
Abstract
The global rise in addictive drug use has become a major public health concern, increasingly linked to the development of depression-a psychiatric disorder affecting over 300 million people worldwide. Emerging evidence indicates that chronic exposure to addictive substances can induce or exacerbate depressive symptoms through reward circuitry alterations, neuroinflammation, altered hypothalamic pituitary adrenal axis function, and mitochondrial dysfunction, thereby establishing a neurobiological milieu that favors drug induced depressive states. This review summarizes how epigenetic mechanisms, such as DNA methylation, histone modifications, non-coding RNAs and RNA modifications, convey the enduring effects of addictive drugs on gene expression related to mood regulation, stress response, and neural plasticity, offering potential biomarkers for early detection and individualized treatment. We further outline major technical constraints and unresolved mechanistic issues and highlight emerging approaches such as cell type resolved epigenomic mapping and locus specific epigenome editing that may enable more causal and translational insights. By integrating neurobiological and epigenetic perspectives, this review offers a clearer framework for understanding addiction and depression comorbidity and points to promising therapeutic opportunities.
Similar content being viewed by others

Introduction
Addiction is a chronic, relapsing systemic disorder characterized by persistent drug craving and compulsive use, despite the emergence of serious adverse consequences. Its high prevalence poses significant social burdens, severely affecting physical and mental health, with rising trends in drug abuse1,2,3,4. Many individuals use addictive substances over extended periods for recreational or medical purposes, with some developing compulsive behaviors. This group is considered susceptible to addiction1,4,5,6. Key brain regions implicated in drug addiction include the ventral tegmental area (VTA), nucleus accumbens (NAc), medial prefrontal cortex (mPFC), hippocampus, among others7. Importantly, prolonged or excessive substance exposure increases vulnerability to negative affective states, including depression, yet the pathophysiological mechanisms bridging addiction and depression remain incompletely understood. This gap highlights the need for mechanistic frameworks that explain how chronic drug exposure interacts with stress and mood-related circuits.
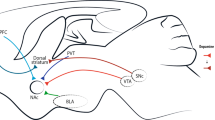
Depression is a complex mental disorder characterized by low mood, anhedonia, worthlessness, suicidal thoughts, psychomotor delay or agitation, and disturbed sleep patterns8,9,10. The World Health Organization predicts that depression will become the leading global health burden by 2030, incurring substantial social and economic costs11. The prevalence of depression is increasing year by year, and it is increasing faster in adolescents than in adults. There is a worrying rise in depression among teenagers, who are 30 times more likely to commit suicide12. The pathogenesis of depression involves environmental, genetic and epigenetic factors, and the etiology is complex and disabling5,13. Both acute and chronic use of addictive substances—such as cocaine, alcohol, and heroin-can result in drug-induced depression5,9,12,14. However, most existing studies tend to address addiction and depression related mechanisms separately, resulting in an insufficiently integrated understanding of the biological basis underlying their comorbidity (Fig. 1).

a By affecting the function of the hypothalamic-pituitary-adrenal axis, it leads to its hypofunction, thereby causing the occurrence of depression. b In the body of patients with depression, the release of neurotransmitters and energy from synapses is reduced, thereby affecting the neural plasticity of synapses. c The elevated levels of pro-inflammatory cytokines in the blood of patients with depression animal models, along with the decreased permeability of the blood-brain barrier, may facilitate monocyte infiltration into the brain, thereby leading to increased inflammation and microglial activation. Stimulate the peripheral immune system and a series of immune cells to generate immune responses, thereby altering the growth and development of neurons. Dopamine(DA), 5-hydroxy tryptamine(5-HT), brain-derived neurotrophic factor(BDNF), adenosine triphosphate(ATP), interleukin-1beta(IL-1β), interleukin-6(IL-6), interleukin-1(IL-10), tumor necrosis factor-α(TNF-α). (Figure was created by Adobe Illustrator 2023).
Epigenetics involves the reversible regulation of gene expression without changes to the DNA sequence. These mechanisms are vital for proper cell development and differentiation15. Epigenetic mechanisms—such as DNA methylation, histone modifications, noncoding RNAs, and RNA editing-contribute to key neural processes, including learning, memory, and emotional regulation, and are increasingly recognized as central to both depression and drug addiction16,17,18. Because epigenetic pathways translate environmental exposures into enduring neural alterations, they offer a unifying framework for understanding the shared biology of addiction and depression.
