
Abstract
The subiculum contains functionally distinct dorsal and ventral subdivisions and is implicated in temporal lobe epilepsy (TLE). Yet, the contribution of ventral subicular glutamatergic neurons (vSubglu) to epileptogenesis remains unclear. Using in vivo fiber photometry and c-Fos mapping in a hippocampal-kindling TLE model, we identified pronounced hyperactivation of vSubglu during seizure development. Optogenetic activation of vSubglu alone recapitulated the kindling process and accelerated kindling progression when applied following electrical stimulation. Conversely, optogenetic or chemogenetic inhibition of vSubglu robustly suppressed seizure development in both the kindling model and the acute KA model and delayed epileptogenesis in the chronic KA model. Circuit-specfic manipulations revealed that the pro-epileptic effects are mediated predominantly through glutamatergic projections from the vSub to the anterior thalamic glutamatergic neurons (ATNglu). This vSubglu-ATNglu circuit exhibited both necessity and sufficiency for epileptogenesis in the kindling model, as evidenced by pathway-specific manipulations. Collectively, our findings uncover a previously unrecognized hippocampo–thalamic circuit that critically governs epileptogenesis in TLE, highlighting vSubglu-ATNglu circuit as a promising target for TLE therapeutic intervention.
Similar content being viewed by others


Introduction
Epilepsy is a circuit-level neurological disorder characterized by recurrent seizures that result from pathological neuronal hyperexcitability, affecting over 70 million individuals worldwide across all age groups1. The epileptogenic process typically begins with seizure initiation at a focal site and propagates through aberrant neural networks, eventually leads to widespread hemispheric or global hyperexcitability2. Although anti-seizure medications (ASMs) remain the first-line therapy, current pharmacological strategies meet major clinical limitations, including drug resistance, side effects and drug-induced disease3,4. Moreover, available ASMs primarily suppress seizures rather than modify the fundamental pathophysiological mechanisms driving epileptogenesis5. This critical gap persists due to the incomplete characterization of the spatiotemporal dynamics within epileptogenic circuits6. Therefore, there is an urgent need to identify circuit-specific targets capable of modulating epileptogenesis itself, rather than merely alleviate its symptoms.
The subiculum proper, as the principal output hub of the hippocampal formation and the primary seizure focus in temporal lobe epilepsy (TLE), comprises functionally segregated subdomains, namely the dorsal subiculum (dSub) and ventral subiculum (vSub)7,8,9,10. These subregions integrate and distribute hippocampal information through topographically organized glutamatergic projections to cortical-subcortical networks11,12. Converging evidence from prior studies and our own work indicates that subicular circuits is involved in both ictogenesis and seizure propagation in TLE11,13,14,15. While research has focused on the dSub, the glutamatergic neurons in the dSub (dSubglu) have been shown as structurally and functionally heterogeneous in TLE16. In contrast, the ventral subiculum - despite its privileged connectivity with limbic and thalamic networks - remains largely unexplored in epilepsy research. This gap presents a critical obstacle to understanding how hippocampal output regulates seizure dynamics.
Viral tracing and single-cell sequencing have revealed the spatial and molecular heterogeneity within the subiculum. Subicular pyramidal neurons exhibit distinct molecular signatures and projection-defined subclasses that are organized along the dorsoventral axis17,18,19,20. Intriguingly, the vSub exhibits a particularly strong epileptogenic potential, as epileptiform discharges often originate from this region in TLE models15,21,22. Paradoxically, while GABAergic interneuron subtypes (parvalbumin-positive, somatostatin-positive, and vasoactive intestinal polypeptide-positive neurons) in vSub have been profiled in TLE23,24,25, its principle glutamatergic neurons (vSubglu) - the primary drivers of vSub excitatory output and presumed mediators of network hyperexcitability—remain poorly understood. Here, combining multiscale interrogation approaches including in vivo optical fiber photometry, optogenetics, chemogenetics, and anterograde viral tracing, we systematically investigated the role of vSubglu in TLE and delineated their associated functional circuits.
Results
Ventral subicular glutamatergic neurons were functionally activated during the kindling-induced epileptogenesis
To characterize the spatiotemporal activation dynamics of vSubglu in TLE, we firstly quantified neuronal activity during epileptogenesis. Immunohistochemical analysis revealed a significant elevation of c-Fos expression in the vSub of epileptic mice compared with non-epileptic controls (Fig. 1a, Fig. S1, Kruskal-Wallis test, n = 5 mice, Con vs. GS, **P < 0.001; Con vs. FS, P > 0.05, ns; GS vs. FS, P > 0.05, ns), indicating that the vSub is hyperactivated during TLE pathogenesis. Given the pivotal role of vSubglu function10,26, we next applied in vivo fiber photometry to monitor calcium dynamics in free-moving mice during kindling-induced epileptogenesis. After stereotaxic injection of the Ca/calmodulin-dependent protein kinase IIα (CaMKIIα) promoter adeno-associated virus, and implantation of an optic fiber into the vSub (Fig. 1b, left), the genetically encoded calcium indicator GCaMP6s was efficiently expressed in vSubglu (CaMKIIα neurons expressed green fluorescent protein, Fig. 1b, right). Then we simultaneously recorded both the seizure activities (as indicated by EEGs recorded from the ventral hippocampal CA3) and the changes in GCaMP6s fluorescence intensities during either focal-onset seizures (FS, Racin score 1–3 stage seizures) or generalized seizures (GS, Racin score 4–5 stage seizures) throughout the kindling process (representative EEGs of FS and GS are shown in Fig. 1c). Quantitative analysis demonstrated marked augmentation of calcium signals (ΔF/F) in vSubglu during both FS and GS (Fig. 1d and e, individual traces for each trial were denoted in Fig. S2–S3, n = 11 trials in FSs, n = 10 trials in GSs, a total of 3 mice were used). The heat map compromised of each trial were shown in Fig. 2f and g for FSs and GSs, respectively. While peak fluorescence intensities can not be differed significantly between seizure severities (Fig. 1h, Unpaired t test, F(1,19) = 1.299, P > 0.05, ns). Because previous studies suggest that vSubglu was activated prior to dSubglu in TLE16. To confirm this activated manner, we next quantified the temporal delay index, which reflects the relative timing or synchrony between neuronal calcium activity and electrographic seizure onset. While no statistical significance was observed (Fig. 1i, Mann–Whitney test, F(1,19) = 9.930, P > 0.05, ns; Fig. 1j, linear regression, R2 = 0.2652 in FSs, R2 = 0.5702 in GSs). These results indicated that the activities of vSubglu exhibit comparable hyperactivity regardless of seizure severities. Collectively, our findings demonstrate that vSubglu were robustly hyperactivated during kindling-induced epileptogenesis in TLE.

a Representative images of c-Fos activation pattern in the vSub (Scale bar = 500 µm; left panel, scale bar =100 µm, right panel). b Scheme of experiments for Ca2+ fiber photometry of vSubglu neurons in hippocampal-kindling induced TLE (left panel), the histological verification of GCaMP6s expression in coronal brain slices in the vSub (Scale bar = 200 µm, right panel). c Representative seizure EEG recoded when FS or GS. d, e Averaged ΔF/F of vSubglu in FSs and GSs. f, g Averaged heatmap of. Ca2+ signals of vSubglu neurons during FSs and GS. h Peak value of Ca2+ signals ΔF/F in FSs and GSs, Unpaired-t test, F(1,19) = 1.299, P = 0.5962, ns. i Temporal delay index in FSs and GSs, Mann–Whitney test, F(1,19) = 9930, P = 0.9725, ns. j Slope distributions of FSs and GSs (R2 = 0.2652, ns, n = 11 in FS, R2 = 0.5702, ns, n = 10 in GS, from 3 mice).

