
Abstract
While evidence supports co-evolution between Mycobacterium tuberculosis and humans, underlying mechanisms remain unclear. We identified PPE50 as a novel subfamily of PE/PPE proteins comprising eight variants. Surveying 387 M. tuberculosis complex (MTBC) strains representing global phylogeography, we found PPE50 variants are lineage-specific and stably associated with geographic regions, defining them as phylogeographically-associated proteins (PAPs). PPE50-381 is the ancestral variant (present in early-branching M. canettii) and the only variant observed in both Ancient and Modern MTBC lineages. Transcriptomic analysis confirmed that ppe50 variant genes are expressed in strains from respective MTBC lineages, but not in all L1 strains and sub-lineages L2.1 and L4.1 where the gene was deleted. In silico analysis revealed significant structural diversity among variants, particularly in C-terminal regions. This strong association of M. tuberculosis protein diversity with phylogeography suggests PPE50 may contribute to MTBC adaptation to different host populations. Further characterization of PPE50 and other PAPs may facilitate improved targeted diagnostics, therapeutics and vaccines.
Similar content being viewed by others


Introduction
Tuberculosis (TB) remains one of the world’s deadliest infectious diseases, with an estimated 10 million cases and 1.3 million deaths annually1. TB is caused by the bacteria of the Mycobacterium tuberculosis complex (MTBC) which is comprised of Mycobacterium tuberculosis and Mycobacterium africanum that cause human disease, and others such as Mycobacterium bovis that cause disease predominantly in animals. In recent years, the explosion of genomic data generation has allowed us to map in detail the genetic diversity of the MTBC, identifying several distinct lineages for M. tuberculosis (L1-L4, L7-L10) and M. africanum (L5-L6), which are geographically stably associated with human host populations globally2,3,4,5,6,7. This phylogeographic pattern raises key questions about the mechanisms driving TB co-evolution and how specific pathogen proteins may facilitate adaptation to diverse human populations.
In this study, we investigated PPE50 (encoded by the Rv3135 gene), a member of the PE/PPE protein family. This protein family is unique to mycobacteria and collectively represents approximately 10% of the M. tuberculosis genome coding capacity8. These proteins, characterized by N-terminal Pro-Glu (PE) or Pro-Pro-Glu (PPE) motifs, are often surface-exposed or secreted and show considerable sequence variation9. While their functions largely remain to be elucidated, evidence suggests roles in antigenic variation, host-pathogen interactions, and immune modulation10. Despite their potential importance, most studies have focused on reference strains (particularly H37Rv), potentially overlooking significant diversity across MTBC lineages.
PPE50 belongs to the PPE sub-lineage IV (SVP subfamily)11, and its genomic location is intriguing, lying in between the Rv3134c gene and Rv3136 which encodes PPE51. Rv3134c is involved in the dosR regulon and biofilm formation12,13, whilst PPE51 has recently been described to form a heterodimer with the PE protein partner PE19, resulting in a cell surface complex involved in nutrient transport14. PPE50 has also been described as an immunogenic antigen in several studies, including during the chronic stages of infection in mice15, as a potent T-cell antigen in latent TB infection (LTBI) cohorts16,17,18, and as a potential vaccine candidate19. PPE50 has also been described as a potential drug target in several studies20,21,22. The potential immunological importance of PPE50 is further supported by studies showing that PPE50 can bind to Toll-like receptor 1 (TLR1) on THP-1 macrophages, upregulating anti-inflammatory responses via interleukin-10 (IL-10) induction23, and that PPE50 peptides are presented via major histocompatibility complex II (MHCII) by THP-1 macrophages infected with M. bovis BCG17. Moreover, PPE50/PPE51 peptides elicit significant interferon gamma (IFNγ) production in peripheral blood mononuclear cells (PBMCs) from both M. tuberculosis-infected individuals and patients with LTBI compared to those with active disease18,19.
Considering PPE50’s likely significance, we used a phylogeographic approach to determine the evolution of this protein in the MTBC to gain further insights into its function. Firstly, we investigated the genetic diversity of the ppe50 locus (Rv3134c-Rv3135-Rv3136) in 18 reference strains from MTBC lineages24, and 6 animal-adapted MTBC strains. We identified significant genetic diversity in the Rv3135 gene compared to neighbouring genes, resulting in eight distinct predicted PPE50 protein variants across the MTBC. Using a well characterised dataset comprising of 387 MTBC strains5, we further showed a stable lineage-specific association among PPE50 protein variants, thus defining them as phylogeographic-associated proteins (PAPs). Furthermore, we show that the ppe50 variant genes are expressed in MTBC strains and through in silico analyses, have also predicted the 3D protein structural characteristics of the PPE50 variants to infer possible functions.
PAPs provide an approach for understanding how pathogens adapt to different host populations and are defined as proteins that show consistent sequence variations that correlate with pathogen phylogeny and geographical distribution, often reflecting adaptation to specific host populations or environments. In Helicobacter pylori, the CagA protein shows population-specific variations that affect host cell signalling and are associated with different gastric cancer rates across regions25,26. Similarly, the SasX protein is associated with specific Asian lineages of methicillin-resistant Staphylococcus aureus (MRSA) and contributes to colonization and virulence, demonstrating geographical clustering of protein variants27. In MTBC, proteins demonstrating PAP-like characteristics include PE_PGRS33, which exhibits sequence variations across lineage-specific clinical strains that elicit differential TLR2 responses, potentially altering host-pathogen interactions28,29. Additionally, the genetic diversity of the PPE38-PPE71 locus across MTBC lineages represents another important example30,31. Notably, PE_PGRS proteins require both the ESX-5 secretion system and functional PPE38 for secretion32. Several virulent M. tuberculosis strains, particularly from the L2 lineage, cannot secrete PE_PGRS and PPE-MPTR proteins due to loss-of-function mutations in the PPE38-PPE71 genetic locus32,33, presenting another potential PAP that may influence host-pathogen interactions in TB.
While other proteins show lineage associations, PPE50 represents, to the best of our knowledge, the first formally characterized PAP in the MTBC, introducing this concept to TB research. PPE50 forms a distinct protein subfamily with clear phylogeographic distribution, potentially playing a fundamental role in host-pathogen co-evolution. This variability across lineages, rather than a limitation, represents a potentially significant approach for therapeutic innovation. It may enable the development of region-specific interventions tailored to local MTBC populations, marking a paradigm shift from traditional approaches targeting only conserved antigens. Furthermore, PAPs may also facilitate the development of antigen ‘cocktail’ approaches that can target all strains of MTBC better than a single conserved antigen. PPE50 thus provides both a prototype for identifying other PAPs and a compelling foundation for developing lineage-specific vaccines and therapeutics that address the global geographical heterogeneity of TB, potentially enhancing diagnostics, prevention and treatment outcomes in specific populations.