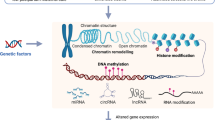
Recent findings indicate that addictive drugs can induce persistent epigenetic remodeling in reward and stress-related pathways, changes that may contribute to the emotional dysregulation frequently observed during and after substance use9,19. Understanding these shared mechanisms may help clarify inconsistencies across studies and reveal new therapeutic opportunities. In this article, we review current evidence on the relationship between depression and drug abuse from an epigenetic perspective, aiming to provide a more integrated understanding of their comorbidity and potential directions for clinical intervention (Fig. 2).

Summarize the common changes in the body caused by addictive drug abuse and depression the mechanism mainly involves the pathophysiological changes resulting from epigenetic modifications. Addictive drugs, such as cannabis, nicotine, and cocaine, as well as depression, affect brain pathways and physiological regulatory processes through various epigenetic mechanisms, including histone modification, DNA modification and noncoding RNA. (Figure was created by Adobe Illustrator 2023).
Addictive drug abuse and depression
The abuse of addictive drugs can lead to serious neurotoxicity, then lead to serious brain damage and mental disorders20. Studies indicate that individuals abusing addictive drugs exhibit elevated depression scale scores, correlating with both frequency of use and injection frequency. Notably, injecting users report more suicide attempts and suicidal ideation20,21,22,23. Treating depression resulting from substance abuse requires a thorough understanding of the underlying mechanisms and interactions, along with the implementation of varied treatment approaches. Although a wide range of neurobiological effects have been described, these mechanisms are often presented independently in the literature, making it difficult to understand how drug exposure produces persistent depressive vulnerability. Integrating these findings into a coherent framework is therefore essential for clarifying the shared pathways linking addiction and depression. This article summarizes amphetamines, opioids, cannabis, and other drugs to elucidate these mechanisms and offer insights for clinical drug use and development.
Amphetamines and their analogs and depression
Amphetamine-type substances (ATS), such as amphetamine, methamphetamine (METH), and methylene dioxy methamphetamine (MDMA), are synthetic central nervous system (CNS) stimulants widely abused worldwide and commonly prescribed for attention deficit hyperactivity disorder20,24. Studies indicate that illegal amphetamine users have a 5.28 times higher risk of developing psychosis compared to nonusers22. Amphetamine-induced depression may be associated with changes in the monoaminergic and glutamatergic systems, the hypothalamic-pituitary-adrenal (HPA) axis, chronic stress, increased inflammation, decreased levels of brain-derived neurotrophic factor (BDNF), and impacts on synaptic function, neuroplasticity, autophagy, and neurogenesis. Research indicates that the dopamine (DA) surge from amphetamine abuse can cause oxidative stress, neuronal death, and morphological changes in the hippocampus, particularly reducing neuron density in the CA1 region and Zn²⁺ concentration25,26,27. Prolonged or repeated exposure to MDMA can lead to long-term abnormalities in the serotonergic system, including decreased levels of serotonin (5-HT) and its major metabolites in the thalamus, striatum, and cortex. 5-hydroxyindoleacetic acid, 5-HT transporter density, and tryptophan hydroxylase (TPH) activity are also decreased21,28,29. DA, 5-HT, BDNF, and Tyrosine Kinase receptor B in the striatum and hippocampus were all reduced over the next 75 days after METH injection in rats30. Amphetamines induced the activation of the endoplasmic reticulum (ER) stress signaling pathway in hippocampal neurons, accompanied by the destruction of ER function and the change of calcium homeostasis31,32. Overactive neuronal autophagy depletes BDNF, impairs adult hippocampal neurogenesis and induces depressive behavior in experimental animals33,34. Both protein kinase B (AKT) and DNA damage-inducing transcript 4 (DDIT4) are mammalian target of rapamycin (mTOR) regulators, which play an important role in the regulation of autophagy35,36. Studies have shown that exposure to METH can increase the expression of DDIT4, and also up-regulate the expression of Beclin-1 and LC3-II35,36,37,38. These changes suggest that the autophagy process in the brain is altered during the administration of METH. These findings suggest that METH may influence cell function and neuronal health by regulating autophagy processes. Amphetamines can also increase the release of excess glutamate from nerve endings of the CNS into the cortex, resulting in an imbalance of cortical signaling. The HPA axis is involved in many psychopathological processes, such as depression, and long-term amphetamine abuse can damage the HPA axis, leading to impaired negative feedback, adrenal enlargement, and hypercortisolemia39,40. Taken together, ATS-induced depressive symptoms arise from the convergence of monoaminergic depletion, ER-stress-related autophagy, glutamatergic imbalance, and HPA-axis disruption. These interacting pathways impair synaptic plasticity and stress resilience, helping explain the persistent emotional vulnerability seen in chronic ATS users. Because key elements of these processes-such as BDNF–TrkB signaling and mTOR-dependent autophagy-are epigenetically regulated41,42, they may represent promising targets for treating ATS-related depression.
