
Abstract
The gating of HCN channels is regulated by both voltage and the binding of cyclic nucleotides to their intracellular domain. However, the molecular determinants underlying this regulation by cyclic nucleotide binding remain unclear and controversial. Here, we combine theoretical and experimental approaches to investigate the binding process in the HCN2 channel. First, molecular dynamics simulations show that the binding of cAMP and cGMP to one HCN2 subunit affects not only the stability of that subunit but also that of neighbouring ones in the absence of any large changes in backbone structure and in a way that is consistent with negative cooperativity. Next, network analysis reveals an inter-subunit communication path that connects cAMP and cGMP binding to the C-linker, which is attached to the pore domain. Finally, experimental analyses confirm that this path is essential for cyclic nucleotide-induced interactions between subunits and high affinity and negatively cooperative binding of ligand that is driven by favourable entropy. Together, these findings provide new insights into the regulatory mechanism of HCN2 gating mediated by cyclic nucleotides and clarify the role of residue E488, which lies on this path and whose mutations are known to cause idiopathic generalized epilepsy.
Similar content being viewed by others

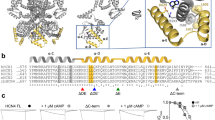
Introduction
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, which are encoded by HCN1-4 genes, regulate membrane potential and mediate electrical pacemaking in specialized cells of the heart and brain1,2,3,4,5. These channels are tetramers with each subunit containing six transmembrane α helices (S1-S6), organized into three main functional domains: the Voltage Sensor Domain (VSD, helices S1-S4), the Pore Domain (PD, helices S5-S6), and a Cyclic Nucleotide-Binding Domain (CNBD) in the C-term which binds the cyclic nucleotides (Supplementary Fig. 1)6,7. The CNBD (Fig. 1a) is connected to the pore by a unique stretch of residues named as the C-linker, which is characterized by four helices (A’ to D’) where helices A’-B’ define the TD motif and C’-D’ form the N3A motif. The CNBD itself contains several conserved structural regions: the Cyclic Nucleotide-Binding Domain (CBD) with eight β-strands (β1 to β8) that directly bind the cyclic nucleotides; the Phosphate-Binding Cassette (PBC), between β6 and helix B, interacts with the phosphate group of the cyclic nucleotides; and the C-terminal region of CNBD that contains the B-C helices (Fig. 1b)8,9. The C-linker and CNBD interact with equivalent regions of the neighbouring subunit, thus resulting into a domain-swapped configuration.

a Side and top view of the structured C-terminal region colored by subunits where the ligands cGMP and cAMP are highlighted. b Single subunit colored by domains: in green, the TD (helices A’-B’); in pink, the N3A (helices C’-D’); in cyan, the Cyclic Nucleotide-Binding Domain (CBD); in lime, the Phosphate-Binding Cassette (PBC); in purple, the B-C helices.
In 1991, DiFrancesco and Tortora found that the range of voltages over which the hyperpolarization-activated funny channel of the sinoatrial node is shifted to less negative voltages by direct binding of cAMP and cGMP to the cytoplasmic side of the plasma membrane10. Functional and structural analyses have since suggested that pore opening is variably inhibited in all four HCN isoforms and that inhibition is relieved by binding of cyclic nucleotides to an intracellular domain11,12,13,14,15,16. A direct comparison of the HCN1 and HCN2 forms showed that the full effect of inhibition and relief of inhibition by cAMP, and the differences in inhibition and its relief between them, require the tetrameric C-linker and CNBD17. In HCN2, the binding of cAMP or cGMP to the CNBD removes this inhibition, which results in an increase in current amplitude and a depolarizing shift of the activation curve8,13. In the absence of the ligands, the inhibition of the pore opening by these C-terminal domains in HCN1 is less than in HCN2. In the absence of cAMP, the position of the activation curve is less negative in cells expressing the HCN1 than in those expressing the HCN2. cAMP determines a smaller maximum depolarizing shift for HCN1 such that the positions of the activation curve for the HCN1 and HCN2 are similar when both are fully bound by ligand. An increase in current amplitude by cAMP has not been reported for the HCN1. Together, the less negative activation curve and, in response to cAMP, the smaller maximum shift and absence an increase in current amplitude suggests that the apo HCN1 behaves as though it is already partly bound by ligand.
Recent structures of HCN1 and HCN4 have shown that the C-linker and CNBD are rotated in a clockwise direction relative to the transmembrane domains when fully bound by cAMP6,7. Presumably, this rotation alleviates the inhibition of the pore opening. However, the atomic mechanism that links the progressive binding of the ligands to this rotation, observed when all sites are bound, remains unclear and controversial18,19,20,21,22,23.
We have measured the cAMP and cGMP binding affinity using isothermal titration calorimetry and a tetrameric version of the HCN2 C-linker and CNBD20,21. Their binding exhibited biphasic thermodynamics, with one high-affinity and three lower-affinity binding events. The high affinity binding was associated with favourable enthalpy and entropy, whereas the low affinity binding with favourable enthalpy but unfavourable entropy. Moreover, the favourable enthalpy of the high affinity binding was smaller than that for low affinity binding, suggesting that the negative cooperativity of the ligand binding is driven by favourable entropy. By contrast, the binding of cAMP to HCN1 occurred with only low affinity and associated with favourable enthalpy but unfavourable entropy. The difference between binding of ligand to HCN1 and HCN2 is consistent with the former behaving as though already partly liganded and activated.
What is the molecular and atomic basis of the entropically-driven and negatively cooperative binding of cyclic nucleotides in the HCN2 channel? Starting from the HCN2 C-linker and CNBD structure in complex with cGMP and cAMP, we run MD simulations showing that the progressive binding of cAMP produces a slightly greater increase in the stability of the bound subunit and alterations in the stability of unbound subunits, than cGMP, but in both cases this occurs without any large changes in backbone structure and in a way that is consistent with negative cooperativity. Subsequent network and experimental analyses, reveal an inter-subunit path linking the CNBD and proximal C-linker and identified E488 in the B’-helix of the C-linker as part of this path which is required for the high affinity and negatively cooperative binding process that is driven by favourable changes in entropy.
Results
cGMP and cAMP binding influences HCN2 structure
To characterize how the binding of the ligands to the CNBD allosterically influences channel gating, the C-linker and CNBD of HCN2 were studied in the presence of different numbers of bound cGMP or cAMP. (see Methods for details). Each system was simulated for 1 μs. RMSD calculations were conducted to compare the equilibrated structures to the experimental ones, i.e., at the beginning of the simulations, using different subsets of backbone atoms corresponding to specific protein domains as a selection to clarify the structural modifications induced by the presence/absence of the cyclic nucleotides. In the absence of cGMP/cAMP, the unbound system whose structure was solved with cAMP (PDB ID 1Q5O) exhibited greater instability, i.e., greater RMSD fluctuations, than the one with cGMP (PDB ID 1Q3E), particularly at the CNBD level (Fig. 2). Although fully bound with ligand, these structures are thought to represent the inactive form of the C-linker and CNBD24.

Panel a shows the systems bound to cGMP while panel b shows those bound to cAMP. Different colors identify different subunits. No ligands are bound in the systems with 0 cGMP/cAMP; in 1 cGMP/cAMP complexes, one ligand is bound to subunit one; in 2 syn cGMP/cAMPs, a ligand is bound to both subunits one and two; in 2 trans cGMP/cAMPs, a ligand is bound to subunit one and three, respectively; in 3 cGMP/cAMPs, a ligand is bound to subunits one, two and three, respectively; in 4 cGMP/cAMPs, each subunit is bound by one ligand. The x-axis shows different regions corresponding to Fig. 1b. Values correspond to the mean over the four independent replicas ± standard deviation.