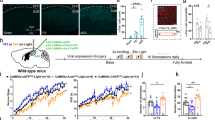
a Left, the diagram of optogenetic activation experiment in hippocampal-kindling induced TLE; right, the representative coronal images for ChR2-EYFP expression in the vSub, scale bar = 100 µm. b Upper, scheme of directly optogenetic activating vSubglu neurons to induce TLE; middle, the first optogenetic stimulation induced a representative focal seizure EEG recorded in hippocampal CA3; bottom, the last optogenetic stimulation induced a representative generalized seizure EEG recorded in hippocampal CA3. c Optogenetic activation of vSubglu directly caused the progression of seizure stages (left, F(2,12) = 163.7) and ADDs (right, F(2,12) = 179.0), n = 5 mice in each group, Two-way ANOVA for repeated measures followed by the Scheffe’s post hoc test compare each group with every other group; CaMKIIα-ChR2-light vs CaMKIIα-ChR2-no light, ***P < 0.001; CaMKIIα-ChR2-light vs CaMKIIα-EYFP-light, ###P < 0.001. d Left-upper, scheme of optogenetic activation of vSubglu during epileptogenesis in hippocampal-kindling induced TLE; bottom, effect of optogenetic activation of vSubglu on the development of seizure stages (F(2,20) = 4409) and ADD (F(2,20) = 6.692), n = 8 mice in CaMKIIα-ChR2- Light OFF and CaMKIIα-ChR2- Light ON group, n = 7 mice in CaMKIIα-EYFP- Light ON, Two-way ANOVA for repeated measures followed by the Scheffe’s post hoc test compare each group with every other group, stage: CaMKIIα-ChR2-Light OFF vs. CaMKIIα-ChR2-Light ON, *P = 0.0451, CaMKIIα-EYFP-Light ON vs. CaMKIIα-ChR2-Light ON, ##P = 0.0030; ADD: CaMKIIα-ChR2-Light off vs CaMKIIα-ChR2-Light ON, *P = 0.0353, CaMKIIα-EYFP-Light ON vs. CaMKIIα-ChR2-Light ON, ##P = 0.0474). e Numbers of stimulations staying in FSs and GSs between the two groups, One-way ANOVA followed by the Scheffe’s post hoc test compare each group with every other group (FS: CaMKIIα-ChR2-Light OFF vs CaMKIIα-ChR2-Light ON, F(1,14) = 1.850, *P = 0.0325, CaMKIIα-EYFP-Light ON vs CaMKIIα-ChR2-Light ON, F(1,13) = 2.138, **P < 0.0079; GS: CaMKIIα-ChR2-Light OFF vs CaMKIIα-ChR2-Light ON, F(1,14) = 1.850, P = 0.4912, ns, CaMKIIα-EYFP-Light ON vs CaMKIIα-ChR2-Light ON, F(1,13) = 2.164, P = 0.5544, ns). f Numbers of kindling stimulations needed to reach fully-kindled, One-way ANOVA followed by the Scheffe’s post hoc test compare each group with every other group (CaMKIIα-ChR2-Light OFF vs CaMKIIα-ChR2-Light ON, F(1,14) = 1.485, *P = 0.0466, CaMKIIα-EYFP-Light ON vs CaMKIIα-ChR2-Light ON, F(1,13) = 1.936, *P = 0.0330). g Representative GS EEGs recorded from ventral hippocampal CA3 in fully-kindled state from mice of the groups. h The scheme of GS expression experiment, compromised by three phases: pre, light and post. i Effects of optogenetic activation of vSubglu on generalized seizure stage, GS-ADDs, and GS-GSDs during GS expression, One-way repeated measures ANOVA with post hoc Dunnett’s test, n = 8 mice, ns.
Optogenetic activation of glutamatergic neurons in the vSub promoted epileptogenesis
To establish a causal link between vSubglu activity and epileptogenesis, we employed optogenetic manipulation to selectively activate Channelrhodopsin-2 (ChR2)-expression CaMKIIα-positive neurons in the vSub during hippocampal kindling process (Fig. 2a, left). Histological verification confirmed viral specificity, with ChR2-EYFP expression restricted to vSub somata (Fig. 2a, right). To test whether activating vSubglu could mimic epileptogenesis in kindling, we applied a repeated optogenetic stimulation protocol (20 Hz, 473 nm, 5 mW, 30 s epochs in 30 min interval, 10 times/day, 30 stimulations) on naïve mice with ChR2 expressed in the vSub (Fig. 2b, top panel). Remarkably, initial optogenetic stimulation in these mice were sufficient to elicited focal seizures with afterdischarges (ADs), demonstrating that direct activation of vSubglu could trigger ictogenesis (Fig. 2b, left). By contrast, identical optogenetic stimulation failed to elicit any epileptic dischargers in vSubEYFP+ mice (Fig. 2b, right). Chronic photostimulation induced progressive epileptogenesis, as evidenced by escalating afterdischarge durations (ADDs) across 30 times of optogenetic stimulations, recapitulating the classic electrical kindling dynamics27. And repetitive optogenetic kindling (30 trials) reliably provoked Racine’s stage 5 seizures in all mice from the CaMKIIα-ChR2-ligh group (Fig. 2c, n = 5 mice in CaMKIIα-ChR2-light, CaMKIIα-ChR2-no light, and CaMKIIα-EYFP-light groups, two-way ANOVA for repeated measures followed by the Scheffe’s post hoc test compare each group with every other group, F(2,14) = 163.7; CaMKIIα-ChR2-light vs. CaMKIIα-ChR2-no light, ***P < 0.001; CaMKIIα-ChR2-light vs. CaMKIIα-EYFP-light, ###P < 0.001), mirroring the electrical kindling progression.
To further examine whether vSubglu hyperactivity potentiates epileptogenesis, we combined optogenetic stimulation with hippocampal electrical-kindling model. Mice received daily electrical kindling (monophasic square-wave pulses, 400 μA, 20 Hz, 1 ms/pulse, 40 pulses/session; 10 sessions/day, 30 min intervals) paired with concurrent vSubglu activation (473 nm, 5 mW, 20 Hz, 30 s photostimulation; Fig. 2d, upper). Mice were divided into three groups: the negative control (n = 7 mice in CaMKIIα-ChR2-Light OFF group, n = 8 mice in CaMKIIα-EYFP Light ON group) and optogenetic activation (n = 8 mice in CaMKIIα-ChR2-Light ON group). Optogenetic activation of vSubglu significantly accelerated the progression of behavioral seizure stages and prolonged the ADDs compared with both control groups (Fig. 2d, two-way ANOVA for repeated measures followed by the Scheffe’s post hoc test compare each group with every other group, CaMKIIα-ChR2-Light OFF vs CaMKIIα-ChR2-Light ON, *P < 0.05, **P < 0.01; CaMKIIα-EYFP-Light ON vs CaMKIIα-ChR2-Light ON, #P < 0.05, ##P < 0.01). This acceleration was reflected as the fewer numbers of stimulations staying in FSs (Fig. 2e, one-way ANOVA followed by the Scheffe’s post hoc test compare each group with every other group; FS: CaMKIIα-ChR2-Light OFF vs. CaMKIIα-ChR2-Light ON, F(1,14) = 1.850, *P < 0.05, CaMKIIα-EYFP-Light ON vs. CaMKIIα-ChR2-Light ON, F(1,13) = 2.138, **P < 0.01; GS: CaMKIIα-ChR2-Light OFF vs. CaMKIIα-ChR2-Light ON, F(1,14) = 1.850, P > 0.05, ns, CaMKIIα-EYFP-Light ON vs. CaMKIIα-ChR2-Light ON, F(1,13) = 2.164, P > 0.05, ns) and reaching fully-kindled state (three consecutively stage-5 seizures) in the CaMKIIα-ChR2-Light ON mice (Fig. 2f, one-way ANOVA followed by the Scheffe’s post hoc test was used to compare each two groups, CaMKIIα-ChR2-Light OFF vs. CaMKIIα-ChR2-Light ON, F(1,14) = 1.485, *P < 0.05, CaMKIIα-EYFP-Light ON vs. CaMKIIα-ChR2-Light ON, F(1,13) = 1.936, *P < 0.05). Fully kindled mice also exhibited a strengthened amplitude of seizure EEGs of in the CaMKIIα-ChR2-LightON group (Fig. 2g). Finally, to test whether optogenetic activation of vSubglu affects the sGS expression, we performed optogenetic stimulation following in fully-kindled mice (Fig. 2h). Optogenetic activation of vSubglu did not alter seizure stages, ADDs or GSDs in this circumstance (Fig. 2i, one-way repeated measures ANOVA with post hoc Dunnett’s test, n = 8 mice, *P < 0.05, ns).
These results implied that optogenetic activation of vSubglu is sufficient to promote epileptogenesis in hippocampal kindling-induced TLE, but has minimal impact on the sGS.
Inhibition of glutamatergic neurons in vSub retarded epileptogenesis
Correspondingly, to evaluate the therapeutic potential of vSubglu inhibition for epileptogenesis, we employed ArchT-mediated optogenetic silencing during hippocampal kindling (Fig. 3a). Mice expressing AAV-CaMKIIα-ArchT-EGFP in vSub received concurrent electrical kindling (as described above) and yellow-light inhibition (589 nm, direct current, 5 mW, 30 s light-stimulations, immediately after each electrical stimulation, Fig. 3b). Similar to the ChR2 experiment, mice were assigned to three groups: the two negative controls (n = 8 mice in CaMKIIα-ArchT-Light Off group and CaMKIIα-EYFP-Light On group) and the optogenetic inhibition (n = 8 mice in CaMKIIα-ArchT light-on group) group. Optogenetic inhibition of vSubglu significantly retarded the progression of behavioral seizure stages and modestly shortened the ADDs compared with the controls (Fig. 3c, Two-way ANOVA for repeated measures followed by the Scheffe’s post hoc test was used to compare every two groups, CaMKIIα-ArchT- Light Off vs CaMKIIα-ArchT-Light On, *P < 0.05, ***P < 0.001; CaMKIIα-EYFP-Light ON vs CaMKIIα-ArchT-Light ON, ##P < 0.01, ###P < 0.01). This retardation was reflected as the more numbers of stimulations staying in FSs and reaching fully-kindled (three consecutively stage-5 seizures) in the CaMKIIα-ArchT-Light On group (Fig. 3d and e, One-way ANOVA followed by the Scheffe’s post hoc test was used to compare every two groups, *P < 0.05, **P < 0.01). Representative seizure EEGs in fully kindled mice of each group were shown in Fig. 3f. Next, we tested whether optogenetic inhibition of vSubglu affected sGS expression in the fully-kindled mice (Fig. 3g). Similarly, optogenetic inhibition of vSubglu had no impact on seizure stages, ADDs or GSDs (Fig. 3h, One-way repeated measures ANOVA with post hoc Dunnett’s test, n = 7 mice, ns).