Synthetic cathinone (SC), the second most common new psychoactive substance, is a β-ketone analog resembling amphetamine and is often called “natural amphetamine” due to its similar chemical structure and effects43,44. There are many SCs, such as 3, 4-methylenedioxypyrrolidone (MDPV), n-ethylpentanone (NEP) and mephedrone. New SC continue to increase, acting primarily by increasing synaptic levels of DA, 5-HT, and norepinephrine (NA) in the brain45. High doses of methcathinone can lead to severe psychiatric symptoms, such as depression. After exposure to NEP, mice may exhibit symptoms of anorexia and depression, with decreased levels of 5-HT and NA in the striatum and mPFC. The tail suspension test indicates that withdrawal from NEP increases the immobility time in mice, suggesting that drug withdrawal can trigger depression-like symptoms46,47. Reduced expression of tyrosine hydroxylase and TPH, biochemical markers of neuronal integrity, was observed after mephedrone administration, along with downregulation of dopamine D2 receptors in the striatum, indicating the presence of damaged nerve endings48,49. Exposure to mephedrone, methylone, and MDPV caused oxidative stress in neuronal cell lines, including dopaminergic neuroblastoma and mouse hippocampal neurons. This included increased reactive oxygen and nitrogen species, reduced glutathione, and elevated oxidized glutathione. Additionally, these substances depleted ATP levels, impaired mitochondrial function, reduced cell proliferation, and induced autophagy, necrosis, and apoptosis50. The mechanism of the toxic action of cathinone is still unclear, and the mechanism of drug-induced depression caused by cathinone abuse needs further study. Overall, SCs appear to induce depression-like effects through overlapping mechanisms—including monoamine imbalance, oxidative stress, and mitochondrial dysfunction—that may converge on persistent epigenetic alterations.
Cannabis and depression
Approximately 3.8% of the global population has used cannabis, primarily due to cannabinoids, which are classified as endocannabinoids, phytocannabinoids, and synthetic cannabinoids. The main active component of cannabis is Δ−9 tetrahydrocannabinol (THC), which causes most psychoactive effects and addiction. Data show that adolescent marijuana use is associated with an increased risk of major depression and suicide in early adulthood51,52. Adolescent cannabis use can lead to anhedonia and anxiety symptoms in adulthood, along with decreased 5-HT levels and increased NA levels53. In addition, adolescents exposed to cannabis over a long period of time have a diminished response of their DA neurons to cannabis, while developing persistent cross-resistance to other substances, such as morphine, cocaine, and amphetamines54. This may be one of the mechanisms by which cannabis abuse triggers depression. The study found that regular marijuana use was associated with decreased frontal white matter integrity and was regulated by the fatty acid amide hydrolase genotype. This decline in white matter integrity is associated with negative emotions and greater symptoms of apathy among cannabis users55. Because cannabinoid receptor 1 (CB1) activation results in inhibition of selective adenylate cyclase activity, THC binding to CB1 receptors may affect perception, memory, and movement, and may trigger depression. Simultaneously, the hippocampus is rich in CB1 receptors. THC and CB1 agonists can reduce the release of presynaptic neurotransmitters and disrupt the long-term plasticity of hippocampal synapses52,56. Some studies suggest that cannabis may have antidepressant effects, but this remains controversial57. Further research is needed to clarify the line between its pharmacological and toxicological effects. As some countries move towards legalizing marijuana, additional studies will help establish a foundation for responsible and informed cannabis use.
Opioids and depression
Prescription opioids are frequently abused, with nearly 30% of chronic pain patients misusing them and about 12% developing opioid use disorder58. While effective for pain relief, long-term opioid use can lead to adverse effects, including immunosuppression and neurohormonal deficiencies59,60. Opioid use is often comorbid with depression and anxiety, affecting around 7.8 million adults with mental disorders61. Mendelian randomization analysis suggests a potential genetic link between increased opioid use and the risk of major depressive disorder (MDD) and anxiety-related disorders62. MTT assay found that fentanyl and remifentanil had cytotoxic effects on neuronal cell lines, which were also found in oxycodone, morphine, tapentadol, and tramadol63,64. Studies have found that fentanyl causes inflammation and increases the levels of total mercaptan, acetylcholine, butyrylcholine, and paraoxonase-1, leading to neuronal cell death, and the level of TNF-α in neuronal cells increased by 2.3 times after fentanyl administration65. After tramadol exposure, the expression of BDNF decreased, and the expression of apoptosis and autophagy genes increased in the striatum and hippocampus66. Overall, opioid-related depressive symptoms appear to result from neuroinflammation, mitochondrial dysfunction, and impaired neurotrophic signaling. These pathways may also interact with stress-related epigenetic regulators, providing a mechanistic basis for the persistent emotional dysregulation seen in opioid misuse.
