
Abstract
Multidisciplinary evidence indicates that the Austroasiatic (AA) language family is the earliest known language in Mainland Southeast Asia (MSEA), dating back to the Neolithic. Yet, the genomic formation and structure of MSEA AA groups remain understudied. Here, we generate genome-wide data for seven AA-speaking and two Sino-Tibetan-speaking populations from Thailand/Laos/Myanmar, which together with published data comprises the largest AA genome-wide dataset to date. We find substantial genetic heterogeneity across both geographic regions and linguistic branches, with the greatest observed in Northern Mon-Khmer highland groups. Analyses with ancient DNA data indicate that northern AA groups exhibit higher East Asian ancestry linking to Iron Age northern Thailand/Cambodia, whereas southern AA groups display additional South Asian ancestry and affinities with Neolithic Laos/Vietnam. Notably, the South Asian-related ancestry is detectable in Neolithic MSEA. Overall, both isolation and contact have together shaped the pronounced genetic heterogeneity observed across linguistic branches of MSEA AA groups.

Similar content being viewed by others

Introduction
Mainland Southeast Asia (MSEA) comprises six present-day countries: Thailand, Laos, Vietnam, Myanmar, Cambodia, and Malaysia. There are five major language families in MSEA: Austroasiatic (AA), Tai-Kadai (TK), Sino-Tibetan (ST), Austronesian (AN) and Hmong-Mien (HM). Among these, AA is likely one of the oldest surviving language families1; AA speakers are primarily distributed across MSEA—especially Vietnam, Cambodia, Laos, Thailand, and Myanmar—as well as parts of northeastern and central India, southern China, and Peninsular Malaysia. The ancestors of AA-speaking populations likely arrived from central or southern regions of the Yangtze River in Southeast Asia, bringing rice agriculture, during the Neolithic period in MSEA (about 4 thousand years ago (kya))2,3. Ancient DNA studies supported this hypothesis by revealing an admixture of local hunter-gatherers and incoming farmers in the Neolithic people of MSEA and their genetic connection to contemporary AA-speaking populations4,5,6.
Subsequently, TK and ST languages spread from the north to MSEA during the Bronze/Iron Age, ~2.5-2 kya7,8. AN languages are also thought to have spread into southern MSEA during the same time, probably from Island Southeast Asia9, while the HM languages probably arrived in the northern hills from China during Historical era10.
Thus, AA speaking groups likely predominated in MSEA during the Neolithic period, but later fragmented due to influences from the expansion of the other language families since the Bronze/Iron Age. Moreover, concurrent with these migrations from East Asia and Island Southeast Asia into MSEA, population contacts between South Asia and MSEA also occurred11,12,13, particularly in several AA groups14,15, possibly facilitated by maritime networks16,17. Cultural and archaeological evidence also indicates notable South Asian influence in MSEA, with ancient DNA studies revealing substantial South Asian ancestry ( ~ 40-50%) in protohistoric samples (1st-3rd centuries CE) from Cambodia18.
These dynamic population movements associated with multiple language families into MSEA suggest a complicated picture of AA-speaking population diversification since the Neolithic. Although previous genome-wide studies have provided some important insights into the genetic background of AA speakers, such as the ancestral relationships within diverse East Asian populations4,5,6,19,20, the correlation of genetic structure with language families in Thailand14, and historical South Asian admixture in several AA groups14,15, various questions remain. These include whether the genetic heterogeneity of MSEA AA groups aligns with the linguistic branches, how much does the genetic heterogeneity reflect the historical diversification versus differential genetic contact, and how the attested East and South Asian cultural influences contributed to the genetic structure of MSEA AA groups. Addressing these questions should shed light on the timeline and processes of genomic formation among ancient and modern MSEA populations.
In this study, we generated genome-wide data for 164 individuals from nine modern populations across Thailand, Laos, and Myanmar: seven AA-speaking populations (Southern Laotian [Lao Tueang], two Kuay groups, Nyeu, Chong, Khmer, Nyahkur) and two ST-speaking populations (Akha and Burmese). The Akha, one of the hill tribes of Thailand, were included to compare interactions between AA groups and the hill tribes. The Burmese were included to understand interactions between AA and South Asian-admixed MSEA groups, providing insights into the history of South Asian admixture into MSEA. We aim to gain an overview on the genomic formation of ancient MSEA populations and present-day AA groups, by analyzing the reported genomic data along with published whole genome data from East Asia, South Asia, and Southeast Asia (SEA).
Results
Genetic heterogeneity within modern AA speakers in MSEA
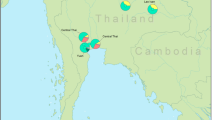
To examine the genetic structure of MSEA AA-speaking populations and interactions with neighboring populations, we first performed Principal Component Analyses (PCA) on the merged dataset comprised 602 individuals from 29 AA-speaking groups (Supplementary Data 1), and observed a clear clustering pattern of MSEA AA groups separating from East Asians and South Asians (Fig. 1). Zooming into MSEA AA speakers, we further observed that AA-speaking individuals formed several distinct genetic groups, some of which aligned with their associated language branches (Fig. 1d; Supplementary Fig. 1). The Aslian-speaking hunter-gatherer Maniq was placed between the MSEA AA groups and the South Asian AA Kharia. The Monic speakers (Mon) formed a cline shifting towards South Asians, whereas the Viet-Muong speakers overlapped with East Asian TK and ST speakers. In contrast, the Northern Mon-Khmer speakers were spread from in-between the Monic and Viet-Muong speakers toward the lower-right corner of the PCA. The Eastern Mon-Khmer and Southern Monic speakers from the Thai-Lao Lowland formed another loose genetic grouping.

AA-speaking groups are distinguished by symbols with colors encoding language branches (according to the classification in Supplementary Fig. 1). Non-AA modern populations are labeled with numbers with colors encoding language family. Ancient groups are denoted by colored symbols. The modern populations sampled in this study are shown in bold in the population legend. PCA was performed using linkage disequilibrium (LD) pruned data, comprising 195,752 unlinked single nucleotide polymorphisms (SNPs) across the whole genome (Methods). Map data were obtained from Natural Earth (https://www.naturalearthdata.com). a Geographical location of the studied modern populations in MSEA. The locations of modern people speaking languages from the Aslian, Northern Mon-Khmer (Khmuic, Palaungic, Mang), Monic, Viet-Muong, Eastern Mon-Khmer (Katuic, Khmeric, Pearic) and Southern Monic language branches, are indicated by dashed line circles; the location of the Munda-speaking Kharia from India is not included. The groups reported in this study are annotated with text. b Geographical location of the analyzed ancient groups. For ancient groups, prefixes indicate the approximate archaeological or chronological context of each individual: Ho for Hoabinhian culture–related; N for Neolithic; BA for Bronze Age; M for Metal Age; IA for Iron Age; and H/Hi for the Historical period. c PCA for populations from South Asia, East Asia, and MSEA (n = 1,575 individuals). d A zoom-in visualization of MSEA clusters in panel c (n = 1,373 individuals), with circles highlighting classified genetic groups (Methods).
To further investigate genetic differentiations among AA-speaking groups, we employed outgroup f3 statistics, for examining allele sharing patterns among the tested populations (Methods). We also annotated the Northern Mon-Khmer (Khmuic, Mang, Palaungic) and Eastern Mon-Khmer (Katuic, Khmeric, Pearic) speakers by their linguistic sub-branches to investigate finer scale differentiations. As shown in the pairwise f3 results among studied AA-speaking groups, Mon, Maniq and Kharia (associated to the Monic, Aslian, and Munda linguistic branches, respectively) share fewer alleles with other AA speakers (Supplementary Figs. 2a-b). Viet-Muong speakers (Kinh and Muong) display more allele-sharing with other MSEA and East Asian groups (Supplementary Fig. 2a). We also noticed some heterogeneity among groups within the same language branch (Supplementary Fig. 2b). For example, Palaung (Palaungic branch) share fewer alleles with other AA speakers, compared to other Palaungic groups. Among Khmuic-speaking groups, the Mlabri, HtinMal, HtinPray, and Lua exhibit strong genetic sharing with each other (and the HtinPray and Lua share even more with each other) but share much less with the Khmu and Khomu. The Multidimensional Scaling (MDS) analysis based on outgroup f3 statistics across MSEA, East Asian and South Asian groups was similar to the PCA (Supplementary Fig. 2c; Fig. 1c).
We then performed ADMIXTURE analysis for K = 2 to K = 20; the lowest cross validation error was at K = 12 (Supplementary Fig. 3). Two hunter-gatherer populations, Mlabri and Maniq, exhibit their own ancestry componentsthat distinguish them from the other AA speakers, likely indicating a strong effect of genetic drift. Most Eastern Mon-Khmer and Southern Monic speaking groups show high frequencies of a yellow component and a small amount of a blue component that is enriched in South Asians. The Northern Mon-Khmer groups show additional heterogeneity. Palaungic groups display high frequencies of the purple component that is at highest frequency in the Karenic-speaking people associated to the ST language family, which are geographically close to the Palaungic groups. Within the Khmuic group, the Khmu and Khomu populations exhibit similar profiles with several ancestry components, while the Lua show very high frequencies of the coral component that is also present in the HtinMal and HtinPray but absent in the Khmu and Khomu. The Viet-Muong populations share similar profiles with several TK populations, while the Mon and Kharia share the yellow component with AA speakers and the purple component with South Asians. Overall, these analyses indicate considerable heterogeneity between and within AA language branches, suggesting population contact and/or language replacement.