a Left, diagram of optogenetic inhibition experiment in hippocampal-kindling induced TLE; right, the representative coronal images for ArchT-EGFP expression in the vSub, scale bar = 100 µm. b The scheme of optogenetic inhibition of vSubglu during epileptogenesis in hippocampal-kindling induced TLE. c Effect of optogenetic inhibition of vSubglu on the development of seizure stages (F(2,21) = 15.43) and ADD (F(2,21) = 15.43), n = 8 mice in CaMKIIα-ArchT- Light Off, CaMKIIα-ArchT- Light On group and CaMKIIα-EYFP- Light On groups, Two-way ANOVA for repeated measures followed by the Scheffe’s post hoc test compare each group with every other group, stage: CaMKIIα-ArchT-Light Off vs. CaMKIIα-ArchT-Light On, ***P < 0.001, CaMKIIα-EYFP-Light On vs. CaMKIIα-ArchT-Light On, ###P < 0.001; ADD: CaMKIIα-ArchT-Light Off vs. CaMKIIα-ArchT-Light On, *P = 0.0150; CaMKIIα-EYFP-Light On vs. CaMKIIα-ArchT-Light On, ##P < 0.0037. d Numbers of stimulations staying in FSs and GSs between the two groups, One-way ANOVA followed by the Scheffe’s post hoc test compare each group with every other group (FS: CaMKIIα-ArchT-Light Off vs. CaMKIIα-ArchT-Light On, F(1,14) = 1.158, *P = 0.0275, CaMKIIα-EYFP-light Off vs CaMKIIα-ArchT-light Off, F(1,14) = 3.851, *P < 0.0220; GS: CaMKIIα-ArchT-Light Off vs. CaMKIIα-ArchT-Light On, F(1,14) = 1.158, P = 0.1621, ns, CaMKIIα-EYFP-light Off vs CaMKIIα- ArchT-light Off, F(1,14) = 3.733, **P = 0.0092). e Numbers of kindling stimulations needed to reach fully-kindled, One-way ANOVA followed by the Scheffe’s post hoc test compare each group with every other group, CaMKIIα-ArchT-Light Off vs. CaMKIIα-ArchT-Light On, F(1,14) = 1.269, **P = 0.0014, CaMKIIα-EYFP-light Off vs CaMKIIα-ArchT-light Off, F(1,14) = 1.625, **P = 0.0015. f Representative GS EEGs recorded from ventral hippocampal CA3 in fully-kindled state from mice of the groups. g The scheme of GS expression experiment, compromised by three phases: pre, light and post. h Effects of optogenetic inhibition of vSubglu on generalized seizure stage, GS-ADDs, and GS-GSDs during GS expression, One-way repeated measures ANOVA with post hoc Dunnett’s te, st, n = 7 mice, ns.
To further confirm the anti-epileptogenesis role of inhibiting vSubglu, we utilized the chemogenetic experiments to achieve sustained silencing on vSubglu. The AAV-CaMKIIα-hM4Di-mCherry was injected in the vSub (Fig. 4a, left), and the immunohistochemistry examination confirmed the potent expression of AAV-CaMKIα-hM4Di-mCherry in vSub (Fig. 4a, right). In hippocampal kindling-induced TLE model, CNO was systematically administrated to inhibit hM4Di-expression vSub neurons (Fig. 4b). Mice were assigned to three groups: CaMKIIα-hM4Di-Saline (n = 11 mice, saline, i.p., 30 min before the first kindling once daily), CaMKIIα-mCherry-CNO and CaMKIIα-hM4Di-CNO (n = 9 mice in CaMKIIα-mCherry-CNO, n = 10mice in CaMKIIα-hM4Di-CNO, 1 mg/kg CNO i.p., in saline vehicle, 30 min before the first kindling once daily) groups. Chemogenetic inhibition of vSubglu markedly retarded the progression of behavioral seizure stages and shortened the ADDs compared with the negative controls (Fig. 4c, Two-way ANOVA for repeated measures followed by the Scheffe’s post hoc test was used to compare every two groups, CaMKIIα-hM4Di-Saline vs CaMKIIα-hM4Di-CNO, ***P < 0.001; CaMKIIα-mCherry-CNO vs CaMKIIα-hM4Di-CNO, #P < 0.05, ##P < 0.01). This retardation was further reflected as the more numbers of stimulations staying in FSs and reaching fully-kindled (three consecutively stage-5 seizures) in mice from the CaMKIIα-hM4Di-CNO group (Fig. 4d and e, One-way ANOVA followed by the Scheffe’s post hoc test was used to compare every other two group, *P < 0.05, **P < 0.01). The shortened duration and alleviated amplitude of seizure EEGs of the CaMKIIα-hM4Di-CNO group can also be observed in representative seizure EEGs in fully kindled mice (Fig. 4f). Mirroring optogenetic findings, chemogenetic silencing lacked anticonvulsant efficacy in fully kindled mice, as CNO administration did not alter GS stages, ADDs, or GSDs (Fig. 4g, Paired t test, n = 10, ns). The temporal specificity indicates that vSubglu activity is selectively involved in epileptogenesis, but not in GS maintenance.

a Left, diagram of chemogenetic inhibition experiment in hippocampal-kindling induced TLE; right, the representative coronal images for hM4Di-mCherry expression in the vSub, scale bar = 100 µm. b The scheme of chemogenetic inhibition of vSubglu during epileptogenesis in hippocampal-kindling induced TLE. c Effect of chemogenetic inhibition of vSubglu on the development of seizure stages (F(2,27) = 5.749) and ADD (F(2,27) = 6.842), n = 11 mice in CaMKIIα-hM4Di-Saline, n = 10 CaMKIIα-hM4Di-CNO, n = 9 mice in CaMKIIα-mCherry-CNO, Two-way ANOVA for repeated measures followed by the Scheffe’s post hoc test compare each group with every other group, stage: CaMKIIα-hM4Di-Saline vs. CaMKIIα-hM4Di-CNO, **P = 0.0098, CaMKIIα-mCherry-CNO vs. CaMKIIα-hM4Di-CNO, ##P = 0.0049; ADD: CaMKIIα-hM4Di-Saline vs. CaMKIIα-hM4Di-CNO, **P = 0.0039, CaMKIIα-mCherry-CNO vs. CaMKIIα-hM4Di-CNO, #P = 0.0116. d Numbers of stimulations staying in FSs and GSs between the two groups, One-way ANOVA followed by the Scheffe’s post hoc test compare each group with every other group, FS: CaMKIIα-hM4Di-Saline vs. CaMKIIα-hM4Di-CNO, F(1,19) = 1.497, *P = 0.0246, CaMKIIα-mCherry-CNO vs. CaMKIIα-hM4Di-CNO, F(1,17) = 3.121, *P = 0.0161; GS: CaMKIIα-hM4Di-Saline vs. CaMKIIα-hM4Di-CNO, F(1,19) = 1.836, P = 0.1763, ns, CaMKIIα-mCherry-CNO vs. CaMKIIα-hM4Di-CNO, F(1,17) = 3.121, ***P = 0.0009. e Numbers of kindling stimulations needed to reach fully-kindled, One-way ANOVA followed by the Scheffe’s post hoc test compare each group with every other group, CaMKIIα-hM4Di-Saline vs. CaMKIIα-hM4Di-CNO, F(1,19) = 1.231, *P = 0.0135, CaMKIIα-mCherry-CNO vs. CaMKIIα-hM4Di-CNO, F(1,17) = 5.710, *P = 0.0225. f Representative GS EEGs recorded from ventral hippocampal CA3 in fully-kindled state from mice of the groups. g Effects of chemogenetic inhibition of vSubglu on generalized seizure stage, GS-ADDs, and GS-GSDs during GS expression, n = 10 mice, Paired t test, GS-stage P = 0.8558, GS-ADD P = 0.3127, GS-GSD P = 0.3564, ns. h Upper, scheme of acute KA-induced seizure model; bottom, representative EEGs of the two groups recorded from ventral hippocampal CA3 in KA-induced seizure model. i Effects of chemogenetic inhibition of vSubglu in KA-induced seizure model on the severity of seizure stages, n = 9 mice in CaMKIIα-hM4Di-Saline, n = 10 mice in CaMKIIα-hM4Di-CNO, Mann–Whitney test, F(1,17) = 4.622, **P = 0.0058. j Effects of chemogenetic inhibition of vSubglu in KA-induced seizure model on the latency to SE, Mann–Whitney test, F(1,17) = 6.338, *P = 0.0233. k Effects of chemogenetic inhibition of vSubglu in KA-induced seizure model on the latency to GS onset, Unpaired t test, F(1,17) = 3.890, **P = 0.0012. l, m Effects of chemogenetic inhibition of vSubglu in KA-induced seizure model on the numbers of FSs and GSs, Unpaired-t test, FS: F(1,17) = 1.394, P = 0.3802, ns; GS, F(1,17) = 1.326, **P = 0.0016. n Upper, scheme of KA-induced epileptogenesis model; bottom, the representative coronal images for hM4Di-mCherry expression in the vSub, scale bar = 100 µm; right, representative EEG signature of interictal epileptic discharge (IED), FS and GS. o Kaplan–Meier curve of progression from KA-induced epiletogenesis to GS, n = 9 mice in CaMKIIα-mCherry-CNO, n = 8 mice in CaMKIIα-hM4Di-CNO, Gehan–Breslow-Wilcoxon test, Chi square = 7.596, **P = 0.0058. p–r Effect of chemogenetic inhibition of vSubglu in KA-induced epileptogenesis on the numbers of IEDs, FSs, and the time of FSs within the first consecutively 7days 8 h-recordings, Two-way ANOVA, IEDs: F(1,15) = 14.64, **P = 0.0017; FS times: F(1,15) = 2.947, P = 0.1066, ns; FSs: F(1,15) = 2.372, P = 0.1444, ns.
