
Abstract
Copy number alteration (CNA) is a major type of cancer genome alteration that drives cancer progression. CNA signature analysis can reveal underlying etiology and provide biomarkers for cancer treatment, and existing CNA signature analyses are all performed using bulk tissue samples. However, CNA usually affects a large proportion of genome, and the CNA profile of bulk sample does not reflect the actual CNA profiles of the individual cancer cells of the sample, especially in tumors with high heterogeneity, such as hepatocellular carcinoma (HCC). Furthermore, the evolutionary trajectory of CNA mutational processes still remains elusive. Here we build a method to comprehensively analyze the CNA signatures of HCC from single-cell and bulk sample perspective, revealing patterns and potential noise signals from the usually performed bulk tissue CNA signature analysis. Single-cell signature analysis delineates the evolutionary trajectory of HCC CNA signatures, and different CNA signatures consistently emerge in different HCC evolution stages. Single-cell CNA signatures show robust performance in patient prognosis and drug sensitivity prediction. This work not only reveals specific considerations in analyzing CNA signature derived from bulk tissue but also depicts CNA evolution process and provides potential biomarkers for the prognosis and treatment of HCC patients.
Similar content being viewed by others


Introduction
Genomic instability is one of the hallmarks of cancer1, encompassing changes ranging from single nucleotide alterations to whole chromosome modifications. Numerous studies have demonstrated the roles of single base substitutions (SBS), small insertions and deletions (INDEL), structural variations (including translocations/inversions), and copy number alterations in the process of cancer development2,3,4. Genomic DNA alteration signatures are recurring genomic patterns that are the imprints of mutagenic processes accumulated over the lifetime of cancer cell5. SBS signature analysis has been extensively studied and represents a prototype for other types of signature study5. While analyzing copy number signature is more intricate than SBS signatures. Pan-cancer CNA signature studies have been carried out6,7,8,9, and these studies indicate that genomic alteration signatures can provide information on mutational processes, and also serve as biomarkers for precision medicine in cancer10,11,12,13,14,15.
Current research on CNA signature is predominantly focused on bulk tissue samples. In the detection of SBS, bulk tissue is capable of preserving the SBS mutation status of the majority of subclones. In contrast, subclone’s CNA state may not be easily inferred from the final CNA profile of bulk tissue. For tumors with low heterogeneity, sequencing of bulk tissue effectively captures the variations in copy numbers. However, for tumors with high heterogeneity, like hepatocellular carcinoma (HCC)16,17,18, sequencing of bulk tissue only reflects the averaged CNA across all tumor subclones19,20, failing to accurately reveal the true patterns of CNA.
Several large-scale studies have explored CNA signatures in human cancers using bulk tissue data. For example, Drews et al. analyzed the distributions of five fundamental copy number features and applied mixture modeling to these distributions to define 43 components across cancer cohorts7. Steele et al. proposed a method using 48 CNA features, incorporating absolute copy number, segment size, and loss of heterozygosity (LOH) status6. While these approaches established foundational frameworks for CNA signature analysis, the selected features are largely limited to static descriptions of CNA segments and do not capture their broader structural context. Our study introduces a comprehensive CNA feature framework. In addition to basic properties like copy number level and segment length, we incorporate segment shape and segment change, which together reflect the local architectural characteristics of CNA events.
The models of tumor evolution have been a contentious subject in tumor biology21,22. Gao et al.’s model of punctuated copy number evolution suggests that copy number aberrations are primarily acquired during brief peaks of genomic crisis, followed by stable clonal expansion23. Guo et al.24 introduced a model of dual-phase copy number evolution in HCC, indicating the coexistence of gradual and punctuated evolution. Although these tumor evolution models have revealed the occurrence rate of CNAs during tumor progression, they have not clarified which CNA patterns or signatures appear in the early stages of a tumor and which appear in the later stages. The evolutionary trajectory of CNA signatures still remains elusive.
In this study, we collected scDNA-seq data from 1222 cells of 10 HCC patients and developed a versatile tool for analyzing CNA signatures that apply to both single-cell and bulk tissue samples (Supplementary Data 1). This tool enabled us to systematically analyze the CNA signatures in HCC at the single-cell level for the first time. Comparing CNA signatures derived from single-cell and those from bulk samples revealed that some CNA signatures observed in bulk tissue samples might not actually exist. Additionally, we have for the first time proposed the evolutionary trajectory of CNA signatures in HCC. These studies enhance our understanding of the mutation processes of HCC and important information for the in-depth study of CNA signatures and their potential clinical applications.
