
Abstract
Dystroglycan is a cell adhesion molecule that localizes to synapses throughout the nervous system. While Dystroglycan is required to maintain inhibitory synapses from cerebellar molecular layer interneurons (MLIs) onto Purkinje cells (PCs) whether initial synaptogenesis during development is dependent on Dystroglycan has not been examined. We show that mice with conditional deletion of Dystroglycan from Purkinje cells prior to synaptogenesis results in impaired MLI:PC synapse formation and function due to reduced presynaptic inputs and abnormal postsynaptic GABAA receptor clustering. Using genetic manipulations that disrupt glycosylation of Dystroglycan or truncate its cytoplasmic domain, we show that Dystroglycan’s role in synapse function requires both extracellular and intracellular interactions, whereas synapse formation requires only extracellular interactions. Together, these findings provide molecular insight into the mechanism of inhibitory synapse formation and maintenance in cerebellar cortex.
Similar content being viewed by others


Introduction
Throughout nervous system development, synapses are formed, refined, and eliminated. Synapse formation begins when a presynaptic axon recognizes potential postsynaptic sites through molecular cues. After initial contact between the pre- and postsynaptic neurons is established, transmembrane and secreted molecules are recruited to the nascent synapse to regulate its maturation and refinement in response to activity1. The wide diversity in cell adhesion molecules (CAMs) present at synapses implies that they can form a “code” for determining synapse type and specificity. While several CAMs have been shown to be involved in the maturation and maintenance of synapses, less is known about the molecular players involved in initial synaptogenesis.
Dystroglycan is a CAM expressed throughout the nervous system in a variety of cellular populations including neurons, astrocytes, oligodendrocytes, vascular endothelial cells, and neuroepithelial cells2,3,4,5,6. Dystroglycan is encoded by a single gene (Dag1) that undergoes post-translational autoproteolytic cleavage to produce two subunits: α- and β-Dystroglycan7,8. The extracellular alpha subunit is extensively glycosylated through a process involving at least 19 different genes, and employs a specific glycan motif referred to as “matriglycan” to bind several extracellular proteins that contain Laminin-G (LG) domains (reviewed in Jahncke and Wright, 2023)9. Mutations that disrupt α-Dystroglycan glycosylation lead to a form of congenital muscular dystrophy referred to as dystroglycanopathy that is frequently accompanied by a wide range of neurodevelopmental abnormalities4. The transmembrane beta subunit of Dystroglycan noncovalently binds the alpha subunit in the extracellular environment, and contains a short C-terminal intracellular domain that binds multiple scaffolding and signaling proteins10. Dystroglycan is the transmembrane component of the Dystrophin Glycoprotein Complex (DGC), which functions to connect the actin cytoskeleton to the extracellular matrix. Dystroglycan and Dystrophin, which interact through Dystroglycan’s intracellular domain, are both central to the DGC, whereas other components of the complex can vary depending on the cellular type.
Early in nervous system development, Dystroglycan plays important roles in neuronal migration, axon targeting, and maintenance of the blood-brain barrier11,12,13,14,15,16. Later in development, as synapses begin to form, Dystroglycan expression increases in several neuronal populations throughout the brain, where it localizes at inhibitory synapses17,18,19,20,21. Dystroglycan is required for the formation and function of subsets of inhibitory synapses onto hippocampal and cortical pyramidal neurons, where it appears to function as a postsynaptic recognition cue for presynaptic CCK+/CB1R+ basket interneuron axons22,23,24. Dystroglycan expression continues past the period of synapse formation in pyramidal neurons and is required to maintain CCK+/CB1R+ basket synapses22. However, whether Dystroglycan plays these same roles at other synapses has not been explored in detail. In the cerebellum, Dystroglycan is present at inhibitory somatic and dendritic synapses onto Purkinje cells (PCs), and deletion of Dag1 from PCs after synaptogenesis results in a gradual reduction of inhibitory synapse number and impaired synaptic function17,18,21. Constitutive loss of Dystrophin results in similar alterations in inhibitory inputs onto PCs, confirming an important role for the DGC at these synapses25,26. However, the molecular mechanisms by which Dystroglycan regulates inhibitory synapses are unclear.
Until recently, tools for the conditional deletion of genes in cerebellar PCs prior to the period of synaptogenesis have been lacking. We identified Calb1Cre as a line that drives recombination in embryonic PCs, allowing us to conditionally delete Dag1 from PCs prior to the onset of synapse formation27,28. Using multiple genetic manipulations, we show that Dystroglycan is required for both inhibitory synapse formation and maintenance in Purkinje neurons. Furthermore, we provide in vivo mechanistic insight by showing that extracellular glycosylation of α-Dystroglycan and intracellular interactions through β-Dystroglycan play distinct roles in synapse formation and function.
Results
Dystroglycan is exclusively localized to inhibitory synapses in the cerebellar cortex
Dystroglycan is associated with populations of inhibitory synapses throughout the brain17,18,22,23,29. To rigorously assess Dystroglycan’s synaptic localization in cerebellar cortex, we conducted analysis of Dystroglycan immunoreactivity colocalization with established markers of synaptic populations. Inhibitory synapses in cerebellar cortex reflect synapses from (1) molecular layer interneurons (MLIs) onto PCs, (2) MLIs onto other MLIs, and (3) PC collaterals onto other PCs. While currently available markers are unable to distinguish between these three populations of synapses, Dystroglycan-positive synapses are presumed to be from MLIs onto PCs18 and potentially reflect a population that is at least partially distinct from the Gephyrin-containing population of synapses20,30. We therefore used two presynaptic inhibitory synapse markers (VGAT and GAD67) to analyze inhibitory MLI:PC synapses. The IIH6 antibody, which specifically recognizes matriglycan disaccharide repeats on α-Dystroglycan, significantly colocalized with both VGAT and GAD67, with ~70% of IIH6 puncta positive for VGAT or GAD67 (Fig. 1A, C). There was a subset of IIH6+ puncta occurring with low incidence that were not associated with the counterstained inhibitory synapse marker, perhaps representing synapses in a dynamic state of formation or elimination. As a control, we flipped the synaptic marker channel (VGAT or GAD67) along the vertical axis to generate a mirror image of the original channel and calculated colocalization between the original IIH6 channel and the mirrored synaptic marker channel. IIH6 colocalized with the original channel significantly more than the mirrored channel for both VGAT and GAD67 (Fig. 1A–D), confirming that Dystroglycan is localized to inhibitory synapses in cerebellar cortex. (See Supplementary Fig. 1 for an outline of the colocalization analysis pipeline employed.)

Cerebellar cortex of lobules V–VI was immunostained with Parvalbumin to show Purkinje cell and MLI morphology and counterstained with IIH6 (glycosylated Dystroglycan) and GAD67 (A) or VGAT (C). Both the merged channels (IIH6, green; GAD67/VGAT, magenta) and colocalized pixels are shown for the original image and for the original IIH6 with the mirrored GAD67/VGAT channel. Images are maximum projections. B, D Quantification of the percent of IIH6 puncta that are colocalized with GAD67/VGAT puncta. Scale bar for (A, C) is 50 μm; scale bar for insets (A’, C’) is 25 μm. GAD67 N = 3 animals, p = 8.87E-04. VGAT N = 4 animals, p = 9.98E-05.
There are two populations of excitatory synapses onto PCs: (1) VGluT1+ parallel fiber inputs from granule cells within the cerebellar cortex and (2) VGluT2+ climbing fiber inputs originating from neurons of the inferior olivary nucleus. Despite the high density of VGluT1+ parallel fibers, Dystroglycan and VGluT1 immunoreactivity were mutually exclusive, such that mirroring the VGluT1 channel increased the incidence of Dystroglycan:VGluT1 overlap (Supplementary Fig. 2A, B). Colocalization between IIH6 and VGluT2 was negligible and was not affected by mirroring the VGluT2 channel, indicating that Dystroglycan does not localize to climbing fiber synapses (Supplementary Fig. 2C, D). Together, this illustrates that Dystroglycan localization is restricted to inhibitory synapses in the cerebellar cortex.