Clustering pattern between modern AA speakers and ancient groups
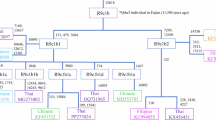
We next examined the relationships of modern samples together with ancient populations from MSEA, South Asia and East Asia. When projecting the ancient individuals onto the PCA of modern samples, we found that the Neolithic MSEA groups are separated from the Bronze/Iron Age groups on PC2, suggesting that the latter probably have additional genetic ancestries (Fig. 1c). In addition, the Neolithic groups mostly overlapped with the Thai Lowland groups (black circle in Fig. 1d) while the Bronze/Iron Age groups were placed in between (and partially overlapped with) the Monic and Northern Mon-Khmer (Palaungic) groupings (Fig. 1d). We further investigated this clustering pattern by DyStruct, which is an ADMIXTURE-like method utilizing the temporal information of ancient populations (Methods; Fig. 2b; Supplementary Fig. 4). Overall, the DyStruct results showed a similar pattern as the ADMIXTURE results, albeit with some discrepancies. For example, the Neolithic group Man Bac was modeled as having a heterogenous profile, with high frequencies of several modern AA group components, such as the pink component shared by the Thai Lowland groups (Eastern Mon-Khmer and Southern Monic), and the orange and yellow components in the Northern Mon-Khmer groups (Khmuic and Mang, respectively).This likely reflects a mismodeled case, where an ancestral population is inferred as a mixture of its highly drifted descendant ancestries21. Still, the heterogeneity among Northern Mon-Khmer speakers observed in the ADMIXTURE analyses was also detected by DyStruct. A turquoise component was largely shared by the Neolithic northern East Asian group (China_YR_MN), MSEA Iron Age groups, and the Palaungic groups, supporting the hypothesis of more East Asian-related ancestry enriched in MSEA Iron Age groups and their connections with the Palaungic speakers. Exceptionally, the Neolithic group Oakaie4 also has high frequencies of this turquoise component, marking them as the earliest East Asian-related group in MSEA, abeit without any detectable indigenous MSEA Hoabinhian-related component. There is also a blue ancestry component present in MSEA Hoabinhian and ancient South Asian populations indicating a relic of an early ancestry preserved in their genetic profile (Fig. 2).

ADMIXTURE and DyStruct analysis were conducted using 142,139 SNPs after additional filtering (Methods). a ADMIXTURE results for modern populations from South Asia, East Asia, and MSEA (n = 1,542 individuals; K = 12). Individuals are color-coded based on their language families and geographic locations. AA-speaking groups are labeled and differentiated by distinct color schemes that correspond to their respective language branches. b DyStruct results for modern AA-speakers, South Asians and ancient individuals from the Neolithic, Iron Age, and Historical era (n = 722 individuals; K = 9). Modern individuals are color-coded by language branches. The modern populations sampled in this study are shown in bold in (a) and (b).
Intra-population and Inter-regional structure among AA speakers and MSEA populations
To further investigate genetic heterogeneity, we analyzed relationships based on the shared Identical-by-Descent (IBD) segments in modern populations (as the data quality of available ancient MSEA samples was insufficient for reliable IBD detection) (Supplementary Note 1; Supplementary Figs. 5-8). Within-population IBD sharing informs about population size, while between-population IBD sharing reflects recent population contact or a recent common ancestor22.
We first inferred the effective population size for each AA-speaking population and language branch group employing hapNe (Methods). For most of the tested MSEA groups aggregated by language branch, we observed a demographic expansion around 125−150 generations ago (Supplementary Fig. 5a), temporally close to the archaeologically hypothesized rice agricultural dispersal around 4 kya23.
To evaluate when interactions with neighboring groups began shaping population structure, we further investigated the temporal genetic patterns via calculating the normalized IBD sharing fraction between individuals from SEA, South Asian and East Asian populations in four categories of IBD length: 1 to 3 cM, 3 to 5 cM, 5 to 10 cM and over 10 cM, which reflects genetic relatedness from 2900, 1160, 653 and 218 years ago respectively when assuming 29 years per generation24,25 (Methods). We found that AA speakers share more IBD with AN/TK/ST speakers than with HM speakers (Fig. 3). Notably, the differential sharing between AA speakers and neighboring AN/TK/ST speakers appeared to be prominent in the length range of 3 to 5 cM. Similarly, we found that the differential sharing with neighboring AA branches emerged in the length range of 3 to 5 cM (Supplementary Fig. 9). AN speakers (Malay, Ede, and Giarai), ST Karen speakers, and TK speakers (Kalueang, LaoIsan, Nyaw, and Phutai), showed more IBD sharing with AA speakers in the range of 5 to 10 cM (Figs. 3c, 3e), indicating their genetic interactions with AA groups remained active until recently. Additionally, the normalized IBD sharing fraction over 10 cM indicates recent genetic interactions between the Khmuic, Viet-Muong, and Monic populations and the Eastern Mon-Khmer and Southern Monic language branches (Supplementary Fig. 9h). Taken together, the interaction between neighboring MSEA AA or other speakers since at least 1160 years ago has played a prominent role in shaping the genetic structure of present-day AA speakers.

The identified IBD blocks were categorized into the range of 1 to 3 cM, 3 to 5 cM, 5 to 10 cM and over 10 cM, in SEA, South Asian and East Asian populations. Populations are color-coded based on their language familes. a−d Corresponding heatmaps showing pairwise IBD sharing among populations (n = 86) for each segment length category. For each pair of populations, we plot the fraction of all pairs of individuals that share at least one IBD block in the size range, which we obtained by dividing the total number of pairs that share such IBD segments by the total number of all possible pairs. e,f Network visualizations for the 5 to 10 cM (n = 88 populations) and over 10 cM (n = 82 populations) categories. In these networks, populations are clustered based on their normalized IBD sharing fraction; populations with higher IBD sharing are located closer to each other.
To understand better the possible driving factors of the observed IBD patterns, such as geography, we visualized the cross-population long IBD (over 10 cM) sharing on the geographic map. The resulting network suggests that differential interaction with other neighboring AA-branches probably shaped the AA sub-group population structure (Supplementary Fig. 9d). For example, the Katuic-speaking Kuay and Nyeu have additional sharing with their geographic neighbor, the Khmeric-speaking Khmer, than do the other Katuic speakers (Supplementary Fig. 9d).
To assess how contact may contribute to AA population substructure, we estimated the population dispersal rate surface across the landscape by the software MAPS, using genome-wide data from all modern-day MSEA populations (Methods). It appears that the mountainous region of northwestern and eastern coastal MSEA has consistently exhibited lower dispersal rates from at least around 2600 years ago (Supplementary Fig. 10). However, the estimated dispersal rate surface and population density varies over time (Supplementary Fig. 10b-h), likely reflecting some complex demographic events that altered the spatial genetic landscape.
South Asian and East Asian admixture in modern AA-speaking groups
To further characterize signals of South Asian and East Asian-related ancestry in ancient and modern MSEA groups, we conducted a SOURCEFIND analysis. We examined the haplotype-based admixture profiles of target AA-speaking populations, given their haplotype sharing with the selected ancestry surrogates. We note that we are not concerned here with the origin/dispersal of AA-speaking populations in South Asia, but rather we are interested in the impact of South Asian-related ancestry in MSEA. We selected Brahmin_Tiwari for South Asian ancestry, Kuay, Kuay1_T and HtinMal for the AA-speaking groups, and Yi for the East Asian ancestry (Methods). We found that Aslian, Eastern Mon-Khmer and Southern Monic groups were mostly surrogated by AA ancestry; the Palaungic and Mang from Northern Mon-Khmer and the Viet-Muong groups were largely surrogated by East Asian ancestry; and the Munda Kharia was completely surrogated by South Asian ancestry (Supplementary Figs. 11-12). The Mon shows all three ancestries (Fig. 4a; Supplementary Fig. 11). As the representative group used for AA ancestry, Kuay likely has some South Asian-related ancestry (Fig. 2), so it is probable that only AA-speaking populations with more South Asian ancestry than that in Kuay would show South Asian ancestry in the SOURCEFIND estimates. In contrast, Lua, which do not have South Asian ancestry (Fig. 2), derive 100% of their ancestry from the surrogate HtinMal, consistent with historical evidence that these two groups are closely related (Supplementary Fig. 11).

Populations (n = 29; Supplementary Data 1) are distinguished by symbols and colors encoding language branches. a SOURCEFIND estimates of ancestry proportions in AA groups. b Biplot of f4(W, Maniq; South Asian/East Asian, Mbuti) statistics, for comparing the excess shared genetic drift between AA-speaking populations with South Asian ancestry (Brahmin Tiwari) or East Asian ancestry (Yi). The red solid line represents a linear fit of the plotted f4 statistics tests for previously published AA-speaking populations4,14,15,20,63,82,83,84, with the gray shading representing the 95% confidence interval. c Date of East Asian/South Asian admixture events inferred by fastGLOBETROTTER. The blue bars represent the admixture times with East Asian, and the pink bars represent the admixture times with South Asian.The box plot displays admixture dates estimated from 100 bootstrap replicates (n = 15; Supplementary Data 2), where the boxes represent the interquartile rage (25−75%) of bootstrap estimates, the line inside the box represents the median, and the whiskers extend to the most extreme points that fall within 1.5 times the interquartile range, and individual points represent outliers. The results correspond to one-date (n = 14; Supplementary Data 3) or one-date-multiway (Paluang; Supplementary Data 3) results, using Yi/Brahmin_Tiwari and Mlabri as surrogates (Methods).
We also carried out allele sharing f4(W, Maniq; South Asian/East Asian, Mbuti) tests for comparing the genetic affinity of AA groups to South Asian and East Asian populations (Fig. 4b). Assuming the African Mbuti is an outgroup, a significantly positive f4 value in this context indicates that, relative to Maniq, population W is genetically closer to the respective South Asian or East Asian group. The results indicate that Viet-Muong speaking groups, Palaungic and Mang from the Northern Mon-Khmer branch exhibit more genetic affinity with East Asian populations than with South Asian populations. In contrast, most of the Eastern Mon-Khmer (except for Soa and SouthernLaotain_T), SouthernMonic, and Monic groups show excess shared genetic drift with South Asian populations. The Northern Mon-Khmer Khmuic groups (HtinMal, HtinPray, Mlabri, Lua) are symmetrically cladal with East Asian and South Asian populations in these two f4 tests (Fig. 4b). Focusing on geographical locations (Fig. 4a), we found that populations shifted towards East Asian populations are all from northern MSEA, while those exhibiting shared genetic drift with South Asian populations are situated in southern MSEA.