Cocaine and depression
Cocaine is a natural stimulant derived from the coca plant, used both recreationally and as a local anesthetic67. Its abuse poses significant public health issues globally, with approximately 0.4% of individuals aged 15–64 reporting use. Cocaine affects the CNS, leading to various physical and psychological issues, including neurodegeneration and seizures. Additionally, repeated use and withdrawal can result in persistent overactivation of the HPA axis, causing elevated cortisol levels and disrupted negative feedback mechanisms68,69,70,71. Long-term withdrawal from cocaine can cause emotional symptoms similar to mental and stress-related illnesses72. The study found an increase in anxiety and depression-like behavior in male rats 24 h after withdrawal. Cocaine reduces motor activity in the rodents’ brains, reduces DA levels and increases γ-aminobutyric acid levels. Acute exposure to cocaine can cause abnormal behavior and damage of the dopaminergic system in zebrafish73. FK506 binding protein 5 (FKBP5) is an auxiliary companion of the glucocorticoid receptor (GR) complex that regulates the sensitivity of GR to cortisol and has been implicated in the pathology of human psychiatric disorders, such as anxiety, depression, and post-traumatic stress disorder. It was found that the mRNA expression of FKBP5 increased significantly in the early stage of chronic cocaine withdrawal74. Current evidence indicates that cocaine withdrawal alters stress-glucocorticoid signaling and FKBP5-related transcriptional pathways, which may partially underlie its depressive consequences. Although causal relationships remain to be fully established, these mechanisms provide a preliminary basis for exploring HPA-axis modulation as a therapeutic approach (Fig. 3).

Addictive drug exposure induces widespread neuroadaptations across multiple brain regions, altering glutamatergic, dopaminergic, and GABAergic signaling and contributing to vulnerability to depressive symptoms. a Depictive summary of the classical neural circuits involved in major depression and stress-related depressive phenotypes, including dysregulation within monoaminergic pathways, neuroinflammatory signaling, and reduced neurotrophic support (e.g., BDNF–TrkB pathways); b schematic representation of the core reward- and motivation-related neurocircuitry underlying addiction. This panel is not intended to represent depression-specific circuits, but rather the canonical pathways involved in reinforcement learning and drug seeking. These addiction circuits partially overlap with depressive circuits (e.g., PFC–NAc), providing a mechanistic basis for comorbidity, while also containing addiction-specific nodes (e.g., LH, VP). Norepinephrine (NA), interleukin-8 (IL-8), tropomyosin receptor kinase B (TrkB), nuclear factor kappa-B (NF-κB), hypothalamus (Hyp), amygdala (Amy), lateral hypothalamic area (LHA), ventral tegmental area (VTA), lateral habenula (LHb), ventral pallidum (VP), basolateral amygdala (BLA), lateral hypothalamus (LH), mediodorsal thalamus (MDT). (Figure created with Adobe Illustrator 2023).
Addictive drug abuse and epigenetics
Beyond these neurobiological outcomes, addictive drugs also exert regulatory effects at the epigenetic level. Repeated exposure can induce epigenetic adaptations within reward-related circuits, contributing to persistent functional changes that support long-term drug-seeking behaviors75. Epigenetic mechanisms provide a regulatory framework that promotes the plasticity required for addiction and persists in the long term after the cessation of drug use76. Studies indicate that repeated drug use can cause histone modification and DNA methylation changes in various brain regions77. Such findings highlight epigenetic regulation as a promising avenue for identifying new therapeutic targets for addiction.
Addictive drug abuse and DNA methylation
DNA methylation was first discovered in CpG regions, known as CpG islands, which are roughly 1000 base-pair long stretches of DNA with high CpG density. Most gene promoters are located within the CpG island78. DNA methylation is a dynamic and reversible process catalyzed by enzymes, such as DNA methyltransferase (DNMT), which covalently adds methyl groups to site 5 of the cytosine nucleotide to produce 5-methylcytosine76,79. This methylation of cytosine regulates transcriptional plasticity of the mammalian genome and is the most stable and well-studied epigenetic modification known to date15. Intermediates of methylation and demethylation may play important roles in disease-related transcriptional plasticity76,80,81.