Results
Structural variation at the ppe50 (Rv3134c-Rv3135-Rv3136) genomic locus
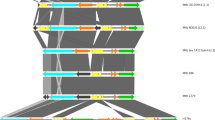
An alignment of reference genomes covering MTBC lineages (L1-L10), M. canettii and other animal adapted strains (M. bovis, M. microti, M. caprae and M. orygis), revealed a remarkable contrast in diversity between the Rv3135 (ppe50) gene and its flanking genes (Supplementary Fig. 1). Genes Rv3134c (dosR regulon) and Rv3136 (ppe51) are highly conserved, with a small number of single nucleotide polymorphisms (SNPs) in their genes and intergenic regions. In contrast, there are large-scale insertions and deletions (indels) across the Rv3135 gene, in reference strains, with respect to M. tuberculosis H37Rv. The Rv3135 gene in all M. canettii and the animal MTBC strains are identical, except for a few SNPs, whilst in the M. tuberculosis reference strains, there is a range of diversity that appears lineage specific (Supplementary Fig. 1). In L1, Rv3135 is completely deleted, whilst there are insertions, with respect to M. tuberculosis H37Rv, in L2, L5, L6 and M. bovis, which were also recently described9 (Supplementary Fig. 1). The variation in L3 was also previously described and was assigned to the principle genetic group 1 phylogenetic classification34. Recently, a comparative genomic study of MTBC strains also found regions of difference (RDs) that corresponded to L1, L2.1, L4.1 and L3 in the Rv3135 genomic locus35. However, in the present study we have also identified RDs at the Rv3135 locus that are linked with L7, L8 and L9 and several sub-lineages in L2 and L4 with respect to M. tuberculosis H37Rv (Supplementary Fig. 1). For each of the MTBC reference strains analyzed, we determined the open reading frames (ORFs) for this locus and found several genes for Rv3135 (Fig. 1a). To better understand the evolution of this locus, all reference MTBC strains were aligned with respect to M. canettii and this revealed RDs with respect to M. canettii (RD50can), showing how the different variants of ppe50 ORFs were formed (Fig. 1b). The extensive sequence diversity we have identified in ppe50 variants has been previously unnoticed, as PPE50 (Rv3135) was primarily known only by the truncated 132-amino acid variant in M. tuberculosis H37Rv, which was presumed to be non-functional36. It is interesting to note that some ppe50 variants have genes with identical stop codon positions (Fig. 1b). Furthermore, the deletions where ppe50 is missing in lineages L1, L2.1 and L4.1 are all distinct in length, showing each to be a unique evolutionary event (Fig. 1b). Curiously, the 3’ end of the ppe50-439 gene also has a 110 bp insertion (of unknown origin) that does not align to the intergenic region between ppe50 and ppe51 in M. canettii and is the only ppe50 member to have this feature (Fig. 1b).

a Predicted open reading frames for ppe50 variants and flanking genes. Genomic structural diversity in the Rv3135 locus has resulted in several ppe50 variant ORFs that are associated with MTBC lineages. ppe50 gene length varies considerably in contrast to the Rv3134c and Rv3136, which are highly conserved. Genomic coordinates are shown with respect to M. tuberculosis H37Rv for ease of comparison. †: Note: ppe50 variant ORFs for lineages L2.2.1 and L4.2.1.1. are shown only. b Regions of difference (RD) that form the ppe50 variants in the MTBC. ppe50 genomic locus for each variant were aligned to the ppe50-381 locus of M. canettii (CIPT140010059). Deletions denoting RDs with respect to M. canettii are shown in black lines for each ppe50 variant. In the ppe50 variants, deletions span part of the ppe50 gene and downstream intergenic region (hash arrows). ABC denotes base pair (bp) positions of the stop codons (downstream from the end of the M. canettii ppe50-381 gene): (A) ppe50-268 and ppe50-345 (position 438); (B) ppe50-387 and ppe50-439 (position 520); (C) ppe50-87, ppe50-132 and ppe50-262 (position 590). ppe50-439 has unknown 110 bp insertion (blue hash). ppe50-deleted 1, 2 and 3 are distinct RDs for lineage 1, 4.1, and 4.2, respectively. †: Note, RDs described in this study and their coordinates are shown in the Supplementary Table 1.
PPE50 is defined by eight distinct variant proteins in the MTBC
The genetic variation described in Rv3135 has resulted in 8 predicted protein variants of PPE50, establishing a new subfamily for this PPE protein member. These protein variants range in size and molecular characteristics (Fig. 2). The shortest variant is PPE50-87, which is 87 amino acids long, followed by PPE50-132, which is 132 amino acids long. Both variants have all of their protein structure in the PPE N-terminal domain. The longest variant is PPE50-439 which is 439 amino acids in length and is comprised of the PPE N-terminal domain and a unique C terminal domain. Variants PPE50-262, PPE50-268, PPE50-345, PPE50-381 and PPE50-387 all have full length PPE N-terminal domains with unique C terminal domains that vary in length and sequence composition (Fig. 2). The size range of PPE50 protein variants is indicative of significant genetic variation, particularly in the C-terminal region. PPE50-87 and PPE50-132 are highly truncated variants, consisting of a partial PPE N-terminal region. Whereas PPE50-262 and PPE50-268 are semi-truncated variants, consisting of a full PPE N-terminal region and a relatively short C-terminal region. PPE50-345, PPE50-381, PPE50-387 and PPE50-439 are full length variants that are classified as PPE-SVP proteins, as they contain the serine-valine-proline (SVP) motif (residues 312-314) in their unique C-terminal domains (Fig. 2). These four additional PPE50-SVP variants increase the number of known PPE-SVP proteins, which are the largest subgroup of PPE proteins11. The presence of this motif is a key feature for substrates of the ESX-5 type VII secretion system36.

The predicted amino acid sequence for the eight PPE50 variants was aligned using MUSCLE. All variants have the WxG motif in their N-terminal domain. With the exception of PPE50-87 and PPE50-132, the remaining variants have the hh motif, a full-length PPE N-terminal domain and diverse C-terminal domains. PPE50-345, PPE50-381, PPE50-387 and PPE50-439 all have SVP motif in their C-terminal region. Residues involved in potential PE protein binding are shown in grey.
Predicted structural diversity and sub-cellular location of PPE50 variant proteins
Three-dimensional structures were predicted for the PPE50 variants (Fig. 3). All variants share the homologous PPE N-terminal domain structure, comprising of 180 amino acids forming 5 α-helices in a helical bundle-like conformation37,38,39,40. The PPE N-terminal domain structure also consists of several features. The WxG motif (residues 57-59 [WWG]) (Fig. 2), located between α-helices 2 and 3, likely forms a composite recognition structure with the PE C-terminal domain feature YXXXD/E, allowing for ESX type VII secretion 41,42. The hh motif or hydrophobic tip (residues 125-126 [LL]) (Fig. 2), is located between α-helices 4 and 5 (Fig. 3), and is essential for binding EspG, a cytosolic chaperone protein involved in the transportation of substrates via the ESX type VII secretion system38,39,43. All variants, except for truncated variants PPE50-87 and PPE50-132, have both the WxG and hh motif. Therefore, these variants are capable of forming, in part, the composite recognition structure needed to bind EspG, both of which are important for ESX type VII secretion over the inner mycobacterial membrane (Fig. 3). Whilst truncated variants PPE50-87 and PPE50-132 consist of only 3 or 4 α-helices, respectively, they do possess the WxG motif. However, it is unclear whether the WxG motif alone is sufficient for secretion.