When one cGMP was bound to the first subunit, the CNBD became more unstable due to a relative instability of the N3A region compared to the unbound system. The second subunit showed slightly enhanced TD stability, while other subunits behaved similarly to the unbound system (Fig. 2a). By contrast, with one cAMP bound, the first subunit’s CNBD became more stable, i.e., lower RMSD values, especially at the CBD, B-C helices and PBC; conversely, the second subunit displayed the highest stability depending on the TD and the B-C helices. The third and fourth subunits showed a more stable CNBD than the second one, due to the greater stability of the TD and B-C helices (Fig. 2b). The system with 2 cGMPs in syn configuration was characterized by higher CNBD instability at the level of the third and fourth subunits which are not bound by the cyclic nucleotide which depends on that of the CBD and B-C helices. Interestingly, the TD of the second subunit was more unstable than that of the other subunits, although this subunit was bound, like the subunit of the unbound system (Fig. 2a). Similarly, with 2 cAMPs in syn configuration, the bound subunits (first and second) had a greater stability of the CNBD than the others which are not bound, especially at the level of the B-C helices (Fig. 2b). In the 2 cGMPs trans configuration (subunits one and three bound), the bound subunits showed a greater stability of the CNBD due to the greater stability of the CBD and the B-C helices. However, the trends of TD and N3A are the opposite: the first subunit which is bound is the most stable while the third, which is also bound, is the most unstable. In contrast, for the N3A domain, the order is reversed: the third subunit is the most stable, and the first is the least stable. Finally, the second subunits not bound showed the greatest instability at most domains (Fig. 2a). A similar trend was found in the 2 cAMPs in trans configuration system, but in this case no unexpected trends have been found: the subunits were generally stabilized by the presence of the ligand (Fig. 2b). In the presence of 3 cGMPs bound, where only subunit four is not bound, the bound subunits showed greater CNBD stability especially at the CBD, B-C helices and PBC than the fourth subunit (Fig. 2a). On the other hand, in the 3 cAMPs system, the major instability was not shown by the unbound subunit as expected, but by the second one which was bound. Again, this depends on the greater instability of the B-C helices (Fig. 2b). Finally, when the complexes were fully bound, all domains exhibited greater stability compared to the unbound complexes (Fig. 2).
Altogether, these results indicate that, in the absence of cyclic nucleotides, the cAMP-solved structure exhibited greater instability than the cGMP-solved one, particularly in the CNBD regions. cAMP binding consistently stabilized bound subunits, especially in CBD, B-C helices, and PBC domains, while cGMP binding produced more complex effects, occasionally destabilizing bound subunits through N3A and TD slight perturbations. Importantly, the partial ligand occupancy provides evidence for allosteric communication between subunits, as the unbound subunits consistently showed stability changes dependent on both the number and spatial configuration (syn vs trans) of the ligand-bound neighbors, rather than behaving like fully unoccupied complexes. This inter-subunit coupling was further demonstrated by an asymmetric response among the unbound subunits and unexpected stability patterns, where the bound subunits exhibited greater instability than their unbound counterparts. The full ligand saturation achieved the maximum stability across all domains, suggesting cooperative binding effects in HCN2 channel regulation, where ligand binding to one subunit allosterically enhances the binding and stabilization of neighboring subunits.
Communication paths between cGMP/cAMP and the C-linker predicted by network analysis
To study the allosteric mechanism by which cGMP and cAMP communicate with the C-linker, each system was represented as a network where nodes coincide with protein residues and edges with the interactions between pairs. Each edge was assigned a weight expressed as \({w}_{{ij}}=-\log {C}_{{ij}}{M}_{{ij}}\), which quantifies the electromechanical coupling in terms of contacts between residues (\({C}_{{ij}}\)) and correlations of their motion (\({M}_{{ij}}\)), computed from MD runs25,26,27. Dijkstra’s algorithm28 was used to determine the shortest paths between the binding pocket of the ligands and the C-linker at the level of the A’ helix which is known to be crucial to couple the movements of the CNBD to the pore of the channels. To be consistent in all the systems, even those completely unbound, the communication paths were studied considering the binding pockets as the source regions, instead of the ligands themselves. Thus, the structural dynamics of the binding pocket was firstly analyzed, and a contact analysis was carried out to reveal the residues that directly interact with cGMP/cAMP. The protein residues with a cGMP/cAMP interaction frequency greater than 80% were selected to form the source region in the network analysis: I545, V564, T566, L574, F580, G581, E582, C584, R591, T592, A593, V595 for HNC2J with cGMP and I545, V564, M572, L574. F580, G581, E582, I583, C584, R591, T592, A593, V595, R632 for HNC2J with cAMP (Supplementary Fig. 2).
Four families of paths that could be either intra- or inter-subunit (Fig. 3a) were identified. In the first family (Fig. 3a), the motion propagated from cGMP/cAMP to the C-linker on the same subunit, following the path β2n → β1n → C’n → A’n. A slight difference on the residues involved in the paths was observed at the level of the C-linker: in presence of cGMP, the path used M485 as a bridge between Y481 and S445, while with cAMP it jumped from Y481 to G483, R446, and finally S445. The second family of path was an inter-subunit one, coupling the cGMP/cAMP of the n subunit to the C-linker of the n + 1 subunit (Fig. 3b). In this case, the motion followed the path β2n → D’n+1 → C’n+1 → A’n+1 and no difference was observed in the presence of different ligands. The communication path between the cGMP/cAMP of the n subunit and the C-linker of the n-1 subunit (Fig. 3c) resembled most of the motion propagation intra-subunit path before going onto the neighboring one. Precisely, it followed the path β2n → β1n → B’n → C’n → A’n-1 with a little difference on the residues involved on the C-linker, as described for the intra-subunit path. Finally, the communication path was studied between opposite subunits (n and n ± 2). In such a case, the motion followed the same path as that described in Fig. 3c, β2n → β1n → B’n → C’n → A’n-1 → A’n±2, with the exception that, once reached Q455 on A’n-1, it jumped onto D443 of A’n±2. In general, no qualitative difference was observed in the communication paths with and without the ligands, meaning that the absence of the cGMP/cAMP does not determine a loss of that allosteric communication between the two regions.

a Intra-subunit paths between β2 of n subunit and the A’ helix of the C-linker of the n subunit; b inter-subunit paths between n and n + 1 subunits; c inter-subunit paths between n and n − 1 subunits. The ligands are represented in licorice the amino acids along the paths in Van der Waals. dmin computed for cGMP (d) and cAMP (e) systems. Values correspond to the mean over the four independent replicates ± standard deviation.
The quantification of information transfer along a path between the source and the sink regions was obtained by computing the minimal path length (dmin) as the sum of the weight values obtained from Eq. 3, along the shortest paths identified by the Dijkstra’s algorithm between the two key regions. Thus, dmin quantifies the communication between the ligands and the C-linker in terms of contacts and correlated motions of the side chains of each pair of residues along the shortest path identified by Dijkstra’s algorithm. Considering the logarithmic nature of this metric, a difference of two units in dmin corresponds to a difference of one order of magnitude in terms of coupling, suggesting that the path with the lowest dmin is characterized by a weaker source-sink communication. In presence of cGMP, the intra-subunit path β2n → A’n and the inter-subunit path β2n → A’n+1 showed similar values of dmin, revealing a similar communication between the ligand and the C-linker (Fig. 3d, see Supplementary Table 1 for dmin of each single subunit). First, the binding of cGMP was not found to affect the dmin of the single subunits with a precise trend. However, considering the dmin of the entire complex, a significant correlation was found with the amount of cGMP: R = 0.98 and R = 0.96 with 2 ligands bound in syn configuration; R = 0.97 and R = 0.92 with 2 ligands bound in trans configuration, respectively (Fig. 3d). This trend revealed that the number of ligands influences the communication between the ligands and the C-linker with positive feedback via a concerted effect on the motion of the entire complex and not just at the level of individual subunits. The inter-subunit paths β2n → A’n-1 and β2n → A’n±2 were significantly less efficient than the others (higher dmin values), thus playing a minor role in the communication between the ligands and the C-linker. However, they exhibited a strong correlation with the number of cGMPs bound, similar to the other path families: R = 0.89 with 2 ligands bound in syn configuration and R = 0.98 with 2 ligands bound in trans configuration for the inter-subunit path β2n → A’n-1; R = 0.90 with 2 ligands bound in syn configuration and R = 0.98 with 2 ligands bound in trans configuration for the inter-subunit path β2n → A’n-1. These results showed that the intra-subunit path β2n → A’n and the inter-subunit path β2n → A’n+1 are the most efficient routes to couple the ligand to the C-linker. Moreover, these results suggest that the communication between ligand and C-linker improves with the number of bound cGMPs.
