
Abstract
Tumor organoids mimicking the tumor microenvironment (TME) are key tools for tumor immunity research and personalized cancer therapy development. We integrated microgravity culture with microfluidic chip technology (Micro-GRA& FLU) to establish a platform for evaluating chimeric antigen receptor (CAR)-γδ T cell efficacy under physiological-like conditions. Patient-derived glioblastoma (GBM) cells were microgravity-cultured into glioblastoma organoids (GBOs). Pathological analysis validated GBO similarity to matched GBM in immune cell phenotypes. Microfluidic chips assessed CAR-γδ T cell cytotoxicity against GBOs. The low-cost, easy-to-operate microgravity system generated viable, uniform GBOs that retained GBM TME features. CAR-γδ T cells showed strong cytotoxicity against GBOs in microfluidic chips; individualized combination therapy enhanced their antitumor activity vs. monotherapy. This study establishes a scalable, physiologically relevant Micro-GRA& FLU platform for evaluating CAR-γδ T cell therapies in GBM organoids.
Similar content being viewed by others


Introduction
With the advancement of tumor immunotherapy, tumor organoids that emulate the tumor microenvironment (TME) have increasingly become a research focus1,2,3,4, offering opportunities for a deeper understanding of tumor immune mechanisms and personalized therapeutic strategies.
Tumor organoids capable of mimicking the TME represent more realistic and reliable models for cancer research5. These models effectively reproduce the complexity of the TME, encompassing key components such as tumor cells, immune cells, and the extracellular matrix6,7. Compared to conventional two-dimensional cell cultures or animal models, tumor organoids more accurately represent the spatial architecture and cellular composition of actual tumors8. Immune cells within tumor organoids, such as T cells and macrophages, interact naturally with tumor cells, thereby reconstituting essential processes such as tumor immune surveillance and immune escape. Consequently, researchers can observe dynamic changes within the TME in vitro, providing an intuitive and reliable model for dissecting mechanisms of tumor immune evasion9,10,11.
With continuous progress in malignant tumor immunotherapy, chimeric antigen receptor (CAR) T cell therapy has demonstrated remarkable efficacy in hematological malignancies12,13. However, several challenges persist in the treatment of solid tumors such as glioblastoma (GBM)14,15,16,17,18,19. The major obstacles include heterogeneous tumor antigens, limited immune cell infiltration, and an immunosuppressive TME20,21. Notably, a suitable model that accurately recapitulates these barriers is lacking22,23,24. Patient-derived tumor organoids represent a promising alternative due to their ability to replicate the complexity and heterogeneity of the human TME. These organoids not only allow for the evaluation of the direct cytotoxic effects of immune cells on tumor cells but also facilitate the analysis of immune cell-mediated modulation of the TME25. Accordingly, patient-derived tumor organoids may enable the selection of the most effective individualized immunotherapy strategies26, thereby advancing the implementation of truly personalized medicine.
Despite the enormous potential of tumor organoids in immuno-oncology research, integrating them with advanced technologies for more efficient evaluation of immunotherapeutic efficacy remains challenging27. Microfluidic chips have emerged as an innovative technological solution with high-throughput capabilities and precise control features28,29,30,31, enabling the co-culture of tumor organoids and immune cells while accurately simulating the dynamic TME32,33. By combining microfluidics with tumor organoids, researchers can achieve precise control of immune cell perfusion and infiltration, as well as real-time monitoring of immune–tumor cell interactions and therapeutic efficacy34. Moreover, microfluidic chips offer an ideal platform for studying individualized combinations of CAR T cell therapy with other immunotherapies, such as immune checkpoint inhibitors or cancer vaccines, allowing simultaneous observation of their combined effects on the TME.
In this study, we explore the potential of our self-developed microgravity culture system for generating glioblastoma organoids (GBOs) that effectively emulate the TME. By integrating microgravity culture with microfluidic chip technology (Micro-GRA& FLU), we establish a platform to evaluate the therapeutic efficacy of CAR-γδ T cells under conditions that approximate physiological environments. This Micro-GRA& FLU system serves as a standardized and scalable exploratory model, capable of predicting clinical outcomes in individualized immunotherapy and accelerating the clinical translation of immunotherapies.
Results
Application of the microgravity organoid culture system
The microgravity organoid culture system designed in this study consists of two main components: the Rotational Bioreactor Drive Unit (RBDU) and a bioreactor (Fig. 1A–B, Supplementary Fig. 1). The RBDU integrates a 24 V power supply, a stepper motor, coupling system, speed control module, and stepper motor driver, enabling stable and adjustable rotational motion. It can be flexibly placed inside an incubator, thereby ensuring a controlled culture environment. Multiple RBDUs can be operated in series or parallel, allowing synchronized, asynchronous, or gradient-linked rotational control (Fig. 1A–B, Fig. 2A, Supplementary Movie 1-2).

A Rotational Bioreactor Drive Unit (RBDU) and personalized bioreactor design. B Schematic diagram of the RBDU control interface.

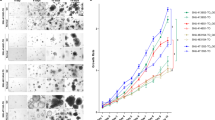
A Workflow of GBO generation using the microgravity culture system. B Macroscopic and microscopic images of fragmented GBM tissues and mature GBOs. C GBO generation efficiency from GBM tissue fragments (n = 4). D Size distribution of GBOs cultured from GBM samples of different patients, demonstrating size consistency. E, F Growth kinetics and quantification of GBOs cultured in the microgravity system. G Viability of GBOs at different growth stages (n = 3). H, I Comparison of GBO size uniformity between different culture methods. J Comparison of cellular viability across culture methods (n = 3). K, L Viability assessment and quantification of GBOs following cryopreservation and thawing (n = 3). Error bars represent standard deviation (SD) of three biological replicates.
The bioreactor is connected to the RBDU via the coupling system, and the speed control module precisely adjusts the rotational speed and direction (clockwise, counterclockwise, or alternating). The stepper motor driver is pre-configured to ensure smooth and stable rotation. The recommended settings for the stepper driver are as follows: SW1: ON, SW2: ON, SW3: ON (Current: 0.2 A); SW4: ON, SW5: OFF, SW6: OFF, SW7: OFF (Pulse setting: 20,000).
