
Abstract
Multi-omics analyses of severe COVID-19 cases are crucial in deciphering the complex interplay between genetic and epigenetic factors. Here, we present an analysis of Expression Quantitative Trait Methylation (eQTM) to investigate the complex interplay of methylation and gene expression pattern during the acute phase of severe COVID-19. We identified 16 differentially expressed genes and 30 nearby differentially methylated CpG sites. Six key genes—SRXN1, FURIN, IL18RAP, FOXO3, GCNT4, and FKBP5—were either up-regulated or down-regulated near hypomethylated CpG sites. These genes are associated with viral infiltration, immune activation, lung damage, and oxidative stress-related multi-organ failure, which are the hallmarks of severe COVID-19. Interestingly, during the recovery phase, methylation and gene expression levels returned to baseline, underscoring the rapid and reversible nature of these molecular changes. These findings provide insight into the dynamics of epigenetic and transcriptomic shifts according to the infectious stage, supporting potential prognostic and therapeutic approaches for severe COVID-19.
Similar content being viewed by others


Introduction
The global impact of the coronavirus disease 2019 (COVID-19) has underscored the urgent need to understand the more precise molecular mechanisms underlying its clinical manifestations, particularly the factors contributing to severe disease outcomes1. The clinical spectrum of COVID-19 ranges from asymptomatic cases to severe respiratory failure, yet the underlying molecular and omics drivers of these varied outcomes remain elusive. COVID-19 can trigger a cytokine storm—an excessive immune response characterized by the overproduction of cytokines—that results in tissue damage and widespread inflammation, which are strongly linked to severe disease2. In the most critical cases, the cytokine storm spreads to multiple organs, ultimately causing multi-organ failure and death3.
In recent years, big-data-driven precision multi-omics approaches have emerged as a powerful tool for dissecting the complex biological interactions that underpin pathologies. By integrating multiple layers of omics data, such as genomics, transcriptomics, and epigenomics, these precision medicine approaches can be utilized to gain a comprehensive understanding of disease mechanisms in instances, such as COVID-19. DNA methylation, as the most common epigenetic modification, plays a crucial role in regulating gene expression and can be influenced by infections, as well as other stressors4,5. Several studies have demonstrated that epigenetic regulation plays a role in the severity of COVID-194,6,7,8,9. Previously, DNA methylation was considered relatively stable than other epigenetic modifications. However, recent studies have revealed that DNA methylation can occur more rapidly than previously thought10. This is particularly evident when cells are exposed to challenging environments, such as infection caused by direct invasion of pathogens. RNA expression analysis is the most informative molecular method measuring the effect and severity of infectious diseases, such as COVID-19, because it directly and rapidly reflects the epigenetic changes caused by viral infection11. Therefore, Expression Quantitative Trait Methylation (eQTM) analysis has been successfully employed in several studies to investigate the relationship between DNA methylation and gene expression12,13,14. However, despite its potential, eQTM approaches have rarely been applied to investigate the connection between DNA methylation and RNA expression in relation to COVID-19 severity.
In this study, we employed a cis-eQTM analysis to investigate the integrated patterns of DNA methylation and RNA expression in 46 patients with varying degrees of COVID-19 severity. Our objective was to determine the molecular mechanisms associated with severe outcomes during the acute phase of the disease and to assess the dynamic changes in multi-omics of these molecular signatures during the recovery phase. These observations underscore the dynamic nature of epigenetic and transcriptomic changes in COVID-19 progression and recovery, providing valuable insights into potential healthcare and therapeutic targets for mitigating severe outcomes.
Results
Dynamics of gene-regulating CpGs and gene expression levels in COVID-19 severity
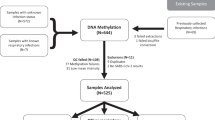
We analyzed blood-based multi-omic differences between severe-critical (SC) and mild-moderate (MM) cases during the acute phase of infection in 46 patients hospitalized with COVID-19 (Fig. 1A). The clinical follow-up period for all patients is presented in Fig. 1B. We confirmed their severity status based on clinical lab values, including blood cell counts and inflammatory biomarker levels. Notably, SARS-CoV-2 viral load, as measured by PCR cycle threshold (Ct) values for multiple gene targets (N, E, and R genes), did not differ significantly between severity groups (Supplementary Fig. 1), suggesting that differences in clinical outcomes were not attributable to viral burden alone but to host-intrinsic factors, including immune and epigenetic responses.

A Overview of blood sampling and analysis design; The marker discovery cohort consisted of hospitalized COVID-19 patients, stratified into Mild-Moderate (MM) (N = 37) and Severe-Critical (SC) (N = 9) groups, and was used to identify severity-associated cis-eQTM markers. The comparison cohort was composed of two independent reference groups: the ‘Convalescent group’ (N = 90) comprising individuals who recovered from COVID-19 and provided a single blood sample 4 to 12 weeks post-infection, and the ‘Healthy Controls’ group (N = 344), consisting of pre-pandemic individuals with no history of SARS-CoV-2 infection. B Clinical follow-up duration of 46 Hospitalized COVID-19 patients. The green and red points indicate MM and SC groups, respectively. Sample C19-C014(*), excluded DEG analysis due to QC failure, was retained in methylation analysis. In panel (A), the DNA, RNA, and blood icons were adapted from Flaticon (https://www.flaticon.com), and the remaining elements were created using icons available in Microsoft PowerPoint.