Dystroglycan is required for the formation and function of inhibitory synapses onto Purkinje cells
While recent work has shown that Dystroglycan is required at CCK+/CB1R+ basket interneuron synapses onto pyramidal neurons in the hippocampus and cortex, our understanding of Dystroglycan’s role at inhibitory cerebellar synapses remains incomplete due to the limited genetic tools available to manipulate cell populations prior to synapse formation in the cerebellum17,22,23. Within the cerebellum, Calbindin (Calb1) expression is restricted to PCs and is expressed in developing PCs as early as E14.531. We used Calb1Cre as a tool for conditional deletion of Dag1 from embryonic PCs, allowing us to investigate Dystroglycan’s role in the initial formation of inhibitory synapses onto PCs27,28. To reduce the amount of Dystroglycan protein that must be turned over, we generated compound mutants with one copy of Dag1 constitutively deleted and the other copy flanked by LoxP sites for conditional deletion by Cre. We crossed Calb1Cre;Dag1+/− mice with a Dystroglycan conditional line (Dag1flox/flox) to generate Calb1Cre;Dag1flox/− conditional knockouts (Calb1Cre;Dag1cKO) and littermate Calb1Cre;Dag1flox/+ controls (Calb1Cre;Dag1Ctrl). Due to the restricted expression of Cre to PCs in the cerebellum, this approach allowed us to investigate the role of postsynaptic Dystroglycan at MLI:PC synapses in a cell-autonomous manner. We previously reported that Calb1Cre;Dag1cKO cerebella show complete loss of synaptic Dystroglycan at the earliest time point examined (P14), though the expression of Cre was confirmed at P0, suggesting that Dag1 is deleted well before synaptogenesis begins28.
In order to assess inhibitory synapse function in Calb1Cre;Dag1Ctrl and Calb1Cre;Dag1cKO cerebella, we recorded mini inhibitory postsynaptic currents (mIPSCs) from PCs at P25–P35. On average, Calb1Cre;Dag1cKO PCs exhibited reduced mIPSC amplitude and frequency compared to littermate controls (Fig. 2A–F). Reduced mIPSC frequency is generally an indicator of a reduction in the number of synapses, whereas reduced amplitude could reflect either (1) changes to postsynaptic receptor clustering resulting in disrupted subsynaptic domains or (2) altered subcellular distribution of synapses driven by a reduction in the number of postsynaptic GABAA receptors. There was no difference in either mIPSC rise or decay time between control and conditional knockout PCs (Table 1), suggesting no substantial changes in the subunit composition of synaptic GABAA receptors between genotypes, though additional analyses are necessary to confirm this hypothesis.

A Average mIPSC event. B Representative 15 s trace of mIPSCs. mIPSC amplitude (C) and frequency (D); error bars are presented as mean + SEM. Cumulative frequency histogram for individual mIPSC amplitudes (E) and inter-event intervals (IEI) (F). Calb1Cre;Dag1Ctrl N = 16 cells, 6 animals; Calb1Cre;Dag1cKO N = 16 cells, 6 animals; pAmp = 0.017, pFreq = 0.018.
To determine whether the synaptic deficits could be explained by fewer MLIs, we immunostained lobules V/VI of cerebellar vermis for Calbindin (PCs) and Parvalbumin (PCs and MLIs) and quantified Purkinje and MLI cell densities. There was no significant difference in PC density, MLI density, or the ratio of MLIs to PCs (Supplementary Fig. 2A, B), suggesting that the changes in synapse function were not due to defects in cellular proliferation or survival.
To evaluate potential structural correlates of the observed reduction in mIPSC frequency and amplitude, we immunostained for the presynaptic inhibitory synapse marker VGAT along with the inhibitory postsynaptic GABAA receptor subunit GABAAα1. Analysis of synaptic puncta in Calb1Cre;Dag1cKO mice using confocal microscopy with linear Wiener filter deconvolution revealed a reduction in both VGAT and GABAAα1 puncta density compared to littermate controls (Fig. 3A–C), supporting a reduction in synapse number in the absence of Dystroglycan and providing a possible explanation for the reduction in mIPSC frequency. While synapse density was reduced in both the dendritic and somatic compartments, the reduction was most striking in the soma (Fig. 3A–C). The near absence of somatic synapses could cause the observed reduction in mIPSC amplitude, as perisomatic events are generally larger in amplitude than events originating further away from the recording pipette as a result of dendritic filtering32,33. We also quantified synaptic puncta size and observed no change in VGAT or GABAAα1 puncta size (Fig. 3D), indicating that changes in the number of postsynaptic receptors are unlikely to explain the observed changes in mIPSC amplitude.

Tissue sections from cerebellar vermis of Calb1Cre;Dag1cKOs (B) and littermate controls (A) were immunostained for Calbindin to visualize Purkinje cells along with the presynaptic marker VGAT (magenta) and postsynaptic GABAA receptor subunit GABAAα1 (green). The first panel shows a low-magnification view of the Calbindin channel. Subsequent panels are magnified views of the yellow outlined region in the first panel. Images are maximum projections. C Quantification of VGAT (left) and GABAAα1 (right) puncta density (per 100 μm3) for dendritic (“Molecular Layer”) and somatic compartments. Error bars are presented as mean + SEM. pVGAT_ML = 0.002, pVGAT_Soma = 2.48E-05, pGABAAα1_ML = 0.003, pGABAAα1_Soma = 0.003. D Quantification of VGAT (left) and GABAAα1 (right) puncta size (in μm3) for the dendritic compartment. pVGAT = 0.778, pGABAAα1 = 0.969. Calb1Cre;Dag1Ctrl N = 5 animals. Calb1Cre;Dag1cKO N = 4 animals.
While the synaptic deficits observed after early developmental deletion of Dag1 suggest that Dystroglycan is required for synapse formation, this finding is inconsistent with earlier work in cultured hippocampal neurons that found Dystroglycan to be dispensable for synapse formation in vitro20. It remains possible that synapses form successfully but disassemble before the P25–P35 experimental time point. To exclude this possibility, we analyzed VGAT and GABAAα1 in Calb1Cre;Dag1cKO mice at P12, a peak synaptogenic time point in MLI:PC development34. We observed a loss of VGAT and GABAAα1 synaptic puncta on the PC soma and a reduction of GABAAα1, but not VGAT, puncta density in the molecular layer (Fig. 4A–C). While there was no change in VGAT puncta volume, there was a reduction in GABAAα1 puncta volume (Fig. 4D). The severity of loss of the postsynaptic element GABAAα1 compared to presynaptic VGAT at this early synaptogenic time point is consistent with Dystroglycan coordinating synapse formation from the postsynaptic side.

Tissue sections from cerebellar vermis of P12 Calb1Cre;Dag1cKOs (B) and littermate controls (A) were immunostained for Calbindin to visualize Purkinje cells along with the presynaptic marker VGAT (magenta) and postsynaptic GABAA receptor subunit GABAAα1 (green). The first panel shows a low-magnification view of the Calbindin channel. Subsequent panels are magnified views of the yellow outlined region in the first panel. Images are maximum projections. C Quantification of VGAT (left) and GABAAα1 (right) puncta density (per 100 μm3) for dendritic (“Molecular Layer”) and somatic compartments. Error bars are presented as mean + SEM. pVGAT_ML = 0.062, pVGAT_Soma = 0.010, pGABAAα1_ML = 0.009, pGABAAα1_Soma = 0.006. D Quantification of VGAT (left) and GABAAα1 (right) puncta size (in μm3) for the dendritic compartment. pVGAT = 0.495, pGABAAα1 = 0.029. Calb1Cre;Dag1Ctrl N = 4 animals. Calb1Cre;Dag1cKO N = 4 animals.
Dystroglycan is required for the long-term maintenance of inhibitory MLI:PC synapses
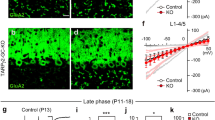
The synaptic phenotypes observed following early deletion of Dag1 from PCs using Calb1Cre suggest that Dystroglycan is required for initial MLI axonal recognition of postsynaptic sites and subsequent synapse formation, similar to what we have reported for CCK+/CB1R+ basket synapses in the hippocampus23. Testing whether Dystroglycan is involved in the maintenance of synapses requires deleting Dag1 after synapse formation. MLI:PC synapses begin forming around P7 and are largely formed by P14, with additional synaptogenesis extending past P2131,35,36. Recent work using L7Cre to delete Dag1 found that Dystroglycan was gradually eliminated from PCs and wasn’t fully absent from all PCs until P90. This prolonged localization of Dystroglycan at synapses in these mice likely reflects its slow rate of protein turnover, as Dystroglycan has a half-life of ~25 days in skeletal muscle37. L7Cre;Dag1cKOs did exhibit impaired MLI:PC synapse function and a reduction in pre- and postsynaptic markers at P180, suggesting that Dystroglycan does indeed play a role in synapse maintenance17. To confirm this result, we used Pcp2Cre mice, in which PCs begin to express Cre gradually between P7-P14, to delete Dag1 after synapse formation has initiated28. Pcp2Cre;Dag1flox/− conditional knockouts (Pcp2Cre;Dag1cKO) showed Cre-mediated recombination in all PCs at P14 and loss of Dystroglycan protein by P30. (See Jahncke and Wright, 202428 for a validation of this genetic manipulation.)