In the systems with cAMP, the inter-subunit paths β2n → A’n-1 and β2n → A’n±2 were longer than the others (higher dmin values) while the intra-subunit path β2n → A’n and the inter-subunit path β2n → A’n+1 were the shortest ones (Fig. 3e, see Supplementary Table 2 for dmin of each single subunit), thus playing a major key role in the communication transfer. As in presence of cGMP, no correlation was found between the individual path length of each subunit and the presence/absence of the ligand. But in all path families, the presence of 1 and 3 ligands correlated with a lower path length of the entire systems, i.e. better communication between cAMP and the C-linker, compared to when 0, 2 or 4 molecules are bound (Fig. 3e). This fluctuating trend (Fig. 3e) suggests that the binding of cAMP negatively influences the binding capacity of subsequent molecules. Indeed, the transition from 0 → 1 cAMP improves all the path families; the transition from 1 → 2 cAMPs significantly worsens the paths, regardless of the binding configuration; the transition from 2 → 3 cAMPs improves the coupling path again; the transition from 3 → 4 cAMPs worsens the paths for the second time.
Role of E488 in oligomerization and cGMP/cAMP binding to HCN2
The network analysis revealed that the communication between cGMP/cAMP and the C-linker can occur through multiple paths, but the most efficient ones are the intra-subunit path β2n → A’n and the inter-subunit path β2n → A’n+1. While the role of the intra-subunit one has been already confirmed29, in the present work we focused on the role of the inter-subunit path β2n → A’n+1. Along it, excluding the residues in the source and sink regions of the network, E488 showed the highest betweenness centrality value (Supplementary Table 3), a quantity defined as the number of times a node lies on a minimal length path between the chosen source and sink regions. So, residues with high betweenness serve as hubs, thus being relevant in the allosteric paths. Based on this evidence, we dissected the molecular role of the inter-subunit path β2n → A’n+1 and E488 by using a combination of mutagenesis, Dynamic Light Scattering (DLS) and Isothermal Titration Calorimetry (ITC).
Firstly, the role of E488 in the oligomerization of HCN2 was studied using DLS. We observed that the estimated molecular weight of the epilepsy-associated E488K mutant increased as the protein concentration rises, suggesting that predominantly tetramers were present at higher concentrations (Fig. 4a).

a Box plots of apparent molecular weight vs concentration of the mutant HCN2 C-linker and CNBD without cAMP or with a saturating concentration of cAMP as determined by DLS. The population of tetramers (~90 kDa) increases with increasing amount of protein. Box shows the range between 25-75%, the error bar shows the range within 1.5IQR, open square is the median and black diamonds are outliers. The number of replicates is 81, 95, 125, 123 and 62 for 25, 50, 100, 200, and 400 μM E488K protein without cAMP. The number of replicates is 82, 88, 124, 116 and 88 for 25, 50, 100, 200, and 400 μM E488K protein with cAMP. The number of replicates is 49, 64, 76, 78 and 26 for 25, 50, 100, 200, and 400 μM E488A protein without cAMP. The number of replicates is 63, 77, 85, 81 and 25 for 25, 50, 100, 200, and 400 μM E488A protein with cAMP. The limited increase in the estimated molecular weight in the presence of cAMP contrasts with our previously reported data on wild type protein showing large increases in molecular weight at lower protein concentrations in the presence of cAMP and cGMP20,30. b Plots of heat upon cumulative injections of cAMP to purified wild type HCN2 protein C-linker and CNBD (400 mM) as measured by ITC. Each inflection in the top plot occurs when cAMP is injected, which decrease in amplitude as the sites for binding saturate. Integration of the area under each peak/inflection in the upper plot are shown in the lower plot versus the ratio of cAMP to protein. The solid line through the values in the lower plot represents a two-independent binding site model, which yielded values for Kd, ΔH, -TΔS and ΔG for both high and low affinity binding events. c Bar overlap plot of dissociation constant (Kd) for high affinity (light grey) and low affinity (dark grey) cAMP binding events. Bars represent mean values. n = 2 replicates. d Bar overlap plot showing the changes in enthalpy (ΔH), entropy (-TΔS) and free energy (ΔG) for the high and low affinity events upon binding of cAMP to wild type HCN2 protein as indicated by the different colours. Note the favourable entropy and unfavourable enthalpy for the high affinity binding event. Bars represent mean and error bars are standard deviation. n = 2 replicates. e Plots of heat upon cumulative injections of cAMP to purified mutant HCN2 protein C-linker and CNBD as measured by ITC. Each inflection in the top plot occurs when cAMP is injected, which decrease in amplitude as the sites for binding saturate. Integration of the area under each peak/inflection in the upper plot are shown in the lower plot versus the ratio of cAMP to protein. The solid line through the values in the lower plot represents a single site binding model, which yielded values for affinity, ΔH, -TΔS and ΔG. f Bar overlap plot of dissociation constant (Kd) for cAMP (light grey) and cGMP (dark grey). Bars represent mean and error bars are standard deviation. For E488K, n = 6 replicates for cAMP and n = 6 replicates for cGMP. For E488A, n = 8 replicates for cAMP and n = 2 replicates for cGMP. g Bar graphs showing the changes in enthalpy (ΔH), entropy (-TΔS) and free energy (ΔG) upon binding of cAMP and cGMP to individual mutant proteins as indicated by the different colours. Bars represent mean and error bars are standard deviation. For E488K, n = 6 replicates for cAMP and n = 6 replicates for cGMP. For E488A, n = 8 replicates for cAMP and n = 2 replicates for cGMP.
This finding is like what we have previously found using the WT protein20. However, in contrast to its effect on the WT, a saturating amount of cAMP did not appear to greatly increase the apparent molecular weight at lower mutant protein concentrations. To reveal the role of the glutamate side chain, E488A was produced and studied using the same protocol. Its molecular weight increased as the concentration of protein, and cAMP did not greatly raise the apparent molecular weight at lower concentrations of the protein (Fig. 4a). This trend was very similar to that obtained for E488K. Taken together, these observations suggest that E488 is necessary for the ligand-induced inter-subunit interaction as suggested by the network analysis, controlling the allosteric effect of cAMP and, ultimately, the gating of the HCN channels.
Then, ITC was employed to determine whether binding of cAMP to the tetrameric form of the HCN2 was impacted by E488 mutations. Using a protein concentration of 400 μM, titration of cAMP produced a pattern of heat release that was best fitted with a single site binding model and yielded a Kd of 2.56 μM in E488K (Fig. 4e–g). This is in sharp contrast with ITC measurements of cAMP binding to WT, which show a clear and prominent biphasic heat response that is best fit by a two-independent-site binding model (Fig. 4a–d). This model indicates negative cooperativity and yields dissociation constants (Kd) of approximately 0.13 μM and 0.86 μM for the high- and low-affinity binding events, respectively. This pattern of binding and values of Kd for the high and low affinity binding events are very similar to those that we reported previously for the WT protein20,21,30.