Ribbon structures were computed using AlphaFold. The N-terminal regions (partial or full-length) show the distinctive known alpha coils and turns of the conserved PPE N-terminal domain. With the exception PPE50-87 and PPE50-132, the disordered unique C-terminal domain is shown for the remaining PPE50 variants. Rainbow coloured gradient denotes N-terminal regions (blue) to C-terminal regions (red). Features are displayed with sticks: white = hh motif, black = WxG motif, and grey = PE binding residues.
Semi-truncated (PPE50-262 and PPE50-268) and full-length (PPE50-345 to PPE50-439) variants show unique C-terminal regions, differing in length, sequence composition, and consequently structure, as indicated by the TM-align scores (Supplementary Fig. 2). However, similarities can still be observed in the C-terminal regions of these variants, including a helix-turn-helix structure near α-helices 2 and 3 of the PPE N-terminal domain structure, except for PPE50-268, which has a single α-helix, and PPE50-387, which has two helix-turn-helix structures (Fig. 3). Another shared structure in the C-terminal regions of these variants, except for PPE50-262 and PPE50-268, is a two-stranded anti-parallel β-sheet near α-helix 2 of the PPE N-terminal domain structure. The C-terminal regions of all variants show a degree of disorder, compared to the ordered PPE N-terminal domain structure (Fig. 3).
Surprisingly, the C-terminal regions of these PPE50 variants also shroud the α-helices 2 and 3 of the PPE N-terminal domain structure, which are essential for binding with a PE family protein to form the canonical PE-PPE heterodimer (Fig. 3). As a result, this suggests that the semi-truncated and full-length PPE50 variants are unlikely to bind with a PE family protein, leaving PPE50-87 and PPE50-132 as the only variants with PE-PPE heterodimeric potential. However, it should be noted that the 3rd α-helix in PPE50-87 is significantly truncated, with only 2/4 PE binding residues (having residues 14[R] and 45[Y] and lacking 72[Y] and 93[A]), which may weaken or prevent dimerization37 (Fig. 2).
Hydrophobicity plots and transmembrane helix topology predictions were performed for the PPE50 variants (Supplementary Fig. 3). PPE50-87 was predicted to have only a cytoplasmic, transmembrane, and extracellular region, whilst all other variants were predicted to have a cytoplasmic, transmembrane, extracellular, and pore-lining helix region. The position of the transmembrane and pore-lining helix region is conserved between PPE50-262 and PPE50-268 (residues 118-133) and PPE50-345, PPE50-381, PPE50-387, and PPE50-439 (residues 237-252, except for PPE50-381, residues 238-253). Interestingly, the position of the N- and C-terminal regions varies between the cytoplasmic and extracellular regions, with PPE50-87, PPE50-345, PPE50-387 and PPE50-439 having a cytoplasmic N-terminal region and an extracellular C-terminal region, and PPE50-132, PPE50-262, PPE50-268 and PPE50-381 having an extracellular N-terminal region and a cytoplasmic C-terminal region.
ppe50 variants are strongly associated with distinct phylogeographic lineages of the MTBC
To investigate the phylogeographic distribution of ppe50 variants in the MTBC, we analyzed an additional 387 well-characterized strains representing the global spread of the MTBC5. We identified the ppe50 variant type for each strain and found a strong association between MTBC strain lineage, geographical location and ppe50 variant (Fig. 4). Distinct predicted PPE50 variants proteins are found within distinct MTBC lineages and sub-lineages and have therefore been designated PAPs. To our knowledge, this is the first MTBC protein to be designated as a PAP. Although there is significant inter-lineage variation in ppe50 variant genes, intra-lineage variation is minute showing high conservation of ppe50 variants within their repective lineage/sub-lineage.

Representative strains spanning MTBC lineages and sublineages were aligned (see Methods) and a tree produced using FastTree2 software96 and viewed on iTOL (minimum of 5 strains per sub-lineage). Maximum Likelihood tree. Bar indicates genetic distance (nucleotide substitutions per site). The type of ppe50 variant for each strain was identified by aligning the Rv3134c-Rv3135-Rv3136 locus (using MUSCLE) and determining the ppe50 ORF. The ppe50 variant was then mapped to each MTBC lineage and sub-lineage. Therefore, this shows a stable phylogeographical association between MTBC lineage/sublineage and ppe50 variant. Details of the MTBC strains studied are shown in Supplementary Data 1.
Following the evolutionary history of the MTBC44,45, the distribution of PPE50 variants between Ancient and Modern MTBC strains is intriguing (Fig. 5). Only PPE50-381 is observed in both Modern and Ancient MTBC lineages, whilst the remaining PPE50 proteins are found exclusively in either Ancient or Modern MTBC (Fig. 5). Our data also shows that PPE50-381 is the ancestral protein of PPE50 subfamily, and is found in all M. canettii strains (an early branching tubercle bacilli)46,47. PPE50-381 is also found in animal-adapted MTBC strains and L8 strains (Fig. 5). M. africanum L5 and M. tuberculosis L7 strains have their own distinct PPE50 variants (PPE50-387, PPE50-268 respectively), whilst L6, L9 and L10 strains have PPE50-345. It is known that L5, L6, L7 and L9 strains are predominantly found in Africa and their PPE50 variants are all closely related to PPE50-381 (Fig. 5). Moreover, PPE50-268 and PPE50-345 have almost identical C-terminal domains, suggesting that they are evolutionarily related, except that PPE50-268 lacks the SVP motif (Fig. 2). In the Modern M. tuberculosis strains, denoted by the TbD1 deletion44, PPE50 variants show the most diversity (truncation), with expansion of several types in lineages L2 and L4 (Fig. 5). In contrast, PPE50-87 is the only PPE50 variant observed in lineage L3. In lineage L4, PPE50-132 is is found exclusively in several sub-lineages L4.3 to L4.10, which include M. tuberculosis strains that have expanded to North, Central and South America from Europe, e.g., the LAM clade48. PPE50-132 is also found in the reference strain M. tuberculosis H37Rv. Additionally in lineage L4, PPE50-439 is exclusively found in sub-lineage 4.2.1.1, whilst PPE50-381 is also observed in sub-lineage 4.2.1. PPE50-439 is the largest protein variant and it’s curious that is it observed only in a sub-lineage of the Modern L4 lineage, since the end of its C-terminal domain is identical to PPE50-387 which occurs in strains from the Ancient lineage 5 (Figs. 2 and 5). In lineage L2, PPE50-262 was exclusively present in sub-lineage 2.2.1, which includes the well-studied M. tuberculosis strain HN878, whilst PPE50-381 was also found in sub-lineage 2.2.2. As mentioned, in strains where ppe50 is deleted (lineage L1, L2.1 and L4.1 strains), these deletions are distinct and have evolved separately. These include other well-studied strains of M. tuberculosis, e.g., CDC1551, Erdman and Haarlem (L4.1) which all have their ppe50 genes deleted.