Leave-one-out (LOO) analysis between SC and MM COVID-19 groups identified 648 hypermethylated and 1296 hypomethylated CpG sites in the severe disease. Cis-eQTM analysis further revealed that 732 of these differentially methylated CpGs (113 hypermethylated CpGs and 619 hypomethylated CpGs) were correlated with 928 genes located within 1 Mbp of the differentially methylated CpGs (DMCs). Transcriptome-wide differential expression analysis identified 297 differentially expressed genes (DEGs) between SC and MM COVID-19 groups that were consistently observed in at least seven LOO iterations. Of these, 283 genes were up-regulated and 14 down-regulated in the severe disease. The up-regulated genes were enriched in pathways related to erythrocyte dynamics, innate immune activation, platelet aggregation, and acetylcholine receptor signaling (Supplementary Fig. 2A), whereas down-regulated genes were predominantly involved in DNA mismatch repair (MMR) pathway (Supplementary Fig. 2B).
Integration of methylation and transcriptomic data through cis-eQTM analysis refined these genes to 16 DEGs that were proximal to 30 DMCs (Supplementary Table 1). Among these, 15 genes were up-regulated and a single gene, GCNT4, was down-regulated. The overlap of these DEGs with cis-eQTM loci indicates their expression is likely regulated by local methylation status (Supplementary Table 1).
From the final set of cis-eQTM genes, six key genes and their ten regulatory CpGs were selected for further analysis based on their previous association with COVID-19 severity (Table 1). These genes are mechanistically linked to viral entry, immune regulation, and oxidative stress–major hallmarks of severe disease. FURIN, SRXN1, and FKBP5, which were up-regulated in the SC group, had strong negative correlations with their associated CpG methylation levels (ρ = –0.602, –0.582 to –0.510, and –0.551, respectively; P = 0.003, –0.004 to 0.021, and 0.009, respectively), indicating that hypomethylation at these loci is linked to increased gene expression. FOXO3 and IL18RAP also showed moderate inverse correlations (ρ = –0.501 and –0.503; P = 0.025 and 0.024), suggesting methylation-dependent regulation. In contrast, GCNT4, the only down-regulated gene among the six, was positively correlated with methylation levels (ρ = 0.528; P = 0.015).
We examined the distributions of DNA methylation and expression of the six prioritized genes according to the infection phase. Mean methylation differences between MM and SC COVID-19 patients were statistically significant for all ten CpGs (Supplementary Table 2). The convalescent group, with samples collected from 4 to 12 weeks post-COVID-19 infection, showed no significant differences in methylation levels compared to the healthy control group (no viral infection at the time of collection). Notably, the convalescent individuals from the MM group during the acute phase presented no significant differences in their methylation profiles across nine of the ten CpGs, with the exception of chr20:1166154 which regulates SRNX1 expression (Fig. 2A, Supplementary Fig. 3). The pattern was mirrored at the gene expression level (Fig. 2B). All six key genes were either up- or down-regulated in the SC group compared to the MM group. Three of the six genes (FURIN, SRXN1, and FKBP5) displayed significant differences in mean expression only between severe patients compared to convalescent individuals, but not between the MM and convalescent group (Supplementary Table 3). As for markers with no previous report on COVID-19 severity, all 24 DMCs showed significantly altered methylated proportions (Supplementary Fig. 3). Among ten DEGs with no past association with COVID-19 severity, nine genes (except GABARAPL2) displayed significant expression changes between MM and SC (Supplementary Fig. 4).

A, C DNA methylation levels (% methylated CpGs) at representative cis-eQTM sites, stratified by clinical group: Healthy controls (n = 310), Mild-Moderate (acute phase; n = 37), Severe-Critical (acute phase; n = 9), and Convalescent individuals (n = 90). Panel A presents five cell-type dependent cis-eQTM CpGs, whereas panel C illustrates a cell-type independent CpG. B, D) Gene expression levels (VST count) for the matched loci are displayed in panels B (cell-type proportion dependent) and D (cell-type proportion independent) across clinical group: Healthy controls (n = 35), Mild-Moderate (acute phase; n = 36), Severe-Critical (acute phase; n = 9), and Convalescent individuals (n = 90). Boxplots display the median, interquartile range (IQR), and individual data points. Statistical significance was evaluated using Welch’s t-test for methylation and the Wilcoxon rank-sum test for gene expression, with nominal P-values. Significance thresholds are indicated as follows: ns (P ≥ 0.05), *(P < 0.05), ** (P < 0.01), *** (P < 0.001), **** (P < 0.0001). VST variance-stabilizing transformation.
Leukocyte-adjusted cis-eQTM analysis reveals FKBP5 methylation as a cell-type proportion-independent marker of severe COVID-19
Acute SARS-CoV-2 infection disrupts circulating leukocyte distributions. We repeated the cis-eQTM analysis while additionally adjusting for neutrophil, lymphocyte, monocyte, eosinophil, and basophil proportions obtained from clinical blood counts. Consistent with this premise, a direct comparison of the measured cell proportions revealed marked baseline differences between severity groups: SC group exhibited a higher neutrophil fraction and a lower monocyte fraction than MM group, whereas lymphocyte, eosinophil and basophil proportions were indistinguishable (Supplementary Fig. 5). Around a week after admission, the distributions of all five cell types converged, and no between-group differences persisted (Supplementary Fig. 5), indicating that the leukocyte imbalance is a transitory feature from acute to recovery of the severe disease. After incorporating these cell-type covariates, 29 of the original 30 CpG-gene pairs lost statistical significance, demonstrating that most apparent methylation-expression associations were confounded by cell-type heterogeneity. The single CpG that retained significance—chr6:36697843—remained inversely associated with the expression of its neighboring gene, FKBP5 (Fig. 2C, D; Supplementary Table 4). The persistence of the chr6:36697843-FKBP5 cis-eQTM after rigorous adjustment establishes FKBP5 as a cell-type proportion-independent epigenetic marker of disease severity.