Results
Single-cell analysis revealed masked CNA profiles compared with bulk tissue CNA analysis
Each point mutation occurs on a single chromosome in a single cell, which gives rise to a lineage of cells bearing the same mutation. If that chromosomal locus is subsequently duplicated, any point mutation on this allele preceding the gain will subsequently be present on the two resulting allelic copies25,26. This process facilitates the accumulation of SBS without interference between mutations. Bulk tissue sequencing enables point mutation detection within a sample, reflecting the cumulative outcome of all subclones (Fig. 1A). However, CNA usually affects a large proportion of genome DNA, when a CNA occurs within a single cell, the subsequent subclonal CNA further modify the original CNA profile, distorting its characteristic signature. Consequently, the CNA observed in bulk tissue is not an accurate reflection of the true CNA occurs in each subclones (Fig. 1B). This phenomenon is particularly pronounced in tumors with high heterogeneity, such as HCC. To accurately explore the true state of CNA, we aggregated data from various HCC databases, covering different sequencing platforms and depths. Our collection comprised 1222 single-cell shallow whole-genome sequencing (WGS) samples24,27, 160 bulk shallow WGS samples28, 178 bulk WGS samples29,30, and 371 bulk SNP array samples31. All raw data were uniformly processed to derive the absolute copy number information of the samples (details provided in the Materials and Methods).

A Schematic diagram of SBS in different cancer subclones. SBS between different subclones do not affect each other at the bulk tissue level. B Schematic diagram of CNA in subclones of different cells. The CNA profile of bulk tissue can not retain some subclonal CNA status.
Development of CNA signature analysis tool for both single-cell and bulk tissue samples
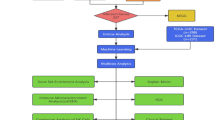
To identify single-cell CNA signatures, we developed a novel method for the extraction of CNA signatures. This method encompasses four principal aspects of CNA: absolute copy number, segment length, segment change, and segment shape (Fig. 2). From these aspects, we further delineated 90 distinct features (Fig. S1 and Supplementary Data 2). These features were selected as hallmarks of previously reported genomic aberrations, including chromothripsis32, large-scale state transitions (LST)33, extrachromosomal circular DNA (ecDNA)34, and tandem duplications35. Following the computation of features for all samples, this feature matrix was processed using non-negative matrix factorization to identify CNA signatures (Fig. 2). This method has previously been employed for extracting signatures of SBS2,5. The number of signatures extracted was determined using two parameters. First is the reconstruction error, and the average Frobenius reconstruction error is reported. Second is the stability of signature extraction and the cosine similarity between the extracted signatures, and average silhouette width. In the single-cell samples, a total of seven CNA signatures were identified. In the analysis of bulk samples, seven CNA signatures were identified with shallow WGS and WGS data. Eight CNA signatures were determined using SNP array data (Fig. S2).

The process of CNA signature extraction. A CNA feature matrix is generated from the absolute copy numbers obtained from different sequencing platforms and sequencing depths, and CNA signatures were extracted via NMF.
Mutational processes for single-cell CNA signatures
Following determining CNA signatures, we initially assessed the similarity among different CNA signatures. The observed similarities between CNA signatures were low (Fig. S3), suggesting that each CNA signature is relatively distinct (Figs. 3A and S4). Further, we compared the CNA signatures extracted from bulk tissue shallow WGS, WGS, and SNP array data, with those obtained from single-cell shallow WGS data. Our results indicate that most CNA signatures are consistently detectable across different data types (Fig. S5), supporting the robustness and general applicability of the proposed CNA signature extraction method. However, specific CNA signatures, such as WGS_Sig5, WGS_Sig6, SNP_Sig3, and SNP_Sig4, did not have similar counterparts in single-cell CNA signatures. This phenomenon may be attributed to several factors. First, the limited number of patients represented in the single-cell samples may have resulted in the exclusion of certain patient-specific CNA signatures. Second, differences in data resolution and signal quality across sequencing platforms may affect the detection of certain CNA signatures. Additionally, these CNA signatures may represent signals specific to bulk tissues rather than single cells. These results highlight the importance of integrating multiple data types for a comprehensive characterization of CNA signatures.

A Distribution of CNA feature contributions across single-cell CNA signatures. The x-axis represents 90 CNA features grouped into four categories: segment length, segment shape, copy number state, and segment change. The y-axis indicates the relative contribution of each feature to the signature. B Representative CNA profile, prominent features, and potential mechanisms for each identified single-cell CNA signature.
Association analysis between CNA signatures and phenotypic data can reveal the underlying clinical and biological information for CNA signatures8,11,15. In the scSig1, we identified numerous genomic fragments smaller than 5 Mb with high copy numbers, consistent with genomic variations induced by ecDNA36,37,38,39. Further associative analyses suggest these variations are attributable to ecDNA (Fig. S6). Based on the analysis of representative samples of CNA signatures, we observed that the scSig3 pattern reflects characteristics of a normal genome, while the genome with the scSig4 pattern exhibits a clear triploid state, aligning with the CNS3 and CNS12 proposed by Tao et al.8 Utilizing these analyses, we have summarized the potential etiologies of each CNA signature in single-cell data from HCC (Fig. 3B).