We estimated the South Asian and East Asian genetic admixture that occurred in AA speaking populations in MSEA using fastGLOBETROTTER (Fig. 4c; Supplementary Data 2), with Brahmin_Tiwari and Yi as the proxy for South Asian and East Asian ancestries and Mlabri as a surrogate for the indigenous MSEA ancestry. Despite the pattern revealed by SOURCEFIND and f4, some of the groups (i.e., Muong, Blang, Bru, HtinMal, HtinPray, Khmu, Khomu, Lua, Mang, Soa, Southern Laotian and Maniq) resulted in unclear admixture signals based on these proxies, likely indicating that the proxies we used here are not proximal sources of the targeted AA speakers. In general, the estimated South Asian admixture times ranged from 800 to 1100 years ago, while the estimated East Asian admixture times were more variable (Fig. 4c). For Kinh and Palaung, using Yi as the surrogate for East Asian ancestry yielded admixture dates between 800 and 1500 years ago. In the Lawa groups, Eastern Lawa showed an admixture date around 2000 years ago, and Western Lawa around 950 years ago.
To account for the potential proximal sources transmitting the East Asian ancestry to the two Lawa groups, we tested alternative proxies based on known demographic history26, using Karen as the proximal surrogate for the retained East Asian ancestry (Mlabri as the other surrogate). fastGLOBETROTTER suggested rather recent contact between MSEA and East Asia at around 300 years ago (Supplementary Data 3). Using DATES with ancient East Asian sources (i.e. Nepal_Chokhopani_2800BP.SG and China_YR_MN), admixture times for the Lawa groups were estimated at 1000 and 1400 years ago, aligning with the estimate for SgawKaren ( ~ 1288 years ago), indicating that the East Asian ancestry in Lawa groups may reflect contact with SgawKaren (Supplementary Data 3).
The genomic formation of ancient MSEA
As AA languages are hypothesized to be the oldest language family in MSEA, dating to 4 – 5 thousand years ago1, we investigated the early genetic landscape of the region by reconstructing the genomic formation of ancient MSEA populations. To dissect the complex admixture process and to infer individual genetic components, we conducted with qpAdm and qpWave analyses to estimate population admixture models by approximating the admixture proportions from three major ancestries in the ancient MSEA populations: indigenous ancient MSEA ancestry; South Asian ancestry, and East Asian ancestry (see Methods for the exact source populations).
We started with one-source models using the indigenous MSEA ancestry as the source, followed by testing 2-sources models by adding one more additional ancestry (Methods, Supplementary Note 2, Supplementary Data 4, Supplementary Fig. 13). We found that ancient MSEA populations preserve a substantial amount of Hoabinhian ancestry in the working 2-sources admixture model (Laos_Hoabinhian.SG and China_YR_MN), ranging from 32% to 53% in Neolithic individuals, 17% to 24% in Iron Age individuals, and 14% to 25% in Historical era individuals (except H-JarengBori, which exhibited 66% of local ancestry) (Supplementary Data 5, 6). The M-LiangBua (Metal Age individual from eastern Indonesia, 2600BP)27 and H-Komodo are better fitted with Laos_Hoabinhian.SG and Liangdao1 as two sources, with local ancestry at 22% and 36% respectively. As Liangdao1 represents an early East Asian lineage possibly associated with MSEA, we noted that the fitted two-sources admixture model likely reflects a simplified distal model for M-Liangbua, as we did not include Papuan populations in this study while the 3-sources model (Papuan, Kankanaey and Mlabri) for ancient Island Southeast Asian (M-LiangBua) populations as reported in the original publication is more proximal27. Overall, we observed a declining trend of indigenous MSEA ancestry from the Neolithic to the Iron Age and Historical era in MSEA (Fig. 5), likely reflecting later population admixture with people from East Asia and South Asia as observed in modern-day MSEA populations4.

a A visualization of the ancestry distribution of ancient groups (n = 22). Inset shows a zoomed-in region of genetically heterogeneous Iron Age groups (n = 5). b Genetic cladality test between ancient groups (n = 25) and modern MSEA populations (n = 43) by qpWave. The colored bars indicate language family and language group for each population. Modern non-AA speaking groups were selected to have little or no admixture from AA groups, i.e. having little or none of the yellow AA-associated component in the ADMIXTURE analysis (Fig. 2a). Gray blocks indicate p values less than 0.05 (Supplementary Data 13). c A visualization of genetic profiles presented in (a) with one standard error bar. The groups from the mainland are labeled in bold. For ancient groups, prefixes indicate the approximate archaeological or chronological context of each individual: Ho for Hoabinhian culture–related; N for Neolithic; BA for Bronze Age; M for Metal Age; IA for Iron Age; and H/Hi for the Historical period.
Subsequently, we conducted qpAdm analyses with a rotating strategy by testing two or three potential sources while adding the other potential sources to the base outgroups28 (Methods). Our findings revealed that most MSEA individuals from the Neolithic era to the Historical era could fit a 2-source admixture using Laos_Hoabinhian.SG and the other East Asian source (either China_YR_MN or Liangdao1) (Fig. 5a, c, Supplementary Data 4, 5). However, N-ManBac, N-MaiDaDieu, BA-NuiNap, and IA-ThamLodCave required a 3-way model with Laos_Hoabinhian.SG, China_YR_MN, and Indus_Periphery sources (Fig. 5a, c, Supplementary Data 7-10). Specifically, N-ManBac and N-MaiDaDieu individuals preserved 20% Hoabinhian ancestry, accompanied with 20% to 22% South Asian ancestry. This finding is further supported by f4 statistics in the form of f4(ancient MSEA population, Maniq; South Asian/East Asian, Mbuti), where IA-VatKomnou and N-ManBac presented extra genetic affinity than other ancient individuals to Indus_Periphery, in comparison to Maniq (Supplementary Fig. 14). To exclude the potential biases caused by ancient DNA damage, we repeated the f4 analysis in the same form using only transversion SNPs (95,924 loci), which confirmed the same signal (Supplementary Fig. 15).
The presence of South Asian ancestry in Neolithic MSEA groups (4291- 2950 BP) suggests that the South Asian ancestry occurs in MSEA earlier than previously thought, and 2000 years earlier than the estimated South Asian admixture in modern-day AA speakers (Fig. 4c). The South Asian ancestry is also prevalent in subsequent time periods, albeit at a lower proportion. BA-NuiNap exhibited 7% local ancestry and 15% South Asian ancestry, while IA-ThamLodCave had 11% local ancestry and 8% South Asian ancestry. Moreover, this signal remains robust when adding India_Harappan into the outgroup list (Methods, Supplementary Data 11), likely indicating a deep ancestral lineage shared by both South Asian groups and ancient MSEA populations.
To further test this hypothesis, we conducted qpGraph analysis, which showed that N-ManBac, N-MaiDaDieu, BA-NuiNap, and IA-ThamLodCave could be modeled as derived ancestral lineage from a northern East Asian–related ancestry lineage and a deeply diverged lineage related to ancient MSEA, southern East Asian and South Asian hunter-gatherers (Supplementary Fig. 16).
In particular, we observed that Hoabinhian ancestry is not required in models that fit the N-Oakaie1, IA-VatKomnou, and IA-LongLongRak individuals (Fig. 5c); they fit models with an East Asian source (China_YR_MN/Longlin) and a South Asian source (Indus_Periphery) (Fig. 5a, c, Supplementary Data 5, 6, 12). This could suggest a signal of complete replacement of indigenous MSEA ancestry in some of the ancient communities in MSEA. Alternatively, considering the wide range of interaction regions at Oakaie 129, the extensive commingling of burials at Vat Komnou30, and the agricultural evidence at Long Long Rak31, these individuals might have been later immigrants who had not yet mixed with the local MSEA populations. Moreover, in this region, we noticed high genetic heterogeneity among the contemporaneous Iron Age groups (Fig. 5a), suggesting complex interactions involving populations with distinct ancestry from East Asia, South Asia and SEA at the time.
The genetic legacy in present-day AA speakers
We next examined the genetic legacy retained in present-day AA speakers to evaluate their connections to ancient ancestries and to assess subsequent gene flow that may have influenced their genomic profiles. The indigenous hunter-gatherers Mlabri (Northern Mon-Khmer speaker) and Maniq (Aslian speaker) harbor substantial drift, as indicated by the long branches in the maximum likelihood tree (Supplementary Fig. 17) and noted in several earlier studies14,32,33,34. These two groups also derive a substantial amount of ancestry from a lineage related to indigenous MSEA Hoabinhian ancestry, as inferred from qpGraph (Methods; Supplementary Fig. 18). The other AA groups are more admixed as a result of multiple waves of admixture events, particularly for Monic speakers and other Northern Mon-Khmer speakers (Supplementary Figs. 18-19). The heterogeneity of the tree structure of various Northern Mon-Khmer speakers (Supplementary Fig. 19), aligned with the heterogeneous clustering pattern from previous analyses (Figs. 1–2).
When comparing present-day AA speakers with ancient samples from MSEA, we noticed that ancient MSEA populations dated to different time periods are genetically close to different AA-speaking populations (Supplementary Data 13). In particular, N−HonHaiCoTien and N−TamPaLing, with substantial Hoabinhian ancestry, fall closely with Eastern Mon-Khmer and Southern Monic speakers on the PCA (Fig. 1), and present genetic cladality with statistical significance (Fig. 5b). By contrast, Iron Age MSEA populations are genetically closer (and geographically-close) to Northern Mon-Khmer groups, as indicated by their close PCA location and their significant genetic cladality in qpWave tests (Figs. 1,5). Viet-Muong speakers are cladal only with Iron Age and Historical era populations, while Monic speakers are most closely related to N-HonHaiCoTien and N-TamPaLing (Fig. 5b). Employing the same qpWave strategy (Methods), we tested ancient populations and modern MSEA non-AA speaking populations for cladality, and found no cladality between Neolithic populations and non-AA speakers (Supplementary Data 13), except for Burmese1_T who was cladal with some Neolithic (N-HonHaiCoTien, N-TamPaLing; N-NamTun) and Iron Age groups (IA-VatKomnou; IA-BanRaiRockshelter) (Supplementary Data 13). Overall, we observed that Neolithic MSEA populations are related to present-day lowland AA speakers, while Iron Age MSEA populations are related to highland AA speakers.