Cyclic AMP binding to E488K was driven by favourable enthalpy (ΔH) and the entropic contribution (-TΔS) was not large (Fig. 4g). By contrast, the high affinity binding of cAMP to the WT, enthalpy was small and unfavourable, but entropy was favourable. For the low affinity event, enthalpy was favourable while entropy was not (Fig. 4d). Thus, as we suggested before for the WT20, the high affinity binding and negative cooperativity is driven by the changes in entropy. The binding affinity of cAMP to E488K was like what we find here and have found for both the low binding affinity of cAMP to the WT protein and to a monomeric version of the HCN2 C-linker and CNBD20. However, the small contribution of entropy to cAMP binding to E488K (Fig. 4g) is most like cAMP binding to the monomeric version. Therefore, the E488K mutation may disrupt the inter-subunit β2n → A’n+1 path between the ligand and the C-linker, thus uncoupling the n and n + 1 subunits. After removal of the glutamate R-group in E488A, the pattern of heat was well fitted with a single binding site model and yielded a Kd of 2.43 μM, almost identical to that obtained for E488K (Fig. 4b–f). The thermodynamics of cAMP binding are also similar, as the binding is driven mainly by enthalpy and the entropic contribution is limited (Fig. 4g). Thus, the contribution of E488 itself is necessary for negative cooperativity of cAMP binding and the favourable role of entropy in this process.
Finally, since in previous works we have shown that cGMP binds to HCN2 and HCN4 channels with negative cooperativity20,31 and our MD simulations showed that the inter-subunit path β2n → A’n+1 and E488 are also important for the communication between cGMP and the C-linker, we examined the cGMP binding to HCN2. As for cAMP, the negatively cooperative binding of cGMP was eliminated by E488K and E488A. By contrast with cAMP, both the enthalpic and entropic contributions of cGMP binding to E488 mutants were favourable which, despite the much higher Kd value, is like the high affinity binding event of cGMP to the WT (Fig. 4f, g). Thus, the mutants may limit the conversion from the high-affinity binding state to the low-affinity binding state upon cGMP binding, rather than disrupting the inter-subunit path and decoupling the subunits.
Contribution of Y459 in stabilizing E488 along the inter-subunit path β2n → A’n+1
E488 was predicted to be a determinant in the communication path β2n → A’n+1 by both computational and experimental analyses. Interestingly, in HCN2 E488 of the n subunit closely interacts with Y459 on the A’ helix of the n + 1 subunit (Supplementary Fig. 3). So, to further dissect the role of E488 in the predicted path, Y459A was studied to remove the R group, disrupt the interaction with E488 and let its side chain to be free of moving far away from the path. The DLS revealed that Y459A mutation affected the oligomerization of HCN2, thus supporting the existence of this inter-subunit interaction between E488 with Y459 (Fig. 5a). Moreover, the pattern of heat was well-fitted with a single binding site model, which yielded a Kd value of ~7.29 μM (Fig. 5b, c). Although this is somewhat larger than the Kd values obtained with the E488K and E488A mutants, the single binding event was similarly driven mainly by enthalpy, and the entropic contribution was again limited (Fig. 5d). These data provide more support for the inter-subunit interaction between E488 with Y459 and its requirement for entropically driven negative cooperativity and associated facilitation of gating by cAMP and cGMP.

a Box plots of apparent molecular weight vs concentration of the mutant HCN2 C-linker and CNBD without cAMP or with a saturating concentration of cAMP as determined by DLS. The population of tetramers (~90 kDa) increases with increasing amount of protein. Box shows the range between 25–75%, the error bar shows the range within 1.5IQR, open square is the median and black diamonds are outliers. The number of replicates is 63, 77, 85, 81, and 25 for 25, 50, 100, 200, and 400 μM Y459A protein without cAMP. The number of replicates is 63, 70, 65, 43 and 9 for 25, 50, 100, 200, and 400 μM Y459A protein with cAMP. The number of replicates is 28, 25, 26, 27 and 23 for 25, 50, 100, 200, and 400 μM E488Y/Y459E protein without cAMP. The number of replicates 24, 25, 9, 24, and 20 for 25, 50, 100, 200, and 400 μM E488Y/Y459E protein with cAMP. b Plots of heat upon cumulative injections of cAMP to purified HCN2 protein C-linker and CNBD as measured by ITC. Each inflection in the top plot occurs when cAMP is injected, which decrease in amplitude as the sites for binding saturate. Integration of the area under each peak/inflection in the upper plot are shown in the lower plot versus the ratio of cAMP to protein. The solid line through the values in the lower plot represents a single site binding model, which yielded values for affinity, ΔH, -TΔS and ΔG. c Bar overlap plot of dissociation constant (Kd) for cAMP (light grey) and cGMP (dark grey). Bars represent mean and error bars are standard deviation. For Y459, n = 5 replicates for cAMP and n = 3 replicates for cGMP. For E488Y/Y459E, n = 2 replicates for cAMP. d Bar overlap plot showing the changes in enthalpy (ΔH), entropy (-TΔS) and free energy (ΔG) binding of cAMP and cGMP to individual mutant proteins. Bars represent mean and error bars are standard deviation. For Y459, n = 5 replicates for cAMP and n = 3 replicates for cGMP. For E488Y/Y459E, n = 2 replicates for cAMP.
Lastly, we reversed E488 and Y459 (E488Y/Y459E) to determine whether the functional, and hence physical, interaction between them could be preserved. By DLS, we found an increase in estimated molecular weight when the protein concentration was increased. cAMP did not appear to increase in molecular weight at lower concentrations of protein (Fig. 5a) as compared to our previously published data using the WT. Moreover, the heat released was fitted best with a single binding site model and yielded a Kd value of ~2.61 μM (Fig. 5b, c). The binding of cAMP was found to be driven completely by enthalpy with a limited entropic contribution (Fig. 5d). Together, the DLS and ITC data show that ligand-driven interaction and entropically driven negatively cooperative binding of cAMP is not preserved when tyrosine and glutamate are substituted for each other. The E488Y-Y459E double mutant may either disrupt the interaction between these residues, or it is unable to recapitulate other, additional interactions that are made by E488 and/or Y459. Indeed, our analysis suggests that Y459 may help to maintain E488 in a position to appropriately interact with F486 and L492 along the β2n → A’n+1 path. Therefore, when the glutamate is mutated to tyrosine at position 488, the interaction with F486 and L492 is altered or lost and the β2n → A’n+1 path is disrupted.
Discussion
The mechanism by which cAMP binds to HCN channels remains a challenging and unresolved issue, with conflicting reports on whether cooperative binding occurs at all and, if so, its precise nature18,19,20,22,23,24,32,33,34,35. By contrast to this variability, data from our laboratory, as well as others, consistently show that cAMP, as well as cGMP and a subset of other cyclic nucleotides, binds to the tetrameric C-linker and cyclic nucleotide-binding domain (CNBD) of HCN2 and HCN4 with negative cooperativity, a process essential for the full inhibition of gating and its reversal upon ligand binding20,30,31. This interaction is primarily driven by favorable entropy, a thermodynamic signature conserved in other cAMP-binding proteins, such as CAP, and seen in diverse ligand–protein systems36,37,38,39,40. Our previously proposed two-step binding model aligns with the functional effects of cAMP on the full HCN2 channel, including an increase in current amplitude that becomes maximal when less than four sites are occupied and depolarizing shift in the activation curve that is at a maximum when all four sites are occupied18,33,41,42. A gating model incorporating negative cooperativity not only explains the depolarizing shifts but also predicts the consequences of single-point mutations within the binding site on those shifts21,43.
