
Abstract
Spatial transcriptomes and Matrix-Assisted Laser Desorption Ionization Mass Spectrometry Imaging (MALDI-MSI) measures mRNA expression and mass-to-charge (m/z) spectra on thousands of spots along with the spatial coordinates. Integrating spatial transcriptomes and MALDI-MSI is challenge due to no shared coordinates or features. We present \({haCCA}\), a workflow to integrate spatial transcriptomes and metabolomes.\(h{aCCA}\) take advantage of modified spatial registration and shared latent space constructed by CCA(Canonical Correlation Analysis)-mediated transfer of high-correlated feature pairs. It enables the simultaneous spatial profiling of metabolites and transcriptome across neighbor tissue section. We tested \({haCCA}\) on pseudo and real data, proving that \(h{aCCA}\) improved the integration accuracy than existing methods. We further applicated \(h{aCCA}\) on a custom dataset from Akt/Yap driven Padi4-/-ICC model which lacks neutrophil extracellular traps(NETs) and revealing the spatial distribution of both mRNA and metabolites,enabled both in situ and in vivo exploration of the metabolic alteration effect of NETs on ICC. A Python package was developed to facilitate its use.

Similar content being viewed by others
Background
Spatial multi-omics techniques allow resolving the spatial molecular profiles at transcript, metabolite, protein or epi-genetic level1,2. Among these techniques, spatial transcriptomes enables the measurement of mRNA expression, while Matrix-Assisted Laser Desorption Ionization Mass Spectrometry Imaging (MALDI-MSI) allows the collection of mass-to-charge (m/z) spectra at defined raster spots across the tissue sections. These two methodologies are among the most frequently applied spatial techniques. Both techniques generate gene expression or peak intensity matrices for sampling spots, with coordinates indicating their spatial locations. The number of spots can vary from thousands to hundreds of thousands, depending on the resolution.
The integration of spatial transcriptomics (ST) and MALDI-MSI to generate multi-modal spatial data provides a more comprehensive understanding and novel insights into the biological states of tissue sections. However, performing such integration remains quite challenging. To date, three categories of integration strategies have been proposed for multi-modal single-cell data3, using shared data features, using shared cells, or using a shared latent space when neither shared cells nor features are accessible. The first two strategies preserve the original resolution at the single-cell or spot level, whereas the last strategy results in loss of resolution. Typically, no shared features can be identified between spatial transcriptomes and MALDI-MSI, nor are there shared spots or cells, which constrains the application of the first two strategies. Fortunately, spatial multi-modal techniques are often applied to adjacent tissue sections, or even the same section4, ensuring the morphological similarity. Therefore, spatial registration—a process of aligning coordinate systems into a common reference frame—which leverages the morphological similarity of spatial coordinates, has been most frequently applied for their integration.5,6,7,8.
This integration strategy has certain limitations. First, there is no benchmark to assess the performance of the integration. Second, as no features are currently leveraged to assist the integration process, the efficiency of integration may be compromised. The use of shared features for alignment has proven effective in integrating single-cell data from different batches, where linear combinations of shared features are used to establish a latent space, allowing cells with the same labels to cluster together into the same group9,10,11. Although no shared features exist between MALDI-MSI and spatial transcriptomes, a few studies revealed the high correlation between gene transcripts and metabolites12,13, inspiring us to explore whether highly correlated gene-metabolite pairs can be utilized for integration.
In this study, we established \(h{aCCA}\), a workflow utilizing high Correlated feature pairs combined with a modified spatial morphological alignment to ensure high resolution and accuracy of spot-to-spot data integration of spatial transcriptomes and metabolomes. We generated a series of benchmarks using spatial transcriptomes and paired pseudo spatial metabolomes, demonstrating that\(h{aCCA}\) outperforms spatial registration methods (for example, STUtility). We tested the performance of \(h{aCCA}\) in assisting multi-modal spatial data analysis using public available 10X Visium and MALDI-MSI data cohort of mouse brain and a home-brew spatial transcriptomes (on BMKMANU S1000 platform) and MALDI-MSI data cohort of ICC (intrahepatic cholangiocarcinoma) model. \(h{aCCA}\) enabled us to capture changes that would not have been fully characterized by either technique alone. Through in situ and in vivo profiling, we investigated the impact of neutrophil extracellular traps (NETs) on ICC, revealing that NETs upregulated Scd1, driving the activation of various metabolic pathways, particularly fatty acid elongation. An easy-to-use Python package has also been developed and made publicly available for implementation.
In summary, we presented an integration workflow named \(h{aCCA}\), which take advantage of modified spatial registration and shared latent space constructed by CCA(Canonical Correlation Analysis)-mediated transfer of high-correlated feature pairs. It enables simultaneous spatial profiling of metabolites and transcriptomes across adjacent tissue sections. By applying \({haCCA}\) to tumor samples, it enhances our understanding of both metabolic and transcriptomic heterogeneity, offering valuable insights into the dynamic crosstalk between genes and metabolites. This crosstalk plays a crucial role in regulating tumor behavior and influencing treatment responses.
Results
The workflow of high correlated feature pairs combined with spatial morphological alignment(haCCA) for multi-modal spatial assay integrating
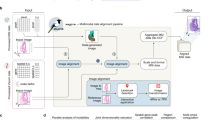
We use a triplet tuple Data(d, f, l)={d, f, l} to represent a spatial assay and m(d, f, l) to represent each spot in Data. For each spot m(d, f, l), f denotes the feature vector, d represents the spatial information and \(l\) represents the cluster label. For two given multi-modal spatial assays \({Dat}{a}_{A}(d,f,l)\) and \({Dat}{a}_{B}(d,f,l)\). \({haCCA}\) integrates multi-modal spatial assay by finding an alignment from \({m}_{a}\) to \({m}_{b}\), noted as \({h{aCCA}}_{A\to B}\). The alignment results combine information from both modified spatial registration \(\left\{d\right\}\) and shared latent feature \(\left\{{f}^{{cca}}\right\}\) constructed from \(f\). After generating data among neighbor sections, \({haCCA}\) workflow is performed through 5 steps: 1: data preparation. We prepare the data by following the standard process with \({Scanpy}\)14(see “Method”). The spatial data in the form of the peak/intensity (MALDI-MSI) or gene expression matrix(spatial transcriptome) are extracted, normalized into the construction of \(m=\{{d}_{m},{f}_{m},{l}_{m}\}\)(Fig. 1A, B). 2: gross alignment and further alignment: Coordinate information \(d\) provides the location of each spot in spatial assay and is crucial for the alignment accuracy. To make the most use of coordinate information, \({haCCA}\) uses a two-stage strategy: gross alignment and further alignment. In the first stage, \(h{aCCA}\) performs gross alignment for the purpose of eliminating the potential shifts and rotations introduced from sampling and section. by default, \({haCCA}\) used manual alignment to introduce prior knowledge from human expert. In manual alignment, three corresponding points, or spatial landmarks in both datasets, sharing high similarity in image characteristics, are manually selected by human experts to generate an affine transformation matrix \({M}_{{manual}}\)., which will be used to align spots between two assays. The detail algorithm for gross alignment can be found in Algorithm 1. After gross alignment, the major part of spots in \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\) will be overlayed to each other, but there might still be a small subset of outlier spots that is not overlayed, which typically occurs near the tissue boundaries and missing parts and are usually introduced during section preparation (the red edge in Fig. 1C). Therefore, in the second stage, we perform further alignment to those outlier spots and its corresponding nearest neighbor in another assay. The outlier spots are identified as the spots which has no corresponding spot within a certain distance in another assay. The detailed algorithm for further alignment can be found in Algorithm 2. 3: Identify anchor spot pairs and high correlated feature pairs. After gross alignment and further alignment, the next step is to find anchor spots pairs from 2 assays, noted as \(\{({m}_{a}^{{anc}h{or}},{m}_{b}^{{anc}h{or}})\}\). Anchor spot pairs are spots sampled from the same or close location in neighbor sections. Anchor spot pairs need to satisfy the following criteria (1) their coordinates are close enough, the distance between them is smaller than a given threshold \({dist\_}\min\), and they are the closest point to each other in two assays and (2) their neighborhood spots are of low variety. i.e., most of their neighbor spots belongs to the same cluster/label. The detailed algorithm to detect anchor spot pairs discovery can be found in Algorithm 3. Once anchor spot pair is identified, we further identify the high correlated feature pairs \(\{{( \, {f}_{A}^{i},{f}_{B}^{j})}_{k}\}\) where \(i\) is the feature index of \({f}_{A}\) and \(j\) is the feature index of \({f}_{B}\). By calculating Pearson correlation matrix on the feature matrix of anchor spot pairs and select the index of the top \(k\) score in that Pearson correlation matrix. 