The observed Pcp2Cre recombination timeline is closely aligned with the synaptogenic period in PCs and, given the time course of protein turnover, is likely deleting Dag1 soon after synapse formation. When we recorded mIPSCs from Pcp2Cre;Dag1cKOs aged P25–P35, they showed no significant difference from littermate controls (Fig. 5A–F), suggesting that synapses remain functional without Dystroglycan in the short term. However, when we recorded mIPSCs from animals aged P55–P65, we observed a reduction in mIPSC amplitude and frequency (Fig. 5G–L). Together with the data from Calb1Cre;Dag1cKOs (Figs. 2–4), these results show that Dystroglycan is required for both the formation and the long-term maintenance of MLI:PC synapses.

A–F mIPSC analysis in Pcp2Cre;Dag1cKO and littermate control Purkinje cells at P25–P35. A Average mIPSC event. B Representative 15 s trace of mIPSCs. mIPSC amplitude (C) and frequency (D); error bars are presented as mean + SEM. Cumulative frequency histogram for individual mIPSC amplitudes (E) and inter-event intervals (IEI) (F). G–L mIPSC analysis in Pcp2Cre;Dag1cKO and littermate control Purkinje cells at P55–P65. G Average mIPSC event. H Representative 15 s trace of mIPSCs. mIPSC amplitude (I) and frequency (J); error bars are presented as mean + SEM. Cumulative frequency histogram for individual mIPSC amplitudes (K) and inter-event intervals (IEI) (L). P25–P35 Pcp2Cre;Dag1Ctrl N = 13 cells, 5 animals; P25–P35 Pcp2Cre;Dag1cKO N = 9 cells, 5 animals; pAmp = 0.794, pFreq = 0.431. P55–P65 Pcp2Cre;Dag1Ctrl N = 12 cells, 5 animals; P55–P65 Pcp2Cre;Dag1cKO N = 14 cells, 4 animals; pAmp = 0.040, pFreq = 1.26E-04.
Extracellular glycosylation of α-Dystroglycan is required for inhibitory synapse formation and function
The observed synaptic phenotypes in the absence of Dystroglycan are likely driven by a combination of (1) extracellular glycan-protein interactions mediated by the matriglycan chains on α-Dystroglycan interacting with binding partners in cis or trans and (2) intracellular protein-protein interactions or signaling cascades through the intracellular C-terminus of β-Dystroglycan. To define which aspects of Dystroglycan’s synaptic function are mediated by these distinct molecular mechanisms, we generated mice with Dystroglycan lacking either (1) extracellular glycosylation or (2) the intracellular domain.
The extracellular alpha subunit of Dystroglycan contains O-mannosyl-linked glycan chains with terminal -3Xylα1-3GlcAβ1-disaccharide repeats termed “matriglycan”, which interacts with LG domain-containing extracellular binding partners38,39. The glycosyltransferase Pomt2 (Protein O-mannosyltransferase 2), in a heterocomplex with Pomt1, is responsible for adding the initial O-mannose to Dystroglycan, and its deletion results in a complete loss of matriglycan chains40. We used Calb1Cre to conditionally delete Pomt2 from PCs, generating Calb1Cre;Pomt2flox/flox conditional knockouts (Calb1Cre;Pomt2cKO) and Calb1Cre;Pomt2flox/+ littermate controls (Calb1Cre;Pomt2Ctrl). Loss of Dag1 glycosylation was confirmed by immunostaining with the IIH6 antibody, which recognizes matriglycan. In contrast to the punctate synaptic staining pattern seen in Calb1Cre;Pomt2Ctrl PCs, Calb1Cre;Pomt2cKO PCs did not show any synaptic IIH6 immunoreactivity (Supplementary Fig. 5A). We tested several potential antibodies against Dystroglycan core protein epitopes and β-Dystroglycan to determine whether loss of the glycan chains affected Dystroglycan’s synaptic localization, but we were unable to find immunostaining conditions in which there was specific labeling in control tissue. While it is feasible that Pomt2 is required for O-mannosylation of synaptic proteins other than Dystroglycan, this is unlikely41,42. Furthermore, we did not observe any phenotypes in Pomt2 conditional knockouts that we also did not observe in Dag1 conditional knockout models.
To assess MLI:PC synapse function in the absence of matriglycan, we conducted mIPSC recordings in P25–P35 Calb1Cre;Pomt2cKOs PCs and littermate controls (Fig. 6A–F). Similar to Dag1 conditional knockouts, Calb1Cre;Pomt2cKOs exhibited a reduced mIPSC frequency compared to controls (Fig. 6A, B, D, F). However, mIPSC amplitude was normal (Fig. 6C, E). Based on the reduced mIPSC frequency, we hypothesized that we would observe a reduction in inhibitory synapse density in Calb1Cre;Pomt2cKOs similar to that observed in Calb1Cre;Dag1cKOs. Indeed, both VGAT and GABAAα1 puncta densities were reduced in both the dendritic and somatic compartments in Calb1Cre;Pomt2cKOs compared to littermate controls (Fig. 7A–C) while there was no change in puncta size for either marker (Fig. 7D). From these data, it is clear that extracellular glycan-protein interactions are involved in regulating the organization of the inhibitory postsynapse.

A–F mIPSC analysis in Calb1Cre;Pomt2cKO and littermate control Purkinje cells at P25–P35. A Average mIPSC event. B Representative 15 s trace of mIPSCs. mIPSC amplitude (C) and frequency (D); error bars are presented as mean + SEM. Cumulative frequency histogram for individual mIPSC amplitudes (E) and inter-event intervals (IEI) (F). G–L mIPSC analysis in Calb1Cre;Dag1cΔICD and littermate control Purkinje cells at P25–P35. G Average mIPSC event. H Representative 15 s trace of mIPSCs. mIPSC amplitude (I) and frequency (J); error bars are presented as mean + SEM. Cumulative frequency histogram for individual mIPSC amplitudes (K) and inter-event intervals (IEI) (L). Calb1Cre;Pomt2Ctrl N = 11 cells, 3 animals; Calb1Cre;Pomt2cKO N = 10 cells, 4 animals; pAmp = 0.426, pFreq = 0.011. Calb1Cre;Dag1WT N = 13 cells, 4 animals; Calb1Cre;Dag1cΔICD N = 14 cells, 4 animals; pAmp = 5.19E-04, pFreq = 7.28E-04.

Tissue sections from cerebellar vermis of Calb1Cre;Pomt2cKOs (B) and littermate controls (A) were immunostained for Calbindin to visualize Purkinje cells along with the presynaptic marker VGAT (magenta) and postsynaptic GABAA receptor subunit GABAAα1 (green). The first panel shows a low-magnification view of the Calbindin channel. Subsequent panels are magnified views of the yellow outlined region in the first panel. Images are maximum projections. C Quantification of VGAT (left) and GABAAα1 (right) puncta density (per 100 μm3) for dendritic (“Molecular Layer”) and somatic compartments. Error bars are presented as mean + SEM. pVGAT_ML = 0.006, pVGAT_Soma = 0.026, pGABAAα1_ML = 0.013, pGABAAα1_Soma = 0.028. D Quantification of VGAT (left) and GABAAα1 (right) puncta size (in μm3) for the dendritic compartment. pVGAT = 0.764, pGABAAα1 = 0.219. Calb1Cre;Pomt2Ctrl N = 3 animals. Calb1Cre;Pomt2cKO N = 3 animals.
The intracellular domain of β-Dystroglycan is required for inhibitory synapse function but not formation
The intracellular domain of Dystroglycan interacts directly with Dystrophin, which mediates interactions with the actin cytoskeleton and other scaffolding/signaling molecules. In a mouse model lacking the relevant brain isoforms of Dystrophin (mdx), PCs show reduced mIPSC frequency and amplitude, reduced GABAAα1 receptor puncta density, and fewer MLI:PC contacts, showing that Dystrophin is required for inhibitory PC synapses in a manner similar to Dystroglycan25,26,43,44,45,46. Mice lacking the cytoplasmic portion of Dystroglycan that interacts with Dystrophin develop muscular dystrophy but exhibit largely normal brain development47. However, cytoplasmic deletion mutants exhibit synaptic deficits at CCK+/CB1R+ basket synapses in the hippocampus23. We therefore hypothesized that, in addition to extracellular Dystroglycan glycosylation, the intracellular domain of Dystroglycan may also be required for MLI:PC synapse function. To investigate the role of the intracellular domain of Dystroglycan specifically at MLI:PC synapses, we generated conditional cytoplasmic domain mutants by crossing Calb1Cre;Dag1flox/+ mice with mice constitutively expressing one copy of truncated Dystroglycan lacking the intracellular domain (Dag1ΔICD/+), generating Calb1Cre;Dag1flox/ΔICD mutants (Calb1Cre;Dag1cΔICD) and Calb1Cre;Dag1+/+ littermate controls (Calb1Cre;Dag1WT). Immunohistochemistry with IIH6 showed that Calb1Cre;Dag1cΔICD mutants maintained synaptic localization of glycosylated PC Dystroglycan in the absence of the intracellular domain (Supplementary Fig. 5B).