Single-cell CNA signature analysis reveals masked patterns in bulk tissue analysis
To further investigate the differences between single-cell CNA signatures and bulk tissue CNA signatures, following the methodology of Drews et al.7 this study simulated CNA events, including early and late whole-genome duplication (early WGD, late WGD)40,41 and LST (associated with homologous recombination deficiency, HRD)33. We observed that single-cell CNA signatures were able to distinguish early WGD from late WGD, whereas bulk-derived signatures showed less resolution in this context (Fig. 4A). This suggests that single-cell data may provide higher sensitivity for capturing subtle differences in CNA evolution under certain conditions. Following the genomic variation simulation method of Bartenhagen et al.42, we simulated the formation of a series of CNA for comparative analysis, and the results also demonstrated that single-cell CNA signatures are more robust than bulk tissue CNA signatures in filtering out such simulated noise (Figs. 4B and S7). These findings highlight the potential of single-cell data in characterizing fine-scale CNA patterns, especially when background noise or subclonal complexity is high.

A Simulation of LST, early WGD, and late WGD events in twenty samples. The similarity between the CNA profile of each sample and the obtained CNA signature is calculated through cosine similarity. B Simulate different levels of CNA noise (0–15%), and calculate the cosine similarity between the CNA profile and the indicated CNA signature under each noise condition. Each noise level is simulated 100 times, and the mean similarity is used for plotting.
We employed the method of Collin Giguere et al.43 to generate bulk tissue genomic sequencing data from corresponding single-cell data, and subsequently, absolute copy numbers were extracted (Fig. S8). Then we fitted the generated bulk tissue CNA profiles with the single-cell CNA signatures. Comparing the differences reveals some inconsistencies between the CNA signatures of bulk tissue and corresponding single cells (Fig. 5A, B). This inconsistency may be attributed to the CNA masking effects of bulk tissue samples. It is also possible that the simulated bulk profiles may not fully reflect the complexity of real tissue samples, suggesting that matched single-cell and bulk WGS data would provide a more reliable means for further validation. In the analysis of CNA patterns in individual samples, such as sample P04, we noted that the scSig4 constituted 96.7% in bulk tissue, whereas in corresponding single-cell samples, most cells predominantly exhibited scSig3 (Figs. 5C, D and S9). This discrepancy underscores the potential for bulk-derived signatures to miss subclonal diversity, particularly in highly heterogeneous cancers such as HCC. These analyses suggest that single-cell and bulk data offer complementary perspectives on CNA architecture. Single-cell data may be more sensitive to subclonal structure and rare events, while bulk data provides consensus profiles that are more robust to sparsity. Integrating single-cell and bulk data may represent a more effective strategy for achieving comprehensive and precise CNA signature characterization in future studies.

A Bulk tissue sample generated from corresponding single cells, and the distribution of scSig as a percentage of the total is presented. B The mean proportion of each scSig across all cells within each sample is calculated, showing the average distribution of scSig within each sample. C At the bulk tissue level, the distribution of scSig in sample P04 and scSig4 accounts for 96.7%. D At the single-cell level, the distribution of scSig in sample P04 and scSig3 accounts for a relatively large proportion. Various scSig are present.
Single cell CNA signature analysis reveals the evolutionary trajectory of HCC CNA signatures
To investigate the evolutionary trajectory of CNA signatures in HCC, we employed three complementary approaches to assess the dynamic changes in the proportion of scSigs across single cells. We first constructed a phylogenetic tree based on the CNA signature composition of each cell. This analysis revealed that scSig7 (characterized by small CNAs) consistently appeared earlier in the inferred timeline, while scSig1 and scSig2 (associated with large-scale CNAs) became increasingly dominant during later tumor progression (Fig. 6A). We then inferred evolutionary transitions by analyzing shared CNA breakpoints between cells. This approach also positioned scSig7 in earlier transition states, with scSig1 and scSig2 emerging at later stages (Fig. 6B). Notably, while the overall trend of early scSig7 and late scSig1/2 was observed across most cases, the specific evolutionary trajectories and timing of signature transitions varied between patients (Figs. S10 and S11), suggesting considerable inter-patient heterogeneity in CNA evolution. Additionally, we applied MEDICC244, a phylogenetic algorithm specifically designed for CNA-based lineage reconstruction. Based on the pseudotemporal ordering of each cell in the evolutionary tree, we analyzed the dynamic changes of scSigs. Consistent with the previous findings, with scSig7 appearing at an earlier stage (Fig. S12). These results elucidate the evolutionary trajectory of tumor genomic CNA signature, demonstrating that amplifications of small DNA fragments occur before large-scale genomic alterations in HCC evolution. The accumulation of these small fragment changes may affect the progression of the cell cycle, leading to chromosomal-level or genome-wide DNA alterations45.

A Evolutionary tree analysis of scSig. Calculate the distance between cells based on scSig, infer the evolutionary relationship based on the distance, and then draw an evolutionary tree. Symbols on each branch represent the most prevalent scSig within that lineage. B Evolutionary trajectory analysis of scSig. The average number of shared breakpoints per cell within the sample is calculated, and cells are ranked according to the number of shared breakpoints. Cells with a higher ratio of shared breakpoints are positioned early in the clonal evolution, as subsequent subclones retain the breakpoints of earlier cells.