This system is compatible with commercial bioreactors such as ClinoReactor and Synthecon, while also supporting customized designs (Fig. 1A, Option 1–2). Key customizable parameters include chamber capacity, semi-permeable membrane structures, low-adsorption materials, multiple outlet configurations, and transparent casings—enhancing the system’s adaptability.
The RBDU can be upgraded with a servo motor, servo drive, and a Programmable Logic Controller (PLC) package to achieve more stable control and complex operations. Additionally, the RBDU system can be expanded through modular design to accommodate a wider range of applications. The expandable modules include, but are not limited to, lighting modules, image/video recording modules, and inertial measurement unit (IMU) (Supplementary Fig. 1B). These expansion modules provide more flexible data monitoring and recording capabilities under various research and experimental conditions.
The real-time adjustable multi-parameter settings ensure that cultured samples consistently experience microgravity conditions, thereby optimizing growth and maintaining physiological relevance.
The microgravity culture system sustains the growth of GBO
Building on previously published studies6 and our prior research35, we established a method for culturing GBOs using a self-designed microgravity bioreactor (Figs. 1A and 2A). By directly seeding freshly resected GBM tissue without single-cell dissociation or the addition of exogenous factors such as epidermal growth factor or fibroblast growth factor, we maximized the preservation of GBM heterogeneity and the native tumor microenvironment (Fig. 2B). Among 52 GBM samples, 37 GBOs were successfully cultured. Most tissue fragments developed into structurally regular, independent GBOs within one to two weeks under microgravity conditions (Fig. 2C). The GBOs exhibited sustained proliferative capacity and high viability throughout the culture period (Fig. 2E–G). Compared to conventional shaker-based culture, GBOs formed in the microgravity system displayed more uniform morphology, whereas shaker-cultured GBOs exhibited greater morphological heterogeneity (Fig. 2D, H–J). This discrepancy may be attributed to shear and frictional forces in shaker cultures, while the microgravity system provides a gentler physical environment. Cryopreservation and recovery experiments further demonstrated that GBO viability returned to pre-freezing levels within one week post-thawing (Fig. 2K–L).
Pathological characterization of the TME in GBOs
Hematoxylin and eosin (H&E) staining revealed that GBOs closely resembled their parental GBM samples in cellular morphology, size, and nuclear chromatin distribution, maintaining a structurally consistent tissue architecture (Fig. 3A). We performed multiplex immunohistochemistry (mIHC) on paired GBM tissues and GBOs (from the same patient sample and identical field of view). Nestin, BLBP, Olig2, and SOX2 were co-stained within this same field of view; the overlap of fluorescent signals reflects the inherent co-localization of these markers within GBM stemness markers. We confirmed that GBOs retained the expression patterns of tumor microenvironment-associated markers (CD31, CD11b, CD3, CD68). (Fig. 3B–C). Consistently, flow cytometry analysis of dissociated single-cell suspensions from GBOs corroborated the immunohistochemical findings (Fig. 3D, Supplementary Fig. 2). Together, these data indicate that GBOs effectively preserve both the structural and biological characteristics of the tumor microenvironment present in the original GBM samples.

A H&E staining of GBM tissues and matched GBO samples. Scale bar: 100 μm. B, C Multiplex immunohistochemistry (mIHC) showing expression patterns and quantification of GBM stemness and immune microenvironment markers in GBOs and corresponding GBM tissues (n = 3). D Flow cytometry analysis comparing the expression of immune markers between GBOs and matched GBM samples (n = 3). Error bars represent standard deviation (SD) of three biological replicates. Note: For the panels of Nestin, BLBP, Olig2, and SOX2 in Figure 3B, the top and bottom panels in this figure correspond to the same patient sample and identical visual field (displayed separately to present different marker combinations). The overlap of fluorescent signals indicates the co-localization of stemness markers in the same cells.
Evaluation of engineered CAR-γδ T cells
Based on our previously reported γδ T cell culture method35, γδ T cells were cultured in a specialized medium supplemented with a T cell activator and multiple cytokines. This protocol generated γδ T cell populations predominantly comprising Vδ1 T cells, which accounted for more than 50% of the total cell population (Supplementary Fig. 4A). Following transduction of the Vδ1 T cells with a lentiviral vector encoding anti-B7-H3 CAR, the CAR-γδ T cells achieved a transduction efficiency of 69.55% (Supplementary Fig. 4B).
The multi-channel microfluidic chip to simulate immune cell therapy for GBOs
To simulate immune cell infiltration into solid tumors via microcirculation-like channels, we developed a specialized microfluidic chip system composed of three parallel channels: two lateral bypass channels for inoculating immune cells (e.g., CAR T cells) and one central channel for seeding patient-derived GBOs (Fig. 4A, B). Specifically, the GBOs were loaded into recessed, honeycomb-shaped regions within the central channel. CAR T cells seeded into the lateral channels migrated through interconnecting microchannels into the central region, where they interacted with and exerted cytotoxic effects on the GBOs. This design recapitulates the in vivo process of CAR T cell infiltration and tumor cell killing through directional fluidic channels. Additionally, the microfluidic chips can be configured in parallel for simultaneous testing of multiple treatments or arranged in serial or parallel arrays to simulate various physiological scenarios (Fig. 4A).

A Schematic diagram and workflow for co-culture of CAR-T cells and GBOs on the microfluidic chip. B Illustration of the microfluidic chip-based co-culture system. C COMSOL simulations of the microfluidic chip under static and dynamic flow conditions. D Effect and quantification of static versus dynamic culture conditions on the viability of GBOs and γδ T cells (n = 3). E Real-time interactions between B7-H3 CAR-γδ T cells and GBOs within the microfluidic chip. Error bars represent standard deviation (SD) of three biological replicates.