4: Feature aid fine alignment. In this step, we calculate \({f}^{{cca}}\), a compressed feature from original feature space by applying Canonical Correlation Analysis (CCA) on high correlated feature pairs \(\{{({f}_{A}^{i},{f}_{B}^{j})}_{k}\}\) for both \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\). \({f}^{{cca}}\) will be used together with coordinate information \(d\) to find a transporting plan \(\Pi\), a \(m* n\) matrix where each entry \({\Pi }_{{ij}}\) represents the probability transport from spot \(i\) to spot \(j\) and \(m\) and \(n\) is the number of spots in \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\) to minimize the \({los}{s}_{{haCCA}}\) where \(d\) contributes to the distance discrepancy and \({f}^{{cca}}\) contributes to the feature discrepancy. In order to resolve \(\Pi\), we propose a few heuristic methods to find its approximate solution using \({ICP}\) or \({FGW}\). The detailed process can be found in Algorithm 4. 5: integrating: the final alignment \(\{\left({m}_{a},{m}_{b}\right)\}\) for \(\forall \,{m}_{a}{in\; Dat}{a}_{A}\) can be inferred for every spot \({m}_{a}\) from transporting plan \(\Pi\) by selecting the index of \({m}_{b}\) with the maximum probability. For each spot \({m}_{{a}_{i}}=\{{d}_{m{a}_{i}},\,{f}_{m{a}_{i}},{l}_{m{a}_{i}}\}\), the data of its alignment spot \({m}_{{b}_{j}}\) in another assay is integrated in order to generate multi-modal data \({m}_{{ai}}^{{integrated}}=\{{d}_{m{a}_{i}},[ \, {f}_{m{a}_{i}},{f}_{m{b}_{j}}],{l}_{m{a}_{i}}\}\).(Fig. 1)

The procedure consist of 5 main steps: Data preparing (A, B): data is normalized into the construction form of \(\{d,f,l\}\). Where \(d\) represent the coordinates, \(f\) represent the feature matrix, \(l\) represents the cluster label. Gross alignment and Further alignment (C, D): Corresponding points were selected based on mutual spatial region(marked by square) with high similarity between \({m}_{a}\) and \({m}_{b}\). (C, upper) After gross alignment, some spots in \({Dat}{a}_{B}\) is not perfectly overlayed with\({Dat}{a}_{A}\). (red edge in C below). After further alignment, a maximum overlap of \({d}_{{m}_{b}}\) and \({d}_{{m}_{a}}\) can be achieved. (C, below) After gross alignment and further alignment the \({d}_{m}\) is noted as \({d}_{m}^{{\prime} }\). The spatial registration of \({haCCA}\) is now finished. Anchor spot pairs and high correlated feature pairs identification (E, F): Anchor spots were shown on \({m}_{a}\) and \({m}_{b}\)(E). After detection of anchor spot pairs, noted as \({anc}h{or}\; {spot}({m}_{a}^{{anc}h{or}},\,{m}_{b}^{{anc}h{or}})\), Pearson correlation matrix was calculated using the feature vector \(f\) among all anchor spot pairs. After Peason correlation matrix was calculated, the index of k-top-most R values are recorded as high-correlated feature pairs\({\{({f}_{{m}_{a}}^{i},\,{f}_{{m}_{b}}^{j})\}}_{k}\). Feature aid fine alignment. G, H \(({f}_{{m}_{a}}^{i},\,{f}_{{m}_{b}}^{j})\) undergo CCA transfer to generate a pair of high-correlated variate \(\{{f}_{{m}_{a}}^{{cca}},\,{f}_{{m}_{b}}^{{cca}}\}\) as a latent space shared by \({m}_{a}\) and \({m}_{b}\). With \(\{{f}_{{m}_{a}}^{{cca}},\,{f}_{{m}_{b}}^{{cca}}\}\) and \(\{{d}_{{m}_{a}}^{{\prime} },{d}_{{m}_{b}}^{{\prime} }\}\), Alignment between 2 assay is carried out by minimizing the \({los}{s}_{h{aCCA}}({{\rm{H}}})\). \(\alpha\) is set by default = 0.5. A 3-D visualization of \(m=\{{d}_{m}^{{\prime} },\,{f}_{m}^{{cca}}\}\) before and after Feature aid fine alignment was shown in G. integrating (I, J): An alignment of \(\{{m}_{{a}_{i}},{m}_{{b}_{j}}\}\) was found For each spot \({m}_{{a}_{i}}=\{{d}_{m{a}_{i}},\,{f}_{m{a}_{i}},\,{l}_{m{a}_{i}}\}\). For each spot \({m}_{{a}_{i}}=\{{d}_{m{a}_{i}},\,{f}_{m{a}_{i}},\,{l}_{m{a}_{i}}\}\), the data of its alignment spot \({m}_{{b}_{j}}\) in another assay is integrated in order to generate multi-modal data \({m}_{{ai}}^{{integrated}}=\{{d}_{m{a}_{i}},\,{f}_{m{a}_{i}},{f}_{m{b}_{j}},\,{l}_{m{a}_{i}}\}\). I shown the label distribution of \({m}_{b}\) in \({m}_{a}^{{integrated}}\).
haCCA perform effective integration on pseudo multi-modal spatial data
We quantitatively evaluated the performance of \(h{aCCA}\) in integrating spatial transcriptome data and pseudo-generated paired MALDI-MSI data. As methods to simultaneously generate both STs and MALDI-MSI data from the exact same spot are still lacking, obtaining precise spot alignment remains challenging, making performance evaluation difficult. To overcome this limitation, we employed a spatial perturbation and sampling strategy on ST data to generate paired pseudo-MALDI-MSI data. These paired datasets served as benchmarks for evaluating the performance of our alignment strategy.
Specifically, pseudo MALDI-MSI was generated by spatial perturbation and resampling the normalized and scaled read counts of spots in the spatial transcriptome dataset. The pseudo-MALDI-MSI data possess several characteristics that recapitulate the differences between MALDI-MSI and ST in real-world scenarios: 1. Small perturbations were added to the spatial coordinates of the pseudo-MALDI-MSI data to reduce morphological similarity with the parent spatial transcriptome data, mimicking the shape distortion observed between adjacent tissue sections. 2. no shared features between pseudo-MALDI-MSI data and parent spatial transcriptome data, mimicking the feature differences between MALDI-MSI and spatial transcriptome. We measure the accuracy of integration using pairwise alignment accuracy (PAA)15, label transfer accuracy (AC) and label transfer adjusted rand index (ARI). PAA is the sum of probabilistic alignment weight over all spots pairs between spatial transcriptome and paired pseudo-MALDI-MSI, label transfer accuracy is the percentage of correctly transferred label (cluster) between spatial transcriptome and paired pseudo-MALDI-MSI. Label transfer ARI measures the similarity between spatial transcriptome and paired pseudo-MALDI-MSI, it provides an objective measure of how well spatial patterns are preserved between modalities. In total, benchmark datasets containing spatial transcriptome and paired pseudo MALDI-MSI data was generated form 3 mouse brain (M1, M3, M4), 1 rat brain(Rat) and 1 human gastric cancer (GC). 15 samples randomly chosen from 5 datasets to form a comprehensive dataset, which was used to evaluate the performance of \(h{aCCA}\).
To evaluate the contribution of each step in \(h{aCCA}\) to the integrating performance in benchmark dataset, we assessed the improvement of 3 metrics (PAA, AC, ARI) at gross alignment, further alignment and feature aid fine alignment in \(h{aCCA}\) workflow (Fig. 2A, B). As shown in representative spatial plot of sample M3, the integration becomes more accurate and achieve high similarity to ground truth after 3 steps of \(h{aCCA}\), along with increasing of metrics. Currently, very few publicly available methods exist for integrating multi-modal spatial data, and most rely on spatial registration. We compared \(h{aCCA}\) with several methodologies applied to multi-modal spatial data integration in the comprehensive dataset. These methods included direct alignment, STUtility, ICP and STalign16. We found that compared with other methods, \(h{aCCA}\) showed a significant increasing of PAA, AC and ARI (Fig. 2C, D), indicating \(h{aCCA}\)’s best performance in integrating multi-modal spatial data. These results demonstrated \(h{aCCA}\) is able to outperform other integrating method in paired spatial transcriptome and pseudo-MALDI MSI data.

A representative spatial plot of groundtruth data (left) compared with results after gross alignment (75% accuracy), further alignment (86% accuracy), and feature-aid fine alignment (98% accuracy) in sample M3. Distribution of clustering labels were shown. Black box indicated zoom in plot. B Quantitative assessment of alignment performance using three metrics: label transfer accuracy (AC), label transfer adjusted rand index (ARI), and pairwise alignment accuracy. Box plots show significant improvements across successive alignment stages (p values indicated above, 15 samples were included in the benchmark dataset). C Comparison of integration results between different methods (directmerge, STUtility, ICP, STAlign, and \({haCCA}\)) against groundtruth. Label transfer accuracy scores demonstrate \({haCCA}\)'s superior performance (95%) compared to other methods (65–85%). Distribution of clustering labels were shown. Black box indicated zoom in plot. D Comprehensive comparison of method performance using multiple metrics. Box plots show \({haCCA}\) consistently outperforms other methods across all evaluation metrics, with statistical significance indicated by p values.
haCCA performs consistent integration of 10X Visium and MALDI-MSI spatial data
To evaluate \({haCCA}\)'s performance on real-world data, we analyzed an integrated dataset comprising 10X Visium and MALDI-MSI data from three mouse brain specimens. Each specimen yielded three spatial datasets from two adjacent sections (Section A generated both 10X Visium ST and MALDI-MSI data, while Section B generated only 10X Visium ST; see Supplementary Data 1). To be noted, 1 run of 10X Visium and 1 run of MALDI-MSI data was performed on the same section, with a novel procedure4. This procedure comprises the following four steps: (1) sectioning nonembedded snap-frozen samples onto noncharged, barcoded gene expression arrays, (2) MSI by MALDI, (3) spatial transcriptome spatial transcriptome. Another run of 10X Visium was performed on the neighbor section.
We evaluated \({haCCA}\)'s integration capabilities in two scenarios: (1) integrating data from the same section, and (2) integrating data from neighboring sections. STUtility, which is applied as integration method in original work4, served as our baseline integration method for same-section data. To assess integration quality, we visualized the distribution of metabolic cluster labels on spatial transcript coordinates.