PPE50 has evolved contrastingly between Ancient and Modern strains of the MTBC. PPE50-381 is the ancestral variant, being observed in M. canettii, and is the only PPE50 to be observed in Ancient and Modern MTBC strains. Recent expansion of Modern M. tuberculosis into lineages L2, L3 and L4 have seen marked diversity of PPE50 variants that are stably distributed in several sub-lineages that have spread globally. Whereas PPE50 variants in Ancient MTBC are mostly associated with Africa, except for L1. *Note: ppe50 is deleted in lineages L1, L4.1, and L4.2 and each are unique event polymorphisms. Maximum Likelihood tree (implemented in IQ-TREE). Bar indicates genetic distance nucleotide substitutions per site for each MTBC lineage/sub-lineage.
ppe50 variant genes are expressed in the MTBC
Transcriptomic analysis of representative MTBC strains from each lineage revealed that the identified ppe50 variant genes are expressed (Fig. 6; transcriptome analysis not performed on ppe50-268). Furthermore, the transcript length corroborates with the length of the ppe50 ORFs that were predicted in this study. The depth of expression in each of the ppe50 variants is comparable in intensity with the expression of the neighbouring genes Rv3134c and Rv3136 (ppe51). Lineages where the ppe50 gene was deleted showed no evidence of expression (Fig. 6). These data therefore suggest that these expression profiles are likely indicative of actual PPE50 proteins variants in MTBC that have been predicted and modelled in our study (Fig. 3 and Supplementary Fig. 3).

Transcriptome RNA-Seq reads from representative MTBC lineages for the Rv3134c, Rv3135 (ppe50) and Rv3136 (ppe51) genes were aligned to a reference locus containing the ppe50-439 gene (lineage L4.2.1.1), since this is the longest ppe50 gene. ppe50 variant genes were shown to be expressed and this matched their sequence length. Transcriptome analysis not performed on ppe50-268 from lineage L7. No expression was observed for all lineages where ppe50 was deleted. Depth: Log scale of counts of transcript reads observed. x-axis scale: 1 kilobase increments for the locus.
Discussion
TB molecular epidemiology has revealed a distinction between globally distributed “Modern” M. tuberculosis strains and geographically constrained “Ancient” endemic strains. The TbD1 deletion, resulting in the loss of MmpS6 and MmpL6 genes, marks this evolutionary divergence and is present in all Modern lineages (L2, L3, L4) but absent in Ancient lineages (L1, L5-L10) and animal MTBC strains. This deletion appears to enhance bacterial fitness under oxidative stress and hypoxia, likely contributing to the successful global spread of Modern strains44. Similarly, we’ve identified PPE50 as a PAP that could also provide insights into TB’s global spread. These eight PPE50 variants show distinct MTBC lineage specificity, suggesting potential involvement in M. tuberculosis adaptation to specific host populations. While PPE50’s function remains unknown, our findings combined with experimental studies may reveal novel host-pathogen co-evolution insights. Notably, ppe50 is deleted in certain lineages (all L1, L4.1, and L2.1). Clinical observations suggest contradictory outcomes: L1 strains are associated with increased pulmonary disease and cavitation49, yet ppe50 deletion has also been linked to extrapulmonary TB50,51, and increased isoniazid and rifampicin tolerance22. This suggests ppe50's loss may enhance virulence and drug resistance, analogous to the TbD1 deletion. Alternatively, ppe50 may be essential only in specific MTBC genetic backgrounds, while lineages with ppe50 deletions likely possess compensatory mechanisms, suggesting adaptive evolution rather than simple dispensability. Further experimental work is needed to test these hypotheses.
Nevertheless, the presence of PPE50 in its different variant forms may also give further insights into its biological role and interactions with the host. PPE50-381 is the ancestral variant protein of the PPE50 subfamily and is likely to have the most evolutionary conserved phenotype out of all the PPE50 variants, since it is the only PPE50 found in both Ancient and Modern MTBC strains. Indeed, PPE50-381 is also found in all divergent strains of M. canettii analysed in our study. Our in silico analysis suggests PPE50-381 is likely to be a cell wall protein capable of host interactions, supported by its presence in M. bovis BCG cell wall fractions52. A study reporting “ppe50 gene replacement” in a meningeal-TB isolate53 actually described what our analysis identifies as a L2.2.2 strain naturally containing ppe50-381. Though this group episomally expressed PPE50-381 in Mycobacterium smegmatis, they observed no enhanced growth in THP-1 macrophages compared to vector controls53. However, it should also be noted that M. smegmatis lacks both PPE50 orthologs and the ESX-5 secretion system, potentially preventing proper PPE50-381 processing.
Most studies on PPE50 have been done on PPE50-132 (H37Rv strain) that is also found exclusively and at high frequency in lineages 4.3 to 4.10. This truncated PPE50, along with PPE50-87, could have a different biological role than PPE50-381 and other semi- and full-length PPE50 proteins, as they maybe more likely to form heterodimers with PE partners. The PPE50 C-terminal domains may shroud the crucial PPE α-helices 2 and 3 preventing PE interaction. Thus the loss of the C-terminal domain may act as “a molecular switch” promoting novel protein-protein binding and altered function. The PE-PPE heterodimeric conformation brings together the PPE WxG motif and the YxxxD/E motif of the PE C-terminal domain, likely forming a composite recognition structure for ESX type VII secretion41,42. Whilst this suggests that semi-truncated and full-length variants may not be secreted, their C-terminal regions are disordered, which may undergo conformational changes to enhance PE binding residue accessibility. Furthermore, other studies have reported type VII secretion system substrates being secreted without both WxG and YxxxD/E motifs and therefore, the three-dimensional structures of these motifs may be more important than the amino acids that define them54. Interestingly, the YxxxD/E has also been described as unstructured37,43. Therefore, semi-truncated and full-length PPE50 variants may not require a canonical PE partner, as the secretion signal may already be present within their disordered C-terminal regions, which also possess the flexibility to bring the motifs in close proximity, similar to the ESX-1 type VII secretion system substrate, EspB55. In either scenario, this would allow the composite recognition structure required for ESX type VII secretion to be formed. From the structural data in this study, all PPE50 variants are likely secreted over the inner mycobacterial membrane, except for PPE50-87 and PPE50-132, due to the absence of a hh motif. Moreover, PPE-SVP proteins are probable substrates of the ESX-5 type VII secretion system, suggesting PPE50 may also be secreted via this route11,36. The ESX type VII secretion system chaperone protein EspG also interacts with the hh motif, a feature that’s also present in all variants except PPE50-87 and PPE50-13243. EspG prevents PE-PPE heterodimer aggregation by binding and shielding the PPE hh motif, as well as stabilizing the complex in a secretion-competent state, but its essentiality has been challenged by the outer membrane/cell surface localization of a PE-PE (PE9/PE10) heterodimer lacking the hh motif56. PPE50 variants with SVP motifs may also be associated with sub-lineage IV ESX-5 secreted PE proteins, analogous to the recent findings for PE19/PPE5114. However, a recent study describes PPE50-132 from H37Rv being transcribed together with PPE51 and forming a proposed PPE-PPE heterodimer23 However, further data is needed given that PPE51 has also been reported to interact with PE1914. Our data suggest all PPE50 variants potentially cross the inner mycobacterial membrane, though the exact secretion mechanism requires further investigation. Structural analysis indicates these proteins likely localize to the cell surface, spanning from cytoplasmic to extracellular regions via a single transmembrane anchor.
