
Abstract
Cardiac hypertrophy is an independent risk factor for heart failure (HF) which often leads to cardiovascular disease-related death worldwide. Here we show that the upregulated expressions of Sorting Nexin 16 (SNX16) are evident in the hypertrophic hearts. Cardiac-specific deletion of SNX16 significantly inhibited AngII-induced cardiac hypertrophy and cardiomyocytic enlargement in male mice. In addition, we observed that both AngII stimulation and SNX16 overexpression markedly enlarged cardiomyocytes and promoted EGFR transactivation, and these effects were almost completely abolished by AZD9291, an inhibitor of the EGFR pathway. SNX16 deficiency significantly inhibited AngII- or EGF-induced recycling of EGFR in endosomal trafficking in cardiomyocytes. Finally, the elevated expression of SNX16 and the phosphorylation of EGFR and Src were further confirmed in heart tissues from patients with cardiac hypertrophy. Therefore, the present study demonstrates that SNX16-mediated transactivation of EGFR plays a key role in AngII-induced cardiac hypertrophy via enhancing the recycling of EGFR.

Similar content being viewed by others

Introduction
Cardiac hypertrophy is an independent risk factor for many serious cardiovascular events1, including heart failure (HF), which is one of the leading causes of cardiovascular disease-related death worldwide2. Angiotensin II (AngII), a well-known hypertrophic stimulus, promotes the expression of hypertrophic genes such as Nppa and Nppb in cardiomyocytes, leading to cardiac hypertrophy and heart remodeling3. Transactivation of epidermal growth factor receptor (EGFR, a receptor tyrosine kinase) mediated by AngII, is considered to play a central role in cardiac hypertrophy. AngII activates EGFR transactivation through the metalloprotease ADAM17-mediated proHB-EGF shedding, in which AngII binds to its G protein-coupled receptor AT1R (AngII type 1 receptor) to activate ADAM17 via Src-mediated intracellular Ca2+/ROS and subsequently leads to EGFR transactivation and activation of downstream signaling pathways, including the Ras/ERK1/2 and Ras/PI3K/Akt/mTOR/S6 kinases, to modulate cellular growth and proliferation4. However, the underlying mechanism of EGFR transactivation in cardiac hypertrophy is still not fully understood.
Sorting nexins (SNXs) are a family of proteins containing phox-homology (PX) domains that are conserved from yeast to mammals, and 34 mammalian family members have been identified5,6. SNXs play critical roles in endocytic trafficking, including endocytosis, endosomal sorting and endosomal signaling, since the PX domain binds to phosphatidylinostil-3-monophosphates (PI3Ps), which are mostly enriched in endosomes7. More importantly, numerous studies have reported that dysfunction of sorting nexins is increasingly associated with many diseases, such as Alzheimer’s disease, carcinomas, hypertension and pathogenic infection6,8.
Sorting nexin 16 (SNX16), a member of the SNX family, contains 343 amino acids containing a PX domain, a coiled-coil (CC) domain and a C-terminal region. The PX domain is associated with membranes via interactions with PI3P in endosomes, and homo-oligomerization and higher-order assembly occur via the CC domain9,10,11. Studies have indicated that SNX16 is distributed in all three different endosomes, including early endosomes9,12,13,14, recycling endosomes12,15, and late endosomes9,13,15, and participates in intracellular protein sorting, such as trafficking of EGFR9,12, recycling of E-cadherin11, and degradation of eEF1A216. Importantly, studies have shown that abnormal SNX16 expression and function might be associated with several human diseases, such as heart failure (HF)17,18, colorectal cancer16 and membranous nephropathy19. Moreover, a large-scale genome-wide investigation revealed that a SNP adjacent to the SNX16 gene was strongly associated with incident heart failure, in which the SNP is located on chromosome 8q21.1, variant G → A, 918.3 kb from SNX1618. It has been reported that the expression of Snx16 mRNA is increased in heart tissues from HF patients17. However, the role of SNX16 in cardiac hypertrophy and heart failure remains to be identified.
This study aimed to investigate the role of SNX16 in cardiac hypertrophy and the underlying mechanisms. Our results showed that SNX16 is upregulated during AngII-induced enlargement of cardiomyocytes both in vitro and in vivo. Heart-specific deletion of Snx16 gene significantly reduced AngII-induced cardiac hypertrophy and EGFR transactivation in mice, whereas SNX16 overexpression aggravated hypertrophy, enhanced EGFR recycling to the cellular membrane, and activated EGFR-mediated Erk1/2 signaling pathways in cardiomyocytes in vitro; these effects were reversed by AZD9291, an EGFR tyrosine kinase inhibitor (TKI). Finally, our study confirmed that SNX16 expression was elevated in cardiac tissues from patients with cardiac hypertrophy. Therefore, we demonstrated that SNX16 is essential for AngII-induced cardiac hypertrophy by promoting EGFR transactivation in cardiomyocytes both in vivo and in vitro.
Results
SNX16 expression was upregulated in cardiac tissues from mice with AngII-induced cardiac hypertrophy
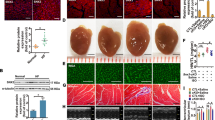
To explore the roles of SNX16 in the heart, we investigated the expression of SNX16 in multiple tissues of mice. The results showed that Snx16 was widely expressed in the mouse heart, liver, brain, spleen, lung, etc. (Supplementary Fig. 2A, B). Additionally, the protein levels of SNX16 gradually increased in mouse hearts with aging (Supplementary Fig. 2C). Next, we induced cardiac hypertrophy in WT mice by TAC or subcutaneous infusion of 2 μg/kg/min AngII for 2 weeks and examined the expression of SNX16. To minimize gender-associated confounding factors and ensure robust experimental reproducibility, male mice were employed as experimental subjects in the present study. As shown in Fig. 1A and Supplementary Fig. 2D, TAC and AngII infusion successfully induced cardiac hypertrophy in mice with significant increases in the HW/BW ratio compared with those in the saline group. In addition, the mRNA and protein expression levels of SNX16 were markedly enhanced in the hypertrophic hearts of the mice (Fig. 1B, C). And we also observed strong correlation of upregulation of SNX16 and enhanced EGFR activation in remodeling hearts with cardiac fibrosis and cardiac dysfunction (Suppementary Fig. S1). To further confirm in vivo results, we examined the SNX16 expression in primary neonatal rat ventricular myocytes (NRVMs) and the rat cardiomyocyte cell line H9c2 after AngII stimulation. The results revealed that AngII stimulation (200 nmol/L for 48 h) led to significant enlargement of cardiomyocytes, accompanied by a notable increase in the expression of SNX16 and hypertrophic genes such as Nppa and Nppb in both NRVMs (Fig. 1D–F) and H9c2 cells (Fig. 1G–I). These results strongly indicated that there was a close association between elevated SNX16 expression and AngII-induced cardiac hypertrophy both in vivo and in vitro, suggesting that SNX16 may play a key role in AngII-induced cardiac hypertrophy.