Discussion
In this study, we generated genome-wide data for 164 individuals associated to 7 AA language branches (together with 2 neighboring ST groups, to investigate potential interactions), which when combined with published data is the largest AA dataset analyzed to date, comprising 28 MSEA groups and 1 South Asian group. Based on genetic clustering, previous MSEA genomic studies of populations from Vietnam and Thailand suggested a good correlation between genetic structure and language families14,20, while a recent study with a broader sampling across MSEA showed that geography played a more important role19. Here, with a denser sampling of AA groups, we integrated modern and ancient genomes, as well as haplotype-based approaches and allele-sharing statistics, to provide a finer-scale and temporally resolved characterization of the genetic structure and population history of MSEA AA groups and the overall genetic structure and history of MSEA. The clustering analysis (Fig. 1d, 2) revealed genetic heterogeneity among AA groups from MSEA that partially corresponds to their linguistic relationships and geographic locations. This suggests that genetic structure, language, and geography are interconnected in these populations to some extent. Groups speaking Aslian, Monic and Northern Mon-Khmer (Khmuic, Palaungic), and Viet-Muong languages formed distinct genetic groupings (with some slight overlaps), while Eastern Mon-Khmer (Katuic, Pearic and Khmeric) and Southern Monic-speaking groups comprised another genetic grouping. Aslian speakers are represented by the Maniq, a negrito group living in the dense rainforests of southern Thailand. Their small census size and extreme isolation, coupled with their traditional practice of endogamy, has intensified genetic drift, inducing marked population differentiation (Figs. 2, 3b; Supplementary Fig. 6, 17)34,35. However, whether the high genetic differentiation of Maniq from other AA speakers could also be a signal of an early split event remains an open question. Further investigation with additional data would be helpful, as we could not exclude the possibility of cultural acculturation, namely the Maniq may have adopted their AA language after splitting off from the ancestor of Mon-Khmer speakers or the ancestor of AA speakers (Supplementary Fig. 18).
To further investigate recent genetic interactions among these AA-speaking populations, we employed haplotype-based analyses, including IBD sharing patterns (see Methods), decoding recent genetic interactions among populations belong to different AA linguistic groups. IBD-sharing analyses (Fig. 3) revealed that AA groups from MSEA tend to share higher levels of IBD with one another than with non-AA groups, with a few exceptions discussed below, suggesting a common origin (or higher levels of contact with each other) for MSEA-AA groups. Some AA groups tend to share high levels of IBD with the ST-speaking Karen groups from northwestern Thailand; this is especially evident for the Western Lawa, who live near the Karen groups. We also observed some high IBD sharing between AA groups and AN groups from MSEA and Borneo (involving some Eastern Mon-Khmer groups, e.g. Pearic, Katuic and Khmeric). The IBD sharing with AN groups probably reflects language shift of AA groups to AN languages in MSEA, as suggested previously14,20,36. The two Viet-Muong groups stand out as exhibiting high levels of IBD sharing with each other but low levels with all other AA groups, which might reflect geographic isolation due to their location in Vietnam and their recent population expansion (Supplementary Figs. 5-7), in line with previous studies19,20. The two Southern Monic groups, Nyakhur and Nyakhur_T, also have high levels of IBD sharing with each other, but every other AA language subgroup represented by more than one population in our study exhibited heterogeneity in the IBD-sharing analyses, as discussed in following paragraphs.
To revisit the important role of geography in shaping the observed structure as suggested by the recent MSEA study using Procrustes statistics to compare the geographic and PCA locations19, we employed a more sophisticated method – MAPS (Methods) – which estimates the potential migration pattern quantitatively across space for the correlation between genetic structure and geographic distribution in MSEA. As shown, the patterns of population isolation and contact described above are further evident in the geographic pattern of effective population migration rates (Supplementary Fig. 10b). It thus appears that geography, in the form of promoting isolation for populations in the highlands versus contact with neighboring groups in the lowlands, can explain some – but not all – of the patterns of the genetic relationships of MSEA-AA groups.
The Khmuic, Palaungic, and Mang groups all belong to the Northern Mon-Khmer branch but show distinct genetic profiles (Fig. 2). Khmuic-speaking populations are dispersed across Laos, Vietnam, China, and Thailand; many of the Khmu people in Thailand are relatively recent immigrants from Laos and Vietnam, having fled the Vietnam War10. Four of the Khmuic groups (Mlabri, Lua, HtinMal, and HtinPray) cluster together based on IBD sharing, while the other two Khmuic groups (Khmu and Khomu) do not exhibit high levels of IBD sharing with them or with each other. The very low levels of genetic diversity in the Mlabri, accounting for their distinctive genetic profile in the ADMIXTURE analysis (Fig. 2), likely reflect an extreme bottleneck and drift in their population history, as some studies based on uniparental markers implied35,37. Despite the extreme bottleneck, intriguingly, the genetic relationship between the Mlabri and the HtinMal and HtinPray is still evident in the IBD sharing analysis (Fig.3; Supplementary Fig. 7), supporting a recent origin of the Mlabri from these Northern Mon-Khmer speakers (or a related group) as indicated by genetic, linguistic and historical sources37,38.
The lowland AA groups from Laos, Northeastern and Eastern Thailand, and Cambodia—including Eastern Mon-Khmer (Katuic, Khmeric, Pearic) and Southern Monic (Nyahkur) speaking populations— broadly cluster together genetically. Historically, these regions share a common history, particularly during the Angkorian Khmer state (802-1431 C.E.) that established its influence over large parts of MSEA17. However, many AA groups experienced a decline followed by an increase in population size during this period, which may reflect (pre)historical sociopolitical factors such as political instability and warfare8,10 (Supplementary Fig. 5). In the 14th century C.E., the Khmer civilization declined and there was migration of TK-speaking Lao people from Northern Laos to these regions. There likely were interactions involving gene flow between Khmeric-speaking populations and both AA and TK groups39, consistent with the historical interactions between neighboring AA branches and other language families, which played a key role in shaping the population structure of AA groups over the last 1200 years. Still, within this broad interaction zone in the lowlands there is genetic heterogeneity: three Katuic groups (Soa, Bru, and SouthernLaotian_T), located further to the north and east in the lowlands, clustered together based on IBD sharing, but the other Katuic groups (Kuay and Nyeu) clustered together with the Khmeric Khmer groups (Khmer and Khmer T), with whom they are in close geographic proximity (Fig. 1a). These results are in line with linguistic evidence for extensive historical language contact between Kuay and Khmer40. The other Khmeric group, Cambodian, is further to the south and does not share high levels of IBD with any other group. Last, the connection between Eastern Mon-Khmer and Southern Monic groups probably involved Indian-related kingdoms as discussed in the next paragraph, which is supported by the South Asian admixture component in both groups.
Under the assumption of geography-driven population contact, we subsequently investigated the potential contact between East Asian and South Asian groups with MSEA-AA groups, and found evidence for substantial East Asian-related ancestry in the Viet-Muong, Mang, and Palaunguic groups, particularly in the Palaung (Fig. 4), which probably contributes to the genetic differentiation of these groups and their relative lack of IBD sharing with other groups. We inferred variable estimates of timing for when East Asian ancestry entered these AA groups, ranging from around 1.5 kya in the Kinh (the northernmost group) to around 200 years ago for the Lawa groups (Fig. 4c; Supplementary Data 3). The older estimated date in the Kinh might be a result of the late phase of the Han Dynasty expansion period from East Asia, ~2-2.5 kya41, while the more recent date in the Lawa groups suggests that their East Asian ancestry might reflect interactions with neighboring ST-speaking Karen groups (Figs. 2, 4a).
The South Asian-related ancestry in lowland AA groups has been previously reported in several populations14,15; here, with additional samples, we propose such signal as a general pattern for AA groups from this region. Moreover, we dated the South Asian admixture to between 800−1200 years ago for all groups, corresponding to the Dvaravati kingdom period, which lasted from the 6th to 11th century C.E., and Angkorian Khmer periods42. However, before the formation of those two early established civilizations in MSEA, there is evidence of cultural contact between MSEA and South Asia via maritime exchange routes during the Iron Age (2.4 kya), which introduced iron working knowledge, exotic ornaments, Hinduism, and Buddhism to Southeast Asia43. Linguistic evidence suggests that the Southern Monic-speaking Nyahkur is a direct descendant of the old Mon language of Dvaravati44 whereas the Monic-speaking Mon groups from Central Thailand migrated from Myanmar during the 16th to 19th centuries C.E45. Our results indicate stronger genetic relatedness between Nyahkur and the lowland AA-groups, especially Khmer, corroborating the previously reported extensive sharing of Y chromosome and mitochondrial DNA haplogroups between the Nyahkur and Khmer40 and archaeological evidence at the Sri Thep site of a shared connection between the Khmer and the Dvaravati Mon17,46.
To gain further insights into the spatio-temporal structure of MSEA-AA groups, we analyzed ancient SEA genomes ranging from Neolithic to Historical era. We found that only modern MSEA-AA groups show cladality with ancient samples from the Neolithic; specifically, the Eastern Mon-Khmer and Southern Monic lowland groups. In contrast, all other language groups show cladality only with samples from the Iron Age or Historical era periods. This supports the general consensus that AA is the most ancient language family in MSEA47. Intriguingly, we identified a signal of ~20% ancient South Asian ancestry (using Indus_Periphery as a proxy, Methods) in the late Neolithic samples MaiDaDieu, ManBac, and Oakaie (Fig. 5), all dating to ~3-4 kya4,5, which has not been reported before. Moreover, we found that this signal remains robust when adding India_Harappan into the outgroup list (Methods, Supplementary Data 11). Such phenomenon has not just occurred to the ancient South Asian group, but also occurs with southern East Asian Longlin and southeastern Asian Hoabinhnian (Supplementary Fig. 14). Together with the qpGraph analysis (Supplementary Fig. 16), these results imply that the ancient South Asian ancestry we detected in ancient MSEA is more likely to be an early deeply diverged lineage relating to ancient South Asia, SEA and East Asia.
