epithelial to mesenchymal transition (EMT) in prostate cancer. J. Biol. Chem. 287, 37732–37744 (2012).
Pearl, L. H., Prodromou, C. & Workman, P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem. J. 410, 439–453 (2008).
Guang, M. H. Z. et al. Targeting proteotoxic stress in cancer: a review of the role that protein quality control pathways play in oncogenesis. Cancers 11, 66 (2019).
Harst, A., Lin, H. Y. & Obermann, W. M. J. Aha1 competes with Hop, p50 and p23 for binding to the molecular chaperone Hsp90 and contributes to kinase and hormone receptor activation. Biochem. J. 387, 789–796 (2005).
Didenko, T., Duarte, A. M., Karagoz, G. E. & Rudiger, S. G. Hsp90 structure and function studied by NMR spectroscopy. Biochim. Et. Biophys. Acta 1823, 636–647 (2012).
Pearl, L. H. & Prodromou, C. Structure, function, and mechanism of the Hsp90 molecular chaperone. Adv. Protein Chem. 59, 157–186 (2001).
Khandelwal, A., Crowley, V. M. & Blagg, B. S. J. Natural product inspired N-terminal Hsp90 inhibitors: from bench to bedside?. Med. Res. Rev. 36, 92–118 (2016).
Zhou, C. et al. Allosteric regulation of Hsp90α‘s activity by small molecules targeting the middle domain of the chaperone. iScience 23, 100857 (2020).
Verma, S., Goyal, S., Jamal, S., Singh, A. & Grover, A. Hsp90: friends, clients and natural foes. Biochimie 127, 227–240 (2016).
Zierer, B. K. et al. Artificial accelerators of the molecular chaperone Hsp90 facilitate rate-limiting conformational transitions. Angew. Chem. 53, 12257–12262 (2014).
D’Annessa, I. et al. Design of allosteric stimulators of the Hsp90 ATPase as new anticancer leads. Chemistry 23, 5188–5192 (2017).
Ferraro, M. et al. Allosteric modulators of HSP90 and HSP70: dynamics meets function through structure-based drug design. J. Med. Chem. 62, 60–87 (2019).
Bassanini, I. et al. Chemo-enzymatic synthesis of (E)-2,3-diaryl-5-styryl-trans-2,3-dihydrobenzofuran-based scaffolds and their in vitro and in silico evaluation as a novel sub-family of potential allosteric modulators of the 90 kDa heat shock protein (Hsp90). Org. Biomol. Chem. 16, 3741–3753 (2018).
Serwetnyk, M. A. & Blagg, B. S. J. The disruption of protein-protein interactions with co-chaperones and client substrates as a strategy towards Hsp90 inhibition. Acta Pharm. Sin. B 11, 1446–1468 (2021).
Singh, J. K. et al. Management of Hsp90-dependent protein folding by small molecules targeting the Aha1 co-chaperone. Cell Chem. Biol. 27, 292–305.e6 (2020).
Hou, Z. H., Ma, A. J. X., Mao, J. L., Song, D. N. & Zhao, X. Overview of the pharmacokinetics and pharmacodynamics of URAT1 inhibitors for the treatment of hyperuricemia and gout. Expert Opin. Drug Metab. Toxicol. 19, 895–909 (2023).
Dong, Y. et al. Novel urate transporter 1 (URAT1) inhibitors: a review of recent patent literature (2016-2019). Expert Opin. Therapeutic Pat. 29, 871–879 (2019).
Jarymowycz, V. A. & Stone, M. J. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem. Rev. 106, 1624–1671 (2006).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Goddard, T. D. & Kneller D. G. SPARKY 3. University of California, San Francisco (2008).
Kneller, D. G. & Kuntz, I. D. UCSF Sparky - an NMR display, annotation and assignment tool. J. Cell. Biochem. 53, 254 (1993).
Min, J. O., Strohaker, T., Jeong, B. C., Zweckstetter, M. & Lee, S. J. Chicago sky blue 6B inhibits α-synuclein aggregation and propagation. Mol. Brain 15, 27 (2022).
Radnai, L., Stremel, R. F., Sellers, J. R., Rumbaugh, G. & Miller, C. A. A Semi-high-throughput adaptation of the NADH-coupled ATPase assay for screening small molecule inhibitors. J. Vis. Exp. 150, e60017 (2019).
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234 (2013).
Harder, E. et al. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 12, 281–296 (2016).
Friesner, R. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 (2004).
Smith, R. H. B., Dar, A. C., Schlessinger, A. PyVOL: a PyMOL plugin for visualization, comparison, and volume calculation of drug-binding sites. bioRxiv, 1–3 (2019).
Bailey, S. The Ccp4 suite - programs for protein crystallography. Acta Crystallogr. Sect. D. 50, 760–763 (1994).
Acknowledgements
The NMR experiments were conducted in the Institutional Center for Shared Technologies and Facilities of SIMM, Chinese Academy of Sciences. We thank the scientists at the Shanghai Synchrotron Radiation Facility (SSRF) for their assistances in collecting the X-ray crystallography data. And we thank the staff members of the Integrated Laser Microscopy System at the National Facility for Protein Science in Shanghai (NFPS), Shanghai Advanced Research Institute, Chinese Academy of Sciences for their help with the flow cytometry analysis-related sample preparation, data collection and analysis. This project was financially supported by the Youth Innovation Promotion Association CAS (2022284), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB1060000), and the National Natural Science Foundation of China (Grant Nos. 32171220, 22477126, and 22107111).
Author information
Authors and Affiliations
Contributions
Yan Zhong, Li Shi, Zhuo Xu, and Chen Zhou performed the experiments and data analysis. Qingyu Ma prepared some of the protein samples used in the study. Junying Tang participated in the crystal structure determination. Muya Xiong and Yechun Xu kindly provided help in generating the model structure of Hsp90-NTD complexed with Benzbromarone. Jing Gao and Hu Zhou participated in the primary discovery of Benzbromarone’s binding to Hsp90. Tianqi Gao and Huixiong Dai participated in the data analysis. Naixia Zhang and Chen Zhou designed the project and participated in the data analysis. Naixia Zhang, Yan Zhong, Li Shi, and Chen Zhou wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Joanna Timmins and Mengtan Xing.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhong, Y., Shi, L., Xu, Z. et al. Benzbromarone interferes with the interaction between Hsp90 and Aha1 by interacting with both of them. Commun Biol 8, 761 (2025). https://doi.org/10.1038/s42003-025-08189-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08189-3