To assess the translational relevance of vSubglu silencing, we extended our investigation to the acute kainic acid (KA)-induced TLE model28,29. Mice expressing hM4Di in vSubglu received systemic CNO (1 mg/kg i.p., n = 10 mice) or saline (n = 9 mice) 30 min prior to intrahippocampal KA microinjection (250 ng, 0.5 μL into ventral CA3, Fig. 4h, upper). In representative EEGs of the two groups, the alleviated seizure severity in hM4Di-CNO group could be observed (Fig. 4h, bottom). The behavioral results indicated that chemogenetic inhibition of vSubglu effectively retarded the seizure progression, as indicated by the reduced highest seizure stages during the observation period (Fig. 4i, Mann–Whitney test, **P < 0.01), the prolonged latency to status epilepticus (SE) and to the onset of first GS (Fig. 4j and k, **P < 0.01, *P < 0.05), and the decreased numbers of GS (Fig. 4m, Unpaired-t test, **P < 0.01). Interestingly, chemogenetic inhibition of vSubglu had no effect on decreasing the numbers of FSs (Fig. 4l, Unpaired t-test, P = 0.3802, ns). To further evaluate the long-term impact of chemogenetic inhibition of vSubglu on epileptogenesis, we established the KA-induced chronic spontaneous seizure model (Fig. 4n). We defined the epileptogenesis latent period as the time from the day of KA insult to the occurrence of the first recorded generalized seizure by conducting 8 h/daily continuous EEG monitoring28,30. Strikingly, chemogenetic inhibition of vSubglu markedly prolonged the latent period (Fig. 4o, n = 9 mice in CaMKIIα-mCherry-CNO, n = 8 mice in CaMKIIα-hM4Di-CNO, Gehan–Breslow-Wilcoxon test, **P < 0.01). Moreover, during the first seven days of EEG recordings, the rates of interictal epileptiform discharges (IEDs) were significantly decreased following chemogenetic inhibition (Fig. 4p, Two-way ANOVA, **P < 0.01), indicating decreased epileptic network hyperexcitability. While chemogenetic manipulation did not significantly alter either the number or duration of FSs during the first seven days recording session (Fig. 4q and r, Two-way ANOVA, ns).
Taken together, these results demonstrated that selective modulation of vSubglu bidirectionally affect epileptogenesis in TLE, represented by accelerated and retarded manners through activation and inhibition.
The glutamatergic neural circuit from vSub-ATN were responsible for the participant of vSub in epileptogenesis
Anatomical mapping studies have established the vSub as the principal output hub of the ventral hippocampus, comprising projection-defined pyramidal neuron subpopulations with distinct molecular and functional heterogeneity7,9,17,18,19,31. To determine the downstream targets of vSubglu that are relevant to epileptogenesis, we performed anterograde circuit tracing using AAV-CaMKIIα-EYFP (Fig. 5a). Histological verification confirmed the viral expression within vSubglu somata (Fig. 5a, bottom). Anterograde tracing results showed an abundant vSubglu terminals distributed in the accumbens nucleus (Acb), bed nucleus of the stria terminalis (BST), lateral septum (LS), anterior thalamic nucleus (AV, anteroventral thalamic nucleus; AM, anteromedial thalamic nucleus, ATN comprised AV and AM), the anterior hypothalamic nucleus (AHN), mammillary nucleus (MM) and basolateral amygdala (BLA) (Fig. 5b). Guided by prior pathophysiological evidence implicating the ATN and BLA in temporal lobe epileptogenesis32,33,34,35, and the negative association of the LS with seizure propagation36,37 we tested these three vSubglu projecting-regions for subsequent functional validation.

a Upper, scheme of virus-assisted anterograde tracing on vSubglu; bottom, representative image of EYFP+ in vSub, scale bar = 200 µm. b Typical EYFP+ vSubglu projecting fibers in the downstream brain regions, distributed in Acb, BST, LS, AV, AM, AHN, MM, and BLA, respectively, scale bar = 200 µm. (Abbrs. vSub ventral subiculum, DG dentate gyrus, LEC lateral entorhinal cortex, Acb nucleus accumbens, BST bed nucleus of the stria terminalis, LS lateral septum, AV anteroventral thalamic nucleus, AM anteromedial thalamic nucleus, AHN anterior hypothalamic nucleus, MM medial mammillary nucleus, BLA basolateral amygdala).
To further elucidate the potential downstream regions mediating the anti-epileptogenesis effect of the vSubglu neurons, we performed projection-specific optogenetic inhibition in the hippocampal kindling model. AAV-CaMKIIα-ArchT was stereotaxically injected into the vSub, with optic fibers positioned to silence efferent terminals in ipsilateral ATN, BLA or LS by 589 nm photostimulation (Fig. 6a1–c1). In the hippocampal kindling model, optogenetic inhibition of the terminals of ATN projected vSubglu successfully retarded the progression of seizure stages (Fig. 6c2, Two-way ANOVA, ***P < 0.001) and shortened the ADDs (Fig. 6c3, n = 7 mice in both light-off and light-on groups, Two-way ANOVA, ***P < 0.001), while optogenetic inhibition of BLA or LS projecting-terminals had no such effect (Fig. 6a2–b5, vSub-BLA: n = 8 mice in Light-OFF and Light-ON; vSub-LS: n = 7 mice in Light-OFF and Light-ON P > 0.05, ns). This retardation caused by inhibiting ATN projecting-terminals was also reflected as more stimulations staying in FSs and reaching fully-kindled state (Fig. 6c4–c5, Unpaired t-test, *P < 0.05) in the light-on group. Notably, terminal inhibition lacked anticonvulsant efficacy in fully kindled mice among the three circuits (Fig. S4a–c, One-way repeated measures ANOVA with post hoc Dunnett’s test, ns, n = 8 mice in vSub-BLA, n = 9 mice in vSub-LS, n = 7 mice in vSub-ATN, P > 0.05, ns), which was in line with the vSubglu inhibition.

a1–c1 Diagram of optogenetic inhibition in BLA, LS, and ATN, left; viral expression and optic fibers in BLA, LS, and ATN, respectively. a2–c2 Effect of optogenetic inhibition of vSubglu-BLA, LS, and ATN on the development of seizure stage, Two-way ANOVA, vSub-BLA: n = 8 mice in Light-OFF and Light-ON, Light ON vs. Light OFF, F(1,14) = 0.04214, P = 0.8403, ns; vSub-LS: n = 7 mice in Light-OFF and Light-ON, Light ON vs. Light OFF, F(1,12) = 0.04998, P = 0.4931, ns; vSub-ATN: n = 7 mice in Light-OFF and Light-ON, F(1,12) = 24.48, ***P = 0.0003. a3–c3 Effect of optogenetic inhibition of vSubglu-BLA, LS, and ATN on the development of ADDs, Two-way ANOVA, vSub-BLA: Light ON vs. Light OFF, F(1,14) = 1.036, P = 0.3260, ns; vSub-LS: Light ON vs. Light OFF, F(1,12) = 0.05663, P = 0.8159, ns; vSub-ATN: F(1,12) = 11.85, **P = 0.0049. a4–c4 Numbers of stimulations needed to fully-kindled, Unpaired t-test, vSub-BLA: F(1,14) = 1.029, P = 0.1236, ns; vSub-LS: Light ON vs. Light OFF, F(1,12) = 1.838, P = 0.5232, ns; vSub-ATN: F(1,12) = 2.773, *P = 0.0146. a5–c5 Comparison of numbers of stimulations staying in FSs and GSs, One-way ANOVA with post hoc Dunnett’s test, vSub-BLA FS: F(1,14) = 2.446, P = 0.7768, ns, GS: F(1,14) = 2.446, P = 0.2906, ns; vSub-LS FS: F(1,12) = 1.866, P = 0.3558, ns, GS: F(1,12) = 2.521, P = 0.9628, ns; vSub-ATN FS: F(1,12) = 6.061, *P = 0.0070, GS: F(1,12) = 5.838, P = 0.3298, ns.