A Models of cardiac hypertrophy were generated in C57Bl/6 mice by infusion of AngII (2 μg/kg/min) for 2 weeks or by TAC surgery, and the heart weight/body weight (HW/BW) ratio was measured in the mice (n = 8–10). These p-values are from unpaired t-tests, ***p < 0.001. B Expression of the SNX16 protein in the hearts of mice was determined by western blot analysis; n = 3–4. These p-values are from unpaired t-tests, **p < 0.01, ***p < 0.001. C Expression of Snx16 mRNA in the hearts of mice was quantitatively determined by RT‒PCR; n = 6. The p-value is from unpaired t-tests, ***p < 0.001. D, G The surface areas of neonatal rat ventricular myocytes (NRVMs) and H9c2 cells were determined by immunofluorescence. The cells were treated with 200 nmol/L AngII or saline for 48 h, stained with an anti-α-actinin antibody (red) and counterstained with DAPI (blue). Scale bar: 20 μm. cells ≥50 from three independent experiments. These p-values are from unpaired t-tests, **p < 0.01. E, H Expression of the SNX16 protein in NRVMs and H9c2 cells stimulated with AngII or saline for 48 h was determined by western blot analysis (n = 4). These p-values are from unpaired t-tests, *p < 0.05, ***p < 0.001. F, I The mRNA expression levels of Nppa, Nppb and Snx16 were determined by RT‒PCR in NRVMs and H9c2 cells stimulated with AngII-stimulation or saline for 48 h (n = 3). These p-values are from unpaired t-tests, *p < 0.05, ***p < 0.001. The data are presented as the means ± SEMs, and three independent experiments were performed.
Cardiac-specific SNX16 deletion attenuated AngII-induced cardiac hypertrophy in mice
To further assess the role of SNX16 in cardiac hypertrophy, we generated cardiac-specific SNX16 knockout (SNX16CKO) mice by mating SNX16fl/fl mice with cardiac-specific MLC2v-Cre mice (Fig. 2A). The results showed that the growth and reproducibility of the SNX16CKO mice were comparable to those of the WT mice, as confirmed by PCR (Fig. 2B) with the primers listed in Supplementary Table 2. Additionally, the mRNA (Fig. 2C) and protein (Fig. 2D, E) expression of Snx16 decreased by 45.5% and 76.3%, respectively, in the heart tissues of the SNX16CKO mice compared with those of the control mice (WT), indicating that SNX16 cardiac-specific knockout mice were successfully constructed. However, SNX16 deficiency significantly protected mice from AngII-induced cardiac hypertrophy, as indicated by decreases in the HW/BW and LW/BW ratios, improvements in cardiac morphology, as shown by HE staining, and reductions in the cell area, as shown by wheat germ agglutinin (WGA) staining (Fig. 2F–K). In addition, SNX16 deficiency markedly improved Ang II infusion-induced cardiac dysfunction, as indicated by changes in the ejection fraction (EF) and fractional shortening (FS) and reduced the mean blood pressure (Supplementary Table 3 and Supplementary Fig. 3). These results indicate that cardiac-specific deletion of SNX16 significantly attenuated AngII-induced cardiac hypertrophy and improved cardiac function in vivo.

A A schematic diagram of generating heart-specific SNX16 knockout mice. B Representative PCR genotyping was determined by PCR with mouse tail genomic DNA. A 400 bp fragment for wild type C57Bl/6, a 458 bp fragment for SNX16Fl/Fl, and a 400 bp fragment for Cre were amplified by PCR. C Expression of Snx16 mRNA in the hearts of WT (f/f-) and SNX16CKO (f/f Cre+) mice was determined by RT‒PCR; n = 4‒5. The p-value is from unpaired t-tests, **p < 0.01. D, E Expression of SNX16 protein in the hearts of WT and SNX16CKO mice was detected by western blot analysis (n = 3). The p-values is from unpaired t-tests, *p < 0.05. F Images of hearts from WT and SNX16CKO mice with or without AngII infusion. Scale bar: 5 mm. G, H Ratios of HW/BW and LW/BW were obtained for WT and SNX16CKO mice, n = 6. These p-values are from unpaired t-tests and one way-ANOVA, *p < 0.05, ***p < 0.001. I Heart tissues with transverse sections were analyzed by H&E staining. Scale bar: 1 mm. n = 3. J, K Representative images and summary data were analyzed with wheat germ agglutinin (WGA) staining. Scale bar: 50 μm. Mice, n = 3; cells ≥50 from three independent experiments. These p-values are from unpaired t-tests and one way-ANOVA, *p < 0.05. The data are presented as the means ± SEMs.
SNX16 deficiency alleviated AngII-induced cardiac hypertrophy by suppressing the EGF/EGFR signaling pathway
AngII-induced transactivation of EGFR is a critical step in the activation of Ras/ERK cascade-mediated cardiac hypertrophy20,21. To determine whether SNX16 modulates EGFR signaling in cardiac hypertrophy, the expression of EGFR and its downstream effectors, such as Erk1/2, was examined in the hearts of AngII-stimulated SNX16CKO mice. As shown in Fig. 3A, B, SNX16 deficiency significantly inhibited AngII-induced autophosphorylation of EGFR (pTyr 1068), phosphorylation of Erk1/2, and expression of hypertrophic gene Nppb in newborn mouse primary cardiomyocytes, suggesting that SNX16 deficiency attenuated AngII-induced cardiac hypertrophy by suppressing EGFR transactivation and its downstream signaling pathways.

A, B The protein expression levels of SNX16, p-EGFR, p-ERK1/2 and BNP in the hearts of WT and SNX16CKO mice treated with or without AngII infusion were determined by western blot analysis (n = 3). These p-values are from unpaired t-tests and one way-ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001. C, D Surface areas of H9c2 cells were determined by immunofluorescence analysis with phalloidin (red) and counterstained with DAPI (blue) with EGF (1 μg/mL, C) or AngII (200 nmol/L, D) stimulation for 24 h with or without pretreatment with AZD9291 (100 nmol/L). Scale bar: 50 μm. Quantitative surface area of H9c2 cells: μM2, cells ≥50. These p-values are from unpaired t-tests and one way-ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001. The data are presented as the means ± SEMs, and three independent experiments were performed.
To further clarify the role of the EGFR signaling pathway in cardiac hypertrophy, the effects of EGF and Ang II on cardiomyocytes were examined in H9c2 cells. Our results revealed that both EGF and AngII effectively induced myocyte enlargement, which was nearly completely abolished by AZD9291, an EGF antagonist (Fig. 3C, D), suggesting that the activation of the EGF/EGFR-mediated cascade might be responsible for AngII-induced hypertrophy.