Our analysis revealed that \({haCCA}\) successfully integrated same-section 10X Visium and MALDI-MSI spatial data, achieving high label transfer accuracies of 87.5%, 79.5%, and 87.6% when using STUtility outcomes as ground truth (Fig. 3A, Supplementary Fig. 4A, C). The metabolic cluster label distribution patterns were well-preserved in \({haCCA}\)-integrated data. Notably, given \({haCCA}\)'s superior performance over STUtility in our previous benchmark using pseudo data (Fig. 2), the actual accuracy of \({haCCA}\) integration may exceed these metrics.

A Integration performance on same-section data from three mouse brain specimens. For each specimen, panels show metabolic clusters from MALDI-MSI (left), transcript clusters from 10X Visium (middle-left), and metabolic cluster distribution after integration using STUtility (middle-right) and \({haCCA}\) (right). \({haCCA}\) maintains consistent spatial patterns. B Integration performance on neighboring-section data. C Spatial distribution patterns of dopamine are shown in MALDI-MSI(left) and \(h{aCCA}\) integrated data (middle), alongside the expression pattern of tyrosine hydroxylase (Th) from spatial transcriptomics(right). Indicated that \({haCCA}\) integration (middle panels) successfully preserves the spatial correspondence between metabolites and their associated genes.
Furthermore, \({haCCA}\) demonstrated robust performance in the more challenging task of integrating data from neighboring sections, where morphological variations are more pronounced. The method successfully maintained metabolic cluster label distribution patterns while achieving high-quality integration (Fig. 3B, Supplementary Fig. 4B, D).
To illustrate the biological relevance of genes and metabolites using our integration approach, we examined the spatial relationships between dopamine and it associated enzymes. Using dopamine, tyrosine hydroxylase (TH; a key enzyme in dopamine metabolism), and 3-MT as examples, we demonstrated \({haCCA}\)'s ability to reconstruct metabolite distributions in the integrated data. Notably, genes associated with dopamine metabolism, such as Th, exhibited overlapping distribution patterns with dopamine (Fig. 3C, Supplementary Fig. 4E), facilitating the identification of metabolite-associated genes.
These results indicated that \({haCCA}\) is a reliable method for integrating ST and metabolomics data in real-world data. The method’s ability to preserve biological patterns and reveal metabolite-gene associations demonstrates its potential as a valuable tool for spatial multi-omics analysis.
haCCA generated multi-modal data allows integrative analysis of transcriptome and metabolome
We applied \({haCCA}\) to integrate spatial transcriptome and metabolome data from mouse brain specimens (M3). The integrated data was analyzed using Weighted Nearest Neighbors analysis to comprehensively characterize tissue heterogeneity. UMAP visualization revealed distinct clustering patterns across three data modalities: metabolome data (5 clusters), transcriptome data (8 clusters), and integrated data (7 clusters), demonstrating that multi-modal integration enables more refined characterization of tissue heterogeneity (Fig. 4A). The relationships between clusters from different modalities were visualized using a Sankey diagram, with each flow proportional to the number of cells (Fig. 4B).

A UMAP visualization of clustering results from three data modalities: metabolic clusters from MALDI-MSI (left), integrated clusters after \({haCCA}\) analysis (middle), and transcript clusters from spatial transcriptomics (right), revealing distinct spatial organization patterns. Cluster labels were used for visualization. B Sankey diagram depicting the relationships between transcript clusters (S0–S6), integrated clusters (C0–C7), and metabolic clusters (M1–M5). Width of connecting lines represents the proportion of shared spots between clusters. C Spatial visualization of cluster distributions demonstrating the preservation of both metabolic and transcriptomic features. Left to right: metabolic clusters, integrated clusters after \({haCCA}\), and transcript clusters. Red boxes highlight regions where metabolic features are predominantly preserved; green box highlight regions where transcript features are predominantly preserved. D Dot plot showing marker metabolites for integrated clusters. Dot size indicates fraction of spots expressing each metabolite, while color intensity represents mean expression level. E Dot plot showing marker gene expression across integrated clusters. Dot size represents fraction of expressing spots, and color intensity indicates mean expression level.
Spatial projection of cluster labels revealed that integrated clusters effectively captured the local heterogeneity patterns from both metabolic and transcriptomic data. Notably, in certain regions (red box), the integrated clusters preserved more features from metabolic clusters while maintaining fewer transcriptomic features. Conversely, in other regions (green box), the integrated clusters retained more transcriptomic characteristics while showing reduced metabolic influence. This selective clusters preservation may help minimize false-positive clustering artifacts introduced during data generation, leading to a more accurate spatial clustering (Fig. 4C).
Further analysis identified distinct marker genes and metabolites for each cluster (Fig. 4D, E). These findings demonstrate that \({haCCA}\) successfully enables integrated analysis of spatial transcriptome and metabolome data, while revealing that spatial heterogeneity is predominantly driven by transcriptomic variation.
haCCA generated profile of NETs induced metabolic alteration in preclinical ICC model
Intrahepatic cholangiocarcinoma (ICC) is a type of highly malignant liver cancer. Neutrophil and NETs consists major components of ICC microenvironment17,18. NETs are web-like structures released by neutrophils, with previous studies highlighting their role in promoting cancer metastasis through various mechanisms, such as alteration inflammation or EMT (epithelial-mesenchymal transition)19,20,21. However, very few studies revealed its capability to alter the metabolic reprogramming of cancer cells22, especially an in situ and in vivo profiling of its metabolic reprogramming capacity. We eliminated NETs formation in ICC model by knocking out Padi4, and comparing the metabolic alteration of tumor zone by \(h{aCCA}\), to profile the NETs effect on metabolic alteration of ICC. \(h{aCCA}\) showed effective merge of spatial transcriptiome and MALDI-MSI, showing the distribution of metabolite derived clusters on the transcriptome background.(Fig. 5A) Notably, in the merged \({haCCA}\) dataset, transcriptional data was used for clustering to determine spatial regions, giving convince of exploring metabolite features among them. This highlights that \({haCCA}\) enabled more precise subregion masking of MALDI-MSI, allowing for finer metabolite profiling. By eliminating NETs, tumor area(cluster 0) of ICC was decreased (1039 spots in KO and 3090 spots in WT), accompanied with previous research. The distribution of tumor area was shown by cluster 0 in Fig. 5B23. We compared the different expressed genes or metabolite (DEG, DEM, scores >15 or <−15 calculated by pyscenic, Supplementary Data 2) between WT and KO tumor area, and found in KO group(without NETs), around 73 genes and 77 metabolites were up-regulated, while in WT group (with NETs), around 130 genes and 20 metabolites were up-regulated(Supplementary Data 2). KEGG annotation of both genes and metabolites by MetaboAnalyst24 revealed in WT groups, many metabolism associated pathways, such as fatty acid metabolism, cholesterol metabolism and glycolysis or gluconeogenesis was enriched(Fig. 5C). Scd1 is a key enzyme to product monounsaturated fatty acid, and is found to induce tumorigenesis in liver cancer model25. Scd1 is the enzyme responsible for oleic acid26, stearate acid or 18:0 saturated fat acid metabolism27. Spatial plot of Scd1, oleic acid, stearic acid and PA(18:0) expression showed a co-localization distribution pattern (Fig. 5D). In cancer area of WT group, those gene/metabolite were also up-regulated compared with KO group. (Fig. 5E) We further joint embedded the genes and metabolites in WT group, and found ~30 co-expression modules (Supplementary Data 3). Amongst them we found a cluster consists of ~45 metabolite and ~130 genes, centralized by Scd1 (Supplementary Fig. 3). Collectively, using \(h{aCCA}\) we perform in vivo and in situ profiling of transcriptome and metabolomes in ICC murine model, indicating a potential mechanism that NETs may alter ICC tumor metabolism by upregulating Scd1. Providing new insights into the dynamic crosstalk between genes and metabolites that regulates the tumor biological behavior.

A spatial transcriptome(left), MALDI-MSI(middle) and \(h{aCCA}\) integrated data(right) of Akt/YapS127A induced ICC model in WT (lower panel)(n = 1) or Padi4 KO(upper panel) mouse(n = 1). Cluster labels were projected as colors of spots. In the left panel, clusters derived from spatial transcriptome were used for projection, while in the middle and right panel, clusters derived from MALDI-MSI were used for projection. B spatial distribution of tumor zone (Cluster 0) in \(h{aCCA}\) integrated data. C KEGG co-annotation of up-regulation gene and metabolites in malignant cell from WT ICC. D Scd1, Oleic acid, Stearic acid and PA distribution in ICC from WT mouse. E Violin plot showed the abundance difference of Scd1, Oleic acid, Stearic acid and PA in malignant cell from KO and WT mouse(P value shown in plot, by t-test).
Discussion
Spatial multi-modal technologies require efficient and accurate integration of spatial data. Although several integration workflows have recently been developed for spatial transcriptome data15,28,29, their application to multi-modal data is limited due to lack of shared features. Here we proposed \(h{aCCA}\), a method that performs highly correlated feature pair-assisted morphological alignment and integration between spatial transcriptome data and MALDI-MSI data. \({{\rm{h}}}{{\rm{aCCA}}}\) is composed of 2 main steps: a modified spatial registration, followed by identifying high correlated feature pairs and turning them into new high correlated variables. Using aligned spatial coordinates and high correlated variable to perform integration with more accuracy than only using spatial registration. While \(h{aCCA}\) employs manual landmark selection during modified spatial registration, we demonstrated that this approach achieves superior and reproducible performance compared to fully automated alternatives. The inter-operator variability in our reproducibility assessment is acceptable (Supplementary Fig. 6). This suggests that expert knowledge in identifying morphologically conserved features provides more value than the potential noise from operator variability. Future developments could explore semi-automated approaches that combine computer vision with expert validation to further enhance scalability while maintaining accuracy.