Building on established role of the ATN as a canonical subicular output hub8,38, our previous study demonstrated that optogenetic inhibition of the dSub-ATN circuit or the ATN glutamatergic neurons (ATNglu) alleviates seizure in both epileptogenesis and sGS expressions16. However, whether ATNglu contribute to the anti-epileptogenic effect mediated by vSubglu is still unclear. Firsly, we performed retrograde tracing using cholera toxin subunit B conjugated with Alexa Fluor-555 in ATN (Fig. S6a). As expected, numerous ATN-projecting neurons were located within the vSub, confirming a direct monosynaptic projection from the vSub to the ATN (Fig. S6b). Thus, to examine the functional relevance of this neural circuit, we injected AAV-CaMKIIα-ChR2 into the vSub and implanted a fiber optic on the ATN to selectively activate vSubglu -projecting terminals. At the same time, AAV-CaMKIIα-hM4Di were injected into ATN for inhibiting ATNglu (Fig. 7a). We named the vSubEYFP-ATN-hM4DiSaline as the negative control group, the vSubChR2- ATN-hM4DiSaline as the positive control group, and the vSubChR2-ATN-hM4DiCNO as the experiment group. CNO (1 mg/kg, i.p., in saline vehicle, 30 min before the first kindling session each day) was administered to inhibit ATNglu, whereas optical stimulation (20 Hz, 473 nm, 5 mW, 30 s light-stimulations, immediately after each electrical stimulation) was applied to activate vSubglu projecting terminals to the ATN. In hippocampal kindling-induced TLE model, optogenetic activation of vSubglu-ATN projecting terminals significantly accelerated the progression of behavioral seizure stages and prolonged the ADDs, while CNO administration reverse that effect (Fig. 7b and c, Two-way ANOVA for repeated measures followed by the Scheffe’s post hoc test was used to compare every two groups, n = 6 mice in EYFP-Saline, ChR2-Saline and ChR2-CNO, EYFP-Saline vs ChR2-CNO, **P < 0.01, *P < 0.05; ChR2-Saline vs ChR2-CNO, ##P < 0.01, #P < 0.05 compared with ChR2-Saline). This effect was further reflected as the reduced numbers of stimulations staying in FSs and reaching the fully-kindled state in the vSubChR2- ATN-hM4DiSaline group, compared with the other two groups (Fig. 7d and e, One-way ANOVA followed by the Scheffe’s post hoc test compare each group with every other group, **P < 0.01, *P < 0.05). While mice experienced CNO administration needed more stimulations to fully-kindled state and staying in FSs. In accordance with previous results, optogenetic activation of vSubglu-ATN and chemogenetic inhibition of ATNglu at the same time did not significantly affect sGS expression (Fig. 7f, One-way repeated measures ANOVA with post hoc Dunnett’s test, n = 8 mice, P > 0.05, ns).

a Scheme of the optogenetic activation of vSubglu and chemogenetic inhibition of ATNglu experiment in kindling-induced TLE. b, c Effect of chemogenetic inhibition on ATNglu and optogenetic activation on vSubglu-ATN on the development of seizure stage and ADDs, Two-way ANOVA for repeated measures followed by the Scheffe’s post hoc test compare each group with every other group, n = 6 mice in EYFP-Saline, ChR2-Saline, and ChR2-CNO groups. Stage: EYFP-Saline vs. ChR2-Saline, F(1,10) = 16.28, **P = 0.0024, ChR2-Saline vs ChR2-CNO, F(1,10) = 16.17, ##P = 0.0024, ChR2-CNO vs. EYFP-Saline, F(1,10) = 2.006, P = 0.1871, ns; ADD: EYFP-Saline vs. ChR2-Saline, F(1,10) = 5.102, *P = 0.0475, ChR2-Saline vs ChR2-CNO, F(1,10) = 9.198, #P = 0.0126, ChR2-CNO vs. EYFP-Saline, F(1,10) = 0.0007, P = 0.9782, ns. d Numbers of stimulations staying in FSs and GSs, One-way ANOVA followed by the Scheffe’s post hoc test compare each group with every other group. FS: EYFP-Saline vs. ChR2-Saline, F(1,10) = 3.685, *P = 0.0178, ChR2-Saline vs ChR2-CNO, F(1,10) = 6.638, **P = 0.0032, ns, ChR2-CNO vs. EYFP-Saline, F(1,10) = 1.801, P = 0.2807, ns; GS EYFP-Saline vs. ChR2-Saline, F(1,10) = 4.455, **P = 0.0041, ChR2-Saline vs ChR2-CNO, F(1,10) = 1.15, P = 0.1041, ns, ChR2-CNO vs. EYFP-Saline, F(1,10) = 3.875, P = 0.1454, ns. e Comparison of numbers of stimulations needed to fully-kindled, One-way ANOVA followed by the Scheffe’s post hoc test compare each group with every other group, EYFP-Saline vs. ChR2-Saline, F(1,10) = 4.455, **P = 0.0041, ChR2-Saline vs ChR2-CNO, F(1,10) = 1.15, P = 0.1041, ns, ChR2-CNO vs. EYFP-Saline, F(1,10) = 3.875, P = 0.1454, ns. f Effect of optogenetic inhibition on vSubglu circuits in seizure stage, GS-ADDs and GS-GSDs during GS, One-way repeated measures ANOVA with post hoc Dunnett’s test, P > 0.05, ns, n = 8 mice in Saline and CNO group.
Taken together, these results indicated that ATN serves as a crucial downstream region of the vSubglu neurons in epileptogenesis.
Discussion
The vSub has been rarely investigated in TLE, while how its neurons contribute to epileptogenesis and its related neural circuits remained unclear. Here, utilizing in vivo calcium imaging, optogenetics, chemogenetics, and anterograde viral tracing in TLE mice models, we have obtained some novel findings: (1) hippocampal-kindling induced TLE innervated hyperactivity of vSubglu neurons; (2) vSubglu neurons bidirectionally modulated epileptogenesis, but not expressed GS in TLE- activation accelerates epileptogenesis while inhibition retards it; (3) the vSubglu-ATN projecting terminals predominantly mediated this anti-epileptogenesis effect. These findings identify the vSubglu-ATN as a potential circuit-level target for therapeutic intervention on epileptogenesis in TLE.
The subiculum serves as a critical epileptogenic hub in TLE, integrating hippocampal hyperexcitability and propagating ictal activity to downstream circuits11,26,39. Physiologically, the dorsal and ventral subiculum compose functionally distinct cell populations, highlighting the heterogeneity of different subicular subregions through the spatial aspect40,41,42. However, previous studies including ours have mainly focused on the dorsal subicular neural circuits in TLE13,16,43,44. Here, we firstly found that direct optogenetic activation of vSubglu reliably mimicked or accelerated the development of hippocampal-kindling induced TLE. This evidence pointed out the critical role of vSubglu in epileptogenesis. Furthermore, we interestingly realized that all effective manipulations on vSubglu focused on FSs during epileptogenesis, as optogenetic or chemogenetic inhibition on vSubglu increased the stimulations staying FSs, but not GSs. These finding suggested that the contribution of vSubglu is restricted in the early stage in epileptogenesis. Resonating with previous findings, in pilocarpine-induced spontaneous seizure model, earliest epileptic activities were most frequently recorded in vSub21. Similarly, long-lasting and specific type of interictal discharges were displayed in vSub before the first seizure occurrence in rats experienced pro-convulsant 4-aminopyridione administration22. Unlike vSub, both activation and inhibition of pyramidal neurons in the dSub would not only influence epileptogenesis, but also attenuate the severities of GS16. These distinct manners may imply that the epileptic activities usually originated from the ventral hippocampus during the epileptogenesis, while the dorsal hippocampus may be more susceptible in the generalization of epileptic activities. Thus, our results demonstrate that vSubglu contribute to epileptogenesis in TLE.
With the great development of neuroscientific tools, cell-type specific modulation emerges as a promising therapy in preventing epilepsy. It seems that pyramidal neurons may be a more ideal target for retarding epileptogenesis than interneurons in vSub. As previous studies proved, different sub-types of vSub interneurons may possess distinct effects in TLE. Chronically inhibition of GABA release from parvalbumin interneurons in the vSub (vSubPV+) induced hyperactivity and spontaneous recurrent seizures, assigned its inhibitory role in epilepsy23. Moreover, selectively inhibiting GABA release from Somatostatin (SST) expressing interneurons in the vSub (vSubSST+) to impair feed-back inhibition on pyramidal neurons promoted interictal hyperactivity and spontaneous seizures, which also indicated the inhibitory role of vSubSST+24. Inversely, the vasoactive intestinal polypeptide interneurons in the vSub (vSubVIP+) showed disinhibitory role in epilepsy, as that activation of vSubVIP+ was enough to increase epileptiform activity25. Although single-cell RNA sequencing and transcriptomic classification have demonstrated that vSubglu also contains different subtypes17,18,19. This heterogeneity within the subiculum may partially explained the diverse effect and innervation in epilepsy or other physiology and pathology36,45. But in TLE, we found that the vSubglu was hyperactivated during seizures and inactivation of the whole vSubglu populations could retard epileptogenesis. Specifically in the KA-induced model of chronic spontaneous seizures, chronic silencing vSubglu sensationally extended the latent phase of epileptogenesis. This highlighted that manipulations targeting on vSubglu may be a potential therapy in interfering the epileptogenesis, ignoring the heterogeneous cell-type within vSubglu. While in lack of genetically-encoded vSub-specific markers, it’s not feasible to selectively label and manipulate the entire anatomically defined vSub. Although the vSub was our intended target region, the viral diffusion or expression spread and transfect into the vSub-adjacent region- such as DG and vCA1, which cannot be entirely expelled these contributions to the effect.