SNX16 overexpression-induced enlargement of cardiomyocytes was associated with EGFR transactivation
To clarify whether SNX16 was associated with EGFR transactivation, the effects of SNX16 overexpression on cardiomyocytes were investigated in vitro. SNX16 was overexpressed in NRVMs and H9c2 cells by transient transfection of the pCMV6-SNX16-GFP (SNX16-OE) or pCMV6-AC-GFP (as the vehicle) plasmid, respectively. The results showed that SNX16 overexpression significantly enlarged both NRVMs and H9c2 cells (Supplementary Fig. 4A, B), elevated the expression of hypertrophic genes such as Nppa and Nppb, and activated the EGFR/ERK1/2 signaling pathway (Fig. 4A–D). Importantly, the hypertrophic effect was markedly reversed by AZD9291, an EGFR antagonist (Fig. 4A, B). Next, the effects of SNX16 overexpression on EGFR transactivation were examined in vitro. The results showed that the expression of SNX16 in the SNX16-OE group was much greater than that in the control group, and the autophosphorylation of EGFR (pTyr1068) and phosphorylation of Erk1/2 were significantly elevated in cardiomyocytes overexpressing SNX16 (Fig. 4C, D); moreover, autophosphorylation/phosphorylation was markedly alleviated by AZD9291 in a dose-dependent manner (Fig. 4E, F). In addition, the interaction between SNX16 and EGFR was further confirmed using SNX16-Flag-overexpressing H9c2 cells stimulated with 200 nmol/L AngII for 6 h (Fig. 4G). These results demonstrated that activation of the EGFR signaling pathway plays a key role in SNX16 overexpression-induced cardiomyocyte hypertrophy, indicating that SNX16-mediated EGFR transactivation is associated with cardiac hypertrophy.

A SNX16 overexpression-induced enlargement of cardiomyocytes was determined by immunofluorescence analysis of H9c2 cells pretreated with or without AZD9291 (100 nmol/L) for 4 h, followed by staining with phalloidin (red) and DAPI (blue), and the surface area of H9c2 cells was quantified. Scale bar: 20 μm, ≥50 cells from three independent experiments. These p-values are from unpaired t-tests and one way-ANOVA, **p < 0.01. B BNP expression induced by SNX16 overexpression or EGF stimulation (1 μg/mL for 24 h) was examined by Western blot analysis in H9c2 cells pretreated with AZD9291 (100 nmol/L) for 4 h or 30 min, respectively. These p-values are from unpaired t-tests and one way-ANOVA, *p < 0.05. C The expression levels of SNX16, p-EGFR, p-ERK1/2, and BNP were determined by western blot analysis in H9c2 cells transfected with SNX16-OE vector for 48 h. n = 3. D Quantification of immunoblots in (C). These p-values are from unpaired t-tests, *p < 0.05, ***p < 0.001. E Phosphorylated and total EGFR and ERK1/2 proteins were detected by western blot analysis in H9c2 cells transfected with SNX16-OE vector for 48 h and then treated with AZD9291 (100 nmol/L) for 4 h. F Quantification of immunoblots in (E). These p-values are from unpaired t-tests and one way-ANOVA, *p < 0.05, **p < 0.01. G Interaction between SNX16 and EGFR in H9c2 cells was determined by a coimmunoprecipitation assay. Three independent experiments were performed. The data are presented as the means ± SEMs.
SNX16 selectively promoted the recycling of EGFR in recycling endosomes in cardiomyocytes
The turnover of the membrane receptor tyrosine kinase (RTK) EGFR is tightly related to its endocytic trafficking22. Rab5, Rab7 and Rab11 are markers of endocytic trafficking in early endosomes, late endosomes and recycling endosomes, respectively. A study showed that SNX16 regulated EGFR trafficking by interacting with its phox domain and phosphatidylinositol-3-phosphate (PI3P) in endosomes12. To determine whether SNX16 plays a role in EGFR endocytic trafficking in cardiomyocytes, the cellular endosomal location of SNX16 was determined in three different endosomes colabeled with Rab5 (an early endosome marker), Rab7 (a late endosome marker) and Rab11 (a recycling endosome marker) in H9c2 cells under EGF stimulation. SNX16 was mainly observed in late endosomes (Rab7) under normal conditions; however, it was enriched in early endosomes (Rab5) and recycling endosomes (Rab11) but scarcely in late endosomes (Rab7) when the cells were treated with 1 μg/mL EGF for 15 min (Fig. 5A–C). Overexpression of SNX16 significantly enhanced autophosphorylation of EGFR (pTyr 1068) in H9c2 cells, and this increase in phosphorylation was markedly abolished by wortmannin, a nonselective inhibitor of the PI3K family (Fig. 5D, E), suggesting that SNX16 may bind to the PI3P of endosomes via its PX domain to further promote the recycling of EGFR. In addition, the fluorescence intensity of p-EGFR was significantly increased around 5 min, then decreased subsequently by 30 min post Ang II stimulation (Supplementary Fig. 5A). In the meantime, SNX16 signals were increased and partially co-localized with EGFR and Rab11 for around 30 min and SNX16 might not co-expressed with Rab7 in cardiomyocytes (Supplementary Fig. 5B, C). These results suggested that SNX16 was involved in AngII-mediated EGFR signaling and EGFR trafficking by promoting the recycling of EGFR in recycling endosomes in hypertrophic cardiomyocytes.

A–C The colocalization of SNX16 and different endosome markers in H9c2 cells treated with EGF (1 μg/mL) or PBS for 15 min was determined by confocal microscopy analysis, and the cells were then stained with anti-SNX16 (green), anti-Rab5 (red), anti-Rab7 (red), and anti-Rab11 (red) antibodies and counterstained with DAPI (blue). Scale bar: 10 pixels. D The expression of EGFR and p-EGFR was detected by immunofluorescence analysis in H9c2 cells transfected with the SNX16-OE vector or vehicle for 48 h and then treated with or without wortmannin (100 nmol/L) for 30 min. The cells were then stained with anti-p-EGFR (red) or anti-EGFR (red) and counterstained with DAPI (blue). Scale bar: 20 μm. E Quantification of the mean fluorescence intensity of p-EGFR in (D). These p-values are from unpaired t-tests and one way-ANOVA, *p < 0.05. The data are presented as the means ± SEMs, and three independent experiments were performed.