Although SEA admixture in South Asian AA groups has been dated to ~2-3.8 kya42, this is considerably older than our inferred time for South Asian admixture in modern MSEA AA groups of 800-1200 ya (Fig. 4). Combined with the discovery of the deeply diverged South Asian, SEA and East Asian-related lineage dating back to at least 4 kya, our results likely indicate the occurrence of multiple pulses of asymmetric population interactions between South Asian and MSEA. South Asian ancestry is also inferred in some prehistorical samples (Fig. 5) whose ages overlap with the admixture time for South Asian ancestry in modern AA-speaking populations, and hence these could represent the early MSEA groups who interacted with South Asians during the early civilization discussed above. However, we caution that most of the ancient samples are not from the same geographic location as the modern populations exhibiting South Asian ancestry today. We also note that Hoabinhian hunter-gatherer ancestry – which shows ancient connections with South Asian - is not necessary to model the ancestry of some of the late Neolithic and Iron Age groups (e.g. Oakaie and Vat Komnou), which means that South Asian ancestry might be overestimated in these samples (or, that they represent incoming groups from East Asia who did not yet admix with local populations). Further ancient DNA studies from MSEA would help elucidate these issues.
In conclusion, using the largest genomic dataset from modern AA groups to date, together with ancient genomes, we have characterized in detail the genetic structure of AA groups, and the spatio-temporal factors that shaped such structure, including isolation within specific groups and contact with other groups in MSEA as well as with populations from East Asia and South Asia across different temporal and spatial scales. We have provided genetic evidence supporting AA as the oldest language family in MSEA, and therefore AA -speaking groups compose the fundamental genetic layer of the present day MSEA population structure. We also reported South Asian ancestry in some Neolithic samples that was not previously detected. Although ancient DNA studies are advancing and ancient samples have revealed valuable insights into the genetic history of MSEA, substantial sampling gaps in MSEA (particularly from the crucial lowland regions) still remain. Overall, our study underscores the importance of incorporating both modern and ancient genomes, as well as including more present-day samples from a broad geographic region, to provide a more comprehensive understanding of the human genetic legacy in MSEA.
Methods
Ethic Statement
Ethical approval for sampling was granted by the Khon Kaen University Ethics Committee (HE622027) and the Institutional Review Board of Naresuan University (P1-0156/2566). All ethical regulations relevant to human research participants were followed. Written informed consent was obtained from all participants, including consent for sample collection, genetic analysis, and long-term storage of biological materials for future research. Participant rights and privacy were protected throughout the study. All samples were anonymized using coded identifiers that could not be used to trace individuals, and no personally identifying information was retained.
Samples and genomic data
DNA samples were from previous studies48,49, with the exception of the samples collected from Burmese. For these samples, we followed the same criteria to recruit participants as in previous studies48,49: we first screened for volunteers unrelated for at least two generations. Buccal samples were then collected using Gentra Puregene Buccal Cell Kits (Qiagen, Germany) and DNA was extracted according to the manufacturer’s directions.
Genotyping was carried out using the Affymetrix Axiom Genome-Wide Human Origins array50 and primary screening used the Affymetrix Genotyping Console v4.2. We used PLINK50 version 1.90b5.251 to exclude loci and individuals with more than 5% missing data and also exclude mtDNA and sex chromosome loci. We further excluded loci which did not pass the Hardy–Weinberg equilibrium test (p value < 0.00005), or had more than 50% missing data, within any population. We checked individual relatedness using KING51 implemented in PLINK50 version 2.0 and excluded one individual from each pair of individuals with first degree kinship. There are in total 164 genotyped individuals with 518,516 loci after these quality control measures (Supplementary Data 1).
The ethnolinguistic classification of AA groups in this study was done according to the Ethnologue52, which comprises two primary branches, Mon-Khmer and Munda. Our merged dataset encompasses the Munda-speaking Kharia for the Munda branch and groups from Aslian, Eastern Mon-Khmer, Monic, Northern Mon-Khmer, Southern Monic and Viet-Muong for the Mon-Khmer branch (Supplementary Fig. 1). For comparative data, we merged published data from 79 ancient and 1378 modern individuals from SEA, East Asia and South Asia with our new data (Supplementary Data 1), and additionally incorporated genetic data of the African Hunter-gatherer Mbuti53 for some analyses requiring an outgroup population. For the clustering analyses (PCA, ADMIXTURE and DyStruct), we performed LD pruning on the merged dataset with PLINK option “-indep-pairwise 200 25 0.4” which excludes one variant from each pair with an r² greater than 0.4 within windows of 200 variants and a step size of 25 variants, ended up with 195,752 unlinked SNPs along the whole genome.
Principal components analysis
We carried out PCA using smartpca v.18140 from the EIGENSOFT package54,55 with the “lsqproject: YES” and “autoshrink: YES” options. 98 modern SEA, East Asian and South Asian populations were used for PC calculation. Individuals AKL117, BR102, and YE127 were removed from subsequent analyses as PCA outliers. The 95% confidence intervals for the clustered groups were calculated using the “stat_ellipse” function from the R package “ggplot2” v.3.4.4, with type = “norm”, and level = 0.95 parameters (Fig. 1).
ADMIXTURE/DyStruct analysis
To further investigate clustering pattern of the merged populations, we used PLINK v.1.90b5.25150 with the parameter “--maf 0.01” to filter SNPs with a minor allele frequency of ≥ 1%, resulting in 142,139 loci. We then ran ADMIXTURE v.1.3.056 with K ranging from 2 to 20, performing 20 replicates using random seeds for each K.
We used DyStruct v.1.1.0 to infer shared genetic ancestry while accounting for archaeological age57. The archaeological date of each ancient group was converted to generations by assuming 29 years per generation24. We performed 10 independent replicates with random seeds, using 2 to 15 ancestral populations (K). A subset of loci (5%) was excluded during training to evaluate the conditional log likelihood and cross-compare estimates for different K values. We employed Laos_Hoabinhian.SG5 as the indigenous MSEA ancient ancestry; Indus_Periphery (Turkmenistan_Gonur_BA_2 and Iran_ShahrISokhta_BA2)58 as the ancient South Asian ancestry; China_YR_MN59 as the ancient northern East Asian ancestry; Liangdao160 as the ancient southern East Asian ancestry; and Longlin61 as a representative for a deep Asian hunter-gatherer ancestry that diverged earlier than Hoabinhian ancestry (Fig. 2b, Supplementary Fig. 4). We used the R package “pophelper” v.2.3.162 to visualize both the ADMIXTURE and DyStruct results.
f-statistics
To evaluate the shared genetic drift between two populations, we employed qp3Pop v.651 from Admixtools packages63 for outgroup-f3 statistic tests in the format of f3(Mbuti; X, Y), where the present central African “Mbuti” was used as an outgroup. The pairwise outgroup-f3 results were then plotted as a heatmap using the R package “pheatmap” v.1.0.12. We used the “cmdscale” function in R to perform MDS analysis based on the matrix of outgroup-f3 statistics (Supplementary Fig. 2).
To assess the genetic affinity of a test group to South Asian and East Asian ancestry, relative to the Maniq hunter-gatherer group (which lacks detectable South Asian or East Asian ancestry), we computed f4-statistics using qpDstat v.980 from Admixtools63 in the format of f4(test group, Maniq; South Asian/East Asian, Mbuti), where South Asian ancestry is represented by Brahmin_Tiwari and East Asian ancestry is represented by Yi. The test groups include all studied AA-speaking populations except the South Asian AA-speaking population Kharia. To confirm the observed affinities in ancient DNA data, we filtered the dataset to retain only transversion SNPs using CONVERTF v.5000 from the EIGENSOFT package54,55 with the parameter “badsnpname: list of transitions SNPs”, resulting in a final dataset of 95,924 SNPs. We then repeated the same f4-statistics analyses using this filtered dataset.
qpWave and qpAdm Analyses
For representing the indigenous ancient MSEA ancestry, we used the 7872 BP MSEA hunter gatherer Laos_Hoabinhian.SG as a proximal source and the 8190 BP Liangdao1 and 11517 BP Longlin from southern East Asia as the distal source of indigenous paleolithic MSEA ancestry, as Hoabinhian is described as a mixture of northern East Asian, southern East Asian and Guangxi ancestry61. We chose the 4015-4221 BP Turkmenistan_Gonur_BA_2 and 4200-5000 BP Iran_ShahrISokhta_BA2 from the Indus_Periphery58 as the proxy for ancient South Asian ancestry by merging the two groups into one (named as “Indus_Periphery” in the analyses), and the 5250 BP China_YR_MN59 from northern East Asia as the proxy for the ancient East Asian ancestry.
To test genetic cladality between modern AA-speaking populations and ancient SEA populations, we applied qpWave v.1520 from Admixtools63 with “allsnps: YES” and “inbreed: NO” parameters. For all qpWave analyses, we used the following populations as outgroups (“right populations”): Mbuti, Palestinians, Iranians, Armenians, Papuans, Nganasan, Kets, Koryaks, Karitiana, and Sardinians. In each qpWave test, we used one modern population and one ancient population/individual as “left populations”.
To test genetic relationships between modern and ancient MSEA populations, we conducted qpAdm v.1520 from Admixtools63 tests starting with choosing the basic outgroups (“right populations”): Mbuti, Russia_Ust_Ishim_HG, China_Tianyuan, Russia_Kostenki14, Iran_GanjDareh_N, Turkey_N, Russia_DevilsCave_N, and Onge. We hypothesized that the following five distinct ancestries might have contributed to the genomic formation of modern and ancient MSEA populations: 1) Laos_Hoabinhian.SG as the proxy for indigenous MSEA ancestry; 2) Indus_Periphery (Turkmenistan_Gonur_BA_2 and Iran_ShahrISokhta_BA2) as the proxy for ancient South Asian ancestry; 3) China_YR_MN as the proxy for northern East Asian ancestry; 4) Liangdao1 as the proxy for southern East Asian ancestry; and 5) Longlin as a proxy for a deep East Asian hunter-gatherer lineage. We employed the strategy of combining any of the two ancestries for evaluating the base outgroup. We also examined the genetic cladality between published Neolithic to Historical era MSEA populations and the five hypothesized ancestry sources.