At the neural circuitry level, we also proved that vSubglu afferent to distinct cortical and subcortical regions, including Acb, LS, AV, AM, AHN, BLA and MM, which is consistent to previous anatomical studies18,19,46. And the vSubglu-ATN circuit mainly contributed to the anti-epileptogenesis efficacy of vSubglu, whereas the vSubglu-BLA circuit showed no effect neither in epileptogenesis nor sGS. This effect was partly similar to dSubglu, as selective activation or inhibition of the dSub-ANT circuit also promotes or alleviates hippocampal seizures in both epileptogenesis and sGS expression states16. This phenomenon may highlight the critical role of the ATN in epilepsy, as epileptic activities arising from both the dorsal and ventral hippocampus may converge in the ATN and then propagate to the whole brain32,47,48,49,50. Indeed, ATN has been considered as the potential ideal target of deep brain stimulation for patients with drug resistant TLE33,51. These findings imply that, in identifying precise therapeutic targets for TLE, attention may need to shift from the hippocampus to secondary propagation regions, including the thalamus and other limbic structures. As for the amygdala, another susceptible brain regions in TLE34,35,52, previous studies have reported the enhanced plasticity from BLA projecting to vSub in pilocarpine-induced TLE mice53. In contrast, our study found that the vSub-BLA circuit does not participate in TLE. One possible explanation for this discrepancy may be the different models used in the two studies. On the other hand, it can be speculated that a feed-back loop exists between vSub and BLA, which may be modulated by epileptic activities but do not directly mediate epileptogenesis. Notably, given the heterogeneity within vSub, future studies should employ genetically encoded markers or Cre-driver mouse lines to dissect vSub circuits more precisely and to clarify their specific roles in epilepsy.
In sum, this research provides the first evidence about vSubglu participate in early epileptogenesis via the downstream ATN circuit in TLE. These findings not only supplement the map of subiculum-related circuitry in epileptogenesis, but also emphasize vSubglu-ATNglu as a potential therapeutic target for intervention epileptogenesis in TLE.
Methods
Animals
In the current study, 8 weeks old male C57/6J (wild-type) mice were used. Mice were house-grouped in a 12 h light/dark cycle and allowed to water and chow ad libitum. All tests were conducted during the light cycle. All procedures were approved by the guidelines of the Animal Advisory Committee of Zhejiang Chinese Medical University (approved number: 20230306-10) and in completely accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Stereotaxic surgeries and electrode/optical fiber implantation
Stereotaxic surgery was performed strictly as previously described16,54. Mice were anesthetized by sodium pentobarbital (50 mg/kg, i.p.) and then head fixed into the stereotaxic apparatus (RWD Life Science, China). Viruses were unilaterally injected into vSub (AP: −3.6 mm; ML: −3.0 mm; DV: 4.7 mm; relative to bregma) using a micro-glass (Sutter Instrument, USA, O.D. 1.5 mm, I.D. 0.86 mm) installed on a 1 μl pipette (Microliter Syringes, China, O.D. 0.2 mm). The pipette was remained in the place for another 5 min after injection, leaving the virus well-absorbed. Next, the scalp was shuttered and the mice were placed on a heated blanket (maintained at 30 °C) for recovery. 3–4 weeks are needed for fully viral expression.
Implantations were applied after viral expression. For electrical kindling and EEG monitoring, twisted-bifilar stainless electrodes (795500, A.M System, USA) were implanted into the right ventral hippocampal CA3 (AP: −2.9 mm; ML: −3.2 mm; DV: −3.2 mm). For optogenetics stimulation, optic fiber cannula (200 μm Core, 6.0 mm length, Inper, China) was located over the vSub (AP: −3.6 mm; ML: −3.0 mm; DV: 4.7 mm; relative to bregma), ATN (AP: −0.5 mm; ML: −0.7 mm; DV: −3.0 mm), BLA (, AP: −1.6 mm; ML: −2.5 mm; DV: −5.0 mm), LS (AP: +1.0 mm; ML: −0.5 mm; DV: −3.5 mm). Then four screws were cemented into the skull, two of them were placed over the cortex and cerebellum as reference and ground electrodes. One-week post-surgery recovery were kept for mice. The site of electrode and viral expression were post-verified in all mice at the end of every experiment. Only the mice with correct placement of electrodes, optic fibers and restricted viral expression were enrolled into analysis.
Viral vectors
For optical fiber photometry in vivo, AAV-CaMKIIα-GCaMP6s (2.82E + 12 vg/ml, Taitool, S0229-9, AAV2/9) was injected into the vSub, injection volume was 200 nL, as 50 nL/min. Two weeks later, an optic fiber (6 mm, Inper) was implanted above the vSub for photometry recording.
For optogenetic manipulation, 100 nL (20 nL/min) AAV-CaMKIIα-ArchT-EGFP (5.5E + 12 vg/ml, BrainVTA, PT0233, AAV2/9) or AAV-CaMKIIa-hChR-EYFP (4.4E + 12 vg/ml, BrainVTA, PT0296, AAV2/9) was injected into the vSub for selectively inhibiting or activating vSubglu respectively. AAV-CaMKII-EYFP (4.6E + 12 vg/ml, BrainVTA, PT0298, AAV2/9) was injected into the vSub for anterograde tracing. Three weeks later, an optic fiber was located 0.01 mm above the vSub, ATN or BLA in different batches of mice.
For chemogenetic manipulation, 100 nL (20 nL/min) AAV-CaMKIIα-hM4Di-mCherry (4.04E + 12 vg/ml, Obio Technology, H5778) was injected into the vSub. After 3 weeks viral transfection, designer receptors exclusively activated by designer drugs (DREADDs) actuator CNO (1 mg/kg, i.p., Abcam, ab141704) was administrated for sustainable silencing vSubglu 30 min before experiments16,27,29,55.
For retrograde tracing afferent connections between ATN and vSub, cholera toxin subunit B conjugated to Alexa-555 (CTB 555, Thermo Fisher, C34776, diluted in PBS solution of 1 µg/µL, 100 nL) was injected into ATN. 7 days after injections, mice were perfused for histology.
All virus were injected into the unilateral brain region (ipsilateral with the kindling focus). Post-verification of viral expression was observed under Olympus VS120.
Optical fiber photometry
Optical fiber photometry was conducted by a genetically-encoded calcium indicator protein GcaMP6 to capture transient neuronal activity during electrical kindling as previously described29. The optic fiber photometry system (Thinkertech, China) includes a 488 nm diode laser (OPIS 488LS; Coherent), a dichroic mirror (MD498, Thorlabs), an optic fiber (0.23 mm Core, 0.37 NA, Inper, China) coupled with a ×10 lens (Olympus) and fiber launch (Thorlabs). The laser intensity was controlled ranging from 0.01 to 0.05 mW. The fluorescence GCaMP6 was bandpass filtered and collected by a photomultiplier tube and converted into the analog voltage signals (sample rate 100 Hz). And the voltage signals were further analyzed in MATLAB. The fluorescence changes (ΔF/F) were calculating (F–F0). In this experiment, we collect a 200 s voltage signals, and kindled at the 100 s. And the F0 was averaged from the 90 to 100 s as baseline signals; the later 100 s signals were defined as F. The data were presented as time-event plot and heatmap. The peak value and time point of peak value in all enrolled trials were read and recorded.
The temporal delay index was calculated as the peak value divided its correspondingly time point. This ratio can be explained as “temporal delay” or “temporal synchrony.” It describes the relative timing between the peak of cellular calcium activity and the onset of epileptic brain activity. Essentially, it reflects the degree of temporal relationship or synchronization between the changes of fluorescent intensity and seizure activity.
Hippocampal kindling mouse model
The hippocampal kindling mouse model was conducted according to our previously studies16,29,43,56. After 1-week post-surgery recovery, the after-discharge threshold (ADT) was determined (monophasic square-wave pulses, 20 Hz, 1 ms/pulse, 40 pulses) by a constant current stimulator (SEN-7203, SS202J; Nihon Kohden, Japan) for each mouse, and the EEG was recorded with a 100 Hz digital amplifier (Neuronscan System, USA). The stimulation intensity began at 40 μA, and increased in 20 μA steps every 1 min till ≥5 s afterdischarge was elicited. This minimal intensity was deemed as the ADT, and only mice with ADTs less or equal than 200 μA were conducted to the following experiments.
All mice received 10 kindling stimulations (monophasic square-wave pulses, 400 μA, 20 Hz, 1 ms/pulse, 40 pulses, 30 min interval). Seizure severity was evaluated by behavioral seizure stage and after-discharge duration (ADD). The seizure stage was measured by a well-trained but unaware of the group observer. Seizure severity was modified according to the Racine’s scale: (1) facial movement; (2) head nodding; (3) unilateral forelimb clonus; (4) bilateral forelimb clonus and rearing; (5) rearing and falling57. Stage 1–3 seizures were indicated as focal seizures (FSs), while stage 4–5 seizures were regarded as generalized seizures (GSs). Once mice exhibited three consecutive stage-5 seizures were regarded as fully kindled. Then, we utilized a suprathreshold current (200 μA, 20 Hz, 1 ms/pulse, 40 pulses, 30 min interval) to induce secondary generalized seizures (sGSs). We classified the first three stimulations as base-state (pre), the later three stimulations as the modulation (optogenetics or chemogenetics) and the last three stimulations as the post-sGS state (post). In sGS expression experiment, seizure stage, ADDs, and generalized after-discharge durations (GSDs) were obtained58,59.