Single-cell CNA signatures predict prognosis and drug response of HCC patients
Bulk tissue CNA signatures have been reported to be associated with cancer clinical outcomes6,7,11,46. To compare the prognostic impact of single-cell CNA signatures with those derived from WGS and SNP array CNA signatures, we analyzed HCC samples with survival data from the PCAWG and TCGA cohorts. We calculated the activity of scSig, WGS_Sig, and SNP_Sig in these samples through signature filtering. Across datasets from different technological platforms, scSig show more robust performance compared with WGS_Sig, and SNP_Sig as reflected in the consistent prognostic outcomes in different datasets (Fig. 7A). In two independent datasets, scSig1 and scSig4 were associated with poorer prognosis, while scSig3 was related to better outcomes in the SNP dataset, with a similar trend observed in the WGS dataset (Figs. 7B and S13). This finding suggests the superiority of single-cell CNA signatures over traditional bulk tissue CNA signatures in terms of HCC prognosis prediction. Multivariable Cox regression identified scSig1 as the only independent prognostic factor (Fig. S14A). While scSig4 and scSig6 were not individually significant in the multivariable model, combined stratification revealed that patients with high levels of both signatures had the poorest overall survival, compared to those with low or intermediate levels (Fig. S14B). This suggests that the combination of these two signatures may offer improved prognostic stratification compared to either alone.

A The prognosis performance of scSig, WGS_Sig, and SNP_Sig in WGS and SNP array data was analyzed. Shown are the HR values of Signature that are significantly related to prognosis (FDR < 0.05), where HR > 1 indicates an association with poor prognosis, and HR < 1 indicates an association with better prognosis. B The prognosis performance of the scSig4 in both WGS and SNP datasets. Consistent prognostic results in two different data sets.
We analyzed the correlations between drug sensitivity and CNA signatures. Our findings indicate that the sensitivity of HCC patients to the commonly used drug sorafenib is negatively correlated with the scSig7. Additionally, certain drugs demonstrated different responses to particular CNA signatures. Inhibitors of protein arginine methyltransferase (PRMT) have been found to impair tumorigenesis in HCC mouse models47,48,49. GSK591, a PRMT5 inhibitor, is regulated by MYC in HCC and has been identified as a direct target gene of MYC50,51. Patients carrying the scSig1 signature exhibited heightened sensitivity to GSK591, whereas they were non-responsive or showed resistance to other drugs (Fig. S15). Importantly, the scSig3 pattern, indicative of normal genomic features, was sensitive to most drugs, suggesting that early-stage tumors without significant CNAs have not yet developed substantial drug resistance. As genomic CNAs increase, tumors gradually acquire drug resistance. These results provide a valuable perspective for using single-cell CNA signatures in studying tumor drug resistance.
Discussion
In this study, we have developed an analytical framework for the extraction of CNA signatures in single cells. This framework is adaptable across various experimental platforms and sequencing depths, effectively extracting CNA signatures. Compared to previous approaches, our framework include not only basic copy number properties but also local structural characteristics.
CNA signatures have demonstrated their clinical value in a multitude of cancer studies11,15. Nevertheless, these existing CNA signature analyzes are all performed using bulk tissue samples, and single-cell CNA signature analysis is needed. We have systematically compared CNA signatures in single cells with those in bulk tissues of HCC. Our findings show that many single-cell CNA signatures correspond to those observed in bulk, supporting their overall concordance. Simulation-based analyses further suggest that single-cell signatures may offer enhanced sensitivity in capturing specific mutational processes and greater robustness to noise. Given the pronounced heterogeneity of HCC, single-cell analysis can better preserve subclonal information and avoid the averaging effects inherent in bulk data. These results highlight the complementary strengths of both data types and suggest that integrating single-cell and bulk approaches may provide a more comprehensive view of CNA signature in HCC.
This study offers insights into the dynamic changes of CNA signatures during HCC progression. We found that CNA signatures with small fragment amplifications, such as scSig7, typically appear in the early stages of HCC development. In contrast, CNA signatures indicative of chromosomal and genomic-level changes, such as scSig1 and scSig2, tend to emerge in the later stages. A reasonable explanation for this pattern is that amplification of small DNA fragments in the early stages disrupts normal cell cycle processes, subsequently leading to large-scale genomic variations, which contribute to the formation of complex large CNA patterns. However, these conclusions still require direct experimental evidence for further validation. Notably, we also observed considerable inter-patient variability in the evolutionary trajectories of CNA signatures, suggesting that different tumors may follow distinct paths during HCC progression. This heterogeneity underscores the need for patient-specific interpretation and warrants further investigation in larger cohorts.
CNA is a complex biological process, and studies focusing on bulk tissues have already demonstrated its potential clinical utility. Nevertheless, the biological significance of certain CNA patterns remains to be elucidated, necessitating further in-depth investigation. In this study, we show that CNA signatures derived from single cells can effectively predict patient prognosis and drug sensitivity. Combining single-cell data with bulk tissue CNA data for integrated analysis could be a promising avenue for future research.