Genome-wide DNA methylation tests reveal altered DNA methylation status in the blood of drug-addicted patients82. Genome-wide analysis found that changes in different hydroxymethylation regions in the brain and increased expression of specific potassium channels may inhibit METH smoking behavior83. METH injection induced differential changes of DNMT1 expression in the NAc region and dorsal striatum of two strains of rats. The expression of DNMT1 in the striatum and NAc was increased in Fisher 344 rats after acute and repeated METH injections, while the corresponding expression was decreased in Lewis rats84. Long-term use of METH inhibits CpG demethylation in the synuclein alpha promoter region and leads to increased expression of alpha-synuclein in striate neurons. Notably, this demethylation persists even during withdrawal from meth use85. Repeated injection of cocaine dramatically reduces levels of the methyltransferase DNMT3a, which continues to increase after cocaine withdrawal, i.e., the increase in DNMT3a may be an epigenetic mechanism that is particularly important for the withdrawal phase of addiction86. DNA hydroxymethylation markers have also been studied after taking cocaine and METH. Overall levels of 5-hydroxymethylcytosine increased after repeated administration of cocaine77,87. Restricting access to cocaine was found to reduce DNA hydroxymethylation levels in the Homer2 promoter region of rats87. Together, the evidence indicates that addictive drugs alter DNA methylation across stress- and reward-related circuits, leading to lasting changes in gene regulation and behavior. Although these methylation shifts are reversible and may hold therapeutic value, their direction and specificity differ across brain regions and cell types, underscoring substantial mechanistic heterogeneity (Table 1).
Addictive drug abuse and histone modification
Histone modification is a key mechanism in epigenetics, particularly the acetylation of lysine residues in histone tails. Gene expression depends on DNA accessibility, as DNA is tightly packaged in chromatin, wrapping around histone octamers composed of H2A, H2B, H3, and H4. The N-terminal “tails” of these histones undergo various modifications-acetylation, phosphorylation, methylation, and ubiquitination—that affect their interaction with DNA, thereby altering chromatin structure and regulating gene expression15,88.
Addictive drug abuse and histone acetylation
Histone acetylation is a dynamic and reversible process occurring on all core histones, catalyzed by histone acetyltransferases and reversed by histone deacetylases (HDACs). It is often associated with an “open” higher-order chromatin structure, increasing nucleosome spacing and allowing easier access for transcription factors and transcription machinery, thereby promoting transcriptional activation76,89,90.
Acute or chronic exposure to psychostimulants, alcohol, or nicotine can elevate total cellular acetylation levels of histones H3 and H4 in the NAc, while reducing HDAC enzymatic activity and disrupting its cellular localization. Chronic cocaine exposure enhances the expression of acetylases in the NAc but decreases the genomic accessibility of class III HDACs, particularly at gene promoters. This limits acetylases’ ability to lower histone acetylation at crucial gene loci91,92,93,94,95. Repeated opioid use increases H3 acetylation in the mesolimbic DA system, notably at H3K9, H3K14, H3K18, and H3K27. Elevated H3K27ac levels were observed in the striatum of humans and rats using heroin, highlighting its importance in opioid dependence. Additionally, the overall high acetylation of H3 in heroin users correlates with the duration of use, resulting from altered functional balance of HAT and HDAC due to the drug75,96,97,98,99,100,101,102. Long-term METH use increases H3 acetylation in the brain, decreasing H3K9ac and H3K18ac while increasing H4K5 and H4K8 acetylation over time. METH use also lowers mRNA levels of HDAC3, HDAC8, and HDAC11, and reduces HDAC1 protein expression in the NAc, while increasing HDAC2 protein levels3,80,103,104,105. Chronic METH exposure reduced acetylated histone H4 levels at the GluA1, GluA2, and GluN1 promoters, while increasing HDAC2 and SIN3A binding at GluA1 and GluA2, suggesting suppressed glutamate receptor expression through epigenetic mechanisms80. In conclusion, addictive drugs remodel histone acetylation in a region- and residue-specific manner, promoting chromatin accessibility and transcription of plasticity-related genes. However, the direction and magnitude of these acetylation changes vary across drugs and experimental paradigms, highlighting the need for more standardized and mechanistically focused investigations.