Intrahippocampal KA model
To evaluate the effect on acute ictogenesis, the acute intrahippocampal KA model was established according to our previous studies29,48,60. Briefly, after viral expression and implantation surgery, freely moving mice were intrahippocampally microinjected with KA (250 ng, 0.5 µL) using an Ultra Micro Pump (160494 F10E, WPI) at a rate of 100 nl/min using a 1 μl micro-glass pipette of 5 min via the guide cannula. The EEG was recorded for 90 min after KA injection using a PowerLab system (filtered with a 50 Hz notch, AD Instruments, Australia) at a sampling rate of 1 kHz. Saline or CNO (1 mg/kg, i.p.) was administrated 30 min before KA injection. Status epilepticus (SE) onset was defined as the time when repetitive and continuous epileptic ictal discharges occur in EEGs28,61. Seizure severity was modified according to the Racine’s scale: (1) facial movement; (2) head nodding; (3) unilateral forelimb clonus; (4) bilateral forelimb clonus and rearing; (5) rearing and falling. Stage 1–3 seizures were indicated as focal seizures (FSs), while stage 4–5 seizures were regarded as generalized seizures (GSs). KA injection induced high-amplitude spike waves in the EEG and gradually elicited FSs, GSs, and SE within tens of minutes. The seizure activity and EEGs were monitored for 90 min.
To better understand the effect on chronic epileptogenesis, chronic intrahippocampal KA model was used, which is characterized by a latent period following the initial KA attack until the appearance of spontaneous recurrent seizures (SRSs)30,62. Firstly, KA (250 ng, 0.5 µL) was stereotaxically injected into the right dorsal hippocampus to induce behavioral seizures and neuropathological lesions. On the next day, EEG was continuously recorded infree-moving mice by PowerLab system (filtered with a 50 Hz notch, AD Instruments, Australia) at a sampling rate of 1 kHz. For chronic epileptogenesis experiments, CNO was administrated in daily drinking water (5 mg/200 mL, with 0.5% saccharine)63,64,65. Interictal discharges was expressed as a fast monophasic electroencephalographic transient, short duration (<250 ms) and high amplitude (>1 mV)56,66,67. The paroxysmal discharge events with duration >10 s, amplitude >3 times versus baseline and spike frequency >2 Hz were defined as nonconvulsive FS. While convulsive motor seizures (typical Racine’s stage 4–5 seizures) with a continuous epileptic event >30 s and post-depression after seizure terminated were termed as GSs. The epileptogenesis period was defined as the time from the day of KA attack to the first seizure recorded28,30. Continuous EEG monitoring was conducted 8 h/day until the first GS was observed in each mouse.
Optogenetics and chemogenetics intervention
Laser (blue light, 473 nm or yellow light, 589 nm) was delivered by a 200 μm diameter optical fiber (0.23 mm Core, 0.37 NA, Inper, China), which was controlled by a Master-8 commutator (AMPI, Israel). The laser intensity was controlled in 5–10 mW. In optogenetic activation section, pulsed blue light stimulation was delivered immediately after 2-s kindling (20 Hz, 10 ms duration, 600 pulses, 30 s). In optogenetic inhibition section, 30 s direct yellow light was delivered immediately light stimulation after 2 s kindling.
Chemogenetics modulation was performed as described in our previous studies16,29,43,68. Briefly, 1 mg/kg clozapine-N-oxide (CNO) was administrated for chemogenetic inhibition in both kindling and acute KA-induced epilepsy models, which can increase intracellular Gi signaling for hm4Di-positive cells and thus decrease neuronal activity55,69. Previous studies have reported that the brain bioavailability of CNO lasts about 5–10 h, sufficient to maintain its efficacy throughout our experiments64,70,71.
Immunohistochemistry
Mice were deeply anesthetized with 100 mg/kg sodium pentobarbital, followed by intracardially perfusion with 0.9% saline and freshly prepared 4% paraformaldehyde (PFA). In c-Fos staining, mice were sacrificed 1.5 h after FS or GS. Dehydrated brains were cut into slices (25 μm) on a freezing microtome (Thermo Scientific, USA). Brain slices were washed 3 time with PBS, then incubated in a blocker solution (1% bovine serum albumin and 3% Triton X-100 in 0.05 M PBS) containing 5% donkey serum kept for 2 h at room temperature. After that, the primary antibody anti-c-Fos (1:1000, ab208942, mouse monoclonal, Abcam) were incubated overnight in a 4 °C refrigerator. In the next day, slices were washed for 3 times in PBS and incubated in a solution containing the secondary conjugated antibody (Alexa Fluor 488, donkey anti-mouse IgG H&L, 1:1000, ab150105, Abcam) for 2 h at room temperature. Last, slices were washed and mounted with DAPI (Vectashield Mounting Media, Vector Labs). Images were captured by a confocal microscopy (SP8, Leica). Images were analyzed in ImageJ software (1.52a version). C-Fos immunactivity-positive cell counts were quantified in corresponding nucleus. Three brain slices were averaged as each mouse.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism software (Version 9.1.1). All data were presented as the mean ± S.E.M. The number of experimental replicates (n) is indicated in figure legend and refers to the number of experimental subjects in each experimental condition. Normal distribution was firstly tested, then matched with parametric or non-parametric tests. The differences between the two groups were analyzed using the student’s t test. For multiple comparisons, the one-way or two-way ANOVA followed by post hoc Tukey test was used. A two-tailed P-value of < 0.05 was considered statistically significant. Survival curves were compared using the Gehan–Breslow–Wilcoxon test. Descriptive statistics and row data for all groups were provided in supplementary data.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. The datasets have been deposited in the Zenodo (https://doi.org/10.5281/zenodo.17785712).
References
Thijs, R. D., Surges, R., O’Brien, T. J. & Sander, J. W. Epilepsy in adults. Lancet 393, 689–701 (2019).
Wang, Y. & Chen, Z. An update for epilepsy research and antiepileptic drug development: Toward precise circuit therapy. Pharmacol. Ther. https://doi.org/10.1016/j.pharmthera.2019.05.010 (2019).
Loscher, W., Klitgaard, H., Twyman, R. E. & Schmidt, D. New avenues for anti-epileptic drug discovery and development. Nat. Rev. Drug Discov. 12, 757–776 (2013).
Kanner, A. M. & Bicchi, M. M. Antiseizure medications for adults with epilepsy: a review. Jama 327, 1269–1281 (2022).
Klein, P., Kaminski, R. M., Koepp, M. & Löscher, W. New epilepsy therapies in development. Nat. Rev. Drug Discov. https://doi.org/10.1038/s41573-024-00981-w (2024).
Bialer, M. & White, H. S. Key factors in the discovery and development of new antiepileptic drugs. Nat. Rev. Drug Discov. 9, 68–82 (2010).
O’Mara, S. The subiculum: what it does, what it might do, and what neuroanatomy has yet to tell us. J. Anat. 207, 271–282 (2005).
Fei, F., Wang, X., Wang, Y. & Chen, Z. Dissecting the role of subiculum in epilepsy: research update and translational potential. Prog. Neurobiol. 201, 102029 (2021).
Fanselow, M. S. & Dong, H.-W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65, 7–19 (2010).
Strange, B. A., Witter, M. P., Lein, E. S. & Moser, E. I. Functional organization of the hippocampal longitudinal axis. Nat. Rev. Neurosci. 15, 655–669 (2014).
de Guzman, P. et al. Subiculum network excitability is increased in a rodent model of temporal lobe epilepsy. Hippocampus 16, 843–860 (2006).
Benini, R. & Avoli, M. Rat subicular networks gate hippocampal output activity in an in vitro model of limbic seizures. J. Physiol. 566, 885–900 (2005).
Zhong, K. et al. Wide therapeutic time-window of low-frequency stimulation at the subiculum for temporal lobe epilepsy treatment in rats. Neurobiol. Dis. 48, 20–26 (2012).
Cohen, I., Navarro, V., Clemenceau, S., Baulac, M. & Miles, R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 298, 1418–1421 (2002).
Huberfeld, G. et al. Glutamatergic pre-ictal discharges emerge at the transition to seizure in human epilepsy. Nat. Neurosci. 14, 627–634 (2011).
Fei, F. et al. Discrete subicular circuits control generalization of hippocampal seizures. Nat. Commun. 13, 5010 (2022).
Cembrowski, M. S. et al. The subiculum is a patchwork of discrete subregions. eLife 7, https://doi.org/10.7554/eLife.37701 (2018).
Wee, R. W. S. & MacAskill, A. F. Biased connectivity of brain-wide inputs to ventral subiculum output neurons. Cell Rep. 30, 3644–3654.e3646 (2020).
Ding, S. L. et al. Distinct transcriptomic cell types and neural circuits of the subiculum and prosubiculum along the dorsal-ventral axis. Cell Rep. 31, 107648 (2020).
Cembrowski, M. S. et al. Dissociable structural and functional hippocampal outputs via distinct subiculum cell classes. Cell 173, 1280–1292.e1218 (2018).
Toyoda, I., Bower, M. R., Leyva, F. & Buckmaster, P. S. Early activation of ventral hippocampus and subiculum during spontaneous seizures in a rat model of temporal lobe epilepsy. J. Neurosci. 33, 11100–11115 (2013).
Lévesque, M., Salami, P., Behr, C. & Avoli, M. Temporal lobe epileptiform activity following systemic administration of 4-aminopyridine in rats. Epilepsia 54, 596–604 (2013).
Drexel, M. et al. Selective silencing of hippocampal parvalbumin interneurons induces development of recurrent spontaneous limbic seizures in mice. J. Neurosci. 37, 8166–8179 (2017).