For anchoring the proximal ancestry model and estimating the exact ancestral proportions in target ancient MSEA populations, we started with 2-sources models using Laos_Hoabinhian.SG as the indigenous base source, and tested another possible ancestry as the other source, while keeping the basic set of outgroups. When a target population had more than two models with a good fit (p < 0.05), we utilized a rotating strategy60 by testing two potential sources while adding the other potential sources to the base outgroups to find the optimal model. For individuals with all the 2-way models rejected, we tried the base and 3-way models to fit the optimized model. Following the same strategy, we modeled the ancestry of modern MSEA populations using the same set of outgroups and potential ancestry sources.
Haplotype Sharing Analyses
To analyze haplotype sharing patterns, we phased the data (consisting of 1542 modern individuals, same as used for PCA analysis, Supplementary Data 1) using SHAPEIT v.4.2.264 with the HapMap phase II b37 recombination map65. We then ran ChromoPainter v.266 on the phased dataset to investigate haplotype sharing and generate the input for SOURCEFIND67 and fastGLOBETROTTER68. Initially, we estimated the mutation (emission) probability and switch rate (recombination rate scaling constant Ne) parameters using 10 iterations of the expectation-maximization (EM) algorithm on a subset of chromosomes (1, 5, 10, and 15). The inferred parameters were averaged by chromosome (accounting for their number of SNPs) and then by individuals. The EM estimation yielded a switch rate at 156.1066 and a global mutation (emission) probability at 0.001099381. These parameters were then fixed for an additional ChromoPainter run across all chromosomes and all individuals.
To identify shared IBD blocks between each pair of individuals, we used Hap-IBD v.1.069 following the recommended data phasing pipeline using Beagle v.5.470,71 with GrCh38 genetic map72. For the overall IBD sharing analysis, we summed the total number and length of all IBD blocks across all chromosomes for each pair of individuals. We then calculated the mean number and length of IBD segments for pairs between and within populations, and plotted the results using the R package “pheatmap” v.1.0.12. To analyze population interactions, we focused on IBD segments with a summed total length greater than 20 cM and employed a network approach using the “kk” cluster algorithm in the R package “ggraph” v.2.1.0, with groups that share more edges clustered closer together.
To calculate the normalized IBD sharing fraction of identified IBD blocks, we generated four datasets based on the identified IBD block lengths: 1–3 cM, 3–5 cM, 5–10 cM, and over 10 cM, reflecting genetic relatedness from approximately 2900 ya, 1160 ya, 653 ya and 218 ya respectively. This is based on the following formula: the expected coalescent time (t, in generations) of IBD segments between length L1 cM and L2 cM is approximately 300/4 (1/L1 + 1/L2) if the effective population size is sufficiently large25. For each pair of groups, we normalized the average IBD sharing by the total number of all possible pairs of individuals: for two different groups of n1 and n2 individuals, there are n1×n2 pairs, while within a group of size n there are n(n-1)/2 pairs. We visualized the IBD sharing fraction on the map using the R packages “ggplot2” v.3.4.4 and “ggmap” v.4.0.0. Heatmaps were plotted with the R package “pheatmap” v.1.0.12. For the network visualization of the 5–10 cM and over 10 cM categories, we used the “kk” cluster algorithm in the R package “ggraph” v.2.1.0. In this network, groups that share more IBD are weighted to cluster closer together.
Given the estimated IBD segments of different lengths shared within populations, we estimated the effective population size (Ne) in the last 200 generations for MSEA populations using HapNe-IBD73. In order to increase the sample sizes for this analysis, we first ran hapNe on each individual population separately, and then aggregated populations by grouping together individuals from the same language branch into a pseudo population group associated with their language branch: Kuay, Bru, Soa, Kuay1_T, Kuay2_T, Southern Laotian and Nyeu for the Katuic-speaking group; Khmer, Cambodian and Khmer_T for the Khmeric-speaking group; Mlabri, HtinMal, HtinPray, Khmu, Lua, and Khomu for the Khmuic-speaking group; Palaung, Blang, Eastern Lawa, and Western Lawa for the Palaungic-speaking group; and Kinh and Muong for the Viet-Muong group. However, the absolute estimates of intensity and timing from hapNe may be biased by population demographic history, and the exact timing of expansions or bottlenecks should be interpreted with caution.
Spatial structure analyses
To investigate the spatial structure of diverse MSEA and South Asian populations, we first ran the software Estimation of Effective Migration surface (EEMS)74, which infers migration barrier via estimating effective migration rate, and also generates demes required by MAPS25.
For EEMS, we set the number of demes to 800 and tuned proposal variances ensuring an optimal balance between exploration and efficiency in the EEMS mathematical model. In this context, the deme is a parameter provided when performing EEMS analysis to divide the Southeast Asian mainland region into a grid. The larger the number of demes, the denser the grid, and the more time-consuming the analysis. When running EEMS, the program groups one or several populations into a single deme based on their geographic location (habitat). In our analysis, 52 populations were divided into 42 demes. Additionally, we adjusted the parameters mEffctProposalS2 and qSeedsProposalS2 so that the proposals were accepted between 10% to 40%, as recommended in the EEMS manual74. For the final run we used 800 demes, with 10 million MCMC, 2 million burn-in, and 10,000 thinning iterations for the determination of the posterior distribution of effective migration and effective diversity rates.
In order to gain further insight into the spatiotemporal structure of MSEA populations, we employed MAPS25, which uses the IBD sharing matrix and different length segments of IBD to estimate the time-dependent dispersal rates and population density surface. We inferred the dispersal rate and population density surface for IBD segments grouped into bins of 1 to 5 cM (after merging 1-3 cM and 3-5 cM IBD segments together), 5 to 10 cM, and longer than 10 cM, reflecting genetic relatedness from at least approximately 2610 ya, 653 ya and 218 ya ago respecitively25. We also used 800 demes from MSEA AA speaking groups, tuned proposal variances based on those that were accepted 10% to 40% of the time, with 5 million MCMC, 1 million burn-in and 10,000 thinning iterations for the determination of the posterior distribution of effective migration and effective diversity rates.
SOURCEFIND and admixture date inferences
We inferred the South Asian and East Asian admixture in AA groups using SOURCEFIND67 with the following settings: 1) allowed up to three surrogates to contribute more than 0% ancestry in each iteration (exp.num.surrogates: 3); 2) ran 200,000 iterations in total (num.iterations: 200,000); 3) discarded the first 50,000 iterations as burn-in (num.burnin: 50,000); and 4) sampled posterior admixture proportions every 5,000 iterations (num.thin: 5,000).
We tested all AA-speaking populations using a simplified set of surrogates as proxies for South Asian, East Asian, and AA ancestries: Brahmin_Tiwari for modern South Asian ancestry, as this group exhibits a high proportion of South Asian genetic components; Yi for East Asian ancestry, due to their high proportion of East Asian genetic components and their geographic proximity to MSEA; and each modern AA group was tested as a proxy for AA ancestry. However, other AA-speaking populations exhibited little or no AA ancestry when we used Khmuic, Palaungic, Viet-Muong, some Katuic (Bru, Soa, and Southern Laotian), Mang, or Maniq groups as surrogates. Thus, we used Kuay, which harbored the least South Asian ancestry among the groups that succeeded, as the AA proxy. To ensure that Kuay could also be represented by AA ancestry, we added Kuay1_T and HtinMal as AA surrogates. Therefore, we used Kuay, Kuay1_T, and HtinMal to represent the AA-speaking groups, ensuring each AA-speaking group was appropriately represented by an AA surrogate.
To further resolve the composition of South Asian–related ancestry in AA-speaking groups, we conducted an additional SOURCEFIND analysis using an expanded set of South Asian surrogates. In this analysis, Brahmin_Tiwari was used as a surrogate for South Asian ancestry, Onge for ASI-related ancestry, and French for Steppe-related ancestry, representing alternative potential sources of South Asian-related components. Yi was included as the East Asian ancestry surrogate, whereas HtinMal, Kuay, and Kuay1_T served as AA ancestry surrogates.
For estimating admixture times in modern-day MSEA populations, we used fastGLOBETROTTER68, ALDER v.1.0375 and DATES v.75376. We ran fastGLOBETROTTER under default settings using Brahmin_Tiwari, Yi, and Mlabri as surrogates to date South Asian and East Asian admixture events in AA-speaking groups. Bru, Soa, and Southern Laotian showed “unknown” results; Muong, Blang, HtinMal, HtinPray, Khmu, Khomu, Lua, Mang, and Maniq showed “unclear signal” results, so these groups were not included. Palaung showed a “one-date-multiway” result, while other AA groups showed “one-date” results. The admixture dates were estimated using 100 bootstrap replicates. As the Western Lawa and Eastern Lawa share a considerable proportion of ancestry with the Karen groups, which might be the proximal source that introduced East Asian ancestry to the Lawa groups, we conducted an additional run using SgawKaren and Mlabri as surrogates in fastGLOBETROTTER runs. For dating admixture events with ALDER v.1.03 and DATES v.753, we used Brahmin_Tiwari as the South Asian ancestral source, Yi, Nepal_Chokhopani_2800BP.SG and China_YR_MN as the modern and ancient East Asian ancestral source, and Khmu or Mlabri as the AA ancestry source, to date South Asian and East Asian admixture events in AA-speaking groups.