Using spatial transcriptome data and pseudo-paired MALDI-MSI data, we demonstrated that \(h{aCCA}\) outperformed other alignment methods, represented by STUtility, by 6–20% across the metrics of label transfer accuracy, label transfer ARI, and PAA. Moreover, the modified spatial registration of spatial coordinates, accomplished through the gross alignment and further alignment steps of \(h{aCCA}\), is able to outperform STUtility, and the feature aid fine alignment is able to further increase the accuracy. It indicated the efficiency of combing alignment strategy using spatial registration and features of \(h{aCCA}\). When testing \(h{aCCA}\) on real world generated data, such as brain and tumor, \(h{aCCA}\) successfully integrated MALDI-MSI data with spatial transcriptome data, and generated gene expression as well as bio-molecule profiling (mainly metabolome). Such multi-modal spatial data allows more sophisticated deciphering to heterogeneity, for example, we identified different metabolic states in tumor region from ICC model. It also facilitates both gene and bio-molecule marker detection for certain clusters.
\({{\rm{haCCA}}}\) can be used in the following aspects: 1. Profile the distribution and abundance of gene as well as metabolite. 2. enable more refined characterization of tissue heterogeneity through multi-modal integration. 3. Identify both distinct marker genes and metabolites for each cluster. 4. co-identify of dys-regulated metabolites and genes, as well as comparison of metabolite and genes in a set of spots.
While \({haCCA}\) demonstrates robust performance in integrating spatial transcriptomes and MALDI-MSI data, several limitations should be acknowledged. First, the current study focuses primarily on the development and validation of the \({haCCA}\) workflow, with biological findings—particularly the metabolic alterations induced by NETs in ICC—requiring further experimental validation. Although our findings reveal that NETs upregulate Scd1 and drive fatty acid elongation pathways, these observations warrant independent validation through targeted metabolomics, functional assays, and intervention studies to confirm the causal relationships and biological significance. Second, the applicability of \({haCCA}\) to other cancer types remains to be systematically evaluated. While we successfully applied \({haCCA}\) to mouse ICC model systems, demonstrating its capacity to characterize metabolic and transcriptomic heterogeneity, the generalizability of this approach to other malignancies—such as colon, breast, and other cancer types with distinct metabolic landscapes—requires further investigation. Third, only spatial transcriptome and spatial metabolome data has been used. \(h{aCCA}\) can expend its capability to integrate more spatial techniques, such as image-based sequencing, spatial proteinomes or histological images. How \(h{aCCA}\) behaves on higher resolution data should be further explored.
Conclusion
To summarize, we presented a workflow named \({{\rm{h}}}{{\rm{aCCA}}}\) that enables the simultaneous spatial profiling of metabolites and transcriptome across neighbor tissue section (graph abstract). Utilizing \({{\rm{haCCA}}}\) is able to: 1. Profile the distribution and abundance of gene as well as metabolite. 2. enable more refined characterization of tissue heterogeneity through multi-modal integration. 3. Identify both distinct marker genes and metabolites for each cluster. 4. co-identify of dys-regulated metabolites and genes, as well as comparison of metabolite and genes in a set of spots.
Method
Data description
Public data
10X Visium and MSI-MALDI data from Parkinson’s disease mouse brain was download from Macro et al’s manuscript4. 6 10X Visium,3 MSI-MALDI data of 6 sections from 3 mouses were included(Supplementary Data 1). Spatial transcriptome data of GC and rat brain were downloaded from SODB database(https://gene.ai.tencent.com/SpatialOmics/Omics).
ICC mouse model data
All animal experiments were approved by the Animal Ethics Committee of Fudan University (approval number: 2025-HSYY-429). We have complied with all relevant ethical regulations for animal use. The present study was performed in accordance with the Declaration of Helsinki for the use of human tissue samples. Approval for the use of human subjects was obtained from the Research Ethics Committee of Huashan Hospital, Fudan University (approval number: KY2023-594), and informed consent was obtained from each individual enrolled in this study. We have complied with all relevant ethical regulations for animal use.
We generated Akt/Yap induced ICC (intrahepatic cholangiocarcinoma) mouse model in WT or Padi4-/- mouse. BMKMANU S1000 ST and MALDI-MSI data of tumor was generated following standard procedure (see below).
6–8 weeks old of male C57BL/6 mouse were purchased from https://www.gempharmatech.com/ and kept in a SPF institute at 25 °C temperature, 50–60% moisture. Pre-clinical ICC model was generated by HDI (hydrodynamic injection) of 20ug pT3-myrAkt-p2a-Yap(S127A) and 5ug S100B plasmid in 6w old wt or Padi4-/- C57 mouse. Padi4 -/- mouse is a kind gift from Prof Chen-De Yang. After 4 weeks, tumors were collected for further experiment (Supplementary Fig. 2A, B).
Efforts were taken to minimize the number of animals used and their suffering. Due to the nature of the hydrodynamic injection (HDI) model, which generates diffuse, multi-focal intrahepatic tumor nodules rather than a single measurable tumor mass, direct in vivo assessment of tumor size was not feasible. Therefore, tumor burden was assessed post-mortem by measuring total liver weight. (Supplementary Fig. 2C, D) The humane endpoint was defined as body weight loss exceeding 20% of initial weight, and mice reaching this threshold were immediately euthanized30. Mice were monitored twice a week for body weight. Euthanasia was performed under isoflurane anesthesia followed by cervical dislocation.
BMKMANU S1000 Spatial transcriptomics
Frozen embedded tissue
After tissue samples was obtained, tissue surfaces were quickly rinsed with a pre-cooled solution of 1X PBS (RNase free) or normal saline to remove residual blood, and sterile gauze was used to blot the surface fluid. The tissue size was required to be suitable, and the tissue should be cut into small pieces(6.8mm2) suitable for subsequent experiments. Small fragments of each tissue were snap-frozen in isopentane pre-chilled with liquid nitrogen and optimum cutting temperature compound (SAKURA,Cat#: 4583) and stored at −80 °C until use.
Slide preparation
ST slides were printed with 1–8 identical 6.8 × 6.8 mm capture areas, each with 2,000,000 spots contain barcoded primers (BMKMANU S1000). The primers are attached to the slide by the 5′ end and contain a cleavage site, a T7 promoter region, a partial read1 Illumina handle, a spot-unique spatial barcode, a unique molecular identifier (UMI), and Poly(dT)VN. The spots have a diameter of 2.5 μm and are arranged in a centered regular hexagonal grid so that each spot has six surrounding spots with a center-to-center distance of 4.8 μm. The frozen tissue was cut in a pre-cooled cryostat at 10 µm thickness and systematically placed on chilled BMKMANU S1000 Tissue Optimization Slides and BMKMANU S1000 Gene Expression Slides, and stored at −80 °C until use.
Tissue optimization
The ST protocol was optimized for tissue according to recommendations. In short, changes were made in the staining procedure by excluding isopropanol, decreasing the incubation time of hematoxylin and bluing buffer, as well as increasing eosin concentration. Moreover, the optimal incubation time for permeabilization was established, and the previously described one-step protocol for tissue removal was altered by using a higher proteinase K:PKD buffer ratio. Once optimal conditions had been established, three cryosections per patient were cut at 10 mm thickness onto spatial slides and processed immediately.
Fixation, staining and imaging
Sectioned slides were incubated at 37 °C for 1 min., fixed in 3.7–3.8% formaldehyde (Sigma-Aldrich) in PBS (Medicago) for 30 min, and then washed in 1x PBS (Medicago). For staining, sections were incubated in Mayer’s hematoxylin (Dako, Agilent, Santa Clara, CA) for 4 min, bluing buffer (Dako) for 30 s, and Eosin (Sigma-Aldrich) diluted 1:5 in Tris-base (0.45 M Tris, 0.5 M acetic acid, pH 6.0) for 30 s. The slides were washed in RNase and DNase free water after each of the staining steps. After air-drying, the slides were mounted with 85% glycerol (Merck Millipore, Burlington, MA) and coverslips (Menzel-Glaser). Bright-field (BF) images were taken at 20× magnification using Metafer Slide Scanning platform (MetaSystems). Raw images were stitched with VSlide software (MetaSystems). The coverslip and glycerol were removed after imaging by immersing slides in RNase and DNase free water. The slides were inserted into slide cassettes to separate the tissue sections into individual reaction chambers (hereinafter wells). For pre-permeabilization, sections were incubated at 37 °C for 20 min with 0.5 U/ml collagenase (ThermoFisher) and 0.2 mg/ml BSA (NEB, Ipswich, MA) in HBSS buffer (ThermoFisher). Wells were washed with 0.1× SSC(Sigma-Aldrich), after which permeabilization was conducted at 37 °C for 7 min in 0.1% pepsin (Sigma-Aldrich) dissolved in 0.1 M HCl (Sigma-Aldrich). After incubation, the pepsin solution was removed and wells washed with 0.1 × SSC.
Reverse transcription, spatial library preparation and sequencing
Reverse transcription (RT), second-strand cDNA synthesis, adapter ligation and a second RT was generated and libraries were constructed according to the performer’s protocol. Sequencing handles and indexes were added in an indexing PCR and the finished libraries were purified and quantified. Sequencing was performed on the Illumina NovaSeq 6000 with a sequencing depth of at least 50,000 reads per sopt(100 μm) and 150 bp (PE150) paired-end reads (performed by Biomarker Technologies Corporation, Beijing, China).
Spot visualization and image alignment
Primer spots were stained by hybridization of fluorescently labeled probes and imaged on the Metafer Slide Scanning platform. The resulting spot image was loaded into the BSTMatrix and BSTViewer along with the previously obtained BF tissue image of the same area. The two images were aligned and the built-in tissue recognition tool was used to extract spots covered by tissue.