Drexel, M., Rahimi, S. & Sperk, G. Silencing of hippocampal somatostatin interneurons induces recurrent spontaneous limbic seizures in mice. Neuroscience 487, 155–165 (2022).
Rahimi, S. et al. The role of subicular VIP-expressing interneurons on seizure dynamics in the intrahippocampal kainic acid model of temporal lobe epilepsy. Exp. Neurol. 370, 114580 (2023).
Aggleton, J. P. & Christiansen, K. The subiculum: the heart of the extended hippocampal system. Prog. Brain Res. 219, 65–82 (2015).
Zhang, X.-F. et al. Ventral subiculum promotes wakefulness through several pathways in male mice. Neuropsychopharmacology 49, 1468–1480 (2024).
Lévesque, M. & Avoli, M. The kainic acid model of temporal lobe epilepsy. Neurosci. Biobehav. Rev. 37, 2887–2899 (2013).
Chen, B. et al. A disinhibitory nigra-parafascicular pathway amplifies seizure in temporal lobe epilepsy. Nat. Commun. 11, 923 (2020).
Williams, P. A. et al. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J. Neurosci. 29, 2103–2112 (2009).
Tang, H. et al. Brain-wide map of projections from mice ventral subiculum. Neurosci. Lett. 629, 171–179 (2016).
Schaper, F. et al. Single-cell recordings to target the anterior nucleus of the thalamus in deep brain stimulation for patients with refractory epilepsy. Int. J. Neural Syst. 29, https://doi.org/10.1142/s0129065718500120 (2019).
Fisher, R. et al. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia 51, 899–908 (2010).
Zeicu, C. et al. Altered amygdala volumes and microstructure in focal epilepsy patients with tonic-clonic seizures, ictal, and post-convulsive central apnea. Epilepsia 64, 3307–3318 (2023).
Aroniadou-Anderjaska, V., Fritsch, B., Qashu, F. & Braga, M. F. Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy. Epilepsy Res. 78, 102–116 (2008).
Mu, M.-D. et al. A limbic circuitry involved in emotional stress-induced grooming. Nat. Commun. 11, https://doi.org/10.1038/s41467-020-16203-x (2020).
Wirtshafter, H. S. & Wilson, M. A. Lateral septum as a nexus for mood, motivation, and movement. Neurosci. Biobehav. Rev. 126, 544–559 (2021).
o’Mara, S. The subiculum what it dose what it might do and whatneuroanatomy has yet to tell us. J. Anat. 207, 271–282 (2005).
Kliot, M. & Poletti, C. E. Hippocampal afterdischarges: differential spread of activity shown by the [14C]deoxyglucose technique. Science 204, 641–643 (1979).
Lipski, W. J. & Grace, A. A. Activation and inhibition of neurons in the hippocampal ventral subiculum by norepinephrine and locus coeruleus stimulation. Neuropsychopharmacology 38, 285–292 (2012).
Bai, T. et al. Learning-prolonged maintenance of stimulus information in CA1 and subiculum during trace fear conditioning. Cell Rep. 42, 112853 (2023).
Torromino, G. et al. Offline ventral subiculum-ventral striatum serial communication is required for spatial memory consolidation. Nat. Commun. 10, 5721 (2019).
Wang, Y. et al. Depolarized GABAergic signaling in subicular microcircuits mediates generalized seizure in temporal lobe epilepsy. Neuron 95, 92–105 e105 (2017).
Xu, C. et al. Subicular pyramidal neurons gate drug resistance in temporal lobe epilepsy. Ann. Neurol. 86, 626–640 (2019).
Cembrowski, M. S. et al. Dissociable structural and functional hippocampal outputs via distinct subiculum cell classes. Cell 174, https://doi.org/10.1016/j.cell.2018.07.039 (2018).
Bienkowski, M. S. et al. Integration of gene expression and brain-wide connectivity reveals the multiscale organization of mouse hippocampal networks. Nat. Neurosci. 21, 1628–1643 (2018).
Jo, H. J. et al. Relationship between seizure frequency and functional abnormalities in limbic network of medial temporal lobe epilepsy. Front. Neurol. 10, https://doi.org/10.3389/fneur.2019.00488 (2019).
Wang, Y. et al. Low-frequency stimulation in anterior nucleus of thalamus alleviates kainate-induced chronic epilepsy and modulates the hippocampal EEG rhythm. Exp Neurol. 276, 22–30 (2016).
Dibue-Adjei, M., Kamp, M. A. & Vonck, K. 30 years of vagus nerve stimulation trials in epilepsy: Do we need neuromodulation-specific trial designs? Epilepsy Res. 153, 71–75 (2019).
Kapustina, M. et al. The cell-type-specific spatial organization of the anterior thalamic nuclei of the mouse brain. Cell Rep. 43, https://doi.org/10.1016/j.celrep.2024.113842 (2024).
van der Vlis, T. et al. Deep brain stimulation of the anterior nucleus of the thalamus for drug-resistant epilepsy. Neurosurg. Rev. 42, 287–296 (2019).
Makhalova, J. et al. Epileptogenic networks in drug-resistant epilepsy with amygdala enlargement: assessment with stereo-EEG and 7 T MRI. Clin. Neurophysiol. 133, 94–103 (2022).
Ma, D. L., Qu, J. Q., Goh, E. L. & Tang, F. R. Reorganization of basolateral amygdala-subiculum circuitry in mouse epilepsy model. Front. Neuroanat. 9, 167 (2015).
Cheng, H. et al. Projection-defined median raphe Pet+ subpopulations are diversely implicated in seizure. Neurobiol. Dis. 189, https://doi.org/10.1016/j.nbd.2023.106358 (2023).
Sternson, S. M. & Roth, B. L. Chemogenetic tools to interrogate brain functions. Annu. Rev. Neurosci. 37, 387–407 (2014).
Lai, N. et al. Interictal-period-activated neuronal ensemble in piriform cortex retards further seizure development. Cell Rep. 41, 111798 (2022).
Racine, R. J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294 (1972).
McNamara, J. O. Kindling model of epilepsy. Adv. Neurol. 44, 303–318 (1986).
Lopes da Silva, F. H., Kamphuis, W., Titulaer, M., Vreugdenhil, M. & Wadman, W. J. An experimental model of progressive epilepsy: the development of kindling of the hippocampus of the rat. Ital J. Neurol. Sci. 16, 45–57 (1995).
Yu, S. et al. High mobility group box-1 (HMGB1) antagonist BoxA suppresses status epilepticus-induced neuroinflammatory responses associated with Toll-like receptor 2/4 down-regulation in rats. Brain Res. 1717, 44–51 (2019).
Kane, N. et al. A revised glossary of terms most commonly used by clinical electroencephalographers and updated proposal for the report format of the EEG findings. Revision 2017. Clin. Neurophysiol. Pract. 2, 170–185 (2017).
Rattka, M., Brandt, C. & Löscher, W. The intrahippocampal kainate model of temporal lobe epilepsy revisited: epileptogenesis, behavioral and cognitive alterations, pharmacological response, and hippoccampal damage in epileptic rats. Epilepsy Res. 103, 135–152 (2013).
Cassataro, D. et al. Reverse pharmacogenetic modulation of the nucleus accumbens reduces ethanol consumption in a limited access paradigm. Neuropsychopharmacology 39, 283–290 (2014).
Alexander, G. M. et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39 (2009).
Jain, S. et al. Chronic activation of a designer G(q)-coupled receptor improves β cell function. J. Clin. Investig. 123, 1750–1762 (2013).
Huneau, C. et al. Shape features of epileptic spikes are a marker of epileptogenesis in mice. Epilepsia 54, 2219–2227 (2013).
de Curtis, M. & Avanzini, G. Interictal spikes in focal epileptogenesis. Prog. Neurobiol. 63, 541–567 (2001).
Wang, Y. et al. Pharmaco-genetic therapeutics targeting parvalbumin neurons attenuate temporal lobe epilepsy. Neurobiol. Dis. 117, 149–160 (2018).
Roth, B. L. DREADDs for neuroscientists. Neuron 89, 683–694 (2016).
Lieb, A., Weston, M. & Kullmann, D. M. Designer receptor technology for the treatment of epilepsy. Ebiomedicine 43, 641–649 (2019).
Raper, J. et al. Metabolism and distribution of clozapine-N-oxide: implications for nonhuman primate chemogenetics. ACS Chem. Neurosci. 8, 1570–1576 (2017).
Acknowledgements
This project was supported by grants from the National Natural Science Foundation of China (No.82173796, 82374064), and the Research Project of Zhejiang Chinese Medical University (2023JKZDZC04).
Author information
Authors and Affiliations
Contributions
Conceptualization: C.X., Z.C., Y.L., Y.W., F.F., and Y.G. Data acquisition: Y.G., S.Z., F.F., and L.H. Data analysis and interpretation: Y.G., S.Z., F.F., L.H., X.G., Y.Y., and M.L. Writing - original draft: Y.G., S.Z., F.F., Y.W., Z.C., and C.X. Writing - review & editing: Y.G., S.Z., F.F., Y.L., Y.W., Z.C., and C.X. Supervision: C.X. and Z.C.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Eliana Scemes and Benjamin Bessieres. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gong, Y., Zhang, S., Fei, F. et al. Ventral subicular pyramidal neurons contribute to epileptogenesis via anterior thalamic circuit. Commun Biol 9, 123 (2026). https://doi.org/10.1038/s42003-025-09402-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09402-z