Hydrodynamic Properties
Computational simulation indicated that, within the defined flow rate range, fluid flow within the microfluidic chip exhibited stable laminar characteristics. The fluid resistance in both lateral channels matched the intended design, allowing effective immobilization of GBOs within the honeycomb recesses during perfusion and preventing displacement due to flow. The diamond-shaped interconnecting channels between the lateral and central channels facilitated uniform fluid distribution and supported homogeneous migration of CAR T cells from the lateral channels into the central region. These flow dynamics created an optimal environment for immune cell infiltration and cytotoxic activity (Fig. 4C). Moreover, nutrients diffused evenly throughout the co-culture region, providing theoretical evidence that supports the chip’s effectiveness in maintaining suitable co-culture conditions.
GBO and γδ T cells maintain high viability in microfluidic chips
After 48 h of static culture within the microfluidic chips, the viability of GBOs significantly decreased, accompanied by a noticeable increase in PI-positive dead cells. In contrast, under dynamic culture conditions, the GBOs maintained stable viability comparable to their initial seeding state. A similar trend was observed for γδ T cells: after 48 h of static culture, γδ T cell viability substantially declined, and the cells failed to aggregate. Conversely, under dynamic conditions, γδ T cells preserved their initial viability and formed distinct cellular aggregates (Fig. 4D, Supplementary Fig. 3). Moreover, in the co-culture setting, γδ T cells exhibited evident proliferation and migratory capacity toward GBOs (Fig. 4E). These findings collectively indicate that the dynamic environment provided by microfluidic chips effectively sustains the high viability of both GBOs and γδ T cells.
Enhanced cytotoxicity of CAR-γδ T cells through combinatorial immunotherapy
Flow cytometric analysis confirmed high expression levels of the B7-H3 target in GBO samples (Supplementary Fig. 4C). Over a 48-h observation period, the γδ T cell group exhibited moderate cytotoxic effects against GBOs, although the overall efficacy remained limited. In contrast, CAR-modified γδ T cells (B7-H3 CAR-γδ T cells) demonstrated markedly increased cytotoxic activity, indicating that CAR modification significantly enhanced their capacity for GBO recognition and killing. Furthermore, combinatorial treatments involving CAR-γδ T cells with either a Programmed Death-1 (PD-1) antibody or a Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) antibody exhibited superior antitumor effects, substantially outperforming the CAR-γδ T cell monotherapy group (Fig. 5A, B). This improved efficacy is likely attributable to PD-1 blockade relieving immune checkpoint suppression or TREM2 antibody-mediated remodeling of the TME, thereby amplifying the functional activity and cytotoxic potential of CAR-γδ T cells.

A Responses of GBOs derived from different patients to various γδ T cell-based immunotherapies. B Quantitative analysis of therapeutic responses of patient-derived GBOs to different γδ T cell treatments (n = 3). Error bars represent standard deviation (SD) of three biological replicates.
Discussion
In the field of malignant tumor research, tumor organoids that emulate the TME represent an advanced in vitro model that closely recapitulates patient-specific tumor characteristics while circumventing ethical concerns36,37. Clinical immunotherapy for GBM is confronted with three core obstacles: heterogeneous tumor antigens, limited immune cell infiltration, and an immunosuppressive TME. These issues render traditional in vivo and in vitro models unable to accurately simulate in vivo therapeutic responses, thereby restricting the efficiency of therapeutic development. To address these challenges, the Micro-GRA&FLU system established in this study offers a targeted solution through an integrated design combining “microgravity culture + microfluidic chip” (Fig. 6). Specifically, the microgravity culture component of the Micro-GRA&FLU system directly preserves the cellular heterogeneity and immunosuppressive microenvironment of patient-derived glioblastoma tissues. This enables a more realistic simulation of the natural distribution characteristics of tumor antigens and the presence of immunosuppressive cell subsets. Additionally, the microfluidic chip, which mimics in vivo circulation, allows for the evaluation of the efficacy of different combined immunotherapeutic regimens. Therefore, the Micro-GRA& FLU model serves as a valuable tool for investigating tumor progression mechanisms and evaluating individualized responses to immunotherapies.

Workflow diagram illustrating the generation of GBOs using a microgravity culture system and their subsequent co-culture on a microfluidic chip (Micro-GRA& FLU) for evaluating the efficacy of CAR-γδ T cell therapy.
Optimized culture protocols can provide a more favorable environment for GBO growth. Our independently developed microgravity culture system utilizes the equilibrium among fluid viscosity, gravity, and rotation-induced lift forces to maintain cells in a continuous free-fall state. Under these conditions, randomized gravitational vectors may directly influence gene expression or indirectly promote cellular autocrine/paracrine signaling, thereby enhancing intercellular communication38,39.
Compared with commercial well-plates (e.g., InSphero) and conventional culture methods (including 2D culture and static 3D culture), the microgravity culture system in this study has two core advantages: First, the starting culture material is small tissue fragments, which eliminates the need for single-cell dissociation. This maximally preserves the cell-cell interaction relationships and microenvironmental components in the original tumor tissue, while avoiding damage to cell phenotypes and microenvironmental structures caused by the single-cell dissociation process. Second, relying on the dynamic culture mode of simulated microgravity, the system can support the growth of GBOs to a relatively large size without the assistance of scaffold materials such as Matrigel. Furthermore, the uniform nutrient distribution effect generated by rotation effectively avoids the central necrosis of organoids that occurs in traditional static culture due to limited nutrient diffusion. That said, the microgravity system also has unique limitations stemming from its inherent design features. Most notably, commercial well-plates (e.g., InSphero’s products) are optimized for high-throughput operations, with standardized protocols that enable simultaneous processing of 96 or more samples. In contrast, our current microgravity setup requires a dedicated rotational bioreactor for each sample; even with a multi-axis configuration, the number of samples that can be run at once is limited to just a few (due to incubator space constraints). Furthermore, additional time is needed to optimize protocols—such as adjusting rotation speeds for tissue fragments of different sizes. Additionally, commercial well-plates often come with pre-validated quality control standards, which effectively reduce experimental variability. However, the performance of our microgravity system depends largely on the operator’s proficiency in assembling the bioreactor and calibrating rotation parameters. Compared to the “ready-to-use” nature of commercial well-plates, new users need more practice to master these operational details, resulting in a relatively steeper learning curve.