A study, published just after the first report on inter-subunit ligand binding cooperativity by our group, used ensemble measurements of current and binding of a fluorescent analog in HEK cells that express the full-length HCN2 channel. Along with global fitting of a ten-state kinetic model, this data suggested a sequence that was positive-negative-positive in open HCN2 channels44. Negative cooperativity was the largest change reported and was thought to occur upon binding of cAMP to the second subunit. By contrast, the same approach in closed HCN2 channels suggested positive cooperativity19. These methods consider all information available from the ensemble of channels in the plasma membrane to fit a model of binding, but they also make it challenging to determine parameters that are unique, or identifiable, for a given model45. Recent single molecule studies using either the full-length or close to full length channel and fluorescent analogs of cAMP yield data without the same issues of parameter identifiability. The first study to use this approach found that cAMP binding was not cooperative in either purified HCN1 or HCN2 channels22. These studies used close to full-length channels (HCN1SM, HCN2SM) that lacked a region of the C-terminus downstream from the C-helix of the CNBD to optimize the expression levels for producing and studying the channels. A second study in which single molecule fluorescence was combined with ensemble measurements suggested that cAMP binds to the full-length HCN2 channel with positive cooperativity. In this study, it was suggested that the differences between the two single molecule studies may be related to a difference in the structure of the fluorescent analog used and its detection23. It is also possible that regions downstream of the HCN2 CNBD, which are present in the latter study but not in the former, might contribute to the differences observed. A very recent report using single molecule florescence and C-terminally truncated channels has also suggested positive cooperativity and, intriguingly, that cooperativity may be regulated by endogenous factors such as lipids46. Our studies have used ITC to measure heat that results from cyclic nucleotide to C-linker and CNBD protein in solution20,21,30,31. These ensemble measurements of heat are fitted to models of binding, which make it a challenge to determine a set of parameters that are unique. In our studies, simple models of binding are used with few parameters that limit identifiability issues, but which may not reflect the true mechanism in detail. For the single site model used to fit the data in this paper, enthalpy is measured directly from the heat measured. Along with the change in enthalpy, binding affinity (Ka) and stoichiometry (n) are estimated from the fitting of one experimental run. The biphasic heat data from the wild type are much different from the heat data from mutants and are not well-fitted with the single site model. They are, however, well-fitted with the two independent binding site model for which we make the simplifying assumption that the stoichiometries indicate the fraction of sites in the tetramer that possess a given affinity. This simple interpretation suggests one ligand binds with high affinity, favoured by changes in entropy, and three bind with a lower affinity, favoured by changes in enthalpy.
Both the ensemble and single molecule studies using fluorescent forms of cAMP described above used full-length or close to full-length channels. These long channel forms provide interactions that influence cAMP binding not only from the C-linker and CNBD, but also from regions outside of these two domains. Regions that have been suggested interacting with the C-linker and CNBD include those that are downstream from the C-helix of the CNBD47, the HCN domain of the N-terminus7,48 and the voltage-sensing transmembrane elements and the pore18,34,49. However, these multiple influences, in addition to those from within the C-linker and CNBD, make it challenging to extract specific information about binding affinity and the nature of cooperativity. Our studies have used tetramers of the C-linker and CNBD, domains found in the C-terminus of the HCN2 channel. This region does not represent the full range of interactions of cAMP with the other parts of the channel that could influence cAMP binding. Nevertheless, using this region offers several advantages. First, it preserves binding and interactions within and between C-linker and CNBD within the tetramer. Second, this region, without any further domains downstream of the C-helix, is required for the full effect of cAMP on the HCN2 channel17, thus providing interactions with ligand that are necessary for the complete facilitation of the gating. Third, the structure of the C-linker and CNBD of the HCN2 channel has been solved for cAMP, as well as other ligands. Fourth, its purification and solubilization are straight forward for the wild type protein, as well as for many mutant versions8,20,21. Because our approach does not require any modification of the ligand, we have been able to examine in detail, as well as cAMP and cGMP, the binding of several analogs and whether they bind with cooperativity in our system30. Lastly, the analysis and interpretation of data obtained by ITC are relatively simple using only this region. A comparison of values for affinity found by the different approaches shows both similarities and differences (Fig. 6).

Previously published equilibrium association constants (Ka) for each ligand binding step (means are plotted). For Kusch et al. 201144 and Thon et al. 201573, these values were determined from ensemble measurements using a fluorescent cAMP analog and the full-length channel expressed in HEK cells at -130 mV and -30 mV, respectively. For White et al. 202122, these values were determined from single molecule measurements using a fluorescent cAMP analog and C-terminally truncated channels expressed in HEK293 cells and purified into detergent micelles. For Chow et al. 201220, these values were determined by ITC using unmodified cAMP and C-linker and CNBD purified from bacteria at a concentration yielding mainly tetramers.
Our approach has yielded high affinity and low affinity values for HCN2 whereas binding to HCN1 was not found to be cooperative, and the affinity was like the low binding affinity for HCN2. For HCN1, the values are higher than those obtained by single molecule fluorescence although that difference may be due to the use of a fluorescent analog in those studies versus cAMP in our studies. Our values for HCN2 are also higher than those found using single molecule fluorescence, but the main difference is that we have suggested binding to the first site is of higher affinity than the other sites. The ensemble fluorescence approach (at -130 mV) yielded positive- negative- positive values with the highest affinity found when bound to the second site and the lowest affinity found when bound to the third site. At -30 mV, the values showed less negative cooperativity and then positive cooperativity.
In this study, we used an integrated computational and experimental approach to investigate, at molecular resolution, how HCN2 subunits interact and communicate upon cyclic nucleotide binding. RMSD calculations from our Molecular Dynamics (MD) simulations showed that the removal of cAMP or cGMP leads to increased structural flexibility in the tetrameric CNBD and C-linker of HCN2, with the extent of these changes varying depending on the binding configuration across subunits. Ligand-bound subunits appear to provide some structural stability to adjacent unbound subunits, which may contribute to the observed reduction in binding affinity upon initial nucleotide binding. This finding suggests a possible negatively cooperative binding mechanism. Biochemical assays of oligomerization and ligand binding in E488A/K and Y459A mutants confirmed that E488 contributes to high-affinity binding and entropically driven negative cooperativity, thereby supporting a two-step binding model: (i) initial high-affinity, entropy-driven binding promotes an increase in current amplitude; and (ii) subsequent lower-affinity, enthalpy-driven binding completes the depolarizing shift in the activation curve. These findings are consistent with both our theoretical predictions and previous evidence that initial cAMP binding induces a dynamic asymmetry across subunits, leading to a non-uniform cooperative pattern. Thus, the initial binding may occur in the absence of a change in the mean positions of the atoms as it has been proposed in other negatively cooperative systems36,39,40,50,51,52. The differences in stabilization of individual subunits observed between cis and trans binding may contribute to differences observed in gating between the syn and trans bound HCN2 channel33,41 as well as to differences in binding and gating observed between different forms of HCN channels.
Furthermore, we showed that the negative cooperativity of cGMP binding is abolished when the inter-subunit communication path coupling cGMP on subunit n to the C-linker of subunit n + 1 is disrupted by E488 and, indirectly, Y459 mutations. Thermodynamic analyses revealed altered entropy contributions and elevated dissociation constants, suggesting that although cAMP and cGMP share this regulatory mechanism, their binding thermodynamics diverge. Our MD simulations indicate that, unlike cAMP, cGMP enhances CNBD–C-linker communication in a concentration-dependent manner which may be related to their binding configuration found in the structures8. Moreover, this difference may contribute to those found in NMR and EPR spectra between the isolated CNBDs when bound to cAMP or cGMP53 and to the much larger EC50 value for the depolarizing effect of cGMP versus cAMP on the activation curve of the full channel, despite no difference in their maximum effects8,43.
To further investigate this dynamic allostery, we applied network analysis, to examine correlated fluctuations between residues during MD simulations. This method has proven effective in identifying long-range communication paths in both membrane-bound and soluble proteins, and in elucidating how these paths are modulated by external molecules25,26,27,54. In HCN2, we identified four distinct families of allosteric communication paths linking the CNBD to the C-linker: one intra-subunit path and three inter-subunit paths involving neighboring subunits. Among these, the intra-subunit path and the inter-subunit path from subunit n to n + 1 were the most efficient. However, the presence of multiple concurrent paths suggests that HCN2 integrates cyclic nucleotide signals through a distributed allosteric network, complicating experimental efforts to isolate specific cooperative mechanisms. Further computational and mutagenesis studies will be necessary to clarify the functional relevance of each path, particularly under conditions in which dominant paths may be compromised.