Resolution of acute-phase epigenetic alterations during COVID-19 recovery
We next examined whether the severity-associated epigenetic alterations observed during the acute phase were reversible during recovery. Specifically, we focused on the 30 CpGs and their corresponding 16 genes identified from the cis-eQTM analysis. Methylation and expression profiles were examined in the hospitalized MM and SC groups, an independent convalescent cohort, and healthy controls. A heatmap of the methylation β-values across all COVID-19 patients revealed a distinct group-wise separation during the acute phase, with SC patients displaying pronounced hyper- or hypo-methylation relative to MM group (Fig. 3A). Notably, this severity-dependent stratification diminished substantially during the recovery phase, and methylation profiles became more similar between the SC and MM groups (Supplementary Fig. 6). To further assess the restoration trajectory, we visualized the dynamics using principal component analysis (PCA). The DNA methylation PCA (Fig. 3B) revealed that while MM group largely overlapped with convalescent and healthy controls, the SC group exhibited only a partial shift, especially along PC2. In the gene expression PCA (Fig. 3C), both SC and MM groups shifted almost completely toward the healthy cluster by 2–3 weeks post-infection, indicating rapid normalization of transcriptional responses (Supplementary Fig. 7). Taken together, although directional recovery was evident, the residual separation of DNA methylation levels from the healthy baseline implies that full epigenetic reversion may require a longer time course, especially for those with severe disease.

A Heatmap depicting DNA methylation profiles of cis-eQTM markers in COVID-19 patients across infection phases (acute vs. recovery) and severity groups (Mild-Moderate vs. Severe-Critical). Differentially methylated CpG sites are indicated by color: hypermethylated sites are shown in red and hypomethylated sites in blue. Infection phases are represented as acute (pink) and recovery (light blue), while severity groups are differentiated by color: Mild-Moderate (dark green) and Severe-Critical (dark red). The β-value scale, ranging from 0 to 100, indicates the proportion of methylation at each CpG site. B Principal component analysis (PCA) of DNA methylation restoration between the acute and recovery phases in COVID-19 patients and healthy controls. C PCA of gene expression restoration between acute and recovery phases in COVID-19 patients. Arrows track group-level transitions, with ellipses denoting 3 standard deviations from the group mean. Methyl. Status Methylation Status.
To examine individual-level changes, we analyzed the methylation difference between the acute and recovery phase in each SC group (Supplementary Fig. 8). Most patients exhibited significant β-value shifts across both hypermethylated and hypomethylated CpGs, consistent with the overall trend of epigenetic normalization. One patient (C19-C058) presented an exception. Unlike other patients in the SC group, this patient showed virtually no methylation change between two time points. Clinical metadata indicated that C19-C058 was classified as “Critical”, had a high Charlson Comorbidity Index (CCI) (CCI = 5), and remained on oxygen therapy at the time of recovery-phase blood collection.
Discussion
Using early‑phase whole blood multi-omics from clinically well-characterized COVID‑19 patients, we mapped CpG‑to‑transcript links with cis‑eQTM analysis and uncovered a focused set of “reactive” CpGs whose methylation shifts reflected both disease severity and subsequent recovery. During the acute phase, coordinated hypo‑ or hyper‑methylation at these sites corresponded to sharp transcriptional changes that distinguished severe‑critical from mild-moderate cases. After correcting for transient leukocyte imbalances, only one CpG–RNA pair, FKBP5, remained independently associated with severity, highlighting it as a core epigenetic signal rather than a by‑product of immune‑cell redistribution. Longitudinal follow‑up demonstrated that both methylation and gene expression at the reactive loci returned to baseline within 4–12 weeks, underscoring the dynamic and largely reversible nature of COVID‑19‑induced epigenetic programming. These findings establish a compact, cell‑type composition–independent CpG-RNA signature that captures the trajectory from acute pathology to convalescence, providing possible prognostic biomarkers with mechanistic insight pertaining to host response and recovery during COVID-19.
By distinguishing between severe and mild cases based on oxygen therapy and intensive care unit (ICU) admission, we explored the molecular underpinnings associated with distinct clinical trajectories. Although there were only ten severe cases in this study, they represented 27% of the data, comparable to the proportion reported in previous large-scale studies. For example, a February 2020 study of 72,314 COVID-19 cases in China found that the majority (81%) of patients experienced mild to moderate symptoms, while 14% suffered from severe respiratory issues, and 5% progressed to critical conditions, including respiratory failure15.