BSTMatrix analysis
Finished libraries were diluted to 4 nM and sequenced on the Illumina Nova 6000 using paired-end sequencing. We completed the upstream analysis through BSTMatrix (v1.0). The mapping was performed to the reference GRCh38_release95 mouse genome.
MALDI-MSI
Matrix coating
Desiccated tissue sections mounted on ITO glass slides were sprayed using an HTX TM sprayer (Bruker) with 10 mg/mL 9AA (9-aminoacridine), dissolved in ethanol-water (7:3, v/v). The sprayer temperature was set to 90 °C, with a flow rate of 0.12 mL/min, pressure of 10 psi. Four passes of the matrix were applied to slides with 10 s of drying time between each pass.
Mass spectrometry imaging
MALDI timsTOF MSI experiments were performed on a prototype Bruker timsTOF flex MS system (Bruker) equipped with a 10 kHz smart beam 3D laser. Laser power was set to 70% and then fixed throughout the whole experiment. The mass spectra were acquired in negative mode. The mass spectra data were acquired over a mass range from m/z 50–1200 Da. The imaging spatial resolution was set to 30 μm for the tissue, and each spectrum consisted of 400 laser shots. MALDI mass spectra were normalized with the Root Mean Square, and the signal intensity in each image was shown as the normalized This experiment was performed at biotree company(www.biotree.cn).
Downstream analysis
Briefly, The downstream analysis of spatial transcriptome and MALDI-MSI was carried out by a standard workflow containing Data Loading and Quality Control (sc.read, sc.pp.calculate_qc_metrics, sc.pp.filter_cells, sc.pp.filter_genes), Data Normalization and Scaling (sc.pp.normalize_total, sc.pp.log1p, sc.pp.highly_variable_genes, sc.pp.scale), Dimensionality Reduction (sc.tl.pca, sc.pp.neighbors, sc.tl.umap, sc.tl.tsne), Clustering and Visualization by scanpy package (sc.tl.leiden, sc.tl.rank_genes_groups, sc.pl.umap, sc.pl.spatial).
Paired spatial transcriptome and pseudo-MALDI-MSI data generation
The use of generated data for evaluating alignment and integration algorithms is well-established in the field, as demonstrated by previous studies15,28. To validate our alignment strategy and evaluating the integration performance using metrics, paired spatial transcriptome and MALDI-MSI data is necessary, which is unavailable in real-world. To compensate that, we simulate MALDI-MSI datasets from parenting spatial transcriptome data.
To generate synthetic spatial transcriptome data \({Data}(d,f,l)=\{{d}_{m}^{2},{{f}_{m}^{n},l}_{m}\}{for}\{m=1,\ldots ,M\}\), where for each data point,\(f\) denotes the feature vector\(,d\) represents the coordinate matrix \(\left({d}_{x},{d}_{y}\right)\), \(l\) represents the cluster label. We resampling from 5 spatial transcriptome data consisting of 3 mouse brain, 1 human GC and 1 rat brain.
The synthetic data generation algorithm operates through a systematic partitioning and augmentation process. Initially, \(n\) datapoints are randomly sampled from the real spatial transcriptome data \({Dat}{a}_{{real}}(d,f,l)\) to create a representative sample set. Then group undergoes further subdivision based on feature dimensions, where the feature vector f is split such that the first part contains the initial \({j}^{{th}}\) dimensions and the second part encompasses the remaining dimensions. This creates feature subsets denoted as \({f}_{A}\), for group \(A\) and \({f}_{B},\) for group \(B\).
To mimic the morphological dissimilarity observed between different tissue sections, we systematically introduce controlled experimental noise to the spatial coordinates of pseudo data. This is achieved through the application of affine transformations that precisely modulate the geometric properties of spot distributions. The transformation parameters are set as below: rotation angles (−10, 10), scaling factors (0.9, 1.1), shearing parameter (0.9, 1.1), and translation (−10, 10). The transformation was performed on a subset of spots and repeated for 1–5 times The final synthetic datasets are assembled into four synthetic datasets \(\left({Dat}{a}_{A},{Dat}{a}_{A}^{{\prime} },{Dat}{a}_{B},{Dat}{a}_{B}^{{\prime} }\right)\) by combining the processed spatial location, split feature parts and constructed labels. Here \({Dat}{a}_{A},{Dat}{a}_{B}\) correspond to the real world datapoints and \({Dat}{a}_{A}^{{\prime} },{Dat}{a}_{B}^{{\prime} }\) serve as validation datapoints to evaluate the performance of aligning \({Dat}{a}_{A}\) to \({Dat}{a}_{B}\) or vice versa.(Supplementary Fig. 5) for validation. \(h{aCCA}\) uses \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\) as inputs, with \({Dat}{a}_{A}\)’‘serving as ground truth for \(h{aCCA}\left({Dat}{a}_{A},{Dat}{a}_{B}\right)\) and \({Dat}{a}_{B}^{{\prime} }\) serving as ground truth for \(h{aCCA}\left({Dat}{a}_{B},{Dat}{a}_{A}\right)\).
Algorithm
pseudo-MALDI-MSI data generation from real data
1:Input:\(i,j\) \({Dat}{a}_{{real}}\left(d,f,l\right)\)
i, jPartition indices
\({Dat}{a}_{{real}}\left(d,f,l\right)\)Real spatial transcriptome
A = R(θ)*S*ShAffine matrix A. θ is rotation angles, S is scaling factors, Sh is shearing factors
ttranslation vector
Output: \({Dat}{a}_{A},{Dat}{a}_{B},{Dat}{a}_{A}^{{\prime} },{Dat}{a}_{B}^{{\prime} }\)
2: \({{Dat}{a}_{{Sample}}(d,{f},l)\leftarrow {random}\; {choice}\; {n}\; {datapoints}\; {from}\; {Dat}{a}_{{real}}(d,{f},l)}\)
3:Initialize spatial coordinates: \(d\leftarrow {Dat}{a}_{{sample}}(d)\)
4:Initialize feature vectors: \(f\leftarrow {Dat}{a}_{{sample}}(f)\)
Initialize label: \(L\leftarrow {Dat}{a}_{{sample}}(l)\)
5:Partition spatial data: \({d}_{A}\leftarrow d\left[:{i}_{A},:\right],\,{d}_{B}\leftarrow d\left[:{i}_{B},:\right]\)
6:Partition feature data: \({{f}_{A}\leftarrow f\left[:{i}_{A},::j\right]{f}_{A}^{{\prime} }\leftarrow f\left[:{i}_{A},{j}:\right]{f}_{B}\leftarrow f\left[:{i}_{B},:j:\right]{f}_{B}^{{\prime} }\leftarrow [:{i}_{B},:j]}\)
7:Add experimental noise: \({d}_{A} \leftarrow A* {d}_{A}+t , \, {d}_{B} \leftarrow {d}_{B}\)
Construct labels: \({l}_{A}\leftarrow L\left[:{i}_{A}\right]{l}_{B}\leftarrow L[:{i}_{B}]\)
8:Construct datasets: \({Dat}{a}_{A}\leftarrow \left({d}_{A},\,{f}_{A},{l}_{A}\right),{Dat}{a}_{A}^{{\prime} }\leftarrow \left({d}_{A},\,{f}_{A}^{{\prime} },\,{l}_{A}\right)\)
9:Construct datasets: \({Dat}{a}_{B}\leftarrow \left({d}_{B},\,{f}_{B},{l}_{B}\right),{Dat}{a}_{B}^{{\prime} }\leftarrow \left({d}_{B},\,{f}_{B}^{{\prime} },\,{l}_{B}\right)\)
10:return \({Dat}{a}_{A},{Dat}{a}_{A}^{{\prime} },{Dat}{a}_{B},{Dat}{a}_{B}^{{\prime} }\)
haCCA workflow
\(h{aCCA}\) is designed for aligning discrete entities.(spots based spatial techniques) The complete \({haCCA}\) workflow contains five steps. In Step 1 & 2, it aligns the spots in \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\) using location information \({d}_{A},\,{d}_{B}\). In Step 3 & 4, it utilizes the location information to identify reliable correspondences from high correlated feature pairs and find low-dimensional latent space \({f}^{{cca}}\) that capture the shared variance between high-correlated feature pairs from \({f}_{A}\) and \({f}_{B}\). In Step 5, \({f}^{{cca}}\) is used as the shared feature between \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\) to generate a more accurate alignment plan, which is used to generate a transport plan \(\Pi\) that minimizes the loss from both feature discrepancy and distance.
Once \(\Pi\) is determined, we can use it to generate the pairwise alignment collections
\(\left\{\left({m}_{a},\,{m}_{b}\right)\right\}\leftarrow {argma}{x}_{{m}_{a},\,{m}_{b}}\Pi\) and produce the final alignment result.
Overview
haCCA workflow
1: Input: DataA (d, f, l), DataB(d, f, l)
2: Output: \({transport\; plan}\Pi \)
3: Step 1: dB←Gross_Alignment(dA, dB, use_manual_alignment, use_icp, use_fgw)
4: Step 2:dB←Further_Alignment (dA, dB, dist_min)
5: Step 3: \({\left\{\left({m}_{a}^{{anchor}},\,{m}_{b}^{{anchor}}\right)\right\}\leftarrow {Anchor\_Spot\_Pairs\_Discovery}({d}_{A},\,{d}_{B},{dist\_}\min ,{\rm{\epsilon }})}\)
6: Step 4: \({\left\{{f}_{A}^{{cca}},\,{f}_{B}^{{cca}}\right\}\leftarrow {High\_Correlated\_Feature\_Discovery}(\left\{\left({m}_{a}^{{anchor}},\,{m}_{b}^{{anchor}}\right),\,{f}_{A},\,{f}_{B},{k},\,{\theta }_{{low}},{\theta }_{{high}}\right.)}\)
7: Step 5: Π, {(ma, mb)}
←Feature_aid_Fine_Alignment (\({d}_{A},\,{d}_{B},\,{f}_{A}^{{cca}},\,{f}_{B}^{{cca}}\), use_icp, use_fgw)
8:return Π, {(ma, mb)}
Step 1: data preparation
Assay was transferred into a triplet tuple Data(d, f, l)={d, f, l} to represent a spatial assay and m(d, f, l) to represent each spot in Data. For each spot m(d,f, l), f denotes the feature vector, d represents the spatial information and \(l\) represents the cluster label.