Addictive drug abuse and histone dopaminination
Histone H3-glutamine 5-dopaminization (H3Q5dop) plays a key role in cocaine induction of midbrain transcriptional plasticity. Researchers collected VTA tissues at different time points after the self-administration of cocaine intravenously in rats. In the VTAs of animals exposed to cocaine for a long time, the level of H3Q5dop was significantly downregulated106. The level of H3Q5dop in NAc significantly increased during the acute withdrawal period and the long-term withdrawal period, while no similar changes were observed in mPFC. This accumulation relies on voluntary drug intake, and natural rewards cannot induce it. By injecting the dominant negative mutant H3.3Q5A, which inhibits the formation of H3Q5dopinto NAc, the cocaine seeking behavior after withdrawal can be significantly reduced. On the contrary, the inhibition of H3Q5dop in mPFC has no effect on behavior, indicating that its effect is region-specific. RNA sequencing revealed that the inhibition of H3Q5dop in NAc led to the differential expression of 112 genes, involving DA metabolism, calcium homeostasis and endogenous opioid pathways. The general downregulation of these genes suggests that H3Q5dop functions by maintaining the cocaine-induced pro-relapse transcriptional program107. Only 9 genes in the mPFC were affected, with no overlap in the NAc. The weakening of H3Q5dop during withdrawal impairs gene expression related to neuronal signaling and dopaminergic function. Expressing the dominant negative mutant H3.3Q5A in the VTA significantly reduced seeking behavior after heroin withdrawal, with behavioral tests showing over a 50% reduction in active responses, highlighting its role in reuptake susceptibility. RNA sequencing revealed that about 50% of gene expression changes from heroin withdrawal (including opioid pathways and DA signaling) could be reversed by inhibiting H3Q5dop. Notably, gene expression changes induced by heroin and cocaine in the VTA overlap significantly, indicating that different drugs regulate similar pathways through H3Q5dop in opposing directions108. Together, current evidence indicates that addictive drugs induce stimulus- and region-specific dopaminylation of histone H3, shaping transcriptional programs involved in reward processing and drug-seeking behavior. However, H3Q5 dopaminylation findings remain heterogeneous across drugs and experimental paradigms, and, notably, no studies to date have established a direct role for this modification in depressive pathology (Table 2).
Addictive drug abuse and noncoding RNA
In recent years, ncRNAs have gained attention for their role in various biological processes and post-transcriptional regulation. NcRNAs include microRNA (miRNA), circular RNA (circRNA), long noncoding RNAs (lncRNA), and other undiscovered small regulatory RNAs. Both miRNA and siRNA inhibit translation and promote mRNA degradation by binding to transcripts76.
MiRNA is a small noncoding RNA molecule, 21–22 nucleotides long, that promotes mRNA degradation by inhibiting translation or binding semi-complementarily to the 3’ untranslated region (UTR), thereby regulating post-transcriptional gene expression109,110. Studies indicate that miRNA regulates gene networks involved in synaptic plasticity. Chronic drug exposure can cause adverse changes in neural networks, particularly affecting synaptic plasticity in the mesocortical limbic DA system, which may contribute to compulsive drug behavior. Research on the plasma miRNA of METH use disorder (MUD) patients revealed that miR-181a, miR-15b, miR-let-7e, and miR-let-7d could serve as potential biomarkers for MUD. Additionally, miR-181a was found to directly negatively regulate the expression of the human glutamate receptor gene GRIA2 in MUD111,112. The study found 78 significantly different miRNAs in the VTA of rats with long-term METH use, of which 71 miRNAs were down-regulated. MiR-26a, miR-29a/b/c, miR-222 and miR-383 are expected to bind to the 3’UTR of Dnmt3B, affecting the expression of Dnmt3B and the vulnerability to addiction113. Cocaine exposure boosts miR-212 expression in the dorsal striatum, which is linked to reduced cocaine intake. This suggests that increased miR-212 acts as a compensatory mechanism to diminish cocaine’s motivational effects114. MiR-495 was rapidly down-regulated after acute cocaine injection. MiR-495 was found in the cocaine model to directly target the 30 UTR of multiple genes known to be increased in substance use disorder, such as BDNF and calmodulin-dependent protein kinase II alpha115. METH and cocaine reduce novel-m009C expression in the NAc via D1R activation. Blocking D1R prevents both novel-m009C suppression and METH-induced sensitization. Conversely, NAc overexpression of novel-m009C diminishes METH reward and lowers Grin1 expression, an effect reversed by NMDA receptor activation. These findings indicate that D1R-dependent repression of novel-m009C relieves inhibition of Grin1, enhancing NMDAR signaling to promote METH reward109. In conclusion, miRNAs play a key role in drug addiction by modulating synaptic plasticity and gene-regulatory programs across reward and stress circuits.