Limited data on ppe50 gene expression regulation (studied only in H37Rv) shows Rv3135 is a commonly upregulated gene during environmental stress responses57. The gene shares putative transcription factor binding sites with Rv3134c and Rv313658 and shows upregulation in SigM mutants59, suggesting involvement in cell surface and secreted molecule regulation during late infection stages. While Rv3135 is not regulated by DevR under hypoxic stress58, a PhoP binding site exists in the intergenic region between Rv3134c and Rv313560. Given PhoP’s role in intracellular survival and persistence60,61, ppe50 expression may be influenced by host immune and antibiotic pressures, potentially resulting in variant-specific phenotypic responses to these stimuli.
PPE50 has also been identified as a potential drug target, with studies predicting interaction with the HIV-1 protease inhibitor Amprenavir20 and the compound GSK14022909A, which also targets proteins in the amino-acyl-tRNA biosynthesis pathway (Rv1640c, Rv3598c, Rv3834c) and peptide chain release factor 2 (Rv3105c)21. Additionally, a G251D mutation in ppe50-381 from M. bovis BCG was associated with Florfenicol resistance, suggesting this residue’s functional importance62. These studies may provide further insights into PPE50 structure and function.
In this study, our phylogeographic approach revealed significant genetic variation in Rv3135, compared to the highly conserved Rv3134c and Rv3136 flanking genes. These findings builds on previous observations showing limited Rv3135 gene variation being associated with distinct principle genetic group 134, and L1, L2.1 and L4.135. Two other studies also identified ppe50 variants in Beijing strains (L2)63,64, whilst a further study found PPE50 to be partially deleted in several M. tuberculosis isolates recovered from the Inuit65. Another study also found ppe50 to be partially deleted in isolates belonging to L3, which were recovered from South Asians living in the United Kingdom66. The reasons for the genetic variation seen in ppe50 are unknown. No evidence of homologous recombination or IS6110 involvement have been reported. The evolutionary timing of the divergence of ppe50 across the MTBC is uncertain. While MTBC evolution estimates vary considerably (70,000-6000 years ago)45,67, archaeological evidence shows TbD1-containing M. tuberculosis strains existed in China and Europe 1000-2200 years ago68,69. This suggests ppe50 divergence in Ancient MTBC likely predated the TbD1 deletion, while most ppe50 diversification in Modern MTBC strains occurred subsequently during neolithic expansion from Africa45. After this, sub-lineages emerged, with MTBC strains L4.1 (ppe50-deleted3), L4.2 (PPE50-381, PPE50-439) and L4.3 to 4.8 (PPE50-132), calculated to have diverged in lineage L4 around the years 800 CE and 1000 CE and 1100 CE, respectively69. Similar findings were reported for L4.2, L4.4 and L4.5 in a recent study in China, in addition to L2.2 (PPE50-381, PPE50-262) which was found to have diverged around the year 806 CE68. Speculatively, these timelines suggest that the emergence of ppe50-deleted or PPE50-truncated types (PPE50-87, PPE50-132, PPE50-262) in MTBC strains may be a determining factor in the geographic spread of TB in densely populated areas driven by human migration over the last 2000 years (Modern L2, L3 and L4 MTBC). Whilst other longer PPE50 variants, (PPE50-345, PPE50-387), found in Ancient MTBC lineages L5, L6, L7, L9 and L10 represent the early evolution of PPE50 that may have contributed to the geographically restricted dissemination of TB in local host populations (e.g. Africa-India). However, the fact that PPE50-381 spans both evolutionary periods suggests that it may have contributed to the most highly adapted and versatile MTBC strains that are found in both animals and human populations that are the most globally distributed. Unlike the other MTBC lineages, lineage 4 strains have the widest global distribution and cause TB to a high frequency70,71. Sublineages 4.3 to 4.10 account for 71% of all L4 strains globally and all have PPE50-132, (with L4.3 accounting for 20.3%)48. Lineage L4.3 (also known as L4.3/LAM) originated in Europe and is the most widespread L4 sublineage spreading to Africa, Asia and the American continents48. The presence of PPE50-132 in a substantial proportion of L4 strains suggests that this PPE50 may be a key determinant of successful M. tuberculosis global spread. However, L4.5 and L4.6 (PPE50-132) have been reported to be more localised in their distribution (China and Central Africa, respectively)48. In contrast, sublineages L4.1 (ppe50-deleted3) and L4.2.1 (PPE50-381, PPE50-439) are present at lower frequencies globally, 19.1% and 4.4% respectively, with L4.1 also having a wide geographical distribution and L4.2.1 being confined to countries in Asia and Africa48. Taken together, it appears that truncation and/or loss of PPE50 in L4 strains maybe associated with increased global dissemination of those strains, although further sampling is needed to test this hypothesis. Lineage 3 M. tuberculosis strains (PPE50-87) originated in South Asia, but have spread elsewhere particularly to East Africa, probably as a result of human migration72. With the exception of Lineage 1 (ppe50-deleted1), all the Ancient MTBC lineage strains are geographically prevalent in Africa. MTBC lineage strains L6 (M. africanum), L10 and L9 are found on the Eastern side of West Africa, Central Africa and East Africa (Somalia) repectively and all have PPE50-345, showing a restricted spread4,7. MTBC lineage 5 (M. africanum) strains (PPE50-387) and lineage 7 strains (PPE50-268) are also geographically restricted, in Western Africa and Ethiopia respectively4,73, with unique PPE50 variants. M. africanum caused half of TB cases in West Africa, with older age, being HIV positive and malnurished as risk factors compared to M. tuberculosis L4 TB cases74. The distribution of full-length PPE50 variants, including PPE50-381 in MTBC lineage 8 from Rwanda6, appears to mirror the phylogeography previously described for Ancient MTBC. While this pattern suggests possible co-evolution with local African human populations, this apparent correlation may be coincidental and requires further investigation.
The evolution and phylogeographic distribution of PPE50 variants may be driven by host immunity, but T cell epitopes in MTBC are highly conserved48,75,76, suggesting PPE50 variation is unlikely to involve immune selection. Nevertheless, studies have demonstrated PPE50 immunological properties: PPE50-132 binds TLR1 on THP-1 macrophages, upregulating anti-inflammatory IL-10 responses23; PPE50-381 peptides are presented via MHCII in BCG-infected macrophages17; and PPE50/PPE51 peptides induce significant IFNγ production in PBMCs from LTBI individuals and TB patients17,18, with higher responses in LTBI. Even with these insights, the specific role of PPE50 variants on the immunopathology of TB remains to be determined.