From a single-cell perspective, we have identified some potential issues with bulk tissue CNA signatures. However, our study also has certain limitations. Although this study included data from 1222 single cells, these data were derived from only 10 patients. The limited sample size might have led to the failure to detect all CNA features. Additionally, due to the lack of publicly available matched datasets, we could only compare platform-specific signatures through computational simulations to explore their differences. Regarding the evolutionary trajectory analysis of CNA signatures, our analysis suggests that certain signatures consistently appear earlier in different samples. However, the complexity of the CNA process makes it challenging to establish an accurate evolutionary timeline and comprehensively describe the detailed evolutionary trajectory of CNA signatures. These analyses provide a direction for future research, and more experimental evidence is required for validation.
Methods
Data collection and processing
We downloaded data from 1222 single-cell shallow whole-genome sequencing (0.4×) of HCC patient samples (totaling 10 patients) from the Genome Sequence Archive (GSA)24,27, under accession numbers HRA000094. Data from 160 HCC bulk samples with shallow whole-genome sequencing (0.5×) were also obtained from the International Cancer Genome Consortium (ICGC) database28 (https://dcc.icgc.org/releases/PCAWG/). Moreover, absolute copy number data of whole-genome sequencing from 178 HCC bulk samples in the Pan-Cancer Analysis of Whole Genomes (PCAWG) cohort were acquired through the UCSC Xena platform29,30 (https://xenabrowser.net/datapages/). Concurrently, SNP array data from 371 HCC bulk samples were sourced from The Cancer Genome Atlas (TCGA) database31 (https://portal.gdc.cancer.gov/).
For genome sequencing raw data (fastq) files, fastp was first used to perform quality control on the data, the BWA MEM algorithm was then applied using hg38 as reference genome52, and finally, the samtools tool was used to convert the SAM files into BAM files53. After obtaining the BAM files for all datasets, use the SortSam and MarkDuplicates commands in the Picard toolkit (https://broadinstitute.github.io/picard/) to sort BAM files and mark duplications for variant and copy number calling. Raw data processing, the codes used can be found at Code availability.
Absolute copy number calling
QDNAseq54 and ACE55 R packages were used to determine the copy number profiles from the low-coverage WGS BAM files. To determine the autosomal copy number profiles, we excluded both chromosome X&Y and mitochondrial DNA, and the ploidy was adjusted using median bin segment value, which was the central assumption of ACE. For single-cell sequencing files, we used a ploidy penalty of 0.5 and lower-cellularity penalty of 1.0 to fit the “squaremodel()” function of ACE. For bulk tissue sequencing files, we used a ploidy penalty of 0.5 and lower-cellularity penalty of 0.5 to fit the “squaremodel()” function of ACE as per the author’s recommendation. ABSOLUTE56 was used to determine the copy number profiles from the SNP array files of TCGA. The code for copy number calling can be found in Code availability.
CNA feature classification
Considering that shallow sequencing data cannot assess the LOH status of the genome, in order to describe the copy number variation of the liver cancer genome more accurately, we classify the copy number segments from four aspects, (1): copy number, which directly reflects the copy number amplification or deletion, divided into 6 states: 0, 1, 2, 3, 4, 5+, where 0 and 1 indicate that the genome fragment is missing, 2 indicates the normal genome state, and 3 and 4 indicate that the corresponding genome fragment is moderately amplified. 5+ is considered highly amplified. (2): segment length, indicating the length of the copy number segment. The segment length of some small genomic variations, such as ecDNA is mostly less than 5 Mb57,58. Since most of the copy number variations produced by large scale transitions are greater than 10MB59, the copy number segment is length is divided into three types: S (5Mb<), M (5–10 Mb), and L (>10 Mb). (3): Segment change: this feature quantifies the difference in copy number between a segment and its immediate downstream segment. It reflects the magnitude of CNA transition at segment boundaries, which may indicate involvement of different mutational mechanisms. Segment change is categorized into two types: AA (absolute copy number change ≤ 2); BB: (absolute copy number change > 2). (4): Segment shape: this feature describes the copy number relationship between a given segment and its two adjacent segments. It is classified into three categories based on the copy number values of the left and right neighboring segments: HH (High–High): both adjacent segments have higher copy number values than the segment itself; LL (Low–Low): both adjacent segments have lower copy number values than the segment itself; OT (Other): the adjacent segments have opposite copy number directions (either Left High–Right Low or Left Low–Right High).
Deriving signatures
Copy number signatures were extracted from individual sample matrices using the gold standard tool SigProfiler v1.0.17 (https://github.com/AlexandrovLab/SigProfilerExtractor) with default parameters. Initially, the non-negative matrix factorization (NMF) algorithm was applied for the decomposition of the patient’s copy number feature matrix. Signature search intervals ranging from 3 to 12 were set, and NMF was run 200 times for each signature number with different random seeds to ensure result stability. For each NMF iteration, initialization was performed using random numbers, and iterations ranged from 10,000 to 1,000,000 until stable results were obtained. Two key parameters determined during this process were the stability of the signatures, as measured by silhouette analysis, and the average Frobenius reconstruction error. Under these criteria, seven signatures were identified as optimal at the single-cell level and were selected for further analysis.