LncRNA is an endogenous noncoding RNA longer than 200 nucleotides, involved in various biological functions by interacting with protein partners. They can also engage with chromatin modification complexes to target specific genomic regions110,116,117,118,119. In the study, high throughput chain-specific complementary DNA sequencing was used to detect the changes of lncRNA expression profile in METH-sensitized mice NAc. In METH sensitized mice, a few lncRNAs were up-regulated, but most lncRNAs were down-regulated. These lncRNAs that react to METH have been identified, but more experiments are needed to study the unique function and precise regulatory mechanism of each candidate lncRNA120.
CircRNA is a ncRNA formed by reverse splicing and covalent fusion, resistant to RNA exonucleases. It inhibits miRNAs and regulates gene expression by absorbing miRNA, thus creating a functional circRNA-miRNA-mRNA regulatory network121,122. The study aimed to screen circRNA expression and analyze its potential functions by culturing primary cortical neurons and using high-throughput RNA filtration. It identified circRNA Homer1 and circRNA Tlk1 as highly correlated with addiction. Homer1 is upregulated in the NAc, VTA, and hippocampus, while Tlk1 is downregulated only in the VTA. Additionally, low circHIPK2 expression significantly inhibited METH-induced astrocyte activation by targeting miR124 and SIGMAR1. CircTmeff-1 promotes morphine craving by sponging miR-541/miR-6934 in the NAc123,124. CircRNA Homer1 may inhibit miR-101-3p and play an important role in the mechanism of meth addiction125. The study found that high concentrations of circ-0015891 blocked miR-129-1-3p during METH treatment, negatively affecting dopaminergic cells. A total of 195 differentially expressed circRNAs were identified in METH-treated mice compared to saline-treated mice, with 88 upregulated and 107 downregulated122. The research of circRNA in the field of addictive drug abuse urgently needs to be strengthened and further elucidated by more studies (Table 3).
Together, current evidence shows that addictive drugs dysregulate multiple classes of ncRNAs—including miRNAs, lncRNAs, and circRNAs—in pathways that control synaptic plasticity, neuroinflammation, and reward-related transcription. However, ncRNA alterations remain highly inconsistent across drugs, brain regions, and experimental paradigms, highlighting the need for more causal, cell-type-specific, and temporally resolved studies (Fig. 4).

DNA methylation and histone modifications both participate in the pathogenesis of addictive drug abuse and depression. Drug abuse and depression-related histone modifications include several types, most prominently acetylation and methylation. Histone acetylation is catalyzed by HATs and reversed by HDACs. Histone methylation is catalyzed by HMTs/KMTs. a DNA methylation. In MDD, aberrant DNA methylation of NR3C1 and FKBP5 was found. Cocaine administration significantly elevated FKBP5 during the early withdrawal phase. Early postpartum alcohol exposure led to persistent hypermethylation of the hippocampal FKBP5 promoter, disrupting HPA axis feedback and thus enhancing susceptibility to depression-like behaviors. In the prefrontal cortex of patients with alcohol use disorder, elevated NR3C1 promoter methylation levels have also been observed; b histone methylation. In addiction research, cocaine enhanced the expression of G9a (an H3K9-specific methyltransferase), leading to increased H3K9me2 in the NAc and enhanced drug reward. Similarly, the antidepressant fluoxetine alleviates depressive behaviors by promoting G9a-mediated restoration of H3K9me2. Both depression and drug addiction are associated with increased H3K4me3. METH reduced the expression of the methyltransferase Mll1 KMT2A and the demethylase Kdm5c in the NAc, leading to elevated H3K4me3 at the Oxtr and Fos promoters and strengthening drug-seeking behavior. In patients with depression—particularly in the prefrontal cortex-elevated H3K4me3 contributes to astrocytic dysfunction, exacerbating depressive symptoms and increasing suicide risk; c histone acetylation. Both addiction and depression exhibit dysregulated histone acetylation. METH exposure and stress-susceptible depressive phenotypes are characterized by reduced H3K9ac. METH and most depressive models show increased HDAC2 activity. The specific mechanisms underlying these changes require further investigation. Histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs/KMTs), major depressive disorder (MDD), FK506 binding protein 5 (FKBP5), nuclear receptor subfamily 3 group C member 1 (NR3C1), nucleus accumbens (NAc), methamphetamine (METH), mixed lineage leukemia 1 (Mll1), lysine-specific methyltransferase 2A (KMT2A), lysine-specific demethylase 5C (Kdm5c), oxytocin receptor (Oxtr). (Figure was created by Adobe Illustrator 2023).