Step 2: gross and further alignment by aligning Data A to Data B using location information d A, d B
Location information \(d\) provides the coordinate information of each spot in \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\) and is crutial for the alignment. To fully utilize spatial coordinate information, we apply two-stage alignment strategies here: gross alignment and further alignment.
In gross alignment, we use manual alignment to perform coordinate transformation and spatial registration in order to eliminate the potential shifts and rotations introduced from sampling and section as much as possible. The process is described as follow:
Algorithm 1
Gross Alignment
1: Input: \({d}_{A},\,{d}_{B},\,{use\_}{{\rm{manual}}}\_{{\rm{alignment}}}\)
2: Output: \({d}_{B}\)
3: if use_manual_alignment then
4: Manually select n alignment pairs from \({d}_{A}\) and \({d}_{B}\) for reference
5: Calculate affine transformation matrix \({M}_{{manual}}\) using the selected paris
6: \({d}_{b}\leftarrow {M}_{{manual}}* {d}_{B}\)
7: end if
In gross alignment process, manual alignment allows human experts to manually select \(n\) alignment pairs from \({d}_{A}\) and \({d}_{B}\) for reference and calculating the affine transformation matrix \({M}_{{manual}}\) using the selected pairs. It is optional and provides an opportunity to introduce prior knowledge to the alignment process. In practice, the \(n\) is usually selected to be 3.
In further alignment, we perform an extra alignment to the outlier spots in \(\{{d}_{B}\}\) and their nearest neighbors in \(\{{d}_{A}\}\) by finding a rotation and transformation matrix \({M}_{{further}}\) to minimize the distance between them. The outlier spots in \(\{{d}_{B}\}\) is defined as the spots where it does not have neighbor within a given threshold (\({dist\_}\min\)) in \(\{{d}_{A}\}\). They typically occur near the tissue boundaries and missing parts and are usually introduced during section preparation. After the outlier spots in \(\{{d}_{B}\}\) are found, we calculate the affine transformation matrix \({M}^{{\prime} }\) using outlier spots in \(\{{d}_{B}\}\) and its nearest neighbor in \(\{{d}_{A}\}\). After further alignment, a maximum overlap of \(\{{d}_{A}\}\) and \(\{{d}_{B}\}\) between can be achieved(see Fig. 1), and their topological relationship can be reserved as only linear transformation is peformed. The further alignment process is described as follows:
Algorithm 2
Further Alignment
1:Input: \({d}_{A},\,{d}_{B},\,{dist\_}\min\)
2:Output: \({d}_{B}\)
3:Calculate the distance matrix \(D\) between \({d}_{A}\) and \({d}_{B}\)
4:Find the outlier spots in \({d}_{B}\) where it does not have neighbor within a given distance \({dist\_}\min\) in \({d}_{A}\), noted as \({d}_{{B\_}{{\rm{outlier}}}}\)
5:Calculate affine transformation matrix \({M}^{{\prime} }\) using outlier spots in \({d}_{{B\_}{{\rm{outlier}}}}\) and its nearest neighbors in \({d}_{A}\)
6:\({d}_{B}\leftarrow {M}^{{\prime} }* {d}_{B}+{d}_{B}\)
7:return \({d}_{B}\)
Parameter: \({dist\_}\min =1\)
Step 3, 4: determine anchor spot pairs \(\{{{\boldsymbol{ancho}}}{{{\boldsymbol{r}}}}_{{{\boldsymbol{A}}}},{{\boldsymbol{ancho}}}{{{\boldsymbol{r}}}}_{{{\boldsymbol{B}}}}\}\) and calculate high correlated feature pairs \(\{{\left({{\boldsymbol{i}}},{{\boldsymbol{j}}}\right)}_{{{\boldsymbol{k}}}}\}\) for \({{\boldsymbol{Dat}}}{{{\boldsymbol{a}}}}_{{{\boldsymbol{A}}}}\) and \({{\boldsymbol{Dat}}}{{{\boldsymbol{a}}}}_{{{\boldsymbol{B}}}}\)
In this step, we first identify anchor spot pairs \(\{{ancho}{r}_{A},{ancho}{r}_{B}\}\) from \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\) where each pair satisfies with the following conditions:
-
The distance between \({ancho}{r}_{A}\) and \({ancho}{r}_{B}\) are smaller than a given threshold \({dist\_}\min\) (default = 1) and they are the closest point to each other.
-
The neighborhood of both \({ancho}{r}_{A}\) and \({ancho}{r}_{B}\) is of low variety. The variety of a neighborhood is evaluated by Simpson index. \(\epsilon\)(default = 0.5).
The Simpson index is calculated using the following formula:
Where:\(\,S={total\; number\; of\; labels}\),\(\,{n}_{i}={number\; of\; spots\; of\; label} \, i\)
The anchor spot pairs discovery process is described as follows:
Algorithm 3
Anchor Spot Pairs Discovery
-
1.
Input: \({d}_{A},\,{d}_{B},\,{dist\_}\min ,{{\rm{\epsilon }}}({{\rm{threshold}}}\; {{\rm{for}}}\; {{\rm{Simpson}}}\; {{\rm{Index}}})\)
-
2.
Output: Anchor spot pair \(\{\left({m}_{a}^{{anchor}},\,{m}_{b}^{{anchor}}\right)\}\)
-
3.
For each spot in \({d}_{A}\), find a set of spots in \({d}_{B}\) where each spot is within a given distance dist_min, noted as \(\{{m}_{a}:\left[{m}_{b}\right]\}\)
-
4.
For each item in \(\{{m}_{a}:\left[{m}_{b}\right]\}\), calculate the Simpson index for each item and remove the items where its Simpson index is higher than a given threshold \(\epsilon\).
-
5.
For the remaining items in \(\{{m}_{a}:\left[{m}_{b}\right]\}\), the final anchor spot pairs \(\{\left({m}_{a}^{{anchor}},\,{m}_{b}^{{anchor}}\right)\}\) are identified as the closest point pair in each item.
-
6.
return \(\{\left({m}_{a}^{{anchor}},\,{m}_{b}^{{anchor}}\right)\}\)
Parameter: \({dist\_}\min =1,\,\epsilon =0.5\)
After anchor spot pair \(\{\left({m}_{a}^{{anchor}},{m}_{b}^{{anchor}}\right)\}\) is identified, we further identify high correlated feature pairs \(\{{\left(i,j\right)}_{k}\}\) where \(i\) is the feature index of \(\{{f}_{A}\}\), \(j\) is the feature index of \(\{{f}_{B}\}\). The high correlated feature pairs discovery process is described as follows: (1) calculate the Pearson correlation matrix on the feature metric of anchor spot pairs \(\{\left({m}_{a}^{{anchor}},{m}_{b}^{{anchor}}\right)\}\), (2) select the index of top \(k\) (default = 100)score of Pearson correlation matrix as the high correlated feature pairs \(\{{\left(i,j\right)}_{k}\}\).
With high correlated feature pairs \({\{(i,j)\}}_{k}\), we then calculate \({f}^{{cca}}\) for both \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\) using canonical variables found by Canonical Correlation Analysis (CCA). The idea behind is to further maximize the correlation and normalize the value between the high correlated feature pairs. After \({f}^{{cca}}\) is calculated, it will be added to \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\) as an individual feature after they are calculated for all the points in \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\). We use \({Dat}{a}_{A}^{{cca}}\) and \({Dat}{a}_{B}^{{cca}}\) to represent the new \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\) after \({f}^{{cca}}\) is added. The high correlated feature pairs discovery process is described below.