Copy number variation simulation
To simulate realistic chromosomal variation events, this study utilized the approach proposed by Drews et al.7. We simulated WGD events by setting a tetraploid background prior to the insertion of other CNAs for early WGD, or by multiplying all copy number states by a factor following the placement of all CNAs for late WGD. Considering that most WGD events are early occurrences, the ratio of early to late WGD samples was set at 4:160. The simulation of LST was based on their real breakpoint characteristics, employing a Poisson distribution for this simulation. Random copy number variations were simulated using the RSVSim42 tool, with hg38 as the reference genome. The simulations aimed to cover 0% to 15% of the genome. The length of each amplified and deleted segment was randomly set between 1 kb and 5 MB, distributed randomly across the entire genome, with all other parameters set to their default values. Calculating the degree of similarity between simulated samples and scSig using cosine similarity, a method widely used for assessing similarities among copy number profiles8.
Phylogenic trees
We utilized the phangorn package in R to calculate the Maximum Parsimony Tree from the scSig matrix using the Parsimony Ratchet algorithm61,62. To ensure the accuracy of our analysis, samples that did not exhibit any CNAs were excluded. The branch lengths between cells were estimated using the ACCTRAN algorithm. The “ggtree” package in R was employed to visualize the phylogenetic trees63, where the predominant CNA signature were marked with distinct colors for easy visual differentiation.
CNA signature evolution
To determine the temporal sequence of emergence for tumor cell subclones, we estimated the likely order by analyzing shared CNA breakpoints. Shared breakpoints refer to genomic CNAs that occur at the same positions across different subclonal cells within the same sample. The rationale is that subclones often accumulate new CNAs on top of existing alterations and, therefore, tend to retain breakpoints from earlier events. This method provides a relative measure of evolutionary proximity, though it may be less informative for large-scale or convergent events such as whole-genome duplication.
where Sj is the frequency of shared break point of cell j in the sample, M is the number of cells in the sample, Lj is the number of shared breakpoints in cell j, and Iij represents the existence of the shared breakpoint I in cell j. Iij is a binary variable. When the breakpoint i exists in the cell j, Iij takes a value of 1; when the breakpoint i does not exist in the cell j, Iij takes a value of 0.
CNA phylogeny inference using MEDICC2
We employed MEDICC2, a phylogenetic tool specifically designed for CNA-based data. As input, we used segmented absolute copy number profiles of individual single cells. MEDICC2 was run with default parameters. The resulting phylogenetic tree was rooted and visualized using the MEDICC2 visualization module. To analyze temporal dynamics, we extracted pseudotime ordering of cells based on their relative positions along the inferred evolutionary tree. CNA signature composition (scSigs) was overlaid onto the tree to examine the temporal distribution of different signature groups.
Survival analysis
In this study, we gathered survival data for samples from the TCGA (SNP array sequencing) and PCAWG (WGS sequencing) projects, encompassing 352 samples from TCGA and 139 samples from PCAWG. R package sigminer15 was utilized to fit each patient’s copy number feature matrix to scSig, SNP_Sig, and WGS_Sig, respectively. The association between CNA feature activity and overall survival was determined using univariate Cox proportional hazards models. For each Cox model, Hazard ratios were calculated and reported to indicate the direction and significance of the survival relationship. The Cox model is given by:
where t is the total survival time, h(t, X) is the hazard function given the presence of a prognostic variable X, and h0(t) represents the baseline hazard. Cox proportional hazards analysis was conducted using the R package ezcox64.
Drug sensitivity analysis
Genomic information and drug sensitivity data for all HCC cell lines were downloaded from the GDSC database65. Following the previously introduced method, copy number variation data were obtained, and copy number feature matrices were calculated. Subsequently, using the R package sigminer, the copy number feature matrices of each cell line were fitted to scSig. Spearman correlation analysis was utilized to assess the relationship between the exposure values of scSig for each sample and IC50. The significance P values obtained were corrected using the FDR method. Finally, the R package ComplexHeatmap66 was employed to draw correlation heatmap.
Statistics and reproducibility
Unless otherwise specified, all statistical analyses and data visualizations were performed in R (v4.1.0). Correlation analyses were conducted using both Pearson’s and Spearman’s correlation coefficients, with P values adjusted for multiple testing using the false discovery rate (FDR) method. Kaplan–Meier survival curves were compared using the log-rank test. Detailed statistical methods and results are provided in the corresponding figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Only publicly available data were used in this study, and data sources and handling of these data are described in the Methods. Supplementary and intermediate data related to this study are available from the corresponding author upon reasonable request.
Code availability
All codes required to reproduce the results reported in this manuscript are freely available at: https://github.com/XSLiuLab/single-cell-CNA-signature. Further information is available from the corresponding author upon request.