To measure the contribution of the intracellular domain of Dystroglycan to MLI:PC synapse function we conducted mIPSC recordings from P25–P35 Calb1Cre;Dag1cΔICD PCs and those of littermate controls and observed a phenotype that resembled that of Calb1Cre;Dag1cKO PC mIPSCs: a reduction in both mIPSC amplitude and frequency (Fig. 6G–L). We therefore conclude that the amplitude component of Dystroglycan-containing MLI:PC synapses is governed by intracellular interactions between Dystroglycan and other postsynaptic proteins (likely including Dystrophin) and/or intracellular signaling initiated by the C-terminus of β-Dystroglycan. The frequency component of this synaptic circuit, however, appears to be influenced by factors dictated by both extracellular glycan-protein interactions and intracellular protein-protein interactions or signaling pathways.
Due to the reduction in mIPSC frequency observed in Calb1Cre;Dag1cΔICD PCs (Fig. 6J, L), we hypothesized that we would see a similar reduction in inhibitory synapse density in Calb1Cre;Dag1cΔICD as that observed in Calb1Cre;Dag1cKO (Fig. 3C) and Calb1Cre;Pomt2cKO (Fig. 7C) PCs. While we still observed a reduction in VGAT and GABAAα1 puncta density on the PC soma, we found no change in the density or size of VGAT or GABAAα1 puncta in the molecular layer (Fig. 8A–D). This suggests that loss of the intracellular domain of Dystroglycan does not affect the ability of presynaptic MLIs to recognize postsynaptic PCs and form synaptic contacts within the molecular layer, but it is required for proper somatic innervation.

Tissue sections from cerebellar vermis of Calb1Cre;Dag1cΔICDs (B) and littermate controls (A) were immunostained for Calbindin to visualize Purkinje cells along with the presynaptic marker VGAT (magenta) and postsynaptic GABAA receptor subunit GABAAα1 (green). The first panel shows a low-magnification view of the Calbindin channel. Subsequent panels are magnified views of the yellow outlined region in the first panel. Images are maximum projections. C Quantification of VGAT (left) and GABAAα1 (right) puncta density (per 100 μm3) for dendritic (“Molecular Layer”) and somatic compartments. Error bars are presented as mean + SEM. pVGAT_ML = 0.575, pVGAT_Soma = 0.018, pGABAAα1_ML = 0.372, pGABAAα1_Soma = 0.044. D Quantification of VGAT (left) and GABAAα1 (right) puncta size (in μm3) for the dendritic compartment. pVGAT = 0.424, pGABAAα1 = 0.719. Calb1Cre;Dag1WT N = 3 animals. Calb1Cre;Dag1cKO N = 3 animals.
As the reduction in somatic synapse number is quite small in comparison to the number of molecular layer synapses, we are left to assume that the reduced mIPSC frequency we observed in Calb1Cre;Dag1cΔICD PCs is due to alterations to the synapse other than simply synapse number. Previous work involving paired recordings between MLI and PC pairs in Dystrophin-deficient mdx mice identified a reduction in the size of the readily releasable pool26. A similar mechanism could explain the reduced mIPSC frequency in Calb1Cre;Dag1cΔICD PCs (Fig. 6J, L). The intracellular domain of Dystroglycan interacts directly with Dystrophin, so a common mechanism between the mdx and Calb1Cre;Dag1cΔICD models is likely48. While additional work will be necessary to determine the details underlying the observed phenotypes, these data show that the intracellular domain of Dystroglycan plays a crucial role in MLI:PC synapse function but is only required for the formation of somatic, not dendritic, synapses.
Discussion
Despite years of study, the molecules required for MLI:PC synaptogenesis remain enigmatic. Several postsynaptic transmembrane proteins, including Neuroligins, Kit, and Dystroglycan, are required for proper MLI:PC synapse maintenance and function17,49,50,51. However, these prior studies were unable to address the role of these proteins in initial synaptogenesis due to the lack of genetic tools that target Purkinje neurons early in development. With the identification of Calb1Cre as a tool for the deletion of genes from PCs early in development27,28, we were able to show for the first time that Dystroglycan is required for initial synapse formation at MLI:PC synapses (Figs. 2–4). Loss of Dystroglycan was accompanied by reductions in both mIPSC amplitude, mIPSC frequency, and inhibitory pre- and postsynaptic marker density, illustrating that synapses are not able to form normally in the absence of Dystroglycan.
Previous work used the L7Cre line to delete Dag1 from PCs after synaptogenesis, which did not achieve complete loss of Dystroglycan protein until P90, limiting the study to the role of Dystroglycan in long-term synapse maintenance17,28. Nevertheless, it was determined that Dystroglycan is indeed required for synapse maintenance, as the L7Cre;Dag1flox/flox PCs showed a decreased inhibitory synapse density and decreased mIPSC frequency and amplitude17. We confirmed these results using a Pcp2Cre mouse line mated with mice in which only one copy of Dag1 needed to be excised (Dag1flox/−), achieving complete loss of Dystroglycan by P3028. We observed similarly impaired synapse function in Pcp2Cre;Dag1flox/− (Pcp2Cre;Dag1cKO) PCs at P55–P65 (Fig. 5G–L), confirming Dystroglycan’s role in MLI:PC synapse maintenance. Curiously, mIPSC recordings in Pcp2Cre;Dag1cKO PCs at P25–P35, in which Dag1 is deleted from most cells and most protein turned over, showed no significant impact on MLI:PC function (Fig. 5A–F). There are two potential explanations for this result: (1) synapses remain stable for some period of time (most likely on the order of days to weeks) after Dystroglycan protein is eliminated, or (2) the amount of Dystroglycan protein remaining at P25–P35 is below the detection threshold for immunohistochemistry, but sufficient to maintain synapse function.
How does Dystroglycan regulate MLI:PC synapse formation and function? The genetic manipulations we used in this study allowed us to distinguish the extracellular (Calb1Cre;Pomt2cKO) and intracellular (Calb1Cre;Dag1cΔICD) functions of Dystroglycan at these synapses. Our experiments using Calb1Cre;Pomt2cKO mice show that extracellular glycosylation of α-Dystroglycan is required for Dystroglycan’s role in both synaptogenesis and synapse function (Figs. 6 and 7), in agreement with our previous work showing that forebrain deletion of Dag1 or Pomt2 results in identical deficits in hippocampal CCK+/CB1R+ basket synapse formation and function23. Removal of Pomt2 results in loss of all O-mannosyl-linked glycosylation on Dystroglycan (Supplementary Fig. 5)40, including the −3Xylα1-3GlcAβ1- disaccharide repeats referred to as matriglycan elongated by the glycosyltransferase LARGE152,53. The length of the matriglycan repeat can serve as a “tunable matrix” capable of binding multiple LG domain-containing proteins simultaneously, allowing large macromolecular complexes to form38,54. PC matriglycan migrates at a significantly higher molecular weight than matriglycan in other neuronal and non-neuronal populations, suggesting PCs may contain longer matriglycan chains with increased binding capacity47.
The intracellular domain of Dystroglycan was long thought to be dispensable for neuronal function, as mice lacking the intracellular domain of Dystroglycan exhibit normal neuronal migration and axon targeting13,23,47. However, we recently found that hippocampal CCK+/CB1R+ basket cells exhibit slightly altered perisomatic axon targeting and impaired synapse function despite normal synapse formation when the intracellular domain is deleted23. In the present study we similarly found impaired synapse function in Calb1Cre;Dag1cΔICD mice (Fig. 6G–L) and reduced somatic innervation despite relatively normal MLI:PC synapse formation (Fig. 8). Not only do these findings confirm the role of the cytoplasmic domain in synapse function, but they also illustrate the cell-autonomous nature of the mechanism as the Calb1Cre mutation is restricted to PCs within the cerebellum. Taken together, the results obtained in Calb1Cre;Pomt2cKO and Calb1Cre;Dag1cΔICD mutants show that both extracellular glycosylation and intracellular scaffolding/signaling by Dystroglycan is required for proper MLI:PC synapse function, while the intracellular domain is required for somatic innervation and is dispensable for the formation of presynaptic contacts onto PC dendrites.