In such simulated microgravity conditions, suspended GBOs grow three-dimensionally in a manner that closely resembles their natural growth patterns. This microgravity system is characterized by its simple structure, low cost, ease of operation, and high flexibility, making it suitable for a wide range of applications. The system’s capacity to support synchronized, asynchronous, and gradient-based control allows it to meet the complex environmental requirements of diverse culture conditions. Its affordability and user-friendly design make it particularly advantageous for startups and novice researchers. Furthermore, the system avoids the use of scaffolds or gels, which could interfere with gene expression, thereby preserving the intrinsic properties of the organoids39. By reducing surface contact and maintaining a low-shear environment, the suspension-based culture minimizes mechanical damage to cells and provides a gentle physical environment conducive to organoid growth. The excellent scalability also provides a foundation for the realization of advanced functions. This system can be used to simulate microgravity or hypergravity environments for studying various cellular processes, including cell growth and proliferation patterns, differentiation, and functional research. It also facilitates the observation of the effects of gravity on cell functions, such as metabolic activity and signal transduction. Additionally, the system is valuable for drug screening, toxicity testing, and research in tissue engineering and regenerative medicine.
However, maintaining low shear forces immediately after tumor fragment inoculation can be disadvantageous. To address this issue, we employed a two-step strategy. Initially, tumor fragments were inoculated onto shaker-based multi-well plates to facilitate the removal of necrotic cells and cellular debris, thereby enabling a smooth transition from the high-shear brain environment to the in vitro setting. Subsequently, the proliferating GBOs were transferred to the microgravity culture system, which effectively prevented physical damage from high shear forces and preserved the structural integrity and stability of the organoids. This environment also promoted nutrient transport through active diffusion. Initially, differences in cell composition and phenotypes among organoids of varying sizes were minimal; however, prolonged culture inevitably amplified these differences. Our two-step cultivation method produced GBOs with significantly improved uniformity compared to the single-step method, resulting in an ideal target cell model for optimizing and evaluating immune cell-based therapies.
Importantly, mIHC and flow cytometry analyses confirmed that GBOs generated under microgravity conditions retained key immune phenotypes, including CD68-positive macrophages and CD3-positive T cells, indicating the preservation of major immune components within the tumor microenvironment. Notably, these results are consistent with previously published literature describing GBM immune cell profiles40,41. These findings suggest that the microgravity culture system is capable of maintaining not only the cellular heterogeneity but also the immune cell constitution of parental GBM samples. Unlike conventional organoid cultures, which often rely on exogenous growth factors or matrix-based scaffolds—conditions that may alter the native immune landscape—our system enables scaffold-free, suspension-based growth under dynamic low-shear conditions. This configuration reduces external perturbation and supports the self-organization of TME components. This advantage is particularly valuable for immunotherapy studies, as it allows for more accurate evaluation of immune cell–tumor interactions, especially in the context of glioblastoma, where immunosuppression and myeloid dominance are hallmark features. Taken together, these observations demonstrate the potential of microgravity-cultured GBOs as a physiologically relevant model for recapitulating GBM immune microenvironments in vitro.
Building upon the successful construction of GBOs that emulate the TME, we further developed a microfluidic co-culture chip for evaluating the efficacy of CAR-γδ T cells. The application of microfluidic technology in tumor immunotherapy has garnered increasing attention in recent years. Microfluidic chips simulate in vivo fluid dynamics, enabling more accurate modeling of cellular interactions within the TME.
Within the microfluidic chip, we designed primary microchannels, bypass microchannels, and connecting channels to mimic the migration pathways of CAR-γδ T cells in the TME, including their movement through blood flow and the microcirculation of tumor tissues. The dynamic culture environment helps maintain the viability of both immune cells and GBOs by ensuring a continuous supply of nutrients and accelerating the removal of metabolic waste. This advantage not only supports cellular metabolic balance but also sustains immune cell activity and promotes the tumor immune response. In contrast, under static culture conditions, cells seeded on the chip rapidly lose viability due to limited nutrient availability and the accumulation of metabolic waste within the microchannels. Therefore, the microfluidic chip provides a reliable model for investigating interactions between immune cells and tumor cells, as well as other immune components, within the TME.
In the microfluidic co-culture system, combination therapy with CAR-γδ T cells and either PD-1 or TREM2 antibodies resulted in enhanced antitumor activity compared to monotherapy. This effect may be attributed to the ability of PD-1 or TREM2 antibodies to enhance CAR-γδ T cell function by inhibiting immune evasion mechanisms or reprogramming the immunosuppressive TME. The superior efficacy observed with combination therapy further validates the microfluidic system as a valuable platform for drug screening and therapeutic efficacy assessment in the context of CAR-γδ T cell-based immunotherapy.
One significant advantage of microfluidic technology is its high degree of customizability, which enables individualized cultivation conditions tailored to patient-specific tumor samples. As a result, microfluidic chips have emerged as an ideal platform for personalized immunotherapy evaluation42,43. In the present study, we integrated patient-derived GBOs—capable of recapitulating the TME—into a microfluidic co-culture model to assess immunotherapeutic responses. This approach facilitates the development of individualized therapeutic strategies for clinical application, ultimately optimizing treatment plans and improving patient outcomes. Moreover, microfluidic chips enable high-throughput screening of immune cell therapies, allowing rapid evaluation of multiple treatment strategies across patient-derived samples44. These chips can also be arranged in parallel or series configurations to simulate diverse physiological conditions, thereby accelerating the clinical translation of immunotherapy.
Although GBO models effectively replicate certain features of the TME—such as immune cell infiltration and cell–cell interactions—they still oversimplify the complexity of actual tumors. GBOs only represent small fragments of the original tumor, random sampling of tumor tissues and expanding the sample size allow them to reflect tumor heterogeneity at the statistical level. A notable limitation is that GBOs cannot simulate the interaction between tumors and distant tissues. To address this, we have initiated patient-derived xenograft (PDX) model validation experiments, and the results will be reported in subsequent studies. Despite the identification of endothelial cell populations within GBOs, these models do not fully recapitulate critical aspects such as tumor angiogenesis, hypoxia, and metabolic dysregulation. In addition, heterogeneity in morphology and cellular composition exists within GBOs and may become amplified over prolonged culture periods. Such variability could result in differential recognition of tumor antigens by CAR-γδ T cells. Moreover, GBOs lack the systemic immune regulation present in vivo. Even within physiologically enhanced microfluidic environments, immune cell behaviors—including migration, tissue penetration, and persistence—may differ significantly from those observed in patients. Therefore, some in vivo immune dynamics may not be fully reproduced in vitro. Despite these limitations, further improvements in chip design, more accurate multi-factor emulation of the TME, and enhanced model stability and reproducibility will undoubtedly strengthen the utility of this approach for immunotherapy evaluation. Integration with in vivo models can also provide valuable complementary data, thereby enhancing the translational relevance of immune cell therapies.