References
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Poulsgaard, G. A., Sørensen, S. G., Juul, R. I., Nielsen, M. M. & Pedersen, J. S. Sequence dependencies and mutation rates of localized mutational processes in cancer. Genome Med. 15, 63 (2023).
Yang, G. et al. Mutational signature and prognosis in adenocarcinoma of the bladder. J. Pathol. 262, 334–346 (2024).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Steele, C. D. et al. Signatures of copy number alterations in human cancer. Nature 606, 984–991 (2022).
Drews, R. M. et al. A pan-cancer compendium of chromosomal instability. Nature 606, 976–983 (2022).
Tao, Z. et al. The repertoire of copy number alteration signatures in human cancer. Brief Bioinform. 24, bbad053 (2023).
Wang, S., Tao, Z., Wu, T. & Liu, X.-S. J. B. Sigflow: an automated and comprehensive pipeline for cancer genome mutational signature analysis. Bioinformatics 37, 1590–1592 (2021).
Wang, S., Jia, M., He, Z. & Liu, X.-S. J. O. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene 37, 3924–3936 (2018).
Macintyre, G. et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 50, 1262–1270 (2018).
Doran, C. G. & Pennington, S. R. J. B. I. M. Copy number alteration signatures as biomarkers in cancer: a review. Biomark Med. 16, 371–386 (2022).
Essers, P. B. et al. Ovarian cancer-derived copy number alterations signatures are prognostic in chemoradiotherapy-treated head and neck squamous cell carcinoma. Int. J. Cancer 147, 1732–1739 (2020).
Yao, H. et al. Copy number alteration features in pan-cancer homologous recombination deficiency prediction and biology. Commun. Biol. 6, 527 (2023).
Wang, S. et al. Copy number signature analysis tool and its application in prostate cancer reveals distinct mutational processes and clinical outcomes. PLoS Genet. 17, e1009557 (2021).
Nguyen, P. H. D. et al. Intratumoural immune heterogeneity as a hallmark of tumour evolution and progression in hepatocellular carcinoma. Nat. Commun. 12, 227 (2021).
Li, L. & Wang, H. J. C. l. Heterogeneity of liver cancer and personalized therapy. Cancer Lett. 379, 191–197 (2016).
Hiley, C., de Bruin, E. C., McGranahan, N. & Swanton, C. J. G. B. Deciphering intratumor heterogeneity and temporal acquisition of driver events to refine precision medicine. Genome Biol. 15, 1–10 (2014).
Lei, H. et al. Tumor copy number deconvolution integrating bulk and single-cell sequencing data. J. Comput. Biol. 27, 565–598 (2020).
Malikic, S. et al. PhISCS: a combinatorial approach for subperfect tumor phylogeny reconstruction via integrative use of single-cell and bulk sequencing data. Genome Res. 29, 1860–1877 (2019).
Graham, T. A. & Sottoriva, A. Measuring cancer evolution from the genome. J. Pathol. 241, 183–191 (2017).
Turajlic, S., Sottoriva, A., Graham, T. & Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 20, 404–416 (2019).
Gao, R. et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 48, 1119–1130 (2016).
Guo, L. et al. Single-cell DNA sequencing reveals punctuated and gradual clonal evolution in hepatocellular carcinoma. Gastroenterology 162, 238–252 (2022).
Gerstung, M. et al. The evolutionary history of 2,658. Cancers 578, 122–128 (2020).
McPherson, A. et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 48, 758–767 (2016).
CNCB-NGDC Members and Partners. Database resources of the national genomics data center, China National Center for Bioinformation in 2023. Nucleic Acids Res. 51, D18–d28, (2023).
Zhang, J. et al. The International Cancer Genome Consortium Data Portal. Nat. Biotechnol. 37, 367–369 (2019).
ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes %. J. Nat. 578, 82–93 (2020).
Goldman, M. J. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38, 675–678 (2020).
Zack, T. I. et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 (2013).
Korbel, J. O. & Campbell, P. J. J. C. Criteria for inference of chromothripsis in cancer genomes. Cell 152, 1226–1236 (2013).
Popova, T. et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 72, 5454–5462 (2012).
Al-Rawi, D. H. & Bakhoum, S. F. Chromosomal instability as a source of genomic plasticity. Curr. Opin. Genet. Dev. 74, 101913 (2022).
Menghi, F. et al. The tandem duplicator phenotype as a distinct genomic configuration in cancer. Proc. Natl. Acad. Sci. USA 113, E2373–E2382 (2016).
Kim, H. et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat. Genet. 52, 891–897 (2020).
Yi, E., Chamorro González, R., Henssen, A. G. & Verhaak, R. G. W. Extrachromosomal DNA amplifications in cancer. Nat. Rev. Genet. 23, 760–771 (2022).
Zhao, X.-K. et al. Focal amplifications are associated with chromothripsis events and diverse prognoses in gastric cardia adenocarcinoma. Nat. Commun. 12, 6489 (2021).
Koche, R. P. et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat. Genet. 52, 29–34 (2020).