SNX16 deficiency inhibited AngII-mediated activation of Src to further alleviate AngII-induced cardiac hypertrophy
Src, a member of the nonreceptor tyrosine kinase family, plays a pivotal role in AngII-induced EGFR transactivation in cardiac hypertrophy, in which Src is not only responsible for ADAM17 (a disintegrin and metalloproteinase 17)-mediated HB-EGF (heparin-binding EGF) shedding and subsequent EGFR transactivation but also directly induces the phosphorylation of EGFR at Tyr845 to activate EGFR cascades23. Therefore, we examined whether SNX16 contributed to the AngII-induced activation of Src and downstream signaling pathways in neonatal mouse ventricular myocytes (NMVMs) from SNX16CKO mice stimulated with AngII. As shown in Fig. 6A, B, AngII significantly enhanced the phosphorylation of Src at Tyr416, a site phosphorylated by Src, and inhibited the phosphorylation of Src at Tyr527, a site phosphorylated by Src, whereas SNX16 deficiency reversed AngII-induced Src activation, although AngII did not alter the mRNA expression of Egfr or Src (Supplementary Fig. 6A, B), suggesting that SNX16 may contribute to AngII-induced Src activation. In addition, the effects of SNX16 overexpression on Src activation were also examined in H9c2 cells. Fluorescence staining revealed that the overexpression of SNX16 did not increase the expression of Src but significantly elevated the phosphorylation of Tyr416 of Src and inhibited the phosphorylation of Tyr527 of Src (Fig. 6C–F). Furthermore, SNX16 deficiency markedly inhibited AngII-induced phosphorylation of EGFR at Tyr845 (Fig. 6G, H), suggesting that SNX16 deficiency-mediated inhibition of AngII-induced Src activation partially protected mice from cardiac hypertrophy by suppressing Src-mediated proHB-EGF shedding and direct phosphorylation of EGFR in cardiomyocytes. These results demonstrated that AngII stimulation or overexpression of SNX16 activated Src, which contributes to EGFR transactivation in cardiac hypertrophy.

A The phosphorylation of p-Src (Y416) and p-Src (Y527) and the expression of Src were determined by Western blot analysis in neonatal mouse cardiomyocytes from WT and SNX16CKO mice under AngII stimulation (200 nmol/L) for 48 h. B Quantification of immunoblots in (A), n = 3. The p-values are from unpaired t-tests and one way-ANOVA, *p < 0.05. Data are presented as the means ± SEMs. C–E The effects of SNX16 overexpression on the expression of Src, p-Src (Y416) and p-Src (Y527) were determined by immunofluorescence analysis in H9c2 cells transfected with vehicle or the SNX16-OE vector for 48 h and then stained with antibodies against SNX16 (red), Src (green), p-Src (Y416) (green), and p-Src (Y527) (green) and counterstained with DAPI (blue). Scale bar: 20 μm. F The mean fluorescence intensities of Src, p-Src (Y416), and p-Src (Y527) were analyzed. G The expression of p-EGFR (Y845) in the heart tissues of WT and SNX16CKO mice treated with or without AngII infusion for 2 weeks was determined by immunohistochemistry (n = 7). These p-values are from unpaired t-tests and one way-ANOVA, ***p < 0.001. H Quantification of p-EGFR (Y845) in (G). These p-values are from unpaired t-tests and one way-ANOVA, ***p < 0.001. The data are presented as the means ± SEMs. I SNX16 expression in heart tissues from patients with cardiac hypertrophy or control people without cardiac hypertrophy was determined by western blot analysis (n = 6). J Expression of p-EGFR, EGFR, p-Src (Y416), p-Src (Y527), Src, and BNP in heart tissues from patients with cardiac hypertrophy or healthy people, n = 6. K, L Quantification of immunoblots in (I and J). These p-values are from unpaired t-tests and one way-ANOVA, * p < 0.05, **p < 0.01. The data are presented as the means ± SEMs.
The expression of SNX16 was upregulated in hypertrophy hearts along with increased EGFR phosphorylation and Src activation
To further confirm the role of SNX16 in cardiac hypertrophy, its clinical relevance was investigated and compared between hypertrophy heart samples from the patients with a ventricular septum thickness of >11 mm and non-hypertrophic ones in which septum thickness is ≤11 mm based on echocardiographic findings. The human myocardium samples were kindly provided by Dr. Ji’s group, in which the heart samples were obtained during human cardiac valve replacement surgeries and the study protocol was approved by the ethics review committee of the First Affiliated Hospital of Nanjing Medical University (approval no. 2019-SR-300)24. As shown in Fig. 6I, K, the protein expression of SNX16 was significantly greater in human hypertrophic cardiac tissues than in control cardiac tissues from patients without cardiac hypertrophy. In addition, consistent with the findings in the animal model, the p-EGFR/EGFR ratio and BNP expression were also enhanced in human hypertrophic cardiac tissues (Fig. 6J, L). Furthermore, the phosphorylation of Src (p-Src-Y527) was significantly lower in hypertrophic tissues than in control heart tissues, although there was no difference in the phosphorylation of Src (p-Src-Y416) between the two groups (Fig. 6J, L), suggesting that Src was activated in hypertrophic tissues in patients with cardiac hypertrophy.
Discussion
This study showed that SNX16 deletion significantly restrained AngII-induced cardiac hypertrophy in SNX16CKO mice. To the best of our knowledge, this is the first study to address the roles and underlying mechanisms of SNX16 in cardiac hypertrophy. SNX16, a type of sorting nexin (SNX), is widely expressed in a variety of tissues and cells9 and plays an important role in various biological processes, such as trafficking of EGFR9,12, recycling of E-cadherin11, and degradation of eEF1A216. GWAS data showed that Snx16 was located in one of the top loci associated with heart failure (HF)18, and the expression of Snx16 mRNA was highly increased in end-stage failing heart tissues17. AngII-induced cardiac hypertrophy is involved in the activation of multiple receptor transactivation cascades, including EGFR, PDGFR and IGF-IR transactivation, among which EGFR transactivation has been revealed to be a central player in mediating the hypertrophic response via GPCR stimulation and activation25. In the present study, we observed that AngII significantly promoted the expression of SNX16 in cardiac tissues and cardiomyocytes both in vivo and in vitro, suggesting that SNX16 plays a role in AngII-induced cardiac hypertrophy. To clarify the role and mechanism of SNX16 in the heart, mouse models of cardiac hypertrophy were induced by AngII infusion in cardiac-specific SNX16 knockout (SNX16CKO) mice. Our results showed that cardiac-specific deletion of SNX16 markedly alleviated AngII-induced cardiac hypertrophy by suppressing autophosphorylation of EGFR and the EGFR signaling cascades in vivo. In addition, the cardiomyocytic hypertrophy induced by both EGF and AngII stimulation was almost completely abolished by AZD9291, an inhibitor of the tyrosine kinase EGFR. Importantly, overexpression of SNX16 increased the surface area of myocytes and promoted EGFR transactivation in NRVMs and H9c2 cells, and these effects were also significantly attenuated by AZD9291. Furthermore, we also showed that SNX16 interacts with EGFR. These results strongly indicated that the inhibitory effect of SNX16 deletion on AngII-induced cardiac hypertrophy was associated with the inhibition of EGFR transactivation.