Admixture graph analyses
To construct a maximum-likelihood tree to reveal population relationships and migration among AA groups, we utilized a pruned dataset containing 56,038 SNPs to run TreeMix v.1.1377. The dataset included modern AA-speaking populations representing their respective language branches, along with selected reference populations. For the AA groups, we used Kharia for the Munda branch, Maniq for Aslian, Mon for Monic, Kinh for Viet-Muong, and Nyahkur for Southern Monic. For Northern Mon-Khmer, we selected Khmu, Lua, and Mlabri for Khmuic, as these three groups displayed distinct genetic profiles in the PCA and ADMIXTURE analyses, alongside Blang for Palaungic and Mang. Eastern Mon-Khmer populations included Bru and Kuay for Katuic, Chong for Pearic, and Khmer for Khmeric. The African Mbuti population was used as an outgroup, while reference populations included Oceanian Papuan, Andamanese Onge, South Asian Brahmin_Tiwari, northern East Asian Oroqen, AN-speaking Mamanwa and Ami, TK-speaking Thai and Dai, ST-speaking Yi and Han, as well as Akha1_T, Burmese1_T, and HM-speaking Hmong and Miao. We performed 10 iterations for migration events ranging from 0 to 10 and identified the optimal tree with 5 migration edges using OptM78.
We utilized the “find_graphs” function in AdmixTools279 and qpGraph v.7580 from Admixtools63 to construct admixture graphs incorporating both modern and ancient populations. To represent the AA language branches, we selected Maniq for Aslian, Mon for Monic, Nyahkur for Southern Monic, Bru for Eastern Mon-Khmer, and Mlabri for Northern Mon-Khmer. The African Mbuti population served as an outgroup. For ancient populations, we used Indus_Periphery as a proxy for ancient South Asians, China_YR_MN for ancient northern East Asians, and Liangdao1 for ancient southern East Asians. We first ran “find_graphs” for admixture events ranging from 0 to 12, incorporating 10 to 12 populations. We accepted the graph as a good fit when the absolute value of the Z-score of the worst f4 statistic output by qpGraph was < 3. Attempts to include Kinh for Viet-Muong and Kharia for Munda failed to produce a model with a good fit. The number of allowed admixture events in each graph was incrementally increased until the best test score was no longer significantly better than the highest-scoring graphs with fewer admixture events. By doing these steps we finalized the analysis using qpGraph with 10 populations and 10 admixture events. The following parameters were applied for qpGraph: outpop: NULL, blgsize: 0.05, forcezmode: YES, diag: 0.0001, bigiter: 6, hires: YES, and lambdascale: 1.
Given the heterogeneity of Northern Mon-Khmer, we attempted to replace Mlabri by another Northern Mon-Khmer group in turn using the same setup of tree typology inferred with Malbri. Based on qpGraph estimation, only Lua fits the same topology as Mlabri with a statistically acceptable fitting Z-score (Supplementary Fig. 19). For other Northern Mon-Khmer speakers (Mang, Blang, Khmu), we then applied “find_graphs” to iteratively search for the best fitting typology tree, and confirmed the robustness of the inferred best-fitting typological tree from “find_graphs” by substituting one Northern Mon-Khmer population by another in an independent qpGraph statistic. Following such iterative strategy, we found that Blang and Mang fit the same alternative topology, while Khmu fit a third distinct topology (Supplementary Fig. 19).
To construct admixture graphs for ancient MSEA populations, we first applied “find_graphs” using a set of representative ancient groups: Laos_Hoabinhian as a proxy for ancient MSEA hunter-gatherers; China_YR_MN for ancient northern East Asians; Liangdao1 for ancient southern East Asians; Longlin for ancient southern East Asian hunter-gatherers; Indus_Periphery_West refers to an Iranian farmer-related individual contributing to the Neolithic South Asian ancestry; India_Harappan corresponds to the ancestry of the Indus Valley Civilization (IVC); Central_Steppe_MLBA represents the Yamnaya-related ancestry component (associated with the Ancestral North Indians, ANI); and Onge stands for Andamanese hunter-gatherer-related Ancestral South Indian (AASI) ancestry. We also included Mbuti as the outgroup to establish a foundational topology including the set of representative ancient groups described above. We tested models with admixture events ranging from 0 to 5, evaluating each topology using qpGraph under consistent parameters and acceptance criteria. Within this scaffold topology, we successfully modeled N-ManBac as an admixed population derived from a China_YR_MN–related northern East Asian source and a deeply diverged lineage related to Laos_Hoabinhian, Longlin, and Onge. Subsequently, we extended the analysis by either replacing N-ManBac with other ancient MSEA groups or incorporating modern AA-speaking populations to test the robustness of the model (Supplementary Fig. 16).
Statistics and Reproducibility
The study includes genome-wide data generated for 164 individuals, together with comparative published data (79 ancient and 1,378 modern individuals). Each individual represents a single biological sample, and no biological replicates were used, which is standard for population genomic studies. All statistical analyses in this study were performed using widely used population genetic frameworks, including principal component analysis, model-based clustering, f-statistics, admixture dating, graph-based modeling and haplotype-based inference.
No statistical methods were used to predetermine sample size; sample numbers were determined prior to analysis based on available specimens and budgetary constraints for genotyping, with the aim of including sufficient samples to support statistical comparisons. As no experimental assays were performed, replication of experimental results is not applicable. All analyses are reproducible using the methods, parameter settings, and genome-wide data described and made publicly available in the associated repositories.
Ethics and Inclusion
This study was conducted in close collaboration with researchers and institutions in the regions where samples were collected. Local researchers participated in project design, sample collection, laboratory work, data generation, data analysis, and interpretation. The study followed ethical and respectful engagement with participating communities, ensured appropriate recognition of local expertise, and avoided extractive research practices. No communities or participant groups were excluded from authorship or acknowledgement where appropriate.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The genome-wide data generated in this study are available from the GSA (https://ngdc.cncb.ac.cn/omix/) under accession number PRJCA046043. The ancient genome data used in this study is publicly available from the Allen Ancient DNA Resource (AADR) v.54.180,81. Source data underlying all figures are provided in the Supplementary Data.
Code availability
All relevant code and software applied in this work have been acknowledged and corresponding citations are provided in the Methods section. No custom code essential for reproducing the results was generated.
References
Sidwell, P. & Blench, R. The austroasiatic urheimat: the Southeastern Riverine hypothesis. In Dynamics of human diversity: the case of mainland Southeast Asia (ed. Enfield, N. J.) 315–343 (Pacific Linguistics, 2011).
Norman, J. & Mei, T.-L. The austroasiatics in ancient south China: some lexical evidence. Monum. Serica 32, 274–301 (1976).
van Driem, G. The ethnolinguistic identity of the domesticators of Asian rice. Comptes Rendus Palevol. 11, 117–132 (2012).
Lipson, M. et al. Ancient genomes document multiple waves of migration in southeast asian prehistory. Science 361, 92–95 (2018).
McColl, H. et al. The prehistoric peopling of Southeast Asia. Science 361, 88–92 (2018).
Wang, T. et al. Prehistoric genomes from Yunnan reveal ancestry related to Tibetans and Austroasiatic speakers. Science 388, eadq9792 (2025).
Moore, R. K. Spoken language processing: Piecing together the puzzle. Speech Commun. 49, 418–435 (2007).
Pittayaporn, P. Layers of Chinese loanwords in protosouthwestern Tai as evidence for the dating of the spread of southwestern Tai. MANUSYA J. Humanit 17, 47–68 (2014).
Sidwell, P. Southeast Asian Mainland: linguistic history. in The Encyclopedia of Global Human Migration Volume 1 Prehistory (ed. Bellwood, P.) 259–268 (Wiley-Blackwell, 2013).
Schliesinger, J. Ethnic Groups of Thailand: Non-Tai-Speaking Peoples. (White Lotus Press, 2000).
Aghakhanian, F. et al. Unravelling the Genetic History of Negritos and Indigenous Populations of Southeast Asia. Genome Biol. Evol. 7, 1206–1215 (2015).
Aghakhanian, F. et al. Sequence analyses of Malaysian Indigenous communities reveal historical admixture between Hoabinhian hunter-gatherers and Neolithic farmers. Sci. Rep. 12, 13743 (2022).
Yew, C.-W. et al. Genomic structure of the native inhabitants of Peninsular Malaysia and North Borneo suggests complex human population history in Southeast Asia. Hum. Genet. 137, 161–173 (2018).
Kutanan, W. et al. Reconstructing the human genetic history of mainland southeast asia: I Thailand and Laos. Mol. Biol. Evol. 38, 3459−3477 (2021).
Changmai, P. et al. Indian genetic heritage in Southeast Asian populations. PLoS Genet 18, e1010036 (2022).
Cœdès, G. The Indianized States of Southeast Asia, Vol. 424 (University of Hawaii Press, 1968).
Higham, C. Early Mainland Southeast Asia: From First Humans to Angkor, Vol. 456 (River Books, 2014).
Changmai, P. et al. Ancient DNA from protohistoric period Cambodia indicates that South Asians admixed with local populations as early as 1st–3rd centuries CE. Sci. Rep. 12, 22507 (2022).
He, Y. et al. Genome diversity and signatures of natural selection in mainland Southeast Asia. Nature 1, 10 (2025).
Liu, D. et al. Extensive ethnolinguistic diversity in vietnam reflects multiple sources of genetic diversity. Mol. Biol. Evol. 37, 2503–2519 (2020).
Lawson, D. J., van Dorp, L. & Falush, D. A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nat. Commun. 9, 3258 (2018).
Palamara, P. F., Lencz, T., Darvasi, A. & Pe’er, I. Length distributions of identity by descent reveal fine-scale demographic history. Am. J. Hum. Genet. 91, 809–822 (2012).
Fuller, D. Q. Pathways to asian civilizations: tracing the origins and spread of rice and rice cultures. Rice 4, 78–92 (2011).
Fenner, J. N. Cross-cultural estimation of the human generation interval for use in genetics-based population divergence studies. Am. J. Phys. Anthropol. 128, 415–423 (2005).
Al-Asadi, H., Petkova, D., Stephens, M. & Novembre, J. Estimating recent migration and population-size surfaces. PLoS Genet 15, e1007908 (2019).