In conclusion, the Micro-GRA& FLU platform offers significant advantages in simulating the TME and evaluating therapeutic efficacy, thereby more accurately reproducing in vivo immune responses. This integrated system serves not only as a precise experimental tool for studying tumor immunology but also as a promising approach for optimizing and personalizing immune cell therapies. Moving forward, critical areas for further investigation include cytokine dynamics during prolonged co-culture, the mechanisms underlying immune cell interactions following combination therapies, and the enhancement of intrinsic CAR-γδ T cell immunotherapeutic activity within the Micro-GRA& FLU system. These efforts will contribute comprehensive theoretical and practical insights, ultimately facilitating the development and clinical translation of personalized immunotherapy strategies for patients with GBM.
Methods
Microgravity organoid culture system
The microgravity organoid culture system consists of two main components: the RBDU and a bioreactor. The RBDU comprises key components, including a 24V–5 A power supply (YEYY, China); a 35 dual-shaft stepper motor (height: 25 mm; front and rear shaft extension: 15 mm; Guangzhou Aofude Precision Electromechanical Co., Ltd., China); a single-disc coupling (outer diameter: 34 mm; length: 32 mm; inner and outer bore diameter: 5 mm; COUP-LINK, China); a stepper speed control module (YMS8-D, YEYY, China); and a step motor driver (DM420, Shanghai Fengxin Transmission Machinery Co., Ltd., China). The coupling mechanism allows flexible attachment to various commercial bioreactors or custom-designed culture chambers via double-sided nano adhesive or magnetic fixation.
Collection of GBM patient tissues
Freshly resected GBM tissues were subjected to rapid pathological examination. Following preliminary pathological confirmation of GBM, the samples were included for collection. Samples were preserved in tissue preservation solution (Miltenyi, USA) at 4 °C and transported to the laboratory for further processing under a laminar flow biosafety cabinet. Each tumor sample was divided into two portions. For GBO culture: Tumor tissues were minced into small fragments (0.5–1 mm in diameter) and washed with culture medium to remove necrotic tissue and cell debris. For histological analysis: Tumor tissues were fixed in 4% formaldehyde for 24 h, dehydrated, and embedded in paraffin to prepare tissue blocks.
Microgravity 3D dynamic culture system for GBOs
A two-step method was employed for the microgravity dynamic culture system. Step 1: Tumor fragments were seeded in ultra-low adhesion 6-well plates with 4 ml of culture medium per well. The culture medium consisted of 50% DMEM:F12 (Basal Media, China); 50% Neurobasal (Basal Media); 1× Glutamax (Gibco, USA); 1× NEAAS (Thermo Fisher Scientific, USA); 1× B27 supplement (Basal Media); 1× N2 supplement (Basal Media); 100 IU ml-1 IL-2 (T&L Biotechnology, China); 30 ng ml-1 TGF-β (T&L Biotechnology); 1 × 2-mercaptoethanol (Thermo Fisher Scientific); and 2.5 mg ml-1 human insulin (Sigma, USA). Plates were incubated on a horizontal shaker at 120 rpm. When cloudiness was observed, 75% of the medium was replaced. This step lasted a total of 3 days. Step 2: The cultured fragments were transferred to a self-developed microgravity 3D dynamic organoid culture reactor, with 10 ml of medium per reactor. The reactor rotated bidirectionally at 25 rpm in an incubator maintained at 37 °C, 5% CO₂, and 90% humidity. Fresh medium was replenished every 3–5 days. As a control, tumor samples were continuously cultured on low-adhesion plates.
Monitoring of GBO growth
To track the growth of GBOs over time, images were periodically captured using an inverted microscope. The diameter of each organoid was quantified using ImageJ software. To assess GBO viability, organoids were stained using acridine orange/propidium iodide (AO/PI) kits. Comprehensive Z-stack imaging of GBOs was performed using fluorescence microscopy, and the percentage of viable cells was calculated with the mixed-cell counting module (Keyence, Japan).
Cryopreservation and recovery of GBO
Upon reaching maturity, GBOs were fragmented into smaller pieces ranging from 100 to 200μm in diameter. After removal of cellular debris, the fragments were incubated in culture medium supplemented with 10 μM Y-27632 (StemCell Technologies, USA) for 1 h. The treated GBO fragments were then resuspended in tissue freezing medium containing 10 μM Y-27632 (Miltenyi, USA). The GBO suspension was transferred into a controlled-rate freezing container (Thermo Fisher Scientific), gradually cooled overnight at −80 °C, and subsequently stored in liquid nitrogen for long-term preservation. For recovery, cryopreserved GBO samples were rapidly thawed in a 37 °C water bath. The thawed GBOs were thoroughly washed to remove residual cryopreservation medium, resuspended in fresh GBO culture medium supplemented with 10 μM Y-27632, and cultured for an additional 24 h. Following a further seven-day culture period, GBO viability was assessed using the AO/PI staining kit, as described previously.
Pathological identification and TME analysis of GBO
Upon maturation, GBOs were embedded in paraffin to prepare histological sections. H&E staining was performed on both GBO sections and the corresponding tumor tissues using standard histopathological protocols.
The mIHC was used to evaluate stemness marker expression and characterize immune cell phenotypes within GBOs and their matched tumor tissues, thereby assessing the fidelity of tumor immune microenvironment reconstruction. Specifically, multiplex fluorescent immunostaining was conducted using the Opal-tyramide signal amplification method (Zen-BioScience, China) in accordance with the manufacturer’s guidelines. Positive and negative controls were included for validation. Two independent pathologists conducted blinded reviews and data analysis. Images were acquired using the Mantra multispectral imaging system (PerkinElmer, USA), and data were analyzed with inForm 2.2.1 software (PerkinElmer, USA).