Despite the extensive genomic sequence data and structural variation documented for the MTBC, a critical challenge now is to determine the functional consequences of these genetic differences. Most studies involving PPE50 have focused solely on the variant found in M. tuberculosis H37Rv, which represents only one chapter in the broader PPE50 narrative. Future investigations must elucidate the phenotypic roles of different PPE50 variants and explain why this PPE protein subfamily exhibits such strong phylogeographic conservation. Our computational analyses, while robust, have inherent limitations. Future experimental work should test several key hypotheses, including whether C-terminal structural differences in PPE50 variants enable lineage-specific host protein interactions and if these variants trigger distinct immune responses, and how certain MTBC lineage strains function without PPE50. These studies would provide functional insights into these phylogeographic variations and potentially illuminate co-evolutionary dynamics between M. tuberculosis and human populations.
Identifying additional PAPs using our approach could reveal further candidates involved in host-pathogen co-evolution in TB. These PAPs represent promising preventative and therapeutic targets, as understanding their global distribution would clarify antigenic and metabolic variations across MTBC strains. Our findings emphasize the importance of studying diverse MTBC lineages rather than focusing solely on reference strains like H37Rv, CDC1551, and Erdman (which all lack PPE50). The discovery of lineage-specific PPE50 variants suggests opportunities for geographically targeted interventions that may overcome limitations of universal approaches like BCG in regions where specific lineages predominate. In conclusion, we’ve identified a subfamily of eight distinct PPE50 variants that are lineage-specific PAPs in the MTBC. While their precise function remains to be characterized, this work provides a template for discovering new PAPs in TB and other infectious diseases.
Methods
Reference genomes and datasets
The ppe50 genomic locus (Rv3134c-Rv3135-Rv3136) was obtained from the M. tuberculosis H37Rv reference genome8. The 18 M. tuberculosis reference genomes (L1-L7) were obtained from a recent paper by Borrel et al.24. Reference genomes from M. bovis (AF2122/97), M. bovis BCG (1173P2), M. microti (OV254), M. caprae (spc-1), M. orygis (51145), and M. canettii (CIPT140010059 (STB-A)77, were obtained from the National Center for Biotechnology Information (NCBI) genomes database. Genomes from a subset of 387 MTBC strains were obtained from a recent paper5, where approximately 5 strains representing all known MTBC lineages and sub-lineages were selected for analysis. MTBC Lineages L8 and L10 reference genomes were also included6,7. Additionally, several more M. canettii strain genomes were also analyzed (CIPT140070010 (STB-K), CIPT140070017 (STB-J), CIPT140070008 (STB-L), CIPT140060008 (STB-D), ET-1291)78,79.
Bioinformatic analysis
Genomic regions (Rv3134c-Rv3135-Rv3136) from reference genomes were aligned using Clustal Omega from EMBL-EBI80, and MUSCLE using MEGA X81. Alignments were visualized using the NCBI MSA Viewer 1.25.0. Phylogenetic reconstruction was performed using the Maximum Likelihood method within MEGA X81. Protein sequences were aligned using MUSCLE82 and visualized using MView from EMBL-EBI83. For large-scale phylogenetic analyses, representative strains for each sub-lineage were selected from the TB-Profiler webserver (tbdr.lshtm.ac.uk)84 (Supplementary Data 1). Raw fastq data for these samples was downloaded from the European Nucleotide Archive (ENA) site and mapped to the H37Rv reference genome using BWA (v0.7.17). Variant calling was performed for each sample individually using gatk HaplotypeCaller in gvcf mode (v4.1.4.1; parameters: -ERC GVCF). Join genotyping was then performed using gatk GenotypeGVCFs to create a single multi-sample vcf file. In-house Python scripts (https://github.com/LSHTMPathogenSeqLab/fastq2matrix/) were then used to transform this into a concatenated SNP fasta file which was used by IQ-TREE (v2.2.2.7; parameters: -m GTR + G + ASC) to perform phylogenetic reconstruction85. The phylogenetic tree was visualized using iTOL86. Assemblies were generated for each sample from raw reads using Shovill (v1.1.0) (https://github.com/tseemann/shovill), and PGAP (2023-10-03.build7061) was used to perform gene annotation87. A custom Python script was used to extract the ppe50 gene from each of the MTBC strains using sequence similarity to the M. tuberculosis H37Rv Rv3135 gene sequence, using BLAST, with a minimum match and identity threshold (https://github.com/jodyphelan/PPE50).
Structural analysis
The tertiary structures of the PPE50 variants were predicted using Colabfold (Alphafold2 using MMseqs2)88 and the resulting PDB files were then visualized using ESPript V3.0 and PyMol 2.588,89. Disordered regions and disordered binding regions were further defined using IUPred3 and ANCHOR290.
Functional and subcellular localization prediction
To predict the subcellular location of PPE50, hydrophobicity plots and transmembrane helix topology predictions were made using MEMSAT-SVM91. To predict the domains and function of PPE50, sequences were analyzed using MOTIF92, and MotifScan93 software tools. The presence of signal peptides was also predicted using SignalP 94.
Gene expression analysis
To establish whether the different ppe50 variant genes found across the sub-lineages are expressed, RNA-seq data in fastq format was downloaded from the ENA site and TB-Profiler and was used to infer sub-lineage for analyses3,84. Representative samples were selected and aligned to the genome assembly which contained the ppe50-439 type (accession: ERR067597). Depth of coverage profiles were calculated with Samtools Depth (v1.12)95.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information. Genome Sequences of the MTBC strains analysed are provided in Supplementary Data 1.
Code availability
A custom Python script was used to extract the ppe50 gene from the MTBC strains. It is deposited at https://github.com/jodyphelan/PPE50.
References
WHO Global Tuberculosis Report. 2023; Available from: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2023.
Brites, D. & Gagneux, S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol. Rev. 264, 6–24 (2015).
Coll, F. et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 5, 4812 (2014).
Coscolla, M. et al. Phylogenomics of Mycobacterium africanum reveals a new lineage and a complex evolutionary history. Microb. Genom. 7, 1–14 (2021).
Napier, G. et al. Robust barcoding and identification of Mycobacterium tuberculosis lineages for epidemiological and clinical studies. Genome Med. 12, 114 (2020).
Ngabonziza, J. C. S. et al. A sister lineage of the Mycobacterium tuberculosis complex discovered in the African Great Lakes region. Nat. Commun. 11, 2917 (2020).
Guyeux, C. et al. Newly Identified Mycobacterium africanum Lineage 10, Central Africa. Emerg. Infect. Dis. 30, 560–563 (2024).
Cole, S. T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 (1998).
Gomez-Gonzalez, P. J. et al. Functional genetic variation in pe/ppe genes contributes to diversity in Mycobacterium tuberculosis lineages and potential interactions with the human host. Front Microbiol 14, 1244319 (2023).
D’Souza, C., Kishore, U. & Tsolaki, A. G. The PE-PPE Family of Mycobacterium tuberculosis: Proteins in Disguise. Immunobiology 228, 152321 (2023).
Gey van Pittius, N. C. et al. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol. Biol. 6, 95 (2006).
Flores-Valdez, M. A. et al. Transcriptional portrait of M. bovis BCG during biofilm production shows genes differentially expressed during intercellular aggregation and substrate attachment. Sci. Rep. 10, 12578 (2020).
Voskuil, M. I. et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198, 705–713 (2003).
Wang, Q. et al. 3rd, PE/PPE proteins mediate nutrient transport across the outer membrane of Mycobacterium tuberculosis. Science 367, 1147–1151 (2020).