Endocytic trafficking is the dynamic process for regulating the signaling of the membrane receptor EGFR, in which the endocytosis and post endocytic sorting of EGFR are dependent upon its internal tyrosine kinase activities and downstream signaling. Studies have indicated that SNX16 is distributed in endosomes, including early endosomes9,12,13,14, recycling endosomes12,15, and late endosomes9,13,15, and plays a critical role in various biological processes, including protein sorting and trafficking and signal transduction. It has been reported that SNX16 regulates EGFR trafficking by interacting with its PX domain and phosphatidylinositol-3-phosphate (PI3P) in endosomes12. The PX-CC unit of SNX16 forms a unique shear-shaped homodimer, and the structure contains a PI3P binding pocket11.
Rab5, Rab7 and Rab11 are markers of endocytic trafficking for three different functional endosomes; Rab5, an early endosome marker, is located in the plasma membrane and early endosomes and functions as a key regulator of vesicular trafficking during early endocytosis26; Rab7, a late endosome marker, is associated with membrane trafficking from early to late endosomes and lysosomes27 and regulates growth receptor endocytic trafficking and degradation28; and Rab11, a recycling endosome marker, functions as a key regulator in the recycling of perinuclear, plasma membrane and Golgi compartment endosomes29. Studies have shown that endocytic internalization of EGF-bound EGFR is significantly accelerated upon EGFR activation, mainly through clathrin-mediated endocytosis (CME) and clathrin-independent endocytosis (CIE). CME is the rate-limiting high-affinity pathway for internalizing active EGFR into clathrin-coated pits (CCPs) that enter into early Rab5+ endosomes22. The recruitment of EGFR to CCPs might be regulated by redundant pathways, either through direct interaction of the clathrin adaptor AP-2 or through Cbl-mediated ubiquitination. Then, endocytic EGFR is further sorted to Rab7+late endosomes via ubiquitinated ILV-MVEs for lysosomal degradation or recycling back to the plasma membrane via Rab4+ or Rab11+recycling endosomes for nonubiquitinated EGFR22. However, the molecular mechanisms of endocytosis and subsequent sorting to recycle or degrade EGFR are not well established30. To clarify the role of SNX16 in EGFR trafficking in cardiac hypertrophy, we detected the cellular localization of SNX16 colabeled with Rab5, Rab7 and Rab11 in cardiomyocytes after EGF stimulation. Our data showed that SNX16 was mainly localized in Rab5+ and Rab11+ areas but scarcely in Rab7+ areas after EGFR activation. Choi et al.12 reported that SNX16 was enriched in the early endosome (EEA1) and recycling endosome (TRF), promoted EGFR degradation and inhibited the activation of ERK1/2 in SNX16-overexpressing COS-7 cells. We observed that overexpression of SNX16 in cardiomyocytes promoted EGFR intracellular recycling and subsequent activation of ERK1/2, strongly suggesting that SNX16 might have different regulatory effects on cells of different origins. In the present study, immunofluorescence staining revealed that SNX16 bound mainly to early and recycling endosomes upon stimulation with EGF but bound to later endosomes in the absence of EGF. These findings suggested that SNX16 might facilitate EGFR trafficking to recycling endosomes for recycling under its activation by EGF in cardiomyocytes.
Class I PI3K is often activated by RTK-EGFR and phosphorylates PtdIns(4, 5)P2 to PtdIns(3, 4, 5)P3 and regulates diverse cellular activities, including cell growth, DNA synthesis, survival, and cytoskeletal organization, in cardiac hypertrophy31. Wortmannin, a nonselective inhibitor of PI3K, abolished the endosomal localization of SNX1612. Our results showed that wortmannin-mediated inhibition of PI3K activity significantly suppressed the internalization of p-EGFR and inhibited the cellular peripheral localization of SNX16, further demonstrating that SNX16 enhanced the EGFR signaling pathway by promoting the endocytic recycling of EGFR in cardiomyocytes. In addition, we observed that overexpression of SNX16 promoted the autophosphorylation of EGFR, which was inhibited by wortmannin, suggesting that SNX16 may bind to PI3P in endosomes via its PX domain to further promote the recycling of EGFR. These results indicated that SNX16 mainly regulated EGFR trafficking by promoting the recycling of EGFR in recycling endosomes in AngII-induced cardiac hypertrophy.
Studies have indicated that AngII-induced EGFR transactivation involves binding to its receptor, GPCR-AT1R, to induce hypertrophic responses such as cardiomyocyte growth, fetal gene expression and/or cytoskeletal reorganization, leading to cardiac hypertrophy32. Src, a member of the nonreceptor tyrosine kinase family, plays a pivotal role in EGFR transactivation in AngII-induced cardiac hypertrophy, in which Src is not only responsible for ADAM17 (a disintegrin and metalloproteinase 17)-mediated HB-EGF (heparin-binding EGF) shedding and subsequent EGFR transactivation33,34 but also directly induces the phosphorylation of EGFR at Tyr845 to activate EGFR cascades23. EGFR exists on the cell surface as a monomer in the absence of ligand, whereas ligand-bound EGFR leads to homo or heterodimerization, internal tyrosine kinase activation, and autophosphorylation of intracytoplasmic tyrosine residues, participating in cell proliferation, survival, differentiation, migration, inflammation, and matrix homeostasis by communicating with downstream effectors such as the EKR, PI3K/AKT and mTOR pathways35,36. Activation of EGFR is essential for normal cardiac development and involves cardiac remodeling, blood pressure regulation, endothelial dysfunction, neointimal hyperplasia, and atherogenesis36. In the present study, we demonstrated that SNX16 deficiency inhibited AngII-induced Src activation, suggesting that SNX16 may contribute to AngII-induced Src activation. In addition, SNX16 deficiency markedly inhibited AngII-induced phosphorylation of EGFR at Tyr845, suggesting that SNX16 deficiency-mediated inhibition of AngII-induced Src activation partially protected mice from cardiac hypertrophy by suppressing proHB-EGF shedding and directly phosphorylating EGFR in cardiomyocytes.
To further confirm the role of SNX16 in cardiac hypertrophy, its clinical relevance was investigated in patients with cardiac hypertrophy. Our study confirmed the elevated expression of SNX16 and the elevated phosphorylation of Src in heart tissues from patients with cardiac hypertrophy, and strong correlation between upregulation of SNX16 and enhanced EGFR activation in remodeling hearts with cardiac fibrosis and cardiac dysfunction in mice. Therefore, it raises concerns in the importance of SNX16 in cardiovascular disease, including cardiac hypertrophy and heart failure.