This study is among the first to apply cis-eQTM analysis for dissecting gene regulatory programs linked to COVID-19 severity. We identified a distinct pattern of hypomethylation and concurrent upregulation of six genes: SRXN1, FURIN, IL18RAP, FOXO3, GCNT4, and FKBP5. These genes are not only epigenetically regulated but also biologically implicated in known pathogenic mechanisms which are recognized as hallmarks of COVID-19 severity. Notably, SRXN1, which was hypomethylated and up-regulated in severe cases across five distinct CpG loci, encodes Sulfiredoxin 1, a critical antioxidant mitigating reactive oxygen species (ROS) damage. Its dysregulation amplifies oxidative stress and has been repeatedly reported to be highly expressed in both blood and lung tissues of severe COVID-19 patients16,17,18,19,20. FURIN, another gene up-regulated in severe cases, facilitates viral entry by cleaving the S1/S2 junction of the SARS-CoV-2 spike protein21. Its expression correlates with disease progression and has been identified as a potential target for antiviral therapy22,23. Our results also revealed significant upregulation of IL18RAP and FOXO3 in severe COVID-19 cases—genes that are mechanistically linked to hyper-inflammatory responses and respiratory dysfunction. IL18RAP, a key mediator of IL-18 signaling, likely contributes to excessive immune activation and cardiopulmonary complications through inflammasome-driven IL-18 release, a pathway implicated in macrophage activation syndrome and multi-organ failure24,25,26. In parallel, FOXO3, which regulates immune balance and oxidative stress, was associated with increased oxygen demand and inflammatory lung injury, suggesting that its overexpression may exacerbate pulmonary damage in severe disease27,28,29.
Interestingly, GCNT4 was the only gene found to be epigenetically downregulated in severe patients. A prior proteomic study reported its elevation in respiratory failure among severe COVID-19 cases30. This discrepancy may reflect differences in regulatory control between the transcriptome and proteome or patient cohort characteristics.
To account for the known shifts in immune cell proportions during acute infection, such as neutrophilia and lymphopenia, we repeated the cis-eQTM analysis while adjusting for estimated immune cell-type proportion. Most CpG–gene associations lost significance following this correction, suggesting that they were biased by inflammation-induced cell redistribution. The persistence of the FKBP5 association after cell-type adjustment suggests that its regulation may reflect a cell-intrinsic epigenetic signal, strongly relevant to the pathophysiology of severe COVID-19. FKBP5 encodes FKBP51, an Hsp90 co‑chaperone that attenuates glucocorticoid‑receptor (GR) signaling31,32. By modulating GR sensitivity it fine‑tunes hypothalamic‑pituitary‑adrenal (HPA) feedback to stress and intersects with NF‑κB/RIG‑I pathways that shape innate‑immune and inflammatory responses33,34. Transcriptomic studies show FKBP5 up‑regulation in corticosteroid‑treated airway epithelium and in brains of fatal COVID‑19 cases35, suggesting that GR‑linked stress and treatment responses converge on FKBP5 during advanced disease, although its causal role in COVID‑19 severity remains inconclusive. While these findings point to a functional association between FKBP5 activity and COVID‑19 pathophysiology, further mechanistic and longitudinal studies are required to clarify whether FKBP5 is a driver of severe COVID-19.
The remaining ten cis-eQTM genes not covered in-depth in this study may still contribute to the broader molecular response to SARS-CoV-2 infection. Further investigation is warranted on elucidating their roles, which could reveal indirect effects on disease severity or related complications. Taken together, the blood-based multi-omic markers discovered here underscore pulmonary lesions and associated clinical symptoms. These markers could help predict respiratory complications and monitor disease severity.
In the recovery phase, spanning two to three weeks post-infection, we observed a relatively fast restoration of both the epigenome and transcriptome to levels akin to those of COVID-19 convalescence, across both severe and mild patient cohorts. The gene regulatory mechanisms observed in severe COVID-19 survivors, who avoided fatal outcomes, did not show significant disparities when compared to those in mild cases. This indicates that the identified CpGs are reactive to both the acute infection and recovery over the relatively short time period, reflecting the rapidly evolving pathological trajectory of the patients. Despite these overall trends, our findings also point to heterogeneity in recovery trajectories, particularly among SC patients. In a subset of individuals, epigenetic reversal was less pronounced, and in one notable case (C19-C058), virtually no change in DNA methylation was observed between the acute and follow-up samples. This individual had a high CCI, remained on oxygen therapy at follow-up, and their recovery-phase sample was taken just 10 days after the acute-phase time point. While these clinical factors may explain the apparent lack of epigenetic normalization, the case highlights that not all patients follow the same recovery timeline—and that molecular reversal may lag behind clinical resolution in more complex or prolonged disease courses.
In conclusion, these observations demonstrate that both cell-intrinsic and extrinsic epigenetic markers are responsive to COVID-19 pathophysiology and capable of tracking the acute-to-recovery transition. However, the variability across individuals also points to the importance of integrating clinical context and extending the observation window to fully understand the persistence, resolution, or relapse of molecular alterations—especially in relation to long-term sequelae, such as long-COVID.
Our investigation encountered several critical limitations. First, the COVID-19 severe cases in this study exhibited variability in the timing and duration of oxygen therapy following hospitalization (Fig. 1B). This discrepancy poses constraints on elucidating the exact association between identified biomarkers and the severity of the condition. Second, the limited number of samples in the severe group presents a challenge in drawing comprehensive general conclusions. Third, the lack of standardized control over the recovery phase, ranging from two to three weeks per patient, introduces variability that influences the interpretation of results. Additionally, the separate sample collection periods in 2021 and 2022 renders complexity. Different SARS-CoV-2 sub-strains—Delta and Omicron—were predominant during these times. The variants differed in transmissibility, virulence, and immune evasion, likely influencing disease severity and treatment response36. Lastly, while cytokine measurements were not performed in this study, the absence of these data limits our ability to directly evaluate the contribution of cytokine-mediated inflammatory responses to disease severity. Given the established role of cytokine storms in severe COVID-19 cases, future studies integrating cytokine profiling with epigenetic and transcriptomic data would offer a more comprehensive understanding of immune dysregulation in acute infection.