Within the inter-subunit path from n to n + 1, residue E488 emerged as a central hub, playing a pivotal role in allosteric signal propagation. Notably, the E488K mutation has previously been associated with idiopathic generalized epilepsy and shown to severely impair HCN2 function55. Here, we provide a molecular explanation for this phenotype: substituting a negatively charged glutamate with a positively charged lysine disrupts critical electrostatic interactions along the inter-subunit β2n → A’n+1 path, effectively abolishing allosteric communication between the CNBD and the channel pore. This interruption blocks the activation of HCN2 channels in response to cyclic nucleotide binding, thereby impairing pacemaker function and contributing to disease pathology.
In summary, our findings demonstrate that cyclic nucleotide binding in HCN2 is mediated by a network of intra- and inter-subunit allosteric paths, with the inter-subunit one between subunits n and n + 1 being essential for cooperative binding. Interestingly, all the residues identified along these paths are conserved between C-linker and CNBD of HCN1 and HCN2, thus suggesting that they can play a role also in these channels (Supplementary Fig. 4). Unlike its binding to the HCN2 C-linker and CNBD, we have found that cAMP binds to the HCN1 channel without cooperativity. This difference in cAMP binding between HCN2 and HCN1 is probably due to the difference in residues between them. The residues found in only one of the two channel forms may uniquely modify the paths originating from cAMP binding to the C-linker. These results not only clarify the molecular and thermodynamic basis of negative cooperativity in HCN2 but also provide a structural framework to understand the impact of disease-associated mutations, of residue differences between isoforms, in other cyclic nucleotide-gated ion channels, and on other cyclic nucleotide analogs, and guide future efforts in modulating channel function.
Methods
Molecular Dynamics simulations
The CBND and the C-linker of HCN2 channels in presence of cGMP and cAMP were produced from the experimentally solved structures (PDB ID 1Q3E and 1Q5O, respectively)8 and the missing loops were modelled using SWISS-MODEL56. The structures of the unbound subunits were produced by manually removing the ligands from the experimental structures. The APBS server57, all aspartates and glutamates were predicted to be ionized; H463, H479 and H559 to be in the δ state while H474 had both δ- and ε-nitrogens protonated. All the systems in presence of a different number of ligands were created using the CHARMM-GUI Membrane Builder58,59, whose characteristics are resumed in Supplementary Table 4. The systems with 2 ligands have been studied in both syn and trans configuration meaning that, in the first case, the ligands are bound to neighbouring subunits while, in the second case, to opposite subunits. A solution of 0.15 KCl was added in all of them with TIP3P water molecules60. After the six steps of equilibration implemented in the CHARMM-GUI Membrane Builder58,59, each system was simulated in the NPT ensemble for 250 ns with four independent replicas, thus totalling 1 μs per system. All simulations were run with NAMD 3.0.161 using the CHARMM36m force field62,63. Pressure was kept at 1.01325 Bar by the Nosé-Hoover Langevin piston method and the temperature was maintained at 303.15 K by a Langevin thermostat with damping coefficient of 1 ps⁻¹64,65. Long-range electrostatic interactions were evaluated with the smooth Particle Mesh Ewald algorithm with a grid space of 1 Å. For short-range non-bonded interactions, a cut-off of 12 Å with a switching function at 10.0 Å was used. The integration time-step was 2 fs. The trajectories were visually inspected using VMD 2.0.0 software66. Root Mean Square Deviation (RMSD) values were computed between the equilibrated structures and the experimental structures at the beginning of the production phase used as a reference conformation. Different subsets of backbone atoms corresponding to different domains were considered. Mean values over the four independent replicas per system were reported ± the standard deviation.
Contact analysis
The contact patterns were identified computing semi-binary contact maps (\({C}_{{ij}}\)) as truncated Gaussian kernels:
where \({d}_{{ij}}\) is the distance between the side chain of the i- amino acid and either the side chain of the j- amino acid or the j- atom of cGMP and cAMP; c is the cut-off distance set to 4.5 Å25,67. The width σ of the Gaussian kernel was chosen so as to attain a negligibly small value of the kernel at \({d}_{{ij}}=10\,{\text{\AA }}\). Specifically, we imposed \({K}({d}_{{cut}})={10}^{-5}\) attaining \(\sigma =1.48\). The final contact map was computed by averaging the value of the kernel over all the frames of the trajectory:
Network analysis
All the systems were represented as a graph where the weight assigned to the edges was:
being \({C}_{{ij}}\) the semi-binary contact map and \({M}_{{ij}}\) the matrix that quantifies the motion correlation of residues.
\({M}_{{ij}}\) was computed as:
where \({d}_{i}\) and \({d}_{j}\) are the displacement of the center of mass of the side chain of the i and j residues with respect to its average position and \({H}_{{ij}}\) is the Shannon entropy68 of those variables.
Dijkstra’s algorithm28 was used to compute the minimal paths between the binding pocket (source) and the A’ helix of the C-linker (sink)8,69 which is known to be crucial to couple the movements of the CNBD to the pore of the channels70,71. The binding pocket encompasses residues I545, V564, T566, L574, F580, G581, E582, C584, R591, T592, A593, V595 for HNC2J with cGMP and I545, V564, M572, L574, F580, G581, E582, I583, C584, R591, T592, A593, V595, R632 for HNC2J with cAMP. \({{\rm{d}}}_{\min }\) corresponds to the sum of the weight values obtained from Eq. 3, along the shortest paths identified by the Dijkstra’s algorithm between the two key regions, computed over the four subunits and the four replicas ± σ where σ is standard deviation. The betweenness centrality of each residue was computed with Brandes’s algorithm as implemented in the NetworkX 3.0 library72. The network analysis was carried out on the concatenated trajectories of each independent replica per system after convergence, as monitored via RMSD calculations, thus resulting into more than 400 ns per system for the analysis (see Supplementary Figs. 5 and 6, Supplementary Table 5).
Cloning and molecular biology
The same protocol used in our previous works was employed20. The C-linker and CNBD of HCN2 were cloned into a modified pEt28 vector. A His-tag, maltose-binding protein, and TEV cleavage site was inserted in the N-terminal side for purification. Site-directed mutagenesis was carried out using the QuikChange protocol (Agilent, Santa Clara, CA, USA). The WT and mutant constructs were expressed in Rosetta DE3 pLacI cells. After OD reached 0.6-0.8, cells were induced by 0.1 M isopropyl-ß-d-thiogalactopyranoside (IPTG) at 25°C and harvested after overnight shaking for 16 h.
Protein expression and purification
Proteins were expressed in E. coli Rosetta (DE3) pLacI cells (MilliporeSigma, Burlington, MA, USA), induced at OD600 of 0.6 with 0.4 M IPTG, and inoculated for 4 h before harvesting, all at 37 degrees. Cell pellets were resolubilized and lysed with sonication in 250 mM KCl and 10 mM HEPES at pH 7.4 (buffer A). Glycerol, 25 μg/mL DNase I, lysozyme, 0.4 mM EDTA, and 10 mM PMSF were also added. The constructs were added to Talon (cobalt-bound-resin) columns, which where then eluted with 250 mM KCl and 500 mM imidazole at pH 7.4. After dialysis with TEV protease in buffer A, the eluent was run on another Talon column and the flow through was collected. Protein was dialyzed against 10 mM KCl + 20 mM MES at pH 6.0 for 2 h and applied to a ResourceS column (GE Healthcare, Chicago, IL, USA) at an increasing KCl concentration from 0 to 50%. SDS-PAGE was used to confirm the presence of protein, which was then dialyzed against 150 mM KCl and 10 mM HEPES at pH 7.4. Using spectrophotometry and the Edelhoch method, protein was concentrated to 20 mg/ml.
Dynamic light scattering
Dynamic Light Scattering (DLS) was used to determine the oligomerization state of the HCN2 C-linker and CNBD, as we have described previously (20). This HCN2 protein was diluted to 25, 50, 100, 200 or 400 μM in a 50 μL either with or without cAMP or cGMP, which was added at a ratio of 10:1 cNMP to protein. This was centrifuged at 6000 rpm for 2 min to remove debris and then added to a 384-multiwell microtiter plate. A DynaPro plate reader II (Wyatt Technology, Santa Barbara, CA, USA) was utilized to determine the hydrodynamic radii at 22 °C. These radii were then converted to molecular weights. Initially, five readings were averaged per acquisition followed by an average of ten acquisitions per well. Acquisitions with values of polydispersity of over 30% were not used.