The CCA is calculated using \({cross\_decomposition}.{CCA}\) in Python package sklearn. The CCA equation is described as follows:
Algorithm 4
High Correlated Feature Pairs Discovery
1: Input: \({f}_{A},\,{f}_{B},\,k,\,{\theta }_{{low}},\,{\theta }_{{high}},\,\{\left({m}_{a}^{{anchor}},\,{m}_{b}^{{anchor}}\right)\}\)
2: Output: \(\{{f}_{A}^{{cca}},\,{f}_{B}^{{cca}}\}\)
3: \({f}_{A}^{{anchor}}\leftarrow [{f}_{{m}_{a}^{{anchor}}}{for}{m}_{a}^{{anchor}}{in}\left\{\left({m}_{a}^{{anchor}},\,{m}_{b}^{{anchor}}\right)\right\}]\)
4:\({f}_{B}^{{anchor}}\leftarrow [{f}_{{m}_{b}^{{anchor}}}{for}{m}_{b}^{{anchor}}{in}\left\{\left({m}_{a}^{{anchor}},\,{m}_{b}^{{anchor}}\right)\right\}]\)
5: Initialize Pearson correlation matrix \(R\leftarrow |{f}_{A}|\times |{f}_{B}|\)
6: for \(i=1\) to \(|{f}_{A}|\) do
7: for \(j=1\) to \(\left|{f}_{B}\right|\) do
8:\({R}_{{ij}}={corr}({f}_{A}^{{anchor}}\left[:,\,i\right],\,{f}_{B}^{{anchor}}\left[:,\,j\right])\)
9: end for
10: end for
11:\(R\leftarrow {\theta }_{{low}}\le R\le {\theta }_{{high}}\)
12:\({top}\_{{\rm{k}}}\_{{\rm{indices}}}\{{\left(i,j\right)}_{k}\}\leftarrow {top}\_{{\rm{k}}}({{\rm{argsort}}}\left(R\right))\)
13:\({f}_{A}^{{\prime} }\leftarrow \left\{\right.{f}_{A}^{i}{for\; i\; in}\left\{{\left(i,j\right)}_{k}\right\},\,{f}_{B}^{{\prime} }\leftarrow \{{f}_{B}^{j}{for\; j\; in}\left\{\right.{\left(i,\,j\right)}_{k}\}\)
14: Find low-dimensional common subspace \({f}^{{cca}}\) using canonical correlation analysis (CCA) on \({f}_{A}^{{\prime} }\) and \({f}_{B}^{{\prime} }\)
15: return \({f}_{{{\rm{A}}}}^{{cca}},\,{f}_{B}^{{cca}}\)
Parameter: \({\theta }_{{low}}=0.5,\,{\theta }_{{high}}=0.9,\,k=100\)
Step 5: minimize the loss using both \(\{{{\boldsymbol{d}}}\}\) and \(\{{{{\boldsymbol{f}}}}^{{{\boldsymbol{cca}}}}\}\) by feature aid fine alignment
Finally, we minimize the \({los}{s}_{{haCC}{A}_{A\to B}}\) with both \(\{d\}\) and \(\{{f}^{{cca}}\}\) using Feature-aid fine alignment. This process returns transportation plan \(\Pi\) which size is \({M}_{A}\times {M}_{B}\) and final alignment collections \(\{\left({m}_{a},{m}_{b}\right)\}\) for \(\forall {m}_{a}\in {Dat}{a}_{A}\). in \(\Pi\), each element \({\Pi }_{{ij}}\) represents the score of how much \({d}_{{m}_{a}}\) in \({Dat}{a}_{A}^{{cca}}\) is aligned to \({d}_{{m}_{b}}\) in \({Dat}{a}_{B}^{{cca}}\). The higher the score, the more similar the two spots are. The process is described as follows:
Algorithm 5
Feature-aid fine alignment
-
1.
Input: \({d}_{A},\,{d}_{B},\,{f}_{A}^{{cca}},\,{f}_{B}^{{cca}},\,{use}\_{{\rm{icp}}},{{\rm{use}}}\_{{\rm{fgw}}}\)
-
2.
Output: \(\Pi ,\,\{\left({m}_{a},\,{m}_{b}\right)\}\)
-
3.
\({f}_{A}^{{\prime} }\leftarrow \{{d}_{A},\,{f}_{A}^{{cca}}\}\)
-
4.
\({f}_{B}^{{\prime} }\leftarrow \{{d}_{B},\,{f}_{B}^{{cca}}\}\)
-
5.
if use_icp then
-
6.
Calculate affine transformation matrix \({M}_{{icp}}\) using ICP algorithm on \({f}_{A}^{{\prime} }\) and \({f}_{B}^{{\prime} }\)
-
7.
\({f}_{B}^{{\prime} }\leftarrow {M}_{{icp}}* {f}_{B}^{{\prime} }\)
-
8.
Calculate \(L2\) distance matrix \(D\) between \({f}_{A}^{{\prime} }\) and \({f}_{B}^{{\prime} }\)
-
9.
Normalize \(D\) to \(\frac{D-\min (D)}{\max \left(D\right)-\,\min (D)}\) and set \(\Pi \leftarrow \,\frac{1}{D+\,\epsilon }\)
-
10.
end if
-
11.
if \({use}\_{{\rm{fgw}}}\) then
-
12.
Generate a transportation plan matrix \({\Pi }_{{fgw}}\) using FGW algorithm on \({f}_{A}^{{\prime} }\) and \({f}_{B}^{{\prime} }\)
-
13.
end if
-
14.
\(\left\{\left({m}_{a},\,{m}_{b}\right)\right\}\leftarrow {argma}{x}_{{m}_{a},\,{m}_{b}}\Pi\)
-
15.
return \(\Pi ,\,\{\left({m}_{a},\,{m}_{b}\right)\}\)
Parameter: \({use\_icp}={True},\,{use\_fgw}={False}\)
cloud mesh alignment algorithm like ICP or FGW enables automatic spatial registration alignment between spots in \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\). The Iterative Closest Point (ICP) algorithm aims to minimize the following error metric and is described as follows:
where:\((R)\) is the rotation matrix, (t) is the translation vector, (pi) are the points in the source point set, (qi) are the corresponding closest points in the target point set, \((N)\) is the number of points.
The FGW algorithm aims to find a transport plan \(\pi\) to resolve the following optimization problem
where \(M=|{f}_{a}^{{cca}}-{f}_{b}^{{cca}}{|}^{2}\), \({C}_{A}\), \({C}_{B}\) is the metric cost matrix for \({Dat}{a}_{A}\) and \({Dat}{a}_{B}\) where \({C}_{i,j}\) is the distance between the \(i\)-th spot and \(j\)-th spot. And \(\alpha\) is the trade-off parameter.
In practice, We use \({numpy}\) library in Python to implement the ICP algorithm and \({PoT}\) library in Python to calculate the FGW algorithm.
Step 6: integrating
After \(\Pi\) is determined, the final alignment result \(\{\left({m}_{a},{m}_{b}\right)\}\) can be determined from \(\Pi\) by selecting the highest score in each row of \(\Pi\) and the corresponding column index. For each spot \({m}_{{a}_{i}}=\{{d}_{m{a}_{i}},\,{f}_{m{a}_{i}},\,{l}_{m{a}_{i}}\}\), the data of its aligment spot \({m}_{{b}_{j}}\) in another assay is integrated in order to generate multi-modal data \({m}_{{ai}}^{{integrated}}=\{{d}_{m{a}_{i}},\,{f}_{m{a}_{i}},{f}_{m{b}_{j}},\,{l}_{m{a}_{i}},{l}_{m{b}_{j}}\}\).
hyper-parameter analysis
Further alignment
The \({dist\_}\min\) parameter serves as a spatial proximity threshold to identify neighborhood spots. Specifically, for any given spot, other spots within the \({dist\_}\min\) distance are considered its neighbors. Spots without any neighbors within this distance are classified as outliers. Higher \({dist\_}\min\) leads to fewer outliers and less transformation by further alignment. By default \({dist\_}\min\) is set as 1
Identification of anchor spot pairs and high correlated feature pairs
\({dist\_}\min\) and \(\epsilon\) were used for identification of anchor spot pairs and high-correlated feature pairs. Their interplay significantly influences the identification process. Higher values of both parameters lead to more anchor spot pairs but fewer high-correlated feature pairs, while lower values result in fewer anchor spot pairs but more high-correlated feature pairs. Through extensive empirical testing, we determined that a \({dis}{{\rm{t}}}\_\min =1\) and \(\epsilon =0.5\) provides an optimal balance for identifying a reliable set of anchor spot pairs. This is evidenced in sample M from our merge benchmark, which demonstrates that while the specific values of these parameters have minimal impact on alignment accuracy once anchor spot pairs are established, setting \({dist\_}\min\) too low may prevent the successful identification of anchor spot pairs altogether(Supplementary Fig. 1). \({\theta }_{{low}}({default}=0.5)\) and \({\theta }_{{high}}({default}=0.9)\) determine the threshold for Pearson correlation R value of feature pairs, \(k({default}=100)\) determines the top k number of high-correlated feature pairs kept for CCA translation. A combination of \({\theta }_{{low}}\), \({\theta }_{{high}}\) and \(k\) makes sure an identification of appropriate number of high-correlated feature pairs. If use_icp is selected, spatial registration of coordinated was performed with Maximum iterations = 100, tolerance = 1 × 10−6. If use_fgw is selected, spatial registration of coordinated was performed with \({alpha}=0.5,{\max }_{{iterations}}=100,{tolerance}=1e-9,{{loss}}_{{function}}={Squared\; Euclidean},\,{regularization}=0.01\).
Evaluation
Pairwise alignment accuracy
PAA provides a smooth measurement of the alignment accuracy compared to the label transfer accuracy. It calculates using the following approach: Given \({\pi }_{{ij}}\) as the alignment matrix produced by \({haCCA}\) where both \(i\), \(j\) are the number of samples in the source and target dataset, respectively. The PAA is defined to be \({\sum }_{{ij}}{\pi }_{{ij}}* {I}_{{ij}}\), where \({I}_{{ij}}\) is the indicator function that is 1 if the label of \(i\)-th sample in the source dataset is the same as the label of \(j\)-th sample in the target dataset, and 0 otherwise.
Label transfer accuracy
Label transfer accuracy measures how well labels from one dataset can be transferred to and correctly applied in another dataset.
Where Ncorrect is the number of correctly transferred labels in the target dataset, Ntotal is the total number of samples in the target dataset.
Label transfer ARI
Label Transfer ARI (Adjusted Rand Index) is a measure of the similarity between two data clusterings or label assignments. It adjusts the Rand Index (RI) to account for chance grouping.
n: Total number of elements in the dataset.
a: Number of pairs of elements that are in the same cluster in both clusterings.
b: Number of pairs of elements that are in different clusters in both clusterings.
c: Number of pairs of elements that are in the same cluster in the first clustering but in different clusters in the second clustering.
d: Number of pairs of elements that are in different clusters in the first clustering but in the same cluster in the second clustering.
\(\left({{{\rm{n}}}}{2}\right)\): Total number of possible pairs of elements, calculated as n(n-1)/2.