Materials and methods
Samples and clinical characteristics
We collected whole blood samples from 46 patients diagnosed with COVID-19 from Ulsan University Hospital (UUH), Ulsan, Republic of Korea. This study has received approval from the Institutional Review Board (IRB) of UUH and Ulsan National Institute of Science and Technology (UNIST) (IRB No.: UUH-2021-04-011-004, UNISTIRB-21-15-A). All ethical regulations relevant to human research participants were followed.
For the hospitalized patient group, whole blood samples were collected twice: once during the acute phase (at the time of hospital admission) and once during the recovery phase (2 to 3 weeks after hospitalization). For the convalescent patient group, whole blood samples were obtained between 4 to 12 weeks after COVID-19 diagnosis. These individuals had been previously hospitalized due to COVID-19 but had fully recovered at the time of sample collection. Our sampling period is divided into two halves. The first was during 2021, which was followed by the second in 2022. The acute phase of infection was defined as the period from hospitalization due to COVID-19 to one week thereafter, while the subsequent period of up to three weeks was defined as the recovery phase. A healthy control group consisted of 344 whole blood samples collected (309 samples for methylation, 34 for gene expression studies, and one sample for both omics) from the Korean Genome Project (KGP), approved by the IRB at UNIST in Ulsan, South Korea (IRB No.: UNISTIRB-21-66-A).
Patient classification of COVID-19 severity
The patients were assigned their severity upon diagnosis according to Food and Drug Administration (FDA) severity categorization and categorized into four groups: “Mild”, “Moderate”, “Severe”, and “Critical” (Supplementary Table 5)37,38. We further categorized the patients upon analysis defining their severity into two discrete groups: MM for mild and moderate categories and SC for severe and critical categories. SC patients suffered from respiratory symptoms with 80% of the patients requiring oxygen therapies, such as nasal prong or high-flow nasal cannula, or both, as compared to 14.6% for the MM group. In addition, 40% of the SC cases were admitted to the ICU, while none in the MM (Table 2).
Clinical information
We collected clinical data from informed and consenting patients alongside whole blood samples. The dataset included routine laboratory measurements, smoking history, and viral load estimates based on PCR Ct values (Supplementary Fig. 1). To investigate clinical correlates of disease severity, we compared these parameters across patient groups stratified by COVID-19 severity. Clinical variables with insufficient sample size (fewer than three observations in any group) were excluded from downstream analyses to ensure statistical robustness.
Target bisulfite sequencing and methylation quantification
Frozen whole blood samples collected in EDTA tubes (Becton Dickinson, #367856) were used for genomic DNA (gDNA) extraction, performed using the DNeasy Blood & Tissue Kit (Qiagen, #69506) according to the manufacturer’s instructions.
Bisulfite-converted DNA libraries were prepared using the SureSelectXT Methyl-Seq Target Enrichment System Kit (Agilent, #G9651), following the manufacturer’s protocol. Sequencing was performed on the NovaSeq 6000 platform (Illumina), generating 150 bp paired-end reads.
Raw sequencing reads were processed using Fastp (v0.23.1) to remove adapters and low-quality bases39, with the following parameters: --cut_front 1 --cut_right 0 --cut_tail 1 --detect_adapter_for_pe --trim_front1 15 --trim_front2 15 --trim_tail1 0 --trim_tail2 0 --cut_mean_quality 20 --n_base_limit 1 --average_qual 20 34. Read quality was assessed before and after filtering reads using FastQC (v0.11.9)40.
Filtered reads were aligned to the human reference genome (GRCh38.p13) using Bismark (v0.23.1), a bisulfite-aware aligner41. Methylation calling and quantification were conducted using the methylKit R package (v1.20.0)42. To annotate CpG sites and associate them with proximal genes or regulatory features, we used the annotatr R package (v1.20.0)43.
mRNA sequencing and expression quantification
Frozen whole blood samples collected in PAXgene® Blood RNA Tubes (PreAnalytiX, #762174) were used for total RNA extraction. RNA integrity and concentration were assessed using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, #Q32866) and the Qubit RNA HS Assay Kit (Thermo Fisher Scientific, #Q32854). For mRNA enrichment, 200 ng of total RNA per sample was processed using the Dynabeads mRNA Purification Kit (Thermo Fisher Scientific, #01152851), which depletes rRNA and isolates polyadenylated transcripts using oligo(dT) beads.
Library preparation was performed using the MGIEasy RNA Directional Library Prep Set (MGI, #MG1000006386), following the manufacturer’s protocol. Fragment size distribution was checked using 4150 TapeStation (Agilent, #G2992A) with the cDNA D1000 ScreenTape (Agilent, #5067-5582). Final libraries were quantitated using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Q32866). RNA sequencing was run on DNBSEQ-T7RS (MGI) platform, generating 150 bp paired-end reads.
RNA-seq reads were trimmed and quality-filtered using Fastp (v0.23.1) with the same parameters as for DNA methylation processing. Read quality was verified using FastQC (v0.11.9).