Isothermal titration calorimetry
Isothermal Titration Calorimetry (ITC) and determination of binding parameters was carried out as has been described previously by our lab (20). Proteins were diluted to 200 μM or 400 μM in dialysis buffer (150 mM KCl + 10 mM HEPES [pH 7.4]). The concentration of cAMP and cGMP was 2 mM and a 1 microL amount was titrated 40 times into the cell. The experiments were performed at 25 °C. The heat required to compensate for the difference in temperature between the sample and reference cell when the ligand was bound was then integrated to obtain kilocalories per mole of injectant. Fitting of the heat data was determined by using either a single site or a two-independent binding site model in Origin 7.0 (MicroCal ITC add-on, Malvern Panalytical, Malvern, UK). The thermodynamics of binding and stoichiometry were determined from the fitting process.
Statistics and reproducibility
All MD simulations were performed in multiple independent replicas with randomized initial atomic velocities.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The initial and final coordinates files, and the simulation input file are deposited in Zenodo https://zenodo.org/records/16788446. The source data for the box plots and bar overlap plots in Figs. 4 and 5 are found in Supplementary data 1 and Supplementary data 2.
Code availability
All software used for this study are available and the links are provided as follows. For the data collection: NAMD, APBS (https://server.poissonboltzmann.org), CHARMM-GUI Membrane Builder (https://www.charmm-gui.org). Accession codes of the structures: CNBD and C-linker of HCN2 in presence of cGMP, PDB ID 1Q3E; CNBD and C-linker of HCN2 in presence of cAMP, PDB ID 1Q5O.
References
Gauss, R., Seifert, R. & Kaupp, U. B. Molecular identification of a hyperpolarization-activated channel in sea urchin sperm. Nature 393, 583–587 (1998).
Ludwig, A., Zong, X., Jeglitsch, M., Hofmann, F. & Biel, M. A family of hyperpolarization-activated mammalian cation channels. Nature 393, 587–591 (1998).
Santoro, B. et al. Identification of a Gene Encoding a Hyperpolarization-Activated Pacemaker Channel of Brain. Cell 93, 717–729 (1998).
DiFrancesco, D. Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol. 55, 455–472 (1993).
Pape, H.-C. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu. Rev. Physiol. 58, 299–327 (1996).
Lee, C.-H. & MacKinnon, R. Structures of the Human HCN1 Hyperpolarization-Activated Channel. Cell 168, 111–120.e11 (2017).
Saponaro, A. et al. Gating movements and ion permeation in HCN4 pacemaker channels. Mol. Cell 81, 2929–2943.e6 (2021).
Zagotta, W. N. et al. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature 425, 200–205 (2003).
Akimoto, M. et al. A Mechanism for the Auto-inhibition of Hyperpolarization-activated Cyclic Nucleotide-gated (HCN) Channel Opening and Its Relief by cAMP. J. Biol. Chem. 289, 22205–22220 (2014).
DiFrancesco, D. & Tortora, P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 351, 145–147 (1991).
Barbuti, A., Baruscotti, M., Altomare, C., Moroni, A. & DiFrancesco, D. Action of internal pronase on the f-channel kinetics in the rabbit SA node. J. Physiol. 520, 737–744 (1999).
Viscomi, C. et al. C Terminus-mediated Control of Voltage and cAMP Gating of Hyperpolarization-activated Cyclic Nucleotide-gated Channels. J. Biol. Chem. 276, 29930–29934 (2001).
Wang, J., Chen, S. & Siegelbaum, S. A. Regulation of Hyperpolarization-Activated Hcn Channel Gating and Camp Modulation Due to Interactions of Cooh Terminus and Core Transmembrane Regions. J. Gen. Physiol. 118, 237–250 (2001).
Stieber, J., Stockl, G., Herrmann, S., Hassfurth, B. & Hofmann, F. Functional expression of the human HCN3 channel. J. Biol. Chem. 280, 34635–34643 (2005).
Wicks, N. L., Wong, T., Sun, J., Madden, Z. & Young, E. C. Cytoplasmic cAMP-sensing domain of hyperpolarization-activated cation (HCN) channels uses two structurally distinct mechanisms to regulate voltage gating. Proc. Natl. Acad. Sci. USA 108, 609–614 (2011).
Peters, C. H. et al. LRMP inhibits cAMP potentiation of HCN4 channels by disrupting intramolecular signal transduction. eLife 12, RP92411 (2024).
Wainger, B. J., DeGennaro, M., Santoro, B., Siegelbaum, S. A. & Tibbs, G. R. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411, 805–810 (2001).
Kusch, J. et al. Interdependence of receptor activation and ligand binding in HCN2 pacemaker channels. Neuron 67, 75–85 (2010).
Benndorf, K., Thon, S. & Schulz, E. Unraveling Subunit Cooperativity in Homotetrameric HCN2 Channels. Biophysical J. 103, 1860–1869 (2012).
Chow, S. S., Van Petegem, F. & Accili, E. A. Energetics of Cyclic AMP Binding to HCN Channel C Terminus Reveal Negative Cooperativity. J. Biol. Chem. 287, 600–606 (2012).
Ng, L. C. T., Zhuang, M., Van Petegem, F., Li, Y. X. & Accili, E. A. Binding and structural asymmetry governs ligand sensitivity in a cyclic nucleotide–gated ion channel. J. Gen. Physiol. 151, 1190–1212 (2019).
White, D. S. et al. cAMP binding to closed pacemaker ion channels is non-cooperative. Nature 595, 606–610 (2021).
Kuschke, S. et al. cAMP binding to closed pacemaker ion channels is cooperative. Proc. Natl. Acad. Sci. USA 121, e2315132121 (2024).
Craven, K. B. & Zagotta, W. N. Salt Bridges and Gating in the COOH-terminal Region of HCN2 and CNGA1 Channels. J. Gen. Physiol. 124, 663–677 (2004).
Westerlund, A. M., Fleetwood, O., Pérez-Conesa, S. & Delemotte, L. Network analysis reveals how lipids and other cofactors influence membrane protein allostery. J. Chem. Phys. 153, 141103 (2020).
Costa, F., Ocello, R., Guardiani, C., Giacomello, A. & Masetti, M. Integrated Approach Including Docking, MD Simulations, and Network Analysis Highlights the Action Mechanism of the Cardiac hERG Activator RPR260243. J. Chem. Inf. Model 63, 4888–4899 (2023).
Bassetto, C. A. Z., Costa, F., Guardiani, C., Bezanilla, F. & Giacomello, A. Noncanonical electromechanical coupling paths in cardiac hERG potassium channel. Nat. Commun. 14, 1110 (2023).
Dijkstra, E. W. A note on two problems in connexion with graphs. Numer. Math. 1, 269–271 (1959).
Pfleger, C. et al. Allosteric signaling in C-linker and cyclic nucleotide-binding domain of HCN2 channels. Biophysical J. 120, 950–963 (2021).
Ng, L. C. T., Putrenko, I., Baronas, V., Van Petegem, F. & Accili, E. A. Cyclic Purine and Pyrimidine Nucleotides Bind to the HCN2 Ion Channel and Variably Promote C-Terminal Domain Interactions and Opening. Structure 24, 1629–1642 (2016).
Ng, L. C. T., Li, Y. X., Van Petegem, F. & Accili, E. A. Altered cyclic nucleotide binding and pore opening in a diseased human HCN4 channel. Biophysical J. 121, 1166–1183 (2022).
DiFrancesco, D. Dual allosteric modulation of pacemaker (f) channels by cAMP and voltage in rabbit SA node. J. Physiol. 515, 367–376 (1999).
Ulens, C. & Siegelbaum, S. A. Regulation of Hyperpolarization-Activated HCN Channels by cAMP through a Gating Switch in Binding Domain Symmetry. Neuron 40, 959–970 (2003).