Primary antibodies used in this study targeted GBM stemness markers (Nestin, BLBP, Olig2, SOX2; Zen-BioScience) and TME-associated markers (CD31, CD11b, CD3, CD68; Zen-BioScience).
Flow cytometry
A Novocyte flow cytometer (Agilent, USA) was used to analyze T cell phenotypes and determine the transduction efficiency of CAR-γδ T cells based on green fluorescent protein (GFP) expression. The following antibodies were used for γδ T cell subcluster and functional analysis: FITC-conjugated anti-human CD3 (BioLegend, USA); PE-conjugated anti-human TCR γ/δ (BioLegend); and APC-conjugated anti-human Vδ1 (Miltenyi).
For flow cytometric analysis of GBOs, single-cell suspensions were prepared using gentleMACS sample preparation technology (Miltenyi). The expression of B7-H3 (CD276) was subsequently evaluated using an APC-conjugated anti-human CD276 antibody (BioLegend). Changes in T cells and macrophages, along with their phenotypes, were analyzed using FITC-conjugated anti-human CD3, and PE-conjugated anti-human CD68 antibodies (all from BioLegend). Antibody staining procedures were strictly conducted according to the manufacturers’ protocols, incorporating Fc receptor blocking reagents and corresponding isotype controls as standard practice. Flow cytometry data were analyzed using NovoExpress 1.5.6 (Agilent). The gating strategy for the flow cytometry analysis was shown in Supplementary Fig. 5.
Isolation and culture of γδ T cells
Peripheral blood mononuclear cells were isolated from healthy volunteers using a standard protocol. γδ T cells were subsequently purified using a human TCR γ/δ T cell isolation kit (Miltenyi), following the manufacturer’s guidelines. The purified γδ T cells were resuspended at a density of 1 × 10⁶cells ml-1 in ALyS705 medium (Baso, China). To activate the γδ T cells, 25 μl of human CD3/CD28/CD2 T cell activator (StemCell Technologies) was added per milliliter of culture medium. Cytokines—including 200 IU ml-1 IL-2, 70 ng ml IL-15-1, 20 ng ml-1 IL-7, 100 ng ml-1 IL-4, and 7 ng ml-1 IL-21—were also supplemented into the culture medium.
Construction of CAR and lentiviral vector production
A clinical-grade lentiviral vector was used to construct the CAR. This CAR structure consisted of a B7-H3 antibody-derived single-chain variable fragment, a hinge region, a CD28 transmembrane domain, the 4-1BB signaling domain as a co-stimulatory molecule, and the CD3-zeta signaling domain derived from the T cell receptor. Lentiviral vectors were produced using a three-plasmid system, involving the co-transfection of Jurkat T cells with the CAR-encoding lentiviral plasmid, the packaging plasmid pSPAX2 (AddGene, plasmid #12260), and the envelope plasmid pMD2.G (AddGene, plasmid #12259). Following co-transfection, the lentiviral supernatant was harvested, filtered, centrifugally concentrated, aliquoted, and stored at −80 °C.
Transduction of γδ T cells
Transduction of γδ T cells with the CAR construct was performed on day 3 after initial activation. The activated γδ T cells were incubated with 1× NATE (InvivoGen, France) at 37 °C for 30 min, followed by co-culture with B7-H3 CAR lentivirus at a multiplicity of infection of 10 for 48. The culture medium was regularly refreshed, and transduction efficiency was periodically assessed by flow cytometry based on GFP expression.
Microfluidic chip design and characteristics
A specialized microfluidic chip was designed to facilitate the co-culture of CAR T cells with GBOs, providing a platform to assess the efficacy of CAR T cell therapy within an emulated TME. The chip consists of a single-layer structure divided into three microfluidic channels separated by connecting channels. The lateral channels (1.2 mm width, 3 mm height) were designed to infuse CAR T cells, whereas the central channel (4.5 mm width, 3 mm height) contains 24 recessed, honeycomb-shaped wells (each 1.5 mm in diameter and 0.1 mm deep) for seeding GBOs. Rhomboid connecting channels link the central channel to the lateral channels, facilitating fluid communication. Inlet and outlet ports were positioned at both ends of the channels to enable fluid exchange. During seeding, GBOs naturally settled into these honeycomb wells. By controlling fluidic resistance through precise flow rates, GBOs remained securely fixed within these regions during perfusion.
Fabrication of microfluidic chips
The microfluidic chips were fabricated using soft lithography techniques with polydimethylsiloxane (PDMS; Dow Corning, USA). Initially, a master mold containing microchannels and connecting structures was produced by patterning photoresist onto a silicon wafer via photolithography, ensuring precise dimensions and structural consistency to meet experimental requirements. PDMS pre-polymer and curing agent were mixed at a 10:1 ratio, degassed to eliminate air bubbles, poured onto the master mold, and cured at 70 °C for 4 to form a 5-mm-thick structured PDMS layer. After curing, the PDMS replica was carefully peeled from the mold and precisely trimmed to the chip dimensions. Both the PDMS and glass surfaces were treated with oxygen plasma (75 W, 0.2 cm³ min-1 O₂; Diener electronic GmbH + Co. KG, Germany) for 24 s to enhance surface activation and adhesion. Subsequently, the PDMS microstructure was precisely aligned and bonded to a glass slide, forming the initial chip structure. Biopsy punches (diameter: 1.6/1.2 mm; World Precision Instruments, Germany) were used to create inlet and outlet ports for cell seeding and medium perfusion. The assembled chips were incubated overnight at 60 °C to reinforce bonding. Finally, the completed chips underwent an additional plasma treatment to sterilize and render the PDMS surfaces hydrophilic.