Filtered reads were aligned to the human reference genome (GRCh38.p13) using STAR (v2.7.10b), a splice-aware aligner, with default settings44. Gene-level quantification was performed using RSEM (v1.3.3) using default options45, with gene annotations from GENCODE v42 (GFF3 format). Raw expression values were derived from the expected counts generated by RSEM.
Batch effect correction of gene expression data
To integrate gene expression data from an independent cohort of 35 healthy individuals, we implemented rigorous batch effect correction to ensure comparability. All samples were processed using an identical bioinformatic pipeline as RNA-seq data for COVID-19 patients to avoid potential in silico batch effects. We applied the ComBat-seq method implemented in the sva R package (v3.54.0)46 to remove batch effect by differences in sequencing platforms (i.e., NovaSeq 6000 vs DNBSEQ-T7RS) while preserving the biological condition (i.e., Healthy vs. COVID-19 status).
Normalization of gene expression data
To account for differences in sequencing depth and library composition, raw expected gene counts were normalized using the DESeq2 R package (v1.38.3)47. A variance stabilizing transformation (VST) was subsequently applied to the normalized counts. VST approximates a log2 transformation while decoupling mean-variance relationships of RNA-seq data, thereby improving the accuracy and interpretability of downstream statistical analyses. VST-transformed values were used for pairwise comparisons stratified by COVID-19 severity, allowing expression differences to be interpreted in fold-change-like units. These transformed values were also used for principal component analysis (PCA) to assess global expression patterns and sample clustering.
Discovery of differential methylation CpGs (DMC) associated with COVID-19 severity
Methylkit (R package; v1.20.0)42 was used to discover the Differentially Methylated CpGs (DMC) for COVID-19 severity, treating age and sex of the patients as covariates. We utilized the leave-one-out (LOO) method for cross-validation, iteratively collecting DMC without a sample for each round of discovery. Methylation sites that suffice the thresholds of absolute methylation difference (|meth.diff | ) >10 and FDR < 0.05 were selected as significant markers of the severity. The adjustment for P-value was done by Benjamini-Hochberg correction. We only selected a set of markers, those of which overlapped over all the folds.
Leukocyte-adjusted discovery of DMCs associated with COVID-19 Severity
We repeated the LOO-based DMC discovery as previously described. Here, we adjusted for age, sex, and blood cell-type composition (%)—including neutrophils, lymphocytes, monocytes, basophils, and eosinophils. This analysis included only samples with available cell count measurements recorded during clinical data collection, comprising 24 MM and 9 SC patients. The significance thresholds and marker selection criteria were identical to those used in the leukocyte-unadjusted analysis.
Discovery of DEGs associated with COVID-19 severity
DEGs associated with COVID-19 severity were identified using the DESeq2 R package (v1.38.3). Given the limited number of SC cases (n = 9), we employed a LOO cross-validation strategy to enhance the robustness of DEG discovery. In each iteration, one SC sample was excluded, and differential expression analysis was performed between the remaining SC and mild cases. This procedure was repeated nine times, each time omitting a different SC sample. Genes were considered significantly differentially expressed if they satisfied the following thresholds: absolute log₂ fold change (|log₂FoldChange | ) >1.3 and false discovery rate (FDR) < 0.05. To define robust DEGs (DEG-LOOs), we retained only those genes that exhibited consistent directionality—either upregulation or downregulation in SC patients—in at least seven out of the nine independent LOO iterations.
Gene ontology enrichment of DMCs and DEGs
To interpret the biological functions associated with differential DNA methylation and gene expression associated with COVID-19 severity, Gene Ontology (GO) enrichment analysis was conducted separately for genes linked to differentially methylated CpGs (DMCs) and DEGs. GO enrichment analysis was performed using ShinyGO v0.82 (https://bioinformatics.sdstate.edu/go82/)48, focusing on the Biological Process (GO:BP) category. The gene universe was defined as all genes expressed in the RNA-seq dataset (for DEGs) or all genes targeted by measured CpGs (for DMCs). Statistical significance was assessed using a hypergeometric test with Benjamini–Hochberg correction. GO terms with FDR < 0.05 were considered significantly enriched.
Prioritization of severity-associated CpG sites via cis-eQTM (expression quantitative trait methylation) analysis
We performed an eQTM analysis to assess the relationship between CpG methylation and gene expression (DESeq2-normalized read count) that have been discovered during previous steps (i.e., DMC and DEG) (Supplementary Fig. 9). Spearman correlation was used to quantify this relationship, with a significance threshold of absolute correlation coefficient > 0.5 using scipy.stats (v1.4.1). Gene coordinates and transcription start sites (TSS) were extracted from GENCODE v42 (GTF format). For cis-association analysis, we restricted the search space to CpG sites located within a ±1Mbp window of the corresponding gene’s TSS. This window was selected to capture local regulatory effects of CpG methylation on gene expression.