In conclusion, our findings revealed an unexplored interaction between SNX16-mediated EGFR transactivation and AngII-induced cardiac hypertrophy (Fig. 6M). We demonstrated that the deletion of SNX16 in the heart reduces cardiac hypertrophy by interrupting the EGFR signaling pathway. Conversely, overexpression of SNX16 enhances the EGFR signaling pathway by promoting endocytic recycling of EGFR, ultimately aggravating cardiac hypertrophy. Targeting SNX16 may be a promising strategy for the prevention and treatment of cardiac hypertrophy and remodeling.
Materials and methods
Animal models
Animal maintenance and experimental procedures were carried out at the Transgenic SPF Animal Center in Institute of Translational Medicine in Nanchang University in accordance with the Guide for the Care and Use of Laboratory Animals in China and were reviewed and approved by the Animal Ethics Committee of Nanchang University (Approval No. NCULAE-20221228062). SNX16flox/flox mice were generated with CRISPR/Cas9 genomic editing technique, in which the exon 2 of Snx16 gene was flanked by loxP sites in C57BL/6 mice. SNX16flox/floxMlc-2vCre (Cardiac-specific SNX16 knockout, termed SNX16CKO) mice were obtained by mating SNX16flox/flox mice with Mlc-2vCre mice (C57BL/6 background), the littermates of SNX16flox/flox were used as wild type controls. Eight to twelve weeks old male mice were used in this study to avoid potential effects of sex differences. The animal experiments were conducted under anesthesia induced by a single administration of 1% isoflurane (RWD, r510-22) via respiration and carbon dioxide inhalation was utilized for euthanasia or specifically indicated. Cardiac hypertrophy was induced by transverse aortic constriction (TAC) for two weeks or AngII infusion with osmotic pumps (Alzet Model 2002) which were implanted in the dorsum subcutaneous pouch in mice, in which the mice were infused with human AngII (MCE, hy-13948) at 2000 ng/kg/day for 14 days or with 0.9% NaCl at a rate of 0.01 μL/min in control mice.
The TAC surgery was carried out according to the protocol37. In brief, the mice were anesthetized with 2–3% isoflurane via inhalation. The aorta was exposed near the left second and third ribs without opening the chest. Then, the aorta was tied with 27G shim needle with a 6-0 nonabsorptive silk thread. After the surgery, the animals were placed in ventilated cages individually and minotored with self-breathing and movements for 24–48 h. Long-term observations were conducted on the TAC mouse models with the observation period extended to 90 days after surgery to ensure cardiac remodeling (Supplementary Fig. 1).
Transthoracic echocardiography was performed in mice at the end of hypertrophy or TAC modeling under isoflurane anesthesia with a Vevo 3100 imaging system (Canada) as reported38. B-mode and M-mode images were obtained, and the parameters of cardiac functions were collected and analyzed to evaluate cardiac function.
At the end of animal modeling, the mice were anesthetized with 1% isoflurane and then the hearts were isolated, weighted and the mRNA and protein were extracted with TRIzol reagent (Thermo Fisher Scientific, USA) or lysis buffer (RIPA), respectively.
Isolation of primary cardiomyocytes
Neonatal ventricular myocytes of rat (NRVMs) or mouse (NMVMs) were isolated from hearts of 1- to 3-day-old Sprague Dawley rats or C57BL/6 mice, respectively, as described39. Notably, mouse or rat neonates were euthanized by decapitation with surgical scissors. Briefly, neonatal hearts were digested with 0.08% trypsin (Gibco, 27250-018) and 0.05% collagenase II (Sigma, C6885) in a shaking water bath (Magnetic Stirrer) at 37 °C, 750 rpm, 10 min for 4–5 times and neutralized with 10% FBS in DMEM. Cardiomyocytes were collected after pre-seeding the isolated cells in an uncoated dish to remove cardiac fibroblasts at 37 °C for 60–90 min in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% FBS, penicillin (100 U/mL, Gibco) and streptomycin (100 μg/mL, Gibco), and 5-bromo-2-deoxyuridine (BrdU, 100 μmol/L, Sigma) were added to prevent overgrowth of rapidly proliferating fibroblasts, at 37 °C with a circumstance of 5% CO2.
Cell culture, transfection and treatment
Rat cardiomyocytes (H9c2) were cultured in DMEM supplemented with 10% FBS (Gibco) and 1% penicillin/streptomycin in humidified chambers at 37 °C, 5% CO2 incubator. pCMV6-AC-GFP vector (vehicle) was purchased from OriGene Technologies, Inc (PS100010) and pCMV6-SNX16-GFP overexpression vector (SNX16-OE) or SNX16-Flag vector was constructed by inserting SNX16-ORF into PCMV6-AC-GFP or pCMV6-Flag vector and confirmed by sequencing. Cardiomyocytes were transfected with the plasmids following Lipofectamine® 3000 reagents’ instructions (Thermo Fisher Scientific) and cultured for 8–12 h, then treated with or without AZD9291 (MCE, HY-15772) for 4 h in serum-free (SF) DMEM and collected after 200 nmol/L AngII (MCE, USA) stimulation or saline for 48 h.
Western blot analysis
Approximately 40 μg of total proteins from mouse heart tissues, and 20 μg from neonatal rat ventricular myocytes (NRVMs), neonatal mouse ventricular myocytes (NMVMs), or H9c2 cells were each separated on a 12% SDS - PAGE gel per lane and then transferred to an NC membrane. Human heart proteins were kindly provided by Dr. Ji’s group and the related information about donors was described24. After blocking with 5% skim milk or 5% BSA, the proteins were incubated with diluted primary antibodies at 4 °C overnight. These included SNX16 (Novus, NBP1 - 85975, 1:1000), Src antibody kit (CST, #9935T, 1:1000), EGFR (Abcam, ab52894, 1:1000), p - EGFR (CST, #3777, 1:1000), p - EGFR845 (Santa Cruz, sc - 57542, 1:1000), ERK1/2 (CST, #4695, 1:1000), p - ERK1/2 (CST, #4370, 1:1000), BNP (Santa Cruz, sc - 67455, 1:1000), Tubulin (Santa Cruz, sc - 398103, 1:1000), and GAPDH (Santa Cruz, sc - 47724, 1:1000). Subsequently, they were incubated with secondary antibodies, such as Goat Anti - Rabbit IgG H&L (HRP) (Abcam, ab6721, 1:5000) or Rabbit Anti - Mouse IgG H&L (HRP) (Abcam, ab6728, 1:5000) at room temperature for 1 h. Immunoblotting images were captured using the Molecular Imager Chemi Doc XR+ System (Bio - Rad).