The identity of Dystroglycan’s presynaptic partner(s) at MLI:PC synapses remains unknown. Matriglycan binds specifically to proteins with LG domains, and there are upwards of 20 LG domain-containing proteins expressed in cerebellar cellular populations that are candidate extracellular binding partners of Dystroglycan. Biochemical isolation of Dystroglycan-containing complexes to identify potential synaptic binding partners is complicated by its expression in many cell types in the brain, with neuronal Dystroglycan representing a minority proportion. Possible binding partners for Dystroglycan at MLI:PC synapses include members of the Neurexin family of presynaptic adhesion molecules. Biochemical studies show that Dystroglycan binds to two of the LG domains present in Neurexins in a glycosylation-dependent manner, and Dystroglycan:Neurexin-3 interactions regulate inhibitory synapse function in the olfactory bulb and medial prefrontal cortex29,55,56. However, whether this interaction occurs at MLI:PC synapses has not yet been examined. All three Neurexins are expressed by MLIs, and these neurons have been difficult to selectively target for conditional deletions57,58.
Neurexins also interact with postsynaptic Neuroligins, and all three Neuroligin isoforms are expressed in PCs. Triple conditional deletion of Neuroligin1/2/3 from PCs causes synaptic alterations similar to those observed when Dystroglycan is conditionally deleted from PCs (Figs. 2 and 3)51. This effect was not observed in conditional Nlgn1/Nlgn3 double knockouts but was seen in Ngln2/Nlgn3 double knockouts. A Nlgn2 constitutive knockout only partially recapitulated these results, suggesting that while Nlgn2 is the primary driver of this phenotype, some degree of molecular compensation can occur in its absence51. Neuroligins are thought to be linked to the intracellular domain of β-Dystroglycan through S-SCAM/MAGI-259, and Nlgn2 puncta density is reduced at somatic MLI:PC synapses in PC Dag1 conditional knockouts17,60. Thus, Dystroglycan can interact directly and indirectly with Neurexins and Neuroligins in a network of pre- and postsynaptic cell adhesion complexes.
Synapse formation occurs in multiple phases: a presynaptic axon must first recognize its postsynaptic target through some sort of recognition cue, after which pre- and postsynaptic molecules are recruited to the nascent synapse. Constitutive or developmentally early deletions of Neurexins or Neuroligins typically result in impaired synapse function; however, whether synapse number is affected by genetic manipulation varies, suggesting that initial postsynaptic partner recognition and synaptogenesis is mediated by other molecules29,51,61,62,63. Dystroglycan, on the other hand, is required for both the initial formation of a synapse and its maintenance; developmentally early deletion of Dag1 results in a loss of synapses and reduced synaptic function (Figs. 2–4)22,23. Similarly, later deletion of Dag1 also impacts synapse function, showing it is also required for subsequent synapse maintenance (Fig. 5)17.
Together with evidence from other Dystroglycan-expressing synapses in the brain22,23,24,29, our data suggest that Dystroglycan acts as a recognition molecule for presynaptic axon targeting of the postsynaptic compartment, after which its role switches to one of postsynaptic molecule recruitment and synapse stabilization. While the glycan-protein interaction between Dystroglycan and Neurexin may be required for the maturation and stabilization of a synapse, the presynaptic binding partner(s) of Dystroglycan during axon targeting may be mediated by other LG domain-containing proteins.
In both the hippocampus23 and the cerebellum (Figs. 6 and 8), the intracellular domain of Dystroglycan is required for inhibitory synapse function, while presynaptic targeting is largely normal. While intracellular interactions with Dystroglycan in the brain remain poorly described, the prevailing interaction contributing to MLI:PC synapse function is likely between Dystroglycan and Dystrophin (Dmd), which interact directly through the intracellular domain of Dystroglycan64,65. There is extensive literature supporting a role for Dystrophin at MLI:PC synapses17,25,26,43,45,46,66. Similar to our data in Calb1Cre;Dag1cΔICD PCs (Fig. 6G–L), Dystrophin-deficient mdx mice exhibit impaired mIPSC frequency and amplitude25,26,43,46. Since mdx mice have a constitutive mutation in Dystrophin, it is not clear if it functions in PCs, MLIs, or both. Dystrophin is predominantly localized to the postsynaptic compartment at MLI:PC synapses67, and its loss results in abnormal GABA receptor clustering in PCs45, suggesting a postsynaptic role. However, loss of Dystrophin also affects presynaptic MLI function. Paired recordings between MLIs and PCs in mdx mice revealed an increase in the failure rate, but no change in paired pulse ratio, suggesting that release probability does not explain the increased failure rate26. Rather, the size of the readily releasable pool and the quantal content were both reduced in mdx MLIs26. One possibility is that Dystroglycan acts as the link between postsynaptic Dystrophin and presynaptic defects in MLI function, possibly through transsynaptic interactions with Neurexins. Studies using MLI:PC paired recordings in Calb1Cre;Dag1cΔICD mice or PC specific deletion of Dystrophin could inform whether this proposed mechanism is at play.
Historically, MLIs have been classified based on their morphological features, with basket cells in the lower third of the molecular layer targeting their axons to PC somata and proximal dendrites, while stellate cells located in the upper third of the molecular layer target their axons to PC distal dendrites. However, recent studies have blurred these distinctions. While transcriptomic classification identified two distinct MLI populations, both types were present throughout the entire molecular layer and did not precisely correspond to the classical morphological distinction of basket and stellate cells57,68. Subsequent functional comparisons showed that the MLI1 transcriptional subtype primarily forms inhibitory inputs onto PCs, while the MLI2 transcriptional subtype primarily forms inhibitory inputs onto MLI1 cells69.
IIH6 immunoreactivity shows Dystroglycan localized at synaptic puncta on PC somata and dendrites throughout the entirety of the molecular layer (Fig. 1). Deletion of Dystroglycan28 or Pomt2 (Supplementary Fig. 5A) from PCs with Calb1Cre results in a complete loss of punctate IIH6 staining in the molecular layer, suggesting Dystroglycan is predominantly localized at MLI1:PC synapses. While loss of Dystroglycan caused a reduction in both VGAT and GABAAα1 puncta throughout the PC layer and molecular layer, the reduction on the surface of the PC soma was particularly striking, especially so for the postsynaptic GABAAα1 receptor subunit (Figs. 3A, B and 4A, B). One unique molecular feature of somatic MLI:PC synapses is that they lack the inhibitory postsynaptic scaffolding molecule Gephyrin, whereas dendritic MLI:PC synapses do contain Gephyrin35. While GABAA receptors are typically thought to be anchored to the postsynapse through Gephyrin, Dystroglycan may play a more pronounced role in scaffolding postsynaptic receptors at Gephyrin-lacking somatic MLI:PC synapses. A complete accounting of the molecular composition of Dystroglycan-containing postsynaptic inhibitory complexes, and how this may be different in distinct subcellular compartments in PCs remains an open question.
Materials and methods
Animal husbandry
All animals were housed and cared for by the Department of Comparative Medicine at Oregon Health and Science University (OHSU), an AAALAC-accredited institution. We have complied with all relevant ethical regulations for animal use. Animal procedures were approved by OHSU Institutional Animal Care and Use Committee (Protocol # IS00000539), adhered to the NIH Guide for the care and use of laboratory animals, and were provided with 24-h veterinary care. Animal facilities are regulated for temperature and humidity and maintained on a 12-h light-dark cycle and were provided food and water ad libitum. Mice were used between the ages of P12 and P65 (as indicated in the text or figure legend). Mice were euthanized by administration of CO2 followed by exsanguination or decapitation.
Mouse strains and genotyping
The day of birth was designated postnatal day 0 (P0). The ages of mice used for each analysis are indicated in the figure and figure legends. Mouse strains used in this study have been previously described and were obtained from Jackson Labs, unless otherwise indicated (Table 2). Breeding schemas are as described in Table 3. Where possible, mice were maintained on a C57BL/6 background. We previously validated the specificity and timing of Calb1Cre and Pcp2Cre for the conditional knockout of Dag1 from PCs (see Jahncke and Wright, 2024)28. The Dag1ΔICD line was outcrossed to a CD-1 background for one generation to increase the viability of mutant pups. The Dag1ΔICD line was then backcrossed to C57BL/6 breeders for 3 generations before performing experiments. Dag1+/− mice were generated by crossing the Dag1flox/flox line to a Sox2Cre line to generate germline Dag1Δ/+ mice, hereafter referred to as Dag1+/− as the resultant transcript is nonfunctional. These mice were thereafter maintained as heterozygotes. Genomic DNA extracted from toe or tail samples using the HotSHOT method70 was used to genotype animals. Primers for genotyping can be found on the JAX webpage or originating article. Dag1+/− mice were genotyped with the following primers: CGAACACTGAGTTCATCC (forward) and CAACTGCTGCATCTCTAC (reverse). For each mouse strain, littermate controls were used for comparison with mutant mice. For all experiments, mice of both sexes were used indiscriminately.