Clemmensen, H. S. et al. Rescuing ESAT-6 Specific CD4 T cells from trminal differentiation is critical for long-term control of murine Mtb infection. Front Immunol. 11, 585359 (2020).
Carpenter, C. et al. A side-by-side comparison of T cell reactivity to fifty-nine Mycobacterium tuberculosis antigens in diverse populations from five continents. Tuberculosis (Edinb.) 95, 713–721 (2015).
Bettencourt, P. et al. Identification of antigens presented by MHC for vaccines against tuberculosis. NPJ Vaccines 5, 2 (2020).
Lewinsohn, D. A. et al. Comprehensive definition of human immunodominant CD8 antigens in tuberculosis. npj Vaccines. 2, 8 (2017).
Monterrubio-Lopez, G. P., Gonzalez, Y. M. J. A. & Ribas-Aparicio, R. M. Identification of novel potential vaccine candidates against tuberculosis based on reverse vaccinology. Biomed. Res. Int. 2015, 483150 (2015).
Anand, P. & Chandra, N. Characterizing the pocketome of Mycobacterium tuberculosis and application in rationalizing polypharmacological target selection. Sci. Rep. 4, 6356 (2014).
Martinez-Jimenez, F. et al. Target prediction for an open access set of compounds active against Mycobacterium tuberculosis. PLoS Comput Biol. 9, e1003253 (2013).
Martini, M. C. et al. Loss of RNase J leads to multi-drug tolerance and accumulation of highly structured mRNA fragments in Mycobacterium tuberculosis. PLoS Pathog. 18, e1010705 (2022).
Mukku, R. P., Poornima, K., Yadav, S. & Raghunand, T. R. Delineating the functional role of the PPE50 (Rv3135) - PPE51 (Rv3136) gene cluster in the pathophysiology of Mycobacterium tuberculosis. Microbes Infect. 23, 105248 (2023).
Borrell, S. et al. Reference set of Mycobacterium tuberculosis clinical strains: A tool for research and product development. PLoS One 14, e0214088 (2019).
Hayashi, T. et al. Differential mechanisms for SHP2 binding and activation are exploited by geographically distinct Helicobacter pylori CagA oncoproteins. Cell Rep. 20, 2876–2890 (2017).
Hatakeyama, M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 93, 196–219 (2017).
Li, M. et al. MRSA epidemic linked to a quickly spreading colonization and virulence determinant. Nat. Med 18, 816–819 (2012).
Talarico, S. et al. Association of Mycobacterium tuberculosis PE PGRS33 polymorphism with clinical and epidemiological characteristics. Tuberculosis (Edinb.) 87, 338–346 (2007).
Talarico, S. et al. Variation of the Mycobacterium tuberculosis PE_PGRS 33 gene among clinical isolates. J. Clin. Microbiol 43, 4954–4960 (2005).
McEvoy, C. R., van Helden, P. D., Warren, R. M. & Gey van Pittius, N. C. Evidence for a rapid rate of molecular evolution at the hypervariable and immunogenic Mycobacterium tuberculosis PPE38 gene region. BMC Evol. Biol. 9, 237 (2009).
McEvoy, C. R., Warren, R. M., van Helden, P. D. & Gey van Pittius, N. C. Multiple, independent, identical IS6110 insertions in Mycobacterium tuberculosis PPE genes. Tuberculosis (Edinb.) 89, 439–442 (2009).
Ates, L. S. et al. Mutations in ppe38 block PE_PGRS secretion and increase virulence of Mycobacterium tuberculosis. Nat. Microbiol 3, 181–188 (2018).
Ates, L. S. et al. RD5-mediated lack of PE_PGRS and PPE-MPTR export in BCG vaccine strains results in strong reduction of antigenic repertoire but little impact on protection. PLoS Pathog. 14, e1007139 (2018).
Musser, J. M., Amin, A. & Ramaswamy, S. Negligible genetic diversity of Mycobacterium tuberculosis host immune system protein targets: evidence of limited selective pressure. Genetics 155, 7–16 (2000).
Liu, Z. et al. Identification of region of difference and H37Rv-related deletion in Mycobacterium tuberculosis complex by structural variant detection and genome assembly. Front Microbiol 13, 984582 (2022).
Ates, L. S. New insights into the mycobacterial PE and PPE proteins provide a framework for future research. Mol. Microbiol 113, 4–21 (2020).
Chen, X. et al. Structural basis of the PE-PPE protein interaction in Mycobacterium tuberculosis. J. Biol. Chem. 292, 16880–16890 (2017).
Ekiert, D. C. & Cox, J. S. Structure of a PE-PPE-EspG complex from Mycobacterium tuberculosis reveals molecular specificity of ESX protein secretion. Proc. Natl. Acad. Sci. USA 111, 14758–14763 (2014).
Korotkova, N. et al. Structure of the Mycobacterium tuberculosis type VII secretion system chaperone EspG5 in complex with PE25-PPE41 dimer. Mol. Microbiol 94, 367–382 (2014).
Strong, M. et al. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 103, 8060–8065 (2006).
Daleke, M. H. et al. General secretion signal for the mycobacterial type VII secretion pathway. Proc. Natl. Acad. Sci. USA 109, 11342–11347 (2012).
Poulsen, C., Panjikar, S., Holton, S. J., Wilmanns, M. & Song, Y. H. WXG100 protein superfamily consists of three subfamilies and exhibits an alpha-helical C-terminal conserved residue pattern. PLoS One 9, e89313 (2014).
Williamson, Z. A., Chaton, C. T., Ciocca, W. A., Korotkova, N. & Korotkov, K. V. PE5-PPE4-EspG(3) heterotrimer structure from mycobacterial ESX-3 secretion system gives insight into cognate substrate recognition by ESX systems. J. Biol. Chem. 295, 12706–12715 (2020).
Bottai, D. et al. TbD1 deletion as a driver of the evolutionary success of modern epidemic Mycobacterium tuberculosis lineages. Nat. Commun. 11, 684 (2020).
Comas, I. et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet 45, 1176–1182 (2013).
Orgeur, M., Sous, C., Madacki, J. & Brosch, R. Evolution and emergence of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 48, fuae006 (2024).
Smith, N. H., Hewinson, R. G., Kremer, K., Brosch, R. & Gordon, S. V. Myths and misconceptions: the origin and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol 7, 537–544 (2009).
Stucki, D. et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat. Genet 48, 1535–1543 (2016).
Du, D. H. et al. The effect of M. tuberculosis lineage on clinical phenotype. PLOS Glob. Public Health 3, e0001788 (2023).
Negrete-Paz, A. M., Vazquez-Marrufo, G., Gutierrez-Moraga, A. & Vazquez-Garciduenas, M. S. Pangenome reconstruction of Mycobacterium tuberculosis as a guide to reveal genomic features associated with strain cinical phenotype. Microorganisms. 11, 1495 (2023).
Negrete-Paz, A. M., Vazquez-Marrufo, G. & Vazquez-Garciduenas, M. S. Whole-genome comparative analysis at the lineage/sublineage level discloses relationships between Mycobacterium tuberculosis genotype and clinical phenotype. PeerJ 9, e12128 (2021).