Depression and epigenetics
Building upon the epigenetic alterations described in drug-exposure models, the following section discusses how similar regulatory layers contribute to depression. The symptoms of depression are usually diagnosed by SIGECAPS mnemonic symbols and the DSM-5 criteria, and are classified into MDD, dysthymia and premenstrual irritability disorder, etc126,127. Rodent depression models mainly include olfactory bulb resection models, mother-child separation and early life stress models, learned helplessness models, repeated restraint stress models, chronic unpredictable mild stress (CUMS), social isolation models, chronic social failure stress (CSDS), witnessed failure models, etc.128.
Epigenetic mechanisms increase the risk of depression following adverse life events, integrating genetic and environmental factors. Research highlights the impact of epigenetic changes on the pathophysiology of MDD, particularly in abnormal stress responses, monoamine dysfunction, and neuroinflammation. These mechanisms alter chromatin structure, modulating gene expression related to neuronal plasticity and responses to stress, depression, and antidepressants9,129. This review summarizes the role of epigenetics in depression, focusing on DNA methylation, RNA modification, and histone modification (Fig. 5).

RNA modification, chromatin modification and noncoding RNA: the generation and mechanism of lncRNA, miRNA, and circRNA. m6A methylation is a common internal mRNA modification in patients with depression; chromatin remodeling disorder is a key mechanism in the pathophysiology of depression; LncRNAs transcribed by RNA polymerase II are involved in the pathophysiological processes of depression through multiple mechanisms: chromatin modification, inhibition of transcriptional activation, sponges and scaffolders; mature miRNAs are produced by a series of cleavage processes of nucleases from longer primary transcripts. miRNAs affect NMDAR and D1R and regulate the expression of regulatory factors by targeting the 3’UTR of regulatory factors; there are various types of circRNAs. They can mediate the expression of regulatory factors by adsorbing miRNAs through sponges, mediate depression, and inhibit cell death through the CEBPB/ZC3H4 axis. Fat mass and obesity-associated protein (FTO), the β2 adrenergic receptor (ADRB2), myelocytomatosis viral oncogene homolog (c-MYC), Silent mating type information regulation 2 homolog-1 (SIRT1), Drosha ribonuclease III (DROSHA), digeorge syndrome critical region 8 (DGCR8), transactivation response element RNA-binding protein (TRBP), argonaute (AGO), CCAAT enhancer binding protein beta (CEBPB), zinc finger CCCH-type containing 4 (ZC3H4), N-methyl-D-aspartic acid receptor (NMDAR). (Figure was created by Adobe Illustrator 2023).
DNA methylation and depression
Studies have found that stress-related MDD is associated with DNA methylation changes in multiple human genes. These include glucocorticoid receptor genes, FK506 binding protein 5 and spindle, centromere-associated complex 2, oxytocin receptor, oligodendrocyte genes and BDNF, among ohters130,131. Acute inhibition of stress-induced DNA methylation with DNMT inhibitors (DNMTi) promotes rapid and sustained antidepressant effects associated with enhanced BDNF-TrkB-mTOR signaling in PFC132. It was found that the BICD2 gene in the peripheral blood of MDD patients and depressed mice showed DNA hypermethylation and mRNA downregulation, while the BICD2 gene in the hippocampus of depressed mice showed DNA hypomethylation and mRNA and protein up-regulation, and the BICD2 protein was also up-regulated in the hippocampus of MDD patients after death. Hippocampal BICD2 knockout in CUMS mouse models of depression shows an antidepressant effect, which may be mediated by increased BDNF expression133. A study found that by examining the postmortem brain tissue of suicidal patients with depression, the astrocyte-specific methylation map in the genome showed that glutamate receptor ionotropic kainate 2 and brain-rich guanosine kinase-related protein, which are associated with depressive psychopathology, were less methylated134. DNMT1 levels were low in the frontopolar cortex, while DNMT3B levels were high. The expression of DNMT1 and DNMT3B in the amygdala decreased, while that of DNMT3B in the paraventricular nucleus increased. In other words, DNMT mRNA expression changes occurred in specific cells in the brain tissue of depressed suicide patients135. CUMS enhanced DNA methylation at specific CpG sites on the glial cell line-derived neurotrophic factor (GDNF) promoter of BALB mice. The use of DNMTi within NAc reversed CUMS-induced depression-like behavior and decreased GDNF expression, and DNA methylation and methyl CpG binding protein 2 binding were also increased in stress-resilient C57BL