Microfluidic chip simulation modeling
COMSOL Multiphysics v6.0 software was employed to simulate the microfluidic chip designed for CAR T cell and GBO co-culture, with the aim of optimizing its structural parameters and performance. The simulation model was constructed within COMSOL to accurately reflect the physical dimensions and characteristics of the actual chip. Multiphysics fields—including fluid flow and mass transport—were incorporated to simulate the hydrodynamic environment of the co-culture system. Specifically, the distribution of nutrient concentration within the microfluidic channels and its temporal evolution were modeled under both static and dynamic conditions, enabling thorough evaluation of the chip’s feasibility and effectiveness.
Influence of microfluidic chip on viability of GBO/γδ T cells
To assess the viability of GBOs and γδ T cells cultured within the microfluidic device, GBOs were seeded into the recessed honeycomb regions of the central channel, while γδ T cells were introduced into the lateral channels. For static conditions, the chip was connected to a culture medium reservoir and placed directly into an incubator. For dynamic conditions, the microfluidic chip was integrated with a perfusion system that maintained a constant flow rate of 10 ml/24 h to simulate physiological fluid dynamics. Cell viability was analyzed at 0 and 48 h post-seeding using Hoechst/PI or AO/PI staining applied directly into the chip system. Images were acquired using an inverted fluorescence microscope, and data analysis was conducted with ImageJ software.
Evaluation of B7-H3 CAR-γδ T cell cytotoxicity in microfluidic co-culture system
Prior to cytotoxicity assays, B7-H3 expression on GBOs was verified via flow cytometry, as previously described. GBOs were then pre-labeled with Hoechst dye (Yeasen Biotechnology, China), while B7-H3 CAR-γδ T cells were labeled with DIO fluorescent dye (Yeasen Biotechnology, China) to facilitate visualization and distinction within the microfluidic system. For large GBOs with a diameter >800 μm, incomplete staining may still occur in the core region due to excessive diffusion distance; therefore, in this study, we selected GBOs with a diameter of approximately 500 μm for experiments to minimize the impact of uneven staining on the results. A single GBO with a diameter of approximately 500 μm contains a total of around 1 × 104 cells. In the central channel of the microfluidic chip, we seeded approximately 10 such GBOs, corresponding to a total cell count of roughly 1 × 105. All staining procedures were performed in strict accordance with the manufacturer’s protocols. Hoechst-labeled GBOs were carefully seeded into the recessed honeycomb structures of the central channel to ensure uniform distribution; subsequently, 1 × 105 DIO-labeled B7-H3 CAR-γδ T cells were introduced into the lateral microfluidic channels. Through the above operations, the effector-to-target (E:T) ratio in the microfluidic chip co-culture system was ultimately achieved at 1:1. Various experimental groups were established, including a γδ T cell-only group, a CAR-γδ T cell group, and combination therapy groups using CAR-γδ T cells with either an anti-human CD279 (PD-1) antibody (BioLegend) or a TREM2 antibody (MedChemExpress, USA). Following co-culture initiation, PI dye (1 μg ml-1) was added to the culture medium at 0, 24, and 48 h to evaluate cell death over time. Fluorescent images across multiple channels were acquired using a fluorescence microscope with Z-stack functionality and reconstructed accordingly. Cell viability and cytotoxicity were quantitatively assessed using a cell-counting module to determine the relative killing efficacy of each experimental group against the GBOs.
Ethics statement
This study was reviewed and approved by the Ethics Committee of the Second Hospital of Shandong University in accordance with the principles of the Declaration of Helsinki (Approval No: KYLL2025036). Written informed consent was obtained from all patients or donors prior to the collection of tumor and blood samples. All patient samples were anonymized before inclusion in the study. All ethical regulations relevant to human research participants were followed.
Statistics and Reproducibility
Statistical analysis was performed using GraphPad Prism software version 7.0. Data are expressed as mean ± standard deviation. Unless otherwise specified, the presented results are representative of at least three independent experiments. Statistical significance was determined using Student’s t-test or analysis of variance. A p-value of less than 0.05 was considered statistically significant. Statistical significance levels are indicated as follows: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; and NS indicates not significant (P > 0.05). Additional details on statistical analyses are provided in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Author's Contribution
G.Z.: Writing–original draft, Visualization, Validation, Supervision, Software, Resources, Formal analysis, Conceptualization. X.S.: Methodology. Y.J.: Methodology. W.Z.: Supervision. L.G.: Resources, Methodology. G.P.: Formal analysis. Y.L.: Methodology. D.T.: Review & editing, Supervision. C.W.: Project administration, Review & editing, Supervision. Z.S.: Funding acquisition, Designed and Conceptualized this study.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Source data can be obtained in Supplementary data 1.
References
Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
Aref, A. R. et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 18, 3129–3143 (2018).
Neal, J. T. et al. Organoid modeling of the tumor immune microenvironment. Cell 175, 1972–88.e16 (2018).
Yuki, K. et al. Organoid models of tumor immunology. Trends Immunol. 41, 652–664 (2020).
Peng, T. et al. Individualized patient tumor organoids faithfully preserve human brain tumor ecosystems and predict patient response to therapy. Cell Stem Cell 32, 652–69.e11 (2025).
Jacob, F. et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 180, 188–204.e22 (2020).
Polak, R. et al. Cancer organoids 2.0: modelling the complexity of the tumour immune microenvironment. Nat. Rev. Cancer 24, 523–539 (2024).
Li, W. et al. 3D Biomimetic Models to Reconstitute Tumor Microenvironment In Vitro: Spheroids, Organoids, and Tumor-on-a-Chip. Adv. Health. Mater. 12, e2202609 (2023).
Huang, L. et al. Engineered exosomes as an in situ DC-primed vaccine to boost antitumor immunity in breast cancer. Mol. Cancer 21, 45 (2022).
Harter, M. F. et al. Analysis of off-tumour toxicities of T-cell-engaging bispecific antibodies via donor-matched intestinal organoids and tumouroids. Nat. Biomed. Eng. 8, 345–360 (2024).
Sun, L. et al. A human mucosal melanoma organoid platform for modeling tumor heterogeneity and exploring immunotherapy combination options. Sci. Adv. 9, eadg6686 (2023).
Zhang, C. et al. Novel CD19 chimeric antigen receptor T cells manufactured next-day for acute lymphoblastic leukemia. Blood Cancer J. 12, 96 (2022).