Dentro, S. C. et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 184, 2239–2254.e2239 (2021).
Davoli, T. & de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 27, 585–610 (2011).
Bartenhagen, C. & Dugas, M. J. B. RSVSim: an R/Bioconductor package for the simulation of structural variations. Bioinformatics 29, 1679–1681 (2013).
Giguere, C. et al. SCSIM: jointly simulating correlated single-cell and bulk next-generation DNA sequencing data. BMC Bioinformatics. 21, 1–10 (2020).
Kaufmann, T. L. et al. MEDICC2: whole-genome doubling aware copy-number phylogenies for cancer evolution. Genome Biol. 23, 241 (2022).
Comaills, V. & Castellano-Pozo, M. J. B. Chromosomal instability in genome evolution: from cancer to macroevolution. Biology 12, 671 (2023).
Maclachlan, K. H. et al. Copy number signatures predict chromothripsis and clinical outcomes in newly diagnosed multiple myeloma. Nat. Commun. 12, 5172 (2021).
Lei, Y., Han, P. & Tian, D. Protein arginine methyltransferases and hepatocellular carcinoma: a review. Transl. Oncol. 14, 101194 (2021).
Gou, Q., He, S. & Zhou, Z. Protein arginine N-methyltransferase 1 promotes the proliferation and metastasis of hepatocellular carcinoma cells. Tumor Biol. 39, 1010428317691419 (2017).
Jeon, J.-Y. et al. Protein arginine methyltransferase 5 is implicated in the aggressiveness of human hepatocellular carcinoma and controls the invasive activity of cancer cells. Oncol. Rep. 40, 536–544 (2018).
Luo, Y. et al. Myelocytomatosis-protein arginine N-methyltransferase 5 Axis defines the tumorigenesis and immune response in hepatocellular carcinoma. Hepatology 74, 1932–1951 (2021).
Dang, C. V. MYC on the path to cancer. Cell 149, 22–35 (2012).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Genomics arXiv preprint arXiv:1303.3997 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Scheinin, I. et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 24, 2022–2032 (2014).
Poell, J. B. et al. ACE: absolute copy number estimation from low-coverage whole-genome sequencing data. Genome Res. 35, 2847–2849 (2019).
Carter, S. L. et al. Absolute quantification of somatic DNA alterations in human cancer. Bioinformatics 30, 413–421 (2012).
Wu, S. et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575, 699–703 (2019).
Wu, T. et al. Extrachromosomal DNA formation enables tumor immune escape potentially through regulating antigen presentation gene expression. Sci. Rep. 12, 3590 (2022).
Schonhoft, J. D. et al. Morphology-predicted large-scale transition number in circulating tumor cells identifies a chromosomal instability biomarker associated with poor outcome in castration-resistant prostate cancer. Cancer Res. 80, 4892–4903 (2020).
Bielski, C. M. et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 50, 1189–1195 (2018).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Schliep, K. P. J. B. phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593 (2011).
Yu, G., Lam, T. T.-Y., Zhu, H. & Guan, Y. Two methods for mapping and visualizing associated data on phylogeny using ggtree. Mol. Biol. Evol. 35, 3041–3043 (2018).
Wang, S., Liu, X.-S., Li, J. & Zhao, Q. Ezcox: an R/CRAN package for cox model batch processing and visualization. https://doi.org/10.48550/arXiv.2110.14232 (2021).
Yang, W. et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961 (2012).
Gu, Z. & Hübschmann, D. J. B. Make interactive complex heatmaps in R. Bioinformatics 38, 1460–1462 (2022).
Acknowledgements
We thank ShanghaiTech University High Performance Computing Public Service Platform for computing services. We thank multi-omics facility, molecular and cell biology core facility of ShanghaiTech University for technical help. This work is supported by National Natural Science Foundation of China (82373149), Shanghai Science and Technology Commission (24J22800700), cross disciplinary Research Fund of Shanghai Ninth People’s Hospital, Shanghai JiaoTong University School of Medicine (JYJC202227), open project fund of the National Health Commission’s key laboratory of individualized diagnosis and treatment of nasopharyngeal cancer (2023NPCCK02), startup funding from ShanghaiTech University.
Author information
Authors and Affiliations
Contributions
C.W. performed all the CNA signature analysis and drafted the manuscript under the supervision of X.-S. L. Z.T., W.C., N.W., J.W., J.S., T.W., D.Q., K.D., X.Z. T.L., L.Z., W.W., X.L., and X.L. participated in cancer genome analysis and discussion. X.X. provided critical materials for this study and participated in critical project discussion and supervision. X.-S.L. conceived this study, supervised this study and wrote the manuscript with C.W.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Ruping Sun and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Dr Kaliya Georgieva and Dr Johannes Stortz. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, C., Tao, Z., Chen, W. et al. Single-cell copy number alteration signature analysis reveals masked patterns and potential biomarkers for cancer. Commun Biol 8, 1582 (2025). https://doi.org/10.1038/s42003-025-08994-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08994-w