Tissue fixation and processing
Mice older than P21 were perfused with either 4% Paraformaldehyde (PFA) in PBS, pH 7 (Fig. 1, Supplementary Figs. 2, 3 and 5) or 9% Glyoxal/8% Acetic acid (GAA) in PBS, pH 4-5 (modified from Konno et al.71) (Figs. 3, 4, 7 and 8). PFA was prepared from powder (Thermo Scientific Chemicals, Cat. No. A1131336). Glyoxal was purchased as a 40% stock solution (Thermo Scientific Chemicals, Cat. No. 156225000), and Glacial Acetic Acid was purchased from Fisher (Cat. No. A38-500). PFA perfusion was used for all immunohistochemical experiments except for those involving anti-GABAAα1, which was only compatible with GAA-fixed tissue. P30 mice were deeply anesthetized using CO2 and transcardially perfused with ice-cold 0.1 M PBS for 2 min to clear blood from the brain, followed by either (1) 15 mL of ice-cold 4% PFA in PBS or (2) 20 mL of ice-cold 9%/8% GAA in PBS, as indicated. After perfusion, brains were dissected and post-fixed in either (1) 4% PFA for 30 min at room temperature or (2) 9%/8% GAA overnight at 4 °C. P12 mice were anesthetized with CO2, followed by swift decapitation; the cerebellum was rapidly excised and immersed in 9%/8% GAA overnight at 4 °C. Brains were rinsed with PBS, embedded in 4% low-melt agarose (Fisher, Cat. No. 16520100), and sectioned at 70 μm using a vibratome (VT1200S, Leica Microsystems Inc., Buffalo Grove, IL) into 24-well plates containing 1 mL of 0.1 M PBS with 0.02% Sodium Azide.
Immunohistochemistry
Immunofluorescence detection of antigens was performed as follows: free-floating vibratome sections (70 μm) were briefly rinsed with PBS, then blocked for 1 h in PBS containing 0.2% Triton-X (PBST) plus 2% normal goat serum. For staining of Dystroglycan synaptic puncta, an antigen retrieval step was performed prior to the blocking step: sections were incubated in sodium citrate solution (10 mM Sodium Citrate, 0.05% Tween-20, pH 6.0) for 12 min at 95 °C in a water bath, followed by 12 min at room temperature. Sections were incubated with primary antibodies (Table 4) diluted in blocking solution at 4 °C for 48–72 h. Following incubation with the primary antibody, sections were washed with PBS three times for 20 min each. Sections were then incubated with a cocktail of secondary antibodies (1:500, Alexa Fluor 488, 546, 647) in blocking solution containing Hoechst 33342 (1:10,000, Life Technologies, Cat. No. H3570) overnight at room temperature. Finally, sections were mounted on slides using Fluoromount-G (SouthernBiotech) and sealed using nail polish.
Microscopy
Imaging was performed on either a Zeiss Axio Imager M2 fluorescence upright microscope equipped with an Apotome.2 module or a Zeiss LSM 980 laser scanning confocal built around a motorized Zeiss Axio Observer Z1 inverted microscope with a Piezo stage. As indicated below (Table 5), some experiments utilizing the LSM 980 confocal, a linear Wiener filter deconvolution step (Zeiss LSM Plus) was used at the end of image acquisition with 1.2X Nyquist sampling. The Axio Imager M2 uses a metal halide light source (HXP 200C), an Axiocam 506 mono camera, and a 20X/0.8 NA Plan-Apochromat objective. The LSM 980 confocal light path has two multi-alkali PMTs and two GaAsP PMTs for four-track imaging. Confocal images were acquired using a 63X/1.4 NA Plan-Apochromat Oil DIC M27 objective. Z-stack images were acquired and analyzed offline in ImageJ/FIJI72 or Imaris 10.2 (Oxford Instruments). Images used for quantification between genotypes were acquired using the same exposure times or laser power. Brightness and contrast were adjusted in FIJI to improve the visibility of images for publication. Figures were composed in Adobe Illustrator 2024 (Adobe Systems).
Image quantification
For imaging experiments, 4–8 images were acquired from 2 to 4 sagittal sections per animal, and at least three animals per genotype were used for analysis. Sections were chosen from the cerebellar vermis, and images were acquired within the cerebellar cortex of lobules V–VI.
For analysis of synaptic colocalization, puncta density, and puncta volume: 0.2 μm z-stacks covering 5μm were acquired using a 63× objective on a Zeiss LSM 980 (as described above) such that the PC layer and molecular layer were captured in the image. The entirety of the imaged region was analyzed, and individual data points represent individual images. Analysis of image stacks was performed in Imaris 10.2 (Oxford Instruments). The Surfaces function was used to reconstruct the PCs using either the Parvalbumin (Fig. 1, Supplementary Fig. 2) or Calbindin (Figs. 3, 4, 7 and 8) channel as illustrated in Supplementary Figs. 1A and 4A. To select for PC signal, the synaptic marker channels were masked to only include information at the surface or inside of the PC surface.
For colocalization analyses, the Surfaces function was used to reconstruct the synaptic puncta volumes of the masked synaptic channels (Supplementary Fig. 1C, D). Surfaces in the IIH6 channel and Surfaces in the marker channel (VGAT, GAD67, VGluT1, or VGluT2) were deemed colocalized if the distance between the two synaptic Surfaces was ≤0 µm (Supplementary Fig. 1E). As a control for random colocalization, the marker channel was mirrored, masked, and reconstructed with the same parameters as the original channel and colocalization with the original IIH6 channel was calculated.
To identify puncta for synaptic density analysis, the Spots function was used to determine the location of synaptic puncta in 3-dimensional space using local contrast. To exclude nonspecific Spots, Spots with a fluorescence standard deviation in the bottom 10th percentile (uniform signal) and a volume less than 0.1 μm3 (noise) were excluded. (Supplementary Fig. 4C). Puncta density was calculated as the total number of Spots in a given compartment, divided by the volume of that compartment as measured by the PC surface reconstruction. Surface reconstruction of synaptic fluorescence was used to measure puncta volume. Surfaces further than 0.7 µm from an identified spot were excluded from analysis (Supplementary Fig. 4D).
For analysis of PC and MLI cell densities: 0.5 μm z-stacks covering 20 μm were acquired using a 20× objective on a Zeiss Axio Imager M2. Maximum projections were used for analysis in FIJI72. Cells were counted using the Multi-Point Tool. The length of the region of cerebellar cortex analyzed was measured using the Freehand Line tool; this value was used to normalize the cell counts to unit length.
Electrophysiology
For acute slice preparation, mice were deeply anesthetized in 4% isoflurane and subsequently injected with a lethal dose of 2% 2, 2, 2-Tribromoethanol in sterile water followed by transcardial perfusion with 10 mL ice-cold cutting solution containing the following (in mM): 100 Choline Chloride, 2.5 KCl, 1.2 NaH2PO4, 25 NaHCO3, 3 3-myo-inositol, 25 glucose, 5 Na Ascorbate, 2 Na Pyruvate, 7 MgSO4, 0.5 CaCl2; pH 7.3, 300–340 mmol/kg. After rapid decapitation, the brain was briefly submerged in ice-cold cut solution bubbled with carbogen (95% oxygen, 5% CO2) and then sectioned into 300 μm sagittal sections (Leica VT1200S vibratome) in bubbled ice-cold cut solution. Slices were recovered in 37 °C recording ACSF, bubbled, for 15 min followed by 1 h in room temperature recording ACSF (in mM: 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 3 KCl, 25 D-Glucose, 2 CaCl2, 1 MgCl2) with an osmolarity of 310–325 mmol/kg and supplemented with 1.5 mM Na Ascorbate, bubbled.
PCs were patched in whole cell configuration using 1.2–2 MΩ borosilicate glass pipettes filled with high chloride internal solution containing the following (in mM): 125 CsCl, 2.5 MgCl2, 0.5 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, 5 QX-314; pH 7.2, 300 mmol/kg. Pipettes were wrapped in parafilm to reduce capacitive currents. Cells were voltage clamped at −70 mV and continuously superfused with 2–3 mL/min bubbled recording ACSF (310–325 mmol/kg) containing 10 μM NBQX to block excitatory transmission and 500 nM TTX to block action potentials. An inline heater was used to maintain the bath temperature at 34 °C. After reaching a stable baseline, 5 min of mIPSCs were recorded. Signals were amplified with an AxoPatch 200B amplifier (Molecular Devices), low-pass filtered at 5 kHz, and digitized and sampled at 10 kHz with a NIDAQ analog-to-digital board (National Instruments). Data were acquired and analyzed using a custom script in Igor Pro 8 (Wavemetrics). A hyperpolarizing step of −10 mV was applied before each sweep to monitor input resistance, series resistance, and measure cell capacitance. Series resistance was not compensated and was maintained below 20 MΩ. Cells were excluded if series resistance changed by more than 25%.