Chen, S., Wang, J., Zhou, L., George, M. S. & Siegelbaum, S. A. Voltage Sensor Movement and cAMP Binding Allosterically Regulate an Inherently Voltage-independent Closed−Open Transition in HCN Channels. J. Gen. Physiol. 129, 175–188 (2007).
Kunzmann, P. et al. Anisotropic Network Analysis of Open/Closed HCN4 Channel Advocates Asymmetric Subunit Cooperativity in cAMP Modulation of Gating. J. Chem. Inf. Model. 64, 4727–4738 (2024).
Popovych, N., Sun, S., Ebright, R. H. & Kalodimos, C. G. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 13, 831–838 (2006).
Li, L., Uversky, V. N., Dunker, A. K. & Meroueh, S. O. A Computational Investigation of Allostery in the Catabolite Activator Protein. J. Am. Chem. Soc. 129, 15668–15676 (2007).
Capdevila, D. A., Braymer, J. J., Edmonds, K. A., Wu, H. & Giedroc, D. P. Entropy redistribution controls allostery in a metalloregulatory protein. Proc. Natl. Acad. Sci. USA 114, 4424–4429 (2017).
Wang, Y. et al. Globally correlated conformational entropy underlies positive and negative cooperativity in a kinase’s enzymatic cycle. Nat. Commun. 10, 799 (2019).
Wankowicz, S. A. & Fraser, J. S. Advances in uncovering the mechanisms of macromolecular conformational entropy. Nat. Chem. Biol. 21, 623–634 (2025).
Sunkara, M. R., Schwabe, T., Ehrlich, G., Kusch, J. & Benndorf, K. All four subunits of HCN2 channels contribute to the activation gating in an additive but intricate manner. J. Gen. Physiol. 150, 1261–1271 (2018).
Xia, J. & Accili, E. A. Regulation of sinoatrial funny channels by cyclic nucleotides: From adrenaline and IK2 to direct binding of ligands to protein subunits. Prog. Biophysics Mol. Biol. 166, 12–21 (2021).
Zhou, L. & Siegelbaum, S. A. Gating of HCN channels by cyclic nucleotides: residue contacts that underlie ligand binding, selectivity, and efficacy. Structure 15, 655–670 (2007).
Kusch, J. et al. How subunits cooperate in cAMP-induced activation of homotetrameric HCN2 channels. Nat. Chem. Biol. 8, 162–169 (2011).
Hines, K. E., Middendorf, T. R. & Aldrich, R. W. Determination of parameter identifiability in nonlinear biophysical models: A Bayesian approach. J. Gen. Physiol. 143, 401–416 (2014).
Idikuda, V. et al. Lipid bilayers determine the allostery but not intrinsic affinity of cAMP binding to pacemaker channels. bioRxiv 2024.12.23.630133 https://doi.org/10.1101/2024.12.23.630133 (2025).
Porro, A. et al. A high affinity switch for cAMP in the HCN pacemaker channels. Nat. Commun. 15, 843 (2024).
Porro, A. et al. The HCN domain couples voltage gating and cAMP response in hyperpolarization-activated cyclic nucleotide-gated channels. Elife 8, e49672 (2019).
Macri, V., Nazzari, H., McDonald, E. & Accili, E. A. Alanine scanning of the S6 segment reveals a unique and cAMP-sensitive association between the pore and voltage-dependent opening in HCN channels. J. Biol. Chem. 284, 15659–15667 (2009).
Cooper, A. & Dryden, D. T. F. Allostery without conformational change: A plausible model. Eur. Biophys. J. 11, 103–109 (1984).
Cui, Q. & Karplus, M. Allostery and cooperativity revisited. Protein Sci. 17, 1295–1307 (2008).
Kalodimos, C. G. Protein function and allostery: a dynamic relationship. Ann. N. Y. Acad. Sci. 1260, 81–86 (2012).
DeBerg, H. A., Brzovic, P. S., Flynn, G. E., Zagotta, W. N. & Stoll, S. Structure and Energetics of Allosteric Regulation of HCN2 Ion Channels by Cyclic Nucleotides. J. Biol. Chem. 291, 371–381 (2016).
Costa, F., Guardiani, C. & Giacomello, A. Molecular dynamics simulations suggest possible activation and deactivation pathways in the hERG channel. Commun. Biol. 5, 165 (2022).
DiFrancesco, J. C. et al. Recessive Loss-of-Function Mutation in the Pacemaker HCN2 Channel Causing Increased Neuronal Excitability in a Patient with Idiopathic Generalized Epilepsy. J. Neurosci. 31, 17327–17337 (2011).
Guex, N. & Peitsch, M. C. SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling. electrophoresis 18, 2714–2723 (1997).
Unni, S. et al. Web servers and services for electrostatics calculations with APBS and PDB2PQR. J. comput. Chem. 32, 1488–1491 (2011).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput Chem. 29, 1859–1865 (2008).
Wu, E. L. et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput Chem. 35, 1997–2004 (2014).
Mark, P. & Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 105, 9954–9960 (2001).
Phillips, J. C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 044130 (2020).
Klauda, J. B. et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. methods 14, 71–73 (2017).
Martyna, G. J., Tobias, D. J. & Klein, M. L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101, 4177–4189 (1994).
Feller, S. E., Zhang, Y., Pastor, R. W. & Brooks, B. R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 103, 4613–4621 (1995).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Yao, X.-Q., Momin, M. & Hamelberg, D. Establishing a Framework of Using Residue–Residue Interactions in Protein Difference Network Analysis. J. Chem. Inf. Modeling 59, 3222–3228 (2019).
Gray, R. M. Entropy and Information Theory. (Springer Science & Business Media, 2011).
Gross, C. et al. Mechanical transduction of cytoplasmic-to-transmembrane-domain movements in a hyperpolarization-activated cyclic nucleotide–gated cation channel. J. Biol. Chem. 293, 12908–12918 (2018).
Dai, G., Aman, T. K., DiMaio, F. & Zagotta, W. N. Electromechanical coupling mechanism for activation and inactivation of an HCN channel. Nat. Commun. 12, 2802 (2021).
Nieves-Cordones, M. & Gaillard, I. Involvement of the S4-S5 linker and the C-linker domain regions to voltage-gating in plant Shaker channels: Comparison with animal HCN and Kv channels. Plant Signal. Behav. 9, e972892 (2014).
Hagberg, A. A., Schult, D. A. & Swart, P. J. Exploring Network Structure, Dynamics, and Function using NetworkX. in 11–15 (Pasadena, California, 2008). https://doi.org/10.25080/TCWV9851.
Thon, S., Schulz, E., Kusch, J. & Benndorf, K. Conformational Flip of Nonactivated HCN2 Channel Subunits Evoked by Cyclic Nucleotides. Biophysical J. 109, 2268–2276 (2015).
Acknowledgements
F.C. acknowledges EuroHPC for accessing to MareNostrum at Barcelona Supercomputing Center, Spain. E.A.A. acknowledges funding from the Canadian Institutes of Health Research and National Sciences and Engineering Research Council of Canada.
Author information
Authors and Affiliations
Contributions
F.C. designed the research, performed the research (simulations) and wrote the paper; L.C.T.N. designed and performed the research (experiments), carried out the analysis and helped to edit the paper; S.S.C. designed and performed the research (experiments), carried out the analysis and helped to edit paper; F.V.P. designed the research and reviewed the paper; E.A.A. designed the research, carried out some analysis and wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Baron Chanda, Marcel P. Goldschen-Ohm and Chen Fan for their contribution to the peer review of this work. Primary Handling Editors: Janesh Kumar and Laura Rodríguez Pérez. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Costa, F., Ng, L.C.T., Chow, S.S. et al. An inter-subunit path is required for entropically-driven and negatively cooperative binding of cyclic nucleotides in the HCN2 channel. Commun Biol 9, 362 (2026). https://doi.org/10.1038/s42003-026-09626-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-026-09626-7