Statistics and reproducibility
Welch’s t-test (a two-sample independent t-test of unequal variances) was performed to find the mean difference of methylation values between different groups of infection stages using scipy.stats (v1.4.1). The normality assumption for the distributions of omics data was checked by the Shapiro-Wilk test using scipy.stats (v1.4.1) (Supplementary Table 6, 7). The P-values were adjusted with the Benjamini-Hochberg approach using statsmodels.stats (v0.13.2). Wilcoxon rank-sum test using scipy.stats (v1.4.1) was performed to test the statistically significant differences in gene expression and clinical values across severities. Principal components for DNA methylation and gene expression were computed using sklearn.decomposition.PCA (v0.23.2). All visualizations were drawn using matplotlib (v3.5.3) and seaborn (v0.11.0). The sample sizes for each analysis are indicated in the figure legends and supplementary tables. No separate replicate experiments were conducted. Instead, we assessed the robustness of our marker selection by applying a LOO cross-validation approach during the identification of DMPs and DEGs. Detailed descriptions of the LOO procedure are provided in the respective sections for DMP and DEG analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
DNA methylation beta values derived from target bisulfite sequencing have been deposited in the GEO under accession number GSE301082. Raw expression values derived from RNA sequencing have been deposited in the GEO under accession number GSE300696. The corresponding raw sequencing data, including bisulfite-seq and RNA-seq reads, are available from the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA1292728. All other data, including raw sequencing data, are available from the corresponding author upon reasonable request. The numerical data underlying all figures and supporting the findings of this study are available in Supplementary Data 1.
Code availability
The codes used to generate data and calculate statistics are openly available in the Github page: https://github.com/korean-genomics-center/Multiomics_COVID19_Severity.
References
Mohandas, S. et al. Immune mechanisms underlying COVID-19 pathology and post-acute sequelae of SARS-CoV-2 infection (PASC). Elife 12, e86014 (2023).
Karki, R. et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci. Immunol. 7, eabo6294 (2022).
Montazersaheb, S. et al. COVID-19 infection: an overview on cytokine storm and related interventions. Virol. J. 19, 92 (2022).
Barturen, G. et al. Whole blood DNA methylation analysis reveals respiratory environmental traits involved in COVID-19 severity following SARS-CoV-2 infection. Nat. Commun. 13, 4597 (2022).
Qin, W., Scicluna, B. P. & van der Poll, T. The role of host cell DNA methylation in the immune response to bacterial infection. Front Immunol. 12, 696280 (2021).
Castro de Moura, M. et al. Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine 66, 103339 (2021).
Bradic, M. et al. DNA methylation predicts the outcome of COVID-19 patients with acute respiratory distress syndrome. J. Transl. Med. 20, 526 (2022).
Li, Y. Y. et al. Cell-free DNA methylation reveals cell-specific tissue injury and correlates with disease severity and patient outcomes in COVID-19. Clin. Epigenetics 16, 37 (2024).
Balnis, J. et al. Blood DNA methylation and COVID-19 outcomes. Clin. Epigenetics 13, 118 (2021).
Pacis, A. et al. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res. 25, 1801–1811 (2015).
Salgado-Albarran, M. et al. Comparative transcriptome analysis reveals key epigenetic targets in SARS-CoV-2 infection. NPJ Syst. Biol. Appl 7, 21 (2021).
Keshawarz, A. et al. Expression quantitative trait methylation analysis elucidates gene regulatory effects of DNA methylation: the Framingham Heart Study. Sci. Rep. 13, 12952 (2023).
Kim, S. et al. Cis- and trans-eQTM analysis reveals novel epigenetic and transcriptomic immune markers of atopic asthma in airway epithelium. J. Allergy Clin. Immunol. 152, 887–898 (2023).
Ruiz-Arenas, C. et al. Identification of autosomal cis expression quantitative trait methylation (cis eQTMs) in children’s blood. Elife 11 (2022).
Wu, Z. & McGoogan, J. M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 cases From the Chinese Center for Disease Control and Prevention. JAMA 323, 1239–1242 (2020).
Saheb Sharif-Askari, N. et al. Upregulation of oxidative stress gene markers during SARS-COV-2 viral infection. Free Radic. Biol. Med 172, 688–698 (2021).
Xia, J. et al. Immune response is key to genetic mechanisms of SARS-CoV-2 infection with psychiatric disorders based on differential gene expression pattern analysis. Front Immunol. 13, 798538 (2022).
Jain, S. K. et al. The potential link between inherited G6PD deficiency, oxidative stress, and vitamin D deficiency and the racial inequities in mortality associated with COVID-19. Free Radic. Biol. Med 161, 84–91 (2020).
Bastin, A. et al. Severity of oxidative stress as a hallmark in COVID-19 patients. Eur. J. Med Res 28, 558 (2023).
Gain, C., Song, S., Angtuaco, T., Satta, S. & Kelesidis, T. The role of oxidative stress in the pathogenesis of infections with coronaviruses. Front Microbiol 13, 1111930 (2022).
Peacock, T. P. et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol 6, 899–909 (2021).
Essalmani, R. et al. Distinctive roles of furin and TMPRSS2 in SARS-CoV-2 Infectivity. J. Virol. 96, e0012822 (2022).
Hossain, M. S. et al. Prediction of the Effects Of Variants And Differential Expression Of Key Host genes ACE2, TMPRSS2, and FURIN in SARS-CoV-2 pathogenesis: an in silico approach. Bioinform Biol. Insights 15, 11779322211054684 (2021).
Liang, S. et al. SARS-CoV-2 spike protein induces IL-18-mediated cardiopulmonary inflammation via reduced mitophagy. Signal Transduct. Target Ther. 8, 108 (2023).
Nasser, S. M. T. et al. Elevated free interleukin-18 associated with severity and mortality in prospective cohort study of 206 hospitalised COVID-19 patients. Intensive Care Med Exp. 11, 9 (2023).