Rise and decay kinetics were calculated on each detected event for a given cell and averaged to a single value for that cell. Rise time was defined as the amount of time between 10% and 90% of the maximum amplitude of a given event. Decay was calculated as the time constant when the decay phase of the event was fit to an exponential curve.
Statistics and reproducibility
Phenotypic analyses were conducted using tissue collected from at least three mice per genotype from at least two independent litters. For microscopy data, multiple samples from the same mouse were averaged into one value used for analysis. For electrophysiology data, cells were used as replicates. The number of mice/cells used for each analysis (“N”) is indicated in the text or figure legends. Two or more technical replicates with unique biological replicates were performed for each experiment. Power analysis was used to determine sample sizes with α = 0.05 and β = 0.80, and the effect size was determined using pilot data. Phenotypes were indistinguishable between male and female mice and were analyzed together. Analyses were performed blind to genotype. For comparisons between two groups, normality was first assessed using a Shapiro–Wilk test. If data points were normally distributed, significance was determined using a two-tailed Student’s t-test. If non-normally distributed, significance was determined using the nonparametric Mann–Whitney U test. Statistical significance was set at α = 0.05 (p < 0.05). Statistical analyses and data visualization were performed in R (version 4.4.2).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Individual data points for all analyses can be found in Supplementary Data 1, and all statistical tests are included in Supplementary Data 2. Electrophysiology data is deposited at https://doi.org/10.6084/m9.figshare.29083331. Microscopy data is deposited at https://doi.org/10.6019/S-BIAD1945.
Code availability
A custom Igor Pro script was used to analyze electrophysiology data. The script can be found at https://github.com/jnjahncke/mini_analysis.
References
Südhof, T. C. Towards an understanding of synapse formation. Neuron 100, 276–293 (2018).
Colognato, H. et al. Identification of dystroglycan as a second laminin receptor in oligodendrocytes, with a role in myelination. Development 134, 1723–1736 (2007).
Nguyen, H. et al. Glial scaffold required for cerebellar granule cell migration is dependent on dystroglycan function as a receptor for basement membrane proteins. Acta Neuropathol. Commun. 1, 58 (2013).
Nickolls, A. R. & Bönnemann, C. G. The roles of dystroglycan in the nervous system: Insights from animal models of muscular dystrophy. DMM Dis. Models Mech. 11, dmm035931 (2018).
Tian, M. et al. Dystroglycan in the cerebellum is a laminin α2-chain binding protein at the glial-vascular interface and is expressed in Purkinje cells. Eur. J. Neurosci. 8, 2739–2747 (1996).
Zaccaria, M. L., Di Tommaso, F., Brancaccio, A., Paggi, P. & Petrucci, T. C. Dystroglycan distribution in adult mouse brain: a light and electron microscopy study. Neuroscience 104, 311–324 (2001).
Holt, K. H., Crosbie, R. H., Venzke, D. P. & Campbell, K. P. Biosynthesis of dystroglycan: Processing of a precursor propeptide. FEBS Lett. 468, 79–83 (2000).
Ibraghimov-Beskrovnaya, O. et al. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355, 696–702 (1992).
Jahncke, J. N. & Wright, K. M. The many roles of dystroglycan in nervous system development and function. Dev. Dyn. 252, 61–80 (2023).
Moore, C. J. & Winder, S. J. Dystroglycan versatility in cell adhesion: a tale of multiple motifs. Cell Commun. Signal. 8, 3 (2010).
Clements, R., Turk, R., Campbell, K. P. & Wright, K. M. Dystroglycan maintains inner limiting membrane integrity to coordinate retinal development. J. Neurosci. 37, 8559–8574 (2017).
Clements, R. & Wright, K. M. Retinal ganglion cell axon sorting at the optic chiasm requires dystroglycan. Dev. Biol. 442, 210–219 (2018).
Lindenmaier, L. B., Parmentier, N., Guo, C., Tissir, F. & Wright, K. M. Dystroglycan is a scaffold for extracellular axon guidance decisions. eLife 8, e42143 (2019).
Menezes, M. J. et al. The extracellular matrix protein laminin α2 regulates the maturation and function of the blood–brain barrier. J. Neurosci. 34, 15260–15280 (2014).
Myshrall, T. D. et al. Dystroglycan on radial glia end feet is required for pial basement membrane integrity and columnar organization of the developing cerebral cortex. J. Neuropathol. Exp. Neurol. 71, 1047–1063 (2012).
Wright, K. M. et al. Dystroglycan organizes axon guidance cue localization and axonal pathfinding. Neuron 76, 931–944 (2012).
Briatore, F. et al. Dystroglycan mediates clustering of essential GABAergic components in cerebellar Purkinje cells. Front. Mol. Neurosci. 13, 164 (2020).
Briatore, F., Patrizi, A., Viltono, L., Sassoe-Pognetto, M. & Wulff, P. Quantitative organization of GABAergic synapses in the molecular layer of the mouse cerebellar cortex. PLoS ONE 5, 8 (2010).
Brünig, I., Suter, A., Knuesel, I., Lüscher, B. & Fritschy, J. M. GABAergic terminals are required for postsynaptic clustering of dystrophin but not of GABAA receptors and gephyrin. J. Neurosci. 22, 4805–4813 (2002).
Lévi, S. et al. Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J. Neurosci. 22, 4274–4285 (2002).
Patrizi, A. et al. Synapse formation and clustering of neuroligin-2 in the absence of GABAA receptors. Proc. Natl. Acad. Sci. USA 105, 13151–13156 (2008).
Früh, S. et al. Neuronal dystroglycan is necessary for formation and maintenance of functional CCK-positive basket cell terminals on pyramidal cells. J. Neurosci. 36, 10296–10313 (2016).
Jahncke, J. N., Miller, D. S., Krush, M., Schnell, E. & Wright, K. M. Inhibitory CCK+ basket synapse defects in mouse models of dystroglycanopathy. eLife 12, RP87965 (2024).
Miller, D. S. & Wright, K. M. Neuronal Dystroglycan regulates postnatal development of CCK/cannabinoid receptor-1 interneurons. Neural Dev. 16, 4 (2021).
Kueh, S. L. L., Head, S. I. & Morley, J. W. GABAA receptor expression and inhibitory post-synaptic currents in cerebellar Purkinje cells in dystrophin-deficient mdx mice. Clin. Exp. Pharmacol. Physiol. 35, 207–210 (2008).
Wu, W. C., Bradley, S. P., Christie, J. M. & Pugh, J. R. Mechanisms and consequences of cerebellar Purkinje cell disinhibition in a mouse model of duchenne muscular dystrophy. J. Neurosci.42, 2103–2115 (2022).
Daigle, T. L. et al. A suite of transgenic driver and reporter mouse lines with enhanced brain-cell-type targeting and functionality. Cell 174, 465–480.e22 (2018).
Jahncke, J. N. & Wright, K. M. Tools for cre-mediated conditional deletion of floxed alleles from developing cerebellar Purkinje cells. eNeuro 11, 6 (2024).
Trotter, J. H., Wang, C. Y., Zhou, P., Nakahara, G. & Südhof, T. C. A combinatorial code of neurexin-3 alternative splicing controls inhibitory synapses via a trans-synaptic dystroglycan signaling loop. Nat. Commun. 14, 1771 (2023).
Uezu, A. et al. Essential role for insyn1 in dystroglycan complex integrity and cognitive behaviors in mice. eLife 8, e50712 (2019).
Morales, D. & Hatten, M. E. Molecular markers of neuronal progenitors in the embryonic cerebellar anlage. J. Neurosci. 26, 12226–12236 (2006).
He, Q. et al. Interneuron- and GABAA receptor-specific inhibitory synaptic plasticity in cerebellar Purkinje cells. Nat. Commun. 6, 7364 (2015).
Stingl, M., Draguhn, A. & Both, M. A dendrite is a dendrite is a dendrite?Dendritic signal integration beyond the “antenna” model. Pflüg. Arch. Eur. J. Physiol. 477, 9–16 (2025).
Sotelo, C. Molecular layer interneurons of the cerebellum: developmental and morphological aspects. Cerebellum 14, 534–556 (2015).
Viltono, L., Patrizi, A., Fritschy, J.-M. & Sassoè-Pognetto, M. Synaptogenesis in the cerebellar cortex: differential regulation of gephyrin and GABAA receptors at somatic and dendritic synapses of Purkinje cells. J. Comp. Neurol. 508, 579–591 (2008).