Nahhas, R. W. Sociolinguistic Survey of Lawa in Thailand. (Survey Unit, Department of Linguistics, Faculty of Humanities, Payap University, 2007).
Oliveira, S. et al. Ancient genomes from the last three millennia support multiple human dispersals into Wallacea. Nat. Ecol. Evol. 6, 1024–1034 (2022).
Harney, É, Patterson, N., Reich, D. & Wakeley, J. Assessing the performance of qpAdm: a statistical tool for studying population admixture. Genetics 217, iyaa045 (2021).
Pryce, T. O. et al. Metallurgical traditions and metal exchange networks in late prehistoric central Myanmar, c. 1000 BC to c. AD 500. Archaeol. Anthropol. Sci. 10, 1087–1109 (2018).
Pietrusewsky, M. & Ikehara-Quebral, R. The bioarchaeology of the Vat Komnou Cemetery, Angkor Borei, Cambodia. Bull. Indo-Pac. Prehistory Assoc. 26, 86–97 (2006).
Shoocongdej, R. The history and practice of archaeology in Thailand. In Handbook of East and Southeast Asian Archaeology (eds. Olsen, J. W., Lape, P. V. & Habu, J.) 97–109 (Springer, 2017).
The HUGO Pan-Asian SNP Consortium et al Mapping human genetic diversity in Asia. Science 326, 1541–1545 (2009).
Xu, S. et al. Genetic evidence supports linguistic affinity of Mlabri - a hunter-gatherer group in Thailand. BMC Genet 11, 18 (2010).
Göllner, T. et al. Unveiling the genetic history of the maniq, a primary hunter-gatherer society. Genome Biol. Evol. 14, evac021 (2022).
Kutanan, W. et al. New insights from Thailand into the maternal genetic history of Mainland Southeast Asia. Eur. J. Hum. Genet. 26, 898–911 (2018).
Thao, D. H. et al. Investigating demic versus cultural diffusion and sex bias in the spread of Austronesian languages in Vietnam. PLoS ONE 19, e0304964 (2024).
Oota, H. et al. Recent origin and cultural reversion of a hunter–gatherer group. PLOS Biol 3, e71 (2005).
Rischel, J. Minor Mlabri: A Hunter-Gatherer Language of Northern Indochina. (Museum Tusculanum, 1995).
Kutanan, W. et al. Contrasting paternal and maternal genetic histories of Thai and Lao populations. Mol. Biol. Evol. 36, 1490–1506 (2019).
Premsrirat, S. Linguistic contributions to the study of the Northern Khmer Language of Thailand in the last two decades. Mon-Khmer Stud. J. 27, 129–136 (1997).
Wen, B. et al. Genetic evidence supports demic diffusion of Han culture. Nature 431, 302–305 (2004).
Tätte, K. et al. The genetic legacy of continental scale admixture in Indian Austroasiatic speakers. Sci. Rep. 9, 3818 (2019).
Higham, C. F. W., Douka, K. & Higham, T. F. G. A New chronology for the Bronze Age of northeastern Thailand and its implications for Southeast Asian prehistory. PLOS ONE 10, e0137542 (2015).
Huffman, F. E. Burmese Mon, Thai Mon, and Nyah Kur: a synchronic comparison. Mon-Khmer Stud. 16, 31–84 (1990).
Ocharoen, S. Mons in Thailand [in Thai]. (Thailand Research Fund, Bangkok, 1998).
Thosarat, R. & Higham, C. Early Thailand: From Prehistory to Sukhothai, Vol. 268 (River Books, 2012).
Sidwell, P. & Reid, L. A. Language macro-families and distant phylogenetic relations in MSEA. In The Languages and Linguistics of Mainland Southeast Asia: A comprehensive guide (eds. Sidwell, P. & Jenny, M.) 261–276 (Walter de Gruyter GmbH & Co KG, 2021).
Prakhun, N. et al. Genetic variability of 23 autosomal STRs in Austroasiatic-speaking populations from Thailand. Mol. Genet. Genomics 299, 80 (2024).
Mawan, A. et al. Autosomal Microsatellite Investigation Reveals Multiple Genetic Components of the Highlanders from Thailand. Genes 12, 383 (2021).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, s13742-015-0047–8 (2015).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinforma. Oxf. Engl. 26, 2867–2873 (2010).
Eberhard, D. M., Simons, G. F. & Fennig, C. D. Ethnologue: Languages of the World. https://www.ethnologue.com/ (2020).
Bergström, A. et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 367, eaay5012 (2020).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Patterson, N., Price, A. L. & Reich, D. Population Structure and Eigenanalysis. PLOS Genet 2, e190 (2006).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Joseph, T. A. & Pe’er, I. Inference of population structure from time-series genotype data. Am. J. Hum. Genet. 105, 317–333 (2019).
Narasimhan, V. M. et al. The formation of human populations in South and Central Asia. Science 365, eaat7487 (2019).
Ning, C. et al. Ancient genomes from northern China suggest links between subsistence changes and human migration. Nat. Commun. 11, 2700 (2020).
Yang, M. A. et al. Ancient DNA indicates human population shifts and admixture in northern and southern China. Science 369, 282–288 (2020).
Wang, T. et al. Human population history at the crossroads of East and Southeast Asia since 11,000 years ago. Cell 184, 3829–3841.e21 (2021).
Francis, R. M. pophelper: An R package and web app to analyse and visualise population structure. Mol. Ecol. Resour. 17, 27–32 (2017).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Delaneau, O., Marchini, J. & Zagury, J.-F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2012).
Frazer, K. A. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–861 (2007).
Lawson, D. J., Hellenthal, G., Myers, S. & Falush, D. Inference of Population Structure using Dense Haplotype Data. PLOS Genet 8, e1002453 (2012).
Chacón-Duque, J.-C. et al. Latin Americans show wide-spread Converso ancestry and imprint of local native ancestry on physical appearance. Nat. Commun. 9, 5388 (2018).
Wangkumhang, P., Greenfield, M. & Hellenthal, G. An efficient method to identify, date, and describe admixture events using haplotype information. Genome Res. 32, 1553–1564 (2022).
Zhou, Y., Browning, S. R. & Browning, B. L. A fast and simple method for detecting identity-by-descent segments in large-scale data. Am. J. Hum. Genet. 106, 426–437 (2020).
Browning, B. L., Tian, X., Zhou, Y. & Browning, S. R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 108, 1880–1890 (2021).
Browning, B. L., Zhou, Y. & Browning, S. R. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 103, 338–348 (2018).
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017).
Fournier, R., Tsangalidou, Z., Reich, D. & Palamara, P. F. Haplotype-based inference of recent effective population size in modern and ancient DNA samples. Nat. Commun. 14, 7945 (2023).
Petkova, D., Novembre, J. & Stephens, M. Visualizing spatial population structure with estimated effective migration surfaces. Nat. Genet. 48, 94–100 (2016).
Loh, P.-R. et al. Inferring admixture histories of human populations using linkage disequilibrium. Genetics 193, 1233–1254 (2013).
Chintalapati, M., Patterson, N. & Moorjani, P. The spatiotemporal patterns of major human admixture events during the European Holocene. eLife 11, e77625 (2022).
Pickrell, J. K. & Pritchard, J. K. Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLOS Genet 8, e1002967 (2012).
Fitak, R. R. OptM: estimating the optimal number of migration edges on population trees using Treemix. Biol. Methods Protoc. 6, bpab017 (2021).
Maier, R. et al. On the limits of fitting complex models of population history to f-statistics. eLife 12, e85492 (2023).
Mallick, S. & Reich, D. The Allen Ancient DNA Resource (AADR): a curated compendium of ancient human genomes. Sci. Data. 11, 182 (2023).
Mallick, S. et al. The allen ancient DNA resource (AADR) a curated compendium of ancient human genomes. Sci. Data 11, 182 (2024).
Lazaridis, I. et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513, 409–413 (2014).
Kampuansai, J. et al. Genetic diversity and ancestry of the Khmuic-speaking ethnic groups in Thailand: a genome-wide perspective. Sci. Rep. 13, 15710 (2023).
Kampuansai, J. et al. Under the name of “Lua”: revisiting genetic heterogeneity and population ancestry of austroasiatic speakers in Northern Thailand through genomic analysis. BMC Genomics 25, 956 (2024).
Acknowledgements
This research is funded by the National Research Council of Thailand (NRCT) and Naresuan University (N42A670264), and the Max Planck Society. W.K. was also funded by the Institute of Suvanabhumi Studies, TASSHA (Thailand Academy of Social Sciences, Humanities and Arts), Ministry of Higher Education, Science, Research and Innovation. W.K., Y.G. and M.Sr. were funded by Naresuan University under global and frontier research university fund (R2566C051). K. W. acknowledges support by the National Science Foundation of China (32370660) and the Shanghai Rising-Star program (23YF1402900), and by 111 project (B25056). D.L. is funded by a Pasteur-Roux-Cantarini Postdoctoral Fellowship. C.S. is supported by the Unit of Excellence 2025 on Biodiversity and Natural Resources Management, University of Phayao.
Author information
Authors and Affiliations
Contributions
W.K., D.L., K.W. conceived the study under supervision by M.S. W.K., N.P., J.K., M.Sr., A.I., P.R., C.K., C.S. and S.L. were involved with sample collection. N.P. and W.K. extracted DNA. W.W., J.R., M.S., W.K. and D.L. were involved with generating data. Y.G. and D.L. analysed preliminary data. Z.Y and K.W. analysed data with critical input and contextualisation from M.S., W.K. and D.L. Z.Y, M.S., W.K., D.L. and K.W. interpreted the analyses. K.W., and Z.Y, D.L., M.S., W.K. wrote the manuscript with input from the remaining authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Cesar Fortes-Lima, Michele Repetto and George Inglis. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yin, Z., Gupta, Y.M., Prakhun, N. et al. Exploring the genomic population structure and history of Austroasiatic speakers in Mainland Southeast Asia. Commun Biol 9, 300 (2026). https://doi.org/10.1038/s42003-025-09471-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09471-0