Korotaeva, A. A. et al. Ratios between the Levels of IL-18, Free IL-18, and IL-1beta-binding protein depending on the severity and outcome of COVID-19. Bull. Exp. Biol. Med 176, 423–427 (2024).
Cheema, P. S., Nandi, D. & Nag, A. Exploring the therapeutic potential of forkhead box O for outfoxing COVID-19. Open Biol. 11, 210069 (2021).
Gimenes-Junior, J. et al. FOXO3a regulates rhinovirus-induced innate immune responses in airway epithelial cells. Sci. Rep. 9, 18180 (2019).
Sullivan, J. A., Kim, E. H., Plisch, E. H. & Suresh, M. FOXO3 regulates the CD8 T cell response to a chronic viral infection. J. Virol. 86, 9025–9034 (2012).
Palmos, A. B. et al. Proteome-wide Mendelian randomization identifies causal links between blood proteins and severe COVID-19. PLoS Genet 18, e1010042 (2022).
Binder, E. B. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 34, S186–S195 (2009).
Wochnik, G. M. et al. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J. Biol. Chem. 280, 4609–4616 (2005).
Hao, W., Wang, L. & Li, S. FKBP5 regulates RIG-I-mediated NF-kappaB activation and influenza A virus infection. Viruses 12, 672 (2020).
Erlejman, A. G. et al. NF-kappaB transcriptional activity is modulated by FK506-binding proteins FKBP51 and FKBP52: a role for peptidyl-prolyl isomerase activity. J. Biol. Chem. 289, 26263–26276 (2014).
Green, R. et al. SARS-CoV-2 infection increases the gene expression profile for Alzheimer’s disease risk. Mol. Ther. Methods Clin. Dev. 27, 217–229 (2022).
Carabelli, A. M. et al. SARS-CoV-2 variant biology: immune escape, transmission and fitness. Nat. Rev. Microbiol 21, 162–177 (2023).
COVID-19: Developing Drugs and Biological Products for Treatment or Prevention. (Food and Drug Administration, 2023).
Toussi, S. S., Hammond, J. L., Gerstenberger, B. S. & Anderson, A. S. Therapeutics for COVID-19. Nat. Microbiol 8, 771–786 (2023).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. (Cambridge, 2010).
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011).
Akalin, A. et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, R87 (2012).
Cavalcante, R. G. & Sartor, M. A. ANNOTATR: genomic regions in context. Bioinformatics 33, 2381–2383 (2017).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 323 (2011).
Zhang, Y., Parmigiani, G. & Johnson, W. E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform 2, lqaa078 (2020).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Ge, S. X., Jung, D. & Yao, R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629 (2020).
Acknowledgements
We thank all voluntary participants for donating their blood and the city of Ulsan for supporting the project. We also appreciate the Ulsan ICT Promotion Agency (UIPA), which provided us with the BioDataFarm system, which supports the storage, analysis, and management of the BioBigData. We are grateful to Asta Blazyte for her assistance in proofreading and refining the English of this manuscript. This work was supported by the Promotion of Innovative Businesses for Regulation-Free Special Zones funded by the Ministry of SMEs and Startups (MSS, Korea) (Grant numbers: P0016195, P0016193 [1425156792, 1425157301; 2.220035.01, 2.220036.01]). It was also supported by the Establishment of Demonstration Infrastructure for Regulation-Free Special Zones fund (MSS, Korea) (Grant number [P0016191] (2.220037.01) (1425157253)) by the Ministry of SMEs and Startups. Additional support was provided by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (Grant numbers: 2022R1F1A1062753 and RS-2023-00227944); the RS-2023-00227944 grant was awarded to Dr. Asaph Young Chun, Principal Investigator at the Seoul National University Institute for Pandemic Sciences AI.celerator. Furthermore, this work was supported by the Ulsan University Hospital Research Grant (UUH-2022-10).
Author information
Authors and Affiliations
Contributions
H.R.: Conceptualization, Project administration, Formal analysis, Data curation, Investigation, Methodology, Results interpretation, Visualization, Writing – original draft, Writing – review and editing; K.A.: Formal analysis, Data curation, Investigation, Methodology, Results interpretation, Visualization, Writing – original draft, Writing – review and editing; Y.K.: Data curation, Formal analysis, Methodology, Writing – review and editing; Y.J.: Writing – review and editing; S.J.: Writing – review and editing; H.C.: Conceptualization, Project administration; Y.J.K.: Writing – review and editing; S.K.: Patient recruitment and sample collection, Clinical phenotyping, Data curation; O.J.S.: Sample processing, Data curation; S.L.: Writing – review and editing; A.Y.C.: Writing – review and editing; E.S.S.: Conceptualization, Results interpretation, Supervision, Writing – original draft, Writing – review and editing; S.W.R.: Conceptualization, Patient recruitment and sample collection, Data curation, Project administration, Conceived and designed the study, Curated clinical phenotyping data, Writing – review and editing; J.B.: Conceptualization, Results interpretation, Supervision, Conceived and designed the study, Writing – review and editing, Funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Markus Weckmann and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Pei Hao and Aylin Bircan, Johannes Stortz. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ryu, H., An, K., Kwon, Y. et al. Identification of reactive CpGs and RNA expression in early COVID-19 through cis-eQTM analysis reflecting disease severity and recovery. Commun Biol 8, 1174 (2025). https://doi.org/10.1038/s42003-025-08609-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08609-4