Robustness of manual alignment
While manual alignment requires expert selection of landmark points, we assessed its reproducibility and impact on integration performance. To evaluate operator variability, we performed 10 independent integration runs on sample M3, with different operators selecting 3 corresponding landmark points for gross alignment. The label transfer accuracy showed minimal variation across runs (94.2% ± 1.8% SD), demonstrating acceptable reproducibility of manual alignment (Supplementary Fig. S6).
Statistics and reproducibility
Statistical analysis
Statistical analyses were performed using R software (version 4.0.4). Paired Student’s t test was used for comparisons between groups (WT vs Padi4-/- ICC samples). For all tests, significance was determined with a 95% confidence interval (ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). P values are reported in the figure legends where applicable.
Sample size and replicates
For benchmark validation, 15 samples were randomly selected from 5 public spatial transcriptome datasets (3 mouse brain specimens [M1, M3, M4], 1 rat brain, and 1 human gastric cancer) to generate paired pseudo-MALDI-MSI data. Each sample underwent independent processing and evaluation.
For real-world validation, publicly available datasets included 6 sections from 3 mouse brain specimens, with each specimen providing both 10X Visium ST and MALDI-MSI data from adjacent sections (detailed in Supplementary Data 1).
For ICC model experiments, biological replicates consisted of n = 1 WT C57BL/6 mouse and n = 1 Padi4-/- mouse (6–8 weeks old, male), with tumors collected at 4 weeks post-hydrodynamic injection. Each tumor sample was processed for both BMKMANU S1000 ST and MALDI-MSI on adjacent tissue sections (10 μm thickness). Technical replicates for sequencing achieved a minimum depth of 50,000 reads per spot.
Performance analysis for \({haCCA}\) workflow
The \({haCCA}\) (hierarchical anchor-guided Canonical Correlation Analysis) algorithm involves multiple computational steps including iterative ICP-like registration, optimal transport computations, and canonical correlation analysis. To assess the practical applicability of our method, we conducted comprehensive performance benchmarks across datasets of varying sizes and complexity.
Experimental setup
Hardware configuration
-
Platform: Windows11
-
Memory: 32GB
-
CPU: i7-13800H
Test datasets
We generated psedo-MALDI_MSI datasets with controlled parameters to systematically evaluate performance.
Configuration | Description |
|---|---|
N = 30, F = 300, i = 10, j = 20 | Minimal test case |
N = 300, F = 3000, i = 100, j = 1000 | Medium test case |
N = 300, F = 10000, i = 100, j = 5000 | High-dimensional feature space |
Performance metrics
For each configuration, we measured
-
Execution Time: Wall-clock time for complete haCCA workflow
-
Memory Usage: Peak memory consumption during execution
Benchmark results
We run the benchmark for 100 times and take the average metric as table below.
Configuration | Execution Time (s) | Peak Memory (MB) |
|---|---|---|
N = 30, F = 300, i = 10, j = 20 | 0.022 | 4.7 |
N = 300, F = 3000, i = 100, j = 1000 | 2.704 | 124.1 |
N = 300, F = 10000, i = 100, j = 5000 | 19.467209 | 261.76 |
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The MALDI mass spectrometry imaging (MALDI-MSI) data and spatial transcriptomics data generated in this study have been deposited in the figshare database (https://doi.org/10.6084/m9.figshare.28320587). Details of the public datasets used in this study are available in the “Methods” section.
Code availability
All code for the haCCA workflow and analyses presented in this manuscript is publicly available without restrictions at GitHub: https://github.com/LittleLittleCloud/haCCA. https://doi.org/10.5281/zenodo.1778597031.
References
Vandereyken, K. et al. Methods and applications for single-cell and spatial multi-omics. Nat. Rev. Genet. 24, 494–515 (2023).
Vicari, M.M. et al. Spatial Multimodal Analysis of Transcriptomes and Metabolomes in Tissues Mendeley Data, V1, https://doi.org/10.17632/w7nw4km7xd.1 (2023).
Argelaguet, R. et al. Computational principles and challenges in single-cell data integration. Nat. Biotechnol. 39, 1202–1215 (2021).
Vicari, M. et al. Spatial multimodal analysis of transcriptomes and metabolomes in tissues. Nat. Biotechnol. 42, 1046–1050 (2023).
Ravi, V. M. et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 40, 639–655 e13 (2022).
Sun, C. et al. Spatially resolved multi-omics highlights cell-specific metabolic remodeling and interactions in gastric cancer. Nat. Commun. 14, 2692 (2023).
Zhi, Y. et al. Spatial transcriptomic and metabolomic landscapes of oral submucous fibrosis-derived oral squamous cell carcinoma and its tumor microenvironment. Adv. Sci. 11, e2306515 (2024).
Zheng, P. et al. Integrated spatial transcriptome and metabolism study reveals metabolic heterogeneity in human injured brain. Cell Rep. Med. 4, 101057 (2023).
Stuart, T. et al. Comprehensive Integration of single-cell Data. Cell 177, 1888–1902 e21 (2019).
Liu, J. et al. Jointly defining cell types from multiple single-cell datasets using LIGER. Nat. Protoc. 15, 3632–3662 (2020).
Argelaguet, R. et al. MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. 21, 111 (2020).
Singh, S. et al. Integrative metabolomics and transcriptomics identifies itaconate as an adjunct therapy to treat ocular bacterial infection. Cell Rep. Med. 2, 100277 (2021).
Iturria-Medina, Y. et al. Unified epigenomic, transcriptomic, proteomic, and metabolomic taxonomy of Alzheimer’s disease progression and heterogeneity. Sci. Adv. 8, eabo6764 (2022).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Zeira, R. et al. Alignment and integration of spatial transcriptomics data. Nat. Methods 19, 567–575 (2022).
Clifton, K. et al. STalign: alignment of spatial transcriptomics data using diffeomorphic metric mapping. Nat. Commun. 14, 8123 (2023).
Dong, L. et al. Proteogenomic characterization identifies clinically relevant subgroups of intrahepatic cholangiocarcinoma. Cancer Cell 40, 70–87 e15 (2022).
Zhang, M. et al. An inflammatory checkpoint generated by IL1RN splicing offers therapeutic opportunity for KRAS-mutant intrahepatic cholangiocarcinoma. Cancer Discov. 13, 2248–2269 (2023).
Yang, L. Y. et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J. Hematol. Oncol. 13, 3 (2020).
Shen, X. T. et al. Pan-cancer analysis reveals a distinct neutrophil extracellular trap-associated regulatory pattern. Front. Immunol. 13, 798022 (2022).
Xiong, S., Dong, L. & Cheng, L. Neutrophils in cancer carcinogenesis and metastasis. J. Hematol. Oncol. 14, 173 (2021).
Yazdani, H. O. et al. Neutrophil extracellular traps drive mitochondrial homeostasis in tumors to augment growth. Cancer Res. 79, 5626–5639 (2019).
van der Windt, D. J. et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 68, 1347–1360 (2018).
Pang, Z. et al. MetaboAnalyst 6.0: towards a unified platform for metabolomics data processing, analysis and interpretation. Nucleic Acids Res. 52, W398–W406 (2024).
Huang, F. et al. Active AKT2 stimulation of SREBP1/SCD1-mediated lipid metabolism boosts hepatosteatosis and cancer. Transl. Res. 268, 51–62 (2024).
Piccinin, E. et al. Role of oleic acid in the gut-liver axis: from diet to the regulation of its synthesis via stearoyl-CoA desaturase 1 (SCD1). Nutrients 11, 2283 (2019).
Caron-Jobin, M. et al. Stearic acid content of abdominal adipose tissues in obese women. Nutr. Diab 2, e23 (2012).
Guo, T. et al. SPIRAL: integrating and aligning spatially resolved transcriptomics data across different experiments, conditions, and technologies. Genome Biol. 24, 241 (2023).
Liu, X., Zeira, R. & Raphael, B. J. Partial alignment of multislice spatially resolved transcriptomics data. Genome Res. 33, 1124–1132 (2023).
De Vleeschauwer, S. I. et al. OBSERVE: guidelines for the refinement of rodent cancer models. Nat. Protoc. 19, 2571–2596 (2024).
Xiaoyun Zhang, B. LittleLittleCloud/haCCA: DOI (v0.0.4-DOI). Zenodo https://doi.org/10.5281/zenodo.17785970 (2025).
Acknowledgements
We sincerely thank Dr Chen-De Yang and Dr Dan-Ye in assisting establishing mouse model, Dr. Yang Zhang in assisting coding and writing improvement. This manuscript was supported by Natural science foundation of Shanghai (No. 24ZR1408800 to L.-Y.Y.), the National Natural Science Foundation of China (No. 82002532 to L.-Y.Y., No 82201445 to Z.-Q.C., No 82503435 to X.-T.S.), China Postdoctoral Science Foundation (2024M750533 to X.-T.S.), Shanghai Anticancer Association Chuying Project (SACA-CY22C10 to X.-T.S.).
Author information
Authors and Affiliations
Contributions
Lu-Yu Yang, Zhou-Qing Chen and Hu-Liang Jia designed and supervised the study and revised the manuscript; Jing Xu, Xiao-Tian Shen and Chen Zhang performed the experiments, Xiao-Yun Zhang and Xiao-Tian Shen write the code and provided helps in experimental techniques as well as data analysis, Xiao-Tian Shen prepared the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Chin Wee Tan and Joe Wandy for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, J., Shen, XT., Zhang, C. et al. haCCA: multi-module Integration of spot-based spatial transcriptomes and metabolomes. Commun Biol 9, 248 (2026). https://doi.org/10.1038/s42003-026-09526-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-026-09526-w