Wang, L. et al. Bispecific CAR-T cells targeting CD19/20 in patients with relapsed or refractory B cell non-Hodgkin lymphoma: a phase I/II trial. Blood Cancer J. 14, 130 (2024).
Ouladan, S. & Orouji, E. Chimeric antigen receptor-T cells in colorectal cancer: pioneering new avenues in solid tumor immunotherapy. J. Clin. Oncol. 43, 994–1005 (2025).
Brown, C. E. et al. Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial. Nat. Med. 30, 1001–1012 (2024).
Monje, M. et al. Intravenous and intracranial GD2-CAR T cells for H3K27M(+) diffuse midline gliomas. Nature 637, 708–715 (2025).
Bagley, S. J. et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Rα2 in recurrent glioblastoma: phase 1 trial interim results. Nat. Med. 30, 1320–1329 (2024).
Vitanza, N. A. et al. Intracerebroventricular B7-H3-targeting CAR T cells for diffuse intrinsic pontine glioma: a phase 1 trial. Nat. Med. 31, 861–868 (2025).
Choi, B. D. et al. Intraventricular CARv3-TEAM-E T cells in recurrent glioblastoma. N. Engl. J. Med. 390, 1290–1298 (2024).
Keshavarz, A. et al. Recent findings on chimeric antigen receptor (CAR)-engineered immune cell therapy in solid tumors and hematological malignancies. Stem Cell Res. Ther. 13, 482 (2022).
Pan, C. et al. Next-generation immuno-oncology agents: current momentum shifts in cancer immunotherapy. J. Hematol. Oncol. 13, 29 (2020).
Kumari, R. et al. Preclinical pharmacology modeling of chimeric antigen receptor T therapies. Curr. Opin. Pharm. 61, 49–61 (2021).
Siegler, E. L. & Wang, P. Preclinical models in chimeric antigen receptor-engineered T-cell therapy. Hum. Gene Ther. 29, 534–546 (2018).
Nukala, U. et al. A systematic review of the efforts and hindrances of modeling and simulation of CAR T-cell therapy. Aaps J. 23, 52 (2021).
Cong, R. et al. Tumor organoids in cancer medicine: from model systems to natural compound screening. Pharm. Biol. 63, 89–109 (2025).
Jiang, Y. et al. Patient-derived bladder cancer organoid model to predict sensitivity and feasibility of tailored precision therapy. Curr. Urol. 17, 221–228 (2023).
Dijkstra, K. K. et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 174, 1586–1598.e12 (2018).
Zhang, K. et al. A dual-functional microfluidic chip for guiding personalized lung cancer medicine: combining EGFR mutation detection and organoid-based drug response test. Lab Chip 24, 1762–1774 (2024).
Lu, Z. et al. Lab-on-a-chip: an advanced technology for the modernization of traditional Chinese medicine. Chin. Med. 19, 80 (2024).
Quintard, C. et al. A microfluidic platform integrating functional vascularized organoids-on-chip. Nat. Commun. 15, 1452 (2024).
Sun, X. et al. Unleashing the potential of urine DNA methylation detection: advancements in biomarkers, clinical applications, and emerging technologies. Curr. Urol. 19, 295–302 (2025).
Li, C. et al. Advances of 3D cell co-culture technology based on microfluidic chips. Biosens. (Basel) 14, 336 (2024).
Wang, Z. & Kelley, S. O. Microfluidic technologies for enhancing the potency, predictability and affordability of adoptive cell therapies. Nat. Biomed. Eng. 9, 803–821 (2025).
Sin, W. X. et al. A high-density microfluidic bioreactor for the automated manufacturing of CAR T cells. Nat. Biomed. Eng. 8, 1571–1591 (2024).
Zhu, G. et al. Rational Design and Organoid-Based Evaluation of a Cocktail CAR-γδ T Cell Therapy for Heterogeneous Glioblastoma. Adv. Sci. (Weinh.) 12, e2501772 (2025).
Marx, V. Closing in on cancer heterogeneity with organoids. Nat. Methods 21, 551–554 (2024).
Zhao, Y. et al. Distinct molecular profiles drive multifaceted characteristics of colorectal cancer metastatic seeds. J. Exp. Med. 221, e20231359 (2024).
Li, L. et al. Effects of simulated microgravity on the expression profiles of RNA during osteogenic differentiation of human bone marrow mesenchymal stem cells. Cell Prolif. 52, e12539 (2019).
Wang, X. et al. Effects of simulated microgravity on human brain nervous tissue. Neurosci. Lett. 627, 199–204 (2016).
Mathewson, N. D. et al. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 184, 1281–1298.e26 (2021).
Pombo Antunes, A. R. et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 24, 595–610 (2021).
Boussommier-Calleja, A. et al. Microfluidics: a new tool for modeling cancer-immune interactions. Trends Cancer 2, 6–19 (2016).
Choi, D. et al. Microfluidic organoid cultures derived from pancreatic cancer biopsies for personalized testing of chemotherapy and immunotherapy. Adv. Sci. (Weinh.) 11, e2303088 (2024).
Maurya, R. et al. Advances in microfluidics devices and its applications in personalized medicines. Prog. Mol. Biol. Transl. Sci. 186, 191–201 (2022).
Acknowledgements
This work was supported by the Key Research and Development Program of Shandong Province (No. 2021CXGC011101), Shandong Province Postdoctoral Innovation Project (No. SDCX-ZG-202303056), the National Natural Science Foundation of China (No. 82103144), China Postdoctoral Science Foundation (No. 2021M691942) and Taishan Scholars of Shandong Province of China (tsqn202312339; tsqn202211324). Partial graphical elements in the flowcharts, schematic diagrams, and graphical abstracts within the article were created using Figdraw.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Dr Shirley Tang and Dr Ophelia Bu.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, G., Shi, X., Jiang, Y. et al. Microgravity-cultured glioblastoma organoids integrated with microfluidic chip for CAR-γδ T evaluation. Commun Biol 8, 1791 (2025). https://doi.org/10.1038/s42003-025-09390-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09390-0