Quantitative RT-PCR analysis
Total tissue RNAs were extracted with TRIzol reagent (Invitrogen) and reversely transcribed into cDNA with Transcriptor First Strand cDNA Synthesis Kit (Roche, Diagnostics, Mannheim, Germany). Relative expressions of specific genes were evaluated using quantitative real-time PCR with SYBR Green (Roche). Expression levels were normalized to GAPDH expressions. Primers of real-time RT-PCR are listed in Supplementary Table 1.
Immunofluorescent staining
Cardiomyocytes were cultured on coverslips and fixed with 4% paraformaldehyde for 10 min, followed by permeabilization with 0.25% Triton X-100 for 5 min. The cells were then blocked with 1–3% BSA in PBS at 37 °C for 1 h. After blocking, they were incubated with primary antibodies (1:50 -1:200 dilution) at 4 °C overnight, followed by incubation with secondary antibodies (1:100 dilution, Alexa Fluor 488, 594 nm) at 37 °C for 1 h. TSA fluorescent dye lablings (AiFang biological, Cat. # AFIHC025) were performed with corresponding TSA dyes as TYR-690 dye for P-EGFR and Rab11, TYR-520 dye for EGFR and Rab5, TYR-570 dye for SNX16, and TYR-620 dye for Rab7. Nuclei were counterstained with DAPI for 1 min. Fluorescent images were captured using an Olympus IX81 fluorescent microscope (Japan) or confocal microscope (ZEISS LSM800). The surface area of over 50 cardiomyocytes per group or the flurrescent signals from at least 5 different photo images were analyzed with Image J sofeware and compared between the AngII treatment and control groups. Each experiment was repeated three times. Primary antibodies include the following: anti-α-actinin (Sigma, A7811), SNX16 (Nouvs, NBP1-85975, 1:100), p-EGFR (CST #3777), EGFR (CST #4267), Rab5 (CST #3547), Rab7 (CST #9367) and Rab11 (CST #5589).
Co-immunoprecipitation
H9c2 cells were transfected with flag-tagged SNX16-OE (4 μg) at around 80% confluency in 10 cm culture dish for 24 h and serum starved for 16 h, then treated with 1 μg/mL EGF for 15 min. The cells were washed with cold PBS and lysed in 1 mL NP-40 buffer (50 mmol/L Tris–HCl, pH 7.5, 150 mmol/L NaCl, 1% Nonidet P-40, 1 mmol/L EDTA, 1% Cocktail, 1% PMSF) for 15 min. The cell lysates were harvested, and about 40–60 μg lysates were incubated anti-Flag beads (Thermo Fisher Scientific, 20169) overnight at 4 °C. Then, the resin beads were washed with NP-40 buffer 3 times and the flag-tagged proteins were eluted with elution buffers. The eluents were boiled in SDS-sample buffer and analyzed by Western blot.
Statistical analysis
All bar graphs and statistical analyses were generated using GraphPad Prism 9. In this study, all cell experiments were performed with three independent replicates, and the number of animals in each group was no less than three. Data were presented as the mean ± SEM. When comparing the means of two groups of data, an unpaired two-sided Student’s t test was used if the data passed the homogeneity of variance test; otherwise, a t-test assuming unequal variance was conducted. When comparing more than two groups of data, the Brown–Forsythe test was used. If the data passed the homogeneity of variance test, a one-way ANOVA was used, followed by a post hoc analysis using the Bonferroni method; otherwise, a Welch ANOVA test was performed, followed by a post hoc analysis using the Tamhane T2 method. The statistical results and corresponding analysis methods were presented in the figure legends. Statistical significance was considered as *P < 0.05.
Graphical abstract: SNX16 participates in AngII-induced cardiac hypertrophy
Sorting nexin 16 (SNX16), a member of the sorting nexin family, is involved in multiple biological processes, including protein sorting, endosomal trafficking, and signal transduction. AngII-mediated transactivation of epidermal growth factor receptor (EGFR) plays a central role in cardiac hypertrophy, in which SNX16 aggravates AngII-induced cardiac hypertrophy via promoting the intracellular recycling of EGFR and activating the Src/EGFR signaling pathway. On the one hand, SNX16 facilitates the recycling of EGFR in endosome trafficking to further sustain AngII-induced transactivation of EGFR, on the other hand, SNX16-induced phosphorylation of Src and EGFR promotes AngII-induced shedding of the metalloprotease ADAM17-mediated proHB-EGF to directly activate EGFR.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data for all graphs and charts such as uncropped, unedited blot/gel images and SNX16-GFP plasmid are deposited in the figshare repository (https://figshare.com/articles/dataset/_/30472484). All other data are available from the corresponding author on reasonable request.
References
Zhang, B. B. et al. Role of GALNT4 in protecting against cardiac hypertrophy through ASK1 signaling pathway. Cell Death Dis. 12, 980 (2021).
Greene, S. J., Failure Society of America Quality of Care et al. Building a Heart Failure Clinic: A Practical Guide from the Heart Failure Society of America. J. Card Fail. 27, 2–19 (2021).
Heineke, J. & Molkentin, J. D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 7, 589–600 (2006).
Smith, N. J., Chan, H. W., Osborne, J. E., Thomas, W. G. & Hannan, R. D. Hijacking epidermal growth factor receptors by angiotensin II: new possibilities for understanding and treating cardiac hypertrophy. Cell Mol. Life Sci. 61, 2695–2703 (2004).
Attar, N. & Cullen, P. J. The retromer complex. Adv. Enzym. Regul. 50, 216–236 (2010).
Teasdale, R. D. & Collins, B. M. Insights into the PX (phox-homology) domain and SNX (sorting nexin) protein families: structures, functions and roles in disease. Biochem. J. 441, 39–59 (2012).
Yang, J., Villar, V. A. M., Rozyyev, S., Jose, P. A. & Zeng, C. The emerging role of sorting nexins in cardiovascular diseases. Clin. Sci. 133, 723–737 (2019).
Liu, C. et al. Increased AT1 receptor expression mediates vasoconstriction leading to hypertension in Snx1-/- mice. Hypertens. Res. 44, 906–917 (2021).
Hanson, B. J. & Hong, W. Evidence for a role of SNX16 in regulating traffic between the early and later endosomal compartments. J. Biol. Chem. 278, 34617–34630 (2003).
Wang, S., Zhao, Z. & Rodal, A. A. Higher-order assembly of Sorting Nexin 16 controls tubulation and distribution of neuronal endosomes. J. Cell Biol. 218, 2600–2618 (2019).
Xu, J. et al. SNX16 regulates the recycling of E-cadherin through a unique mechanism of coordinated membrane and cargo binding. Structure 25, 1251–1263 e1255 (2017).