Chen, Y., Danelishvili, L., Rose, S. J. & Bermudez, L. E. Mycobacterium bovis BCG surface antigens expressed under the granuloma-like conditions as potential inducers of the protective immunity. Int J. Microbiol 2019, 9167271 (2019).
Lam, J. T. et al. Truncated Rv2820c enhances mycobacterial virulence ex vivo and in vivo. Micro Pathog. 50, 331–335 (2011).
Garrett, S. R. et al. A type VII-secreted lipase toxin with reverse domain arrangement. Nat. Commun. 14, 8438 (2023).
Korotkova, N. et al. Structure of EspB, a secreted substrate of the ESX-1 secretion system of Mycobacterium tuberculosis. J. Struct. Biol. 191, 236–244 (2015).
Tiwari, B., Ramakrishnan, U. M. & Raghunand, T. R. The Mycobacterium tuberculosis protein pair PE9 (Rv1088)-PE10 (Rv1089) forms heterodimers and induces macrophage apoptosis through Toll-like receptor 4. Cell Microbiol 17, 1653–1669 (2015).
Skvortsov, T. A., Ignatov, D. V., Majorov, K. B., Apt, A. S. & Azhikina, T. L. Mycobacterium tuberculosis transcriptome profiling in mice with genetically different susceptibility to tuberculosis. Acta Nat. 5, 62–69 (2013).
Chauhan, S. & Tyagi, J. S. Cooperative binding of phosphorylated DevR to upstream sites is necessary and sufficient for activation of the Rv3134c-devRS operon in Mycobacterium tuberculosis: implication in the induction of DevR target genes. J. Bacteriol. 190, 4301–4312 (2008).
Raman, S. et al. Mycobacterium tuberculosis SigM positively regulates Esx secreted protein and nonribosomal peptide synthetase genes and down regulates virulence-associated surface lipid synthesis. J. Bacteriol. 188, 8460–8468 (2006).
Vashist, A., Malhotra, V., Sharma, G., Tyagi, J. S. & Clark-Curtiss, J. E. Interplay of PhoP and DevR response regulators defines expression of the dormancy regulon in virulent Mycobacterium tuberculosis. J. Biol. Chem. 293, 16413–16425 (2018).
Gonzalo-Asensio, J. et al. PhoP: a missing piece in the intricate puzzle of Mycobacterium tuberculosis virulence. PLoS One 3, e3496 (2008).
Kanvatirth, P., Jeeves, R. E., Bacon, J., Besra, G. S. & Alderwick, L. J. Utilisation of the Prestwick Chemical Library to identify drugs that inhibit the growth of mycobacteria. PLoS One 14, e0213713 (2019).
Mokrousov, I. et al. Phylogenetic reconstruction within Mycobacterium tuberculosis Beijing genotype in northwestern Russia. Res. Microbiol. 153, 629–637 (2002).
Ghebremichael, S. et al. Drug resistant Mycobacterium tuberculosis of the Beijing genotype does not spread in Sweden. PLoS One 5, e10893 (2010).
Lee, R. S. et al. Population genomics of Mycobacterium tuberculosis in the Inuit. Proc. Natl. Acad. Sci. USA 112, 13609–13614 (2015).
Menendez, M. C. et al. Genome analysis shows a common evolutionary origin for the dominant strains of Mycobacterium tuberculosis in a UK South Asian community. Tuberculosis (Edinb.) 87, 426–436 (2007).
Bos, K. I. et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature 514, 494–497 (2014).
Liu, Q. et al. China’s tuberculosis epidemic stems from historical expansion of four strains of Mycobacterium tuberculosis. Nat. Ecol. Evol. 2, 1982–1992 (2018).
Kay, G. L. et al. Eighteenth-century genomes show that mixed infections were common at time of peak tuberculosis in Europe. Nat. Commun. 6, 6717 (2015).
Coscolla, M. & Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol. 26, 431–444 (2014).
Demay, C. et al. SITVITWEB–a publicly available international multimarker database for studying Mycobacterium tuberculosis genetic diversity and molecular epidemiology. Infect. Genet Evol. 12, 755–766 (2012).
Shuaib, Y. A. et al. Origin and global expansion of Mycobacterium tuberculosis cmplex lineage 3. Genes (Basel). 13, 990 (2022).
Nebenzahl-Guimaraes, H. et al. Genomic characterization of Mycobacterium tuberculosis lineage 7 and a proposed name: ‘Aethiops vetus. Micro Genom. 2, e000063 (2016).
de Jong, B. C., Antonio, M. & Gagneux, S. Mycobacterium africanum–review of an important cause of human tuberculosis in West Africa. PLoS Negl. Trop. Dis. 4, e744 (2010).
Comas, I. et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet 42, 498–503 (2010).
Coscolla, M. et al. M. tuberculosis T cell epitope analysis reveals paucity of antigenic variation and identifies rare variable TB antigens. Cell Host Microbe 18, 538–548 (2015).
van Soolingen, D. et al. A novel pathogenic taxon of the Mycobacterium tuberculosis complex, Canetti: characterization of an exceptional isolate from Africa. Int J. Syst. Bacteriol. 47, 1236–1245 (1997).
Supply, P. et al. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat. Genet 45, 172–179 (2013).
Yenew, B. et al. A smooth tubercle bacillus from Ethiopia phylogenetically close to the Mycobacterium tuberculosis complex. Nat. Commun. 14, 7519 (2023).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004).
Brown, N. P., Leroy, C. & Sander, C. MView: a web-compatible database search or multiple alignment viewer. Bioinformatics 14, 380–381 (1998).
Phelan, J. E. et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med 11, 41 (2019).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44, 6614–6624 (2016).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 (1999).
Meszaros, B., Erdos, G. & Dosztanyi, Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res 46, W329–W337 (2018).
Nugent, T. & Jones, D. T. Transmembrane protein topology prediction using support vector machines. BMC Bioinforma. 10, 159 (2009).
Kanehisa, M., Goto, S., Kawashima, S. & Nakaya, A. The KEGG databases at GenomeNet. Nucleic Acids Res 30, 42–46 (2002).
Pagni, M. et al. MyHits: improvements to an interactive resource for analyzing protein sequences. Nucleic Acids Res 35, W433–W437 (2007).
Almagro Armenteros, J. J. et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423 (2019).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Acknowledgements
TGC is funded by the UKRI (BBSRC BB/X018156/1; MRC MR/X005895/1; EPSRC EP/Y018842/1).
Author information
Authors and Affiliations
Contributions
AGT conceived the study, analysed the data and wrote the manuscript. CDS performed the genetic diversity, phylogenetic and structural analyses. JEP, TGC and AGT performed the phylogeographic analyses. JEP, PJGG and JT performed the gene expression analyses. All authors contributed to revisions of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
D’Souza, C., Phelan, J.E., Gomez-Gonzalez, PJ. et al. PPE50 variants as novel phylogeographic signatures of host-pathogen co-evolution in tuberculosis. Commun Biol 8, 1024 (2025). https://doi.org/10.1038/s42003-025-08383-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08383-3