Wizeman, J. W., Guo, Q., Wilion, E. M. & Li, J. Y. Specification of diverse cell types during early neurogenesis of the mouse cerebellum. eLife 8, e42388 (2019).
Novak, J. S. et al. Interrogation of dystrophin and dystroglycan complex protein turnover after exon skipping therapy. J. Neuromuscul. Dis. 8, S383–S402 (2021).
Goddeeris, M. M. et al. LARGE glycans on dystroglycan function as a tunable matrix scaffold to prevent dystrophy. Nature 503, 136–140 (2013).
Yoshida-Moriguchi, T. & Campbell, K. P. Matriglycan: a novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology 25, 702–713 (2015).
Manya, H. et al. Demonstration of mammalian protein O-mannosyltransferase activity: Coexpression of POMT1 and POMT2 required for enzymatic activity. Proc. Natl. Acad. Sci. USA 101, 500–505 (2004).
Larsen, I. S. B. et al. Discovery of an O-mannosylation pathway selectively serving cadherins and protocadherins. Proc. Natl. Acad. Sci. USA114, 11163–11168 (2017).
Larsen, I. S. B. et al. Mammalian O-mannosylation of cadherins and plexins is independent of protein O-mannosyltransferases 1 and 2. J. Biol. Chem. 292, 11586–11598 (2017).
Anderson, J. L., Head, S. I. & Morley, J. W. Altered inhibitory input to Purkinje cells of dystrophin-deficient mice. Brain Res. 982, 280–283 (2003).
Grady, R. M., Wozniak, D. F., Ohlemiller, K. K. & Sanes, J. R. Cerebellar synaptic defects and abnormal motor behavior in mice lacking α- and β-dystrobrevin. J. Neurosci. 26, 2841–2851 (2006).
Knuesel, I. et al. Altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice). Eur. J. Neurosci. 11, 4457–4462 (1999).
Kueh, S. L. L., Dempster, J., Head, S. I. & Morley, J. W. Reduced postsynaptic GABAA receptor number and enhanced gaboxadol induced change in holding currents in Purkinje cells of the dystrophin-deficient mdx mouse. Neurobiol. Dis. 43, 558–564 (2011).
Satz, J. S. et al. Distinct functions of glial and neuronal dystroglycan in the developing and adult mouse brain. J. Neurosci. 30, 14560–14572 (2010).
Jung, D., Yang, B., Meyer, J., Chamberlain, J. S. & Campbell, K. P. Identification and characterization of the dystrophin anchoring site on β-dystroglycan (*). J. Biol. Chem. 270, 27305–27310 (1995).
Liu, Z. et al. Deletion of Calsyntenin-3, an atypical cadherin, suppresses inhibitory synapses but increases excitatory parallel-fiber synapses in cerebellum. eLife 11, e70664 (2022).
Zaman, T. et al. Kit Ligand and Kit receptor tyrosine kinase sustain synaptic inhibition of Purkinje cells. eLife 12, RP89792 (2024).
Zhang, B. et al. Neuroligins sculpt cerebellar Purkinje-cell circuits by differential control of distinct classes of synapses. Neuron 87, 781–796 (2015).
Briggs, D. C. et al. Structural basis of laminin binding to the LARGE glycans on dystroglycan. Nat. Chem. Biol. 12, 810–814 (2016).
Inamori, K. I. et al. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 335, 93–96 (2012).
Sheikh, M. O. et al. Cell surface glycan engineering reveals that matriglycan alone can recapitulate dystroglycan binding and function. Nat. Commun. 13, 3617 (2022).
Reissner, C. et al. Dystroglycan binding to α-Neurexin competes with neurexophilin-1 and neuroligin in the brain. J. Biol. Chem. 289, 27585–27603 (2014).
Sugita, S. et al. A stoichiometric complex of neurexins and dystroglycan in brain. J. Cell Biol. 154, 435–445 (2001).
Kozareva, V. et al. A transcriptomic atlas of mouse cerebellar cortex comprehensively defines cell types. Nature 598, 214–219 (2021).
Saunders, A. et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16 (2018).
Sumita, K. et al. Synaptic scaffolding molecule (S-SCAM) membrane-associated guanylate kinase with inverted organization (MAGI)-2 is associated with cell adhesion molecules at inhibitory synapses in rat hippocampal neurons. J. Neurochem. 100, 154–166 (2007).
Nguyen, T. & Südhof, T. C. Binding properties of neuroligin 1 and neurexin 1β reveal function as heterophilic cell adhesion molecules*. J. Biol. Chem. 272, 26032–26039 (1997).
Liang, J. et al. Conditional neuroligin-2 knockout in adult medial prefrontal cortex links chronic changes in synaptic inhibition to cognitive impairments. Mol. Psychiatry 20, 850–859 (2015).
Missler, M. et al. α-Neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423, 939–948 (2003).
Varoqueaux, F. et al. Neuroligins determine synapse maturation and function. Neuron 51, 741–754 (2006).
Rosa, G., Ceccarini, M., Cavaldesi, M., Zini, M. & Petrucci, T. C. Localization of the dystrophin binding site at the carboxyl terminus of β-dystroglycan. Biochem. Biophys. Res. Commun. 223, 272–277 (1996).
Suzuki, A. et al. Molecular organization at the glycoprotein-complex-binding site of dystrophin: Three dystrophin-associated proteins bind directly to the carboxy-terminal portion of dystrophin. Eur. J. Biochem. 220, 283–292 (1994).
Gao, Q. & McNally, E. M. The Dystrophin Complex: structure, function and implications for therapy. Compr. Physiol. 5, 1223–1239 (2015).
Lidov, H. G. W., Byers, T. J., Watkins, S. C. & Kunkel, L. M. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature 348, 725–728 (1990).
Wang, W. X. & Lefebvre, J. L. Morphological pseudotime ordering and fate mapping reveal diversification of cerebellar inhibitory interneurons. Nat. Commun. 13, 3433 (2022).
Lackey, E. P. et al. Specialized connectivity of molecular layer interneuron subtypes leads to disinhibition and synchronous inhibition of cerebellar Purkinje cells. Neuron 112, 2333–2348.e6 (2024).
Truett, G. E. et al. Preparation of PCR-quality mouse genomic dna with hot sodium hydroxide and tris (HotSHOT). BioTechniques 29, 52–54 (2000).
Konno, K., Yamasaki, M., Miyazaki, T. & Watanabe, M. Glyoxal fixation: An approach to solve immunohistochemical problem in neuroscience research. Sci. Adv. 9, eadf7084 (2023).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Zhang, X. M. et al. Highly restricted expression of Cre recombinase in cerebellar Purkinje cells. Genesis 40, 45–51 (2004).
Cohn, R. D. et al. Disruption of Dag1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell 110, 639–648 (2002).
Hu, H. et al. Conditional knockout of protein O-mannosyltransferase 2 reveals tissue-specific roles of O-mannosyl glycosylation in brain development. J. Comp. Neurol. 519, 1320–1337 (2011).
Satz, J. S. et al. Visual impairment in the absence of dystroglycan. J. Neurosci. 29, 13136–13146 (2009).
Hayashi, S., Lewis, P., Pevny, L. & McMahon, A. P. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev. 119, S97–S101 (2002).
Acknowledgements
We acknowledge expert technical assistance by staff in the OHSU Advanced Light Microscopy shared resource (RRID: SCR_009961) and the OHSU Department of Comparative Medicine. This work was funded by NIH Grants R01NS091027 (K.M.W.), R01NS126247 (E.S.) CureCMD (K.M.W.), F31NS120649 (J.N.J.), P30NS061800 (OHSU ALM), VA I01-BX004938 (E.S.), Department of Defense W81XWH-18-1-0598 (E.S.). The contents of this manuscript do not represent the views of the US Department of Veterans Affairs or the US government.
Author information
Authors and Affiliations
Contributions
J.N.J., E.S., and K.M.W. contributed to the study design. J.N.J. performed all the experiments, data collection, data analysis, and data visualization. E.S. and K.M.W. both held supervisory roles. J.N.J. and K.M.W. wrote and edited the manuscript. J.N.J., E.S., and K.M.W. discussed and reviewed the manuscript together at all stages.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Jaewon Ko and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editor: Benjamin Bessieres. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jahncke, J.N., Schnell, E. & Wright, K.M. Distinct functional domains of Dystroglycan regulate inhibitory synapse formation and maintenance in cerebellar Purkinje cells. Commun Biol 8, 878 (2025). https://doi.org/10.1038/s42003-025-08323-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08323-1