Choi, J. H. et al. Sorting nexin 16 regulates EGF receptor trafficking by phosphatidylinositol-3-phosphate interaction with the Phox domain. J. Cell Sci. 117, 4209–4218 (2004).
Blanc, I. L. et al. Endosome-to-cytosol transport of viral nucleocapsids. Nat. Cell Biol. 7, 653–664 (2005).
Zhang, L., Qin, D., Hao, C., Shu, X. & Pei, D. SNX16 negatively regulates the migration and tumorigenesis of MCF-7 cells. Cell Regen. 2, 3 (2013).
Brankatschk, B., Pons, V., Parton, R. G. & Gruenberg, J. Role of SNX16 in the dynamics of tubulo-cisternal membrane domains of late endosomes. PLoS One 6, e21771 (2011).
Shen, Z. et al. SNX16 activates c-Myc signaling by inhibiting ubiquitin-mediated proteasomal degradation of eEF1A2 in colorectal cancer development. Mol. Oncol. 14, 387–406 (2020).
Li, J. et al. SNX13 reduction mediates heart failure through degradative sorting of apoptosis repressor with caspase recruitment domain. Nat. Commun. 5, 5177 (2014).
Smith, N. L. et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ. Cardiovasc. Genet. 3, 256–266 (2010).
Sui, W. G. et al. ChIP-seq analysis of histone H3K9 trimethylation in peripheral blood mononuclear cells of membranous nephropathy patients. Braz. J. Med. Biol. Res. 47, 42–49 (2014).
Forrester, S. J. et al. Epidermal growth factor receptor transactivation: mechanisms, pathophysiology, and potential therapies in the cardiovascular system. Annu. Rev. Pharm. Toxicol. 56, 627–653 (2016).
Peng, K. et al. Novel EGFR inhibitors attenuate cardiac hypertrophy induced by angiotensin II. J. Cell Mol. Med. 20, 482–494 (2016).
von Zastrow, M. & Sorkin, A. Mechanisms for regulating and organizing receptor signaling by endocytosis. Annu. Rev. Biochem. 90, 709–737 (2021).
Sato, K. Cellular functions regulated by phosphorylation of EGFR on Tyr845. Int. J. Mol. Sci. 14, 10761–10790 (2013).
Zhang, Y. et al. HINT1 (Histidine Triad Nucleotide-Binding Protein 1) attenuates cardiac hypertrophy via suppressing HOXA5 (Homeobox A5) expression. Circulation 144, 638–654 (2021).
Higuchi, S. et al. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin. Sci. 112, 417–428 (2007).
Zerial, M. & McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117 (2001).
Feng, Y., Press, B. & Wandinger-Ness, A. Rab 7: an important regulator of late endocytic membrane traffic. J. Cell Biol. 131, 1435–1452 (1995).
Meresse, S., Gorvel, J. P. & Chavrier, P. The rab7 GTPase resides on a vesicular compartment connected to lysosomes. J. Cell Sci. 108, 3349–3358 (1995).
Chen, W., Feng, Y., Chen, D. & Wandinger-Ness, A. Rab11 is required for trans-golgi network-to-plasma membrane transport and a preferential target for GDP dissociation inhibitor. Mol. Biol. Cell 9, 3241–3257 (1998).
Wilde, A. et al. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell 96, 677–687 (1999).
Aoyagi, T. & Matsui, T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 17, 1818–1824 (2011).
Lorenz, K., Schmitt, J. P., Vidal, M. & Lohse, M. J. Cardiac hypertrophy: targeting Raf/MEK/ERK1/2-signaling. Int. J. Biochem. Cell Biol. 41, 2351–2355 (2009).
Ohtsu, H. et al. ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler Thromb. Vasc. Biol. 26, e133–e137 (2006).
Ohtsu, H., Dempsey, P. J. & Eguchi, S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am. J. Physiol. Cell Physiol. 291, C1-10 (2006).
Makki, N., Thiel, K. W. & Miller, F. J. Jr The epidermal growth factor receptor and its ligands in cardiovascular disease. Int. J. Mol. Sci. 14, 20597–20613 (2013).
Conte, A. & Sigismund, S. Chapter six - the ubiquitin network in the control of EGFR endocytosis and signaling. Prog. Mol. Biol. Transl. Sci. 141, 225–276 (2016).
W, C, Zaw, A. M., Law, H. K. & Chow, B. K. Minimally invasive transverse aortic constriction in mice. J. Vis. Exp. 14, 55293 (2017).
Li, Z. et al. Long noncoding RNA myocardial infarctionassociated transcript is associated with the microRNA1505p/P300 pathway in cardiac hypertrophy. Int. J. Mol. Med. 42, 1265–1272 (2018).
Hauselmann, S. P. et al. Sustained activation of p42/p44 mitogen-activated protein kinase during recovery from simulated ischaemia mediates adaptive cytoprotection in cardiomyocytes. Biochem. J. 350, 891–899 (2000).
Acknowledgements
The authors thank Dr. Wei Wang for hopeful discussion of the manuscript revision, and we would also like to thank the members at SPF transgenic facility of The Institute of Translational Medicine in Nanchang University, and the laboratory members of Drs. Hong-Bo Xin and Ke-Yu Deng for assistance with the experiments and animal care. This study was supported by the National Key Research and Development Program of China (2022YFA1104300), the Natural Science Foundation of China (81970256, 81760140, 82270302, 82000354, 82470454 and 82460078), the Natural Science Foundation of Jiangxi Province (20142BCB24001, 20224BAB216015, 20212BCJ23043 and 20212BAB206087), the Jiangxi Province Key Laboratory of Bioengineering Drugs (No.2024SSY07061) and the Science and technology project of Health Commission of Jiangxi Province (202410005).
Author information
Authors and Affiliations
Contributions
Ke-Yu Deng and Hong-Bo Xin led the project and contributed to the conception and design of the study, data analysis and interpretation, and manuscript revision. Lin Xie, Ke Wen, Guan-Hui Yu and Xin Li performed most of the experiments and data analysis. Yu-Ye Zeng, Xu-Hui Qiao, Bing-Qian Lu, Lu Chen, Ying-Jiong Lin, Xiao-Yong Ren, Xin Wu, and Qi-Kun Yang generated the animal models and performed the cell experiments. Lin Xie wrote the manuscript draft. Xiao-Lei Wangand Jing He provided advice. Yong Ji and Lu-Lu Hu provided the human heart tissue samples and advice. All the authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Hui-Hua Li and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Dario Ummarino. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, L., Wen, K., Yu, GH. et al. SNX16 aggravates AngII-induced cardiac hypertrophy in mice via EGFR transactivation. Commun Biol 9, 14 (2026). https://doi.org/10.1038/s42003-025-09243-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09243-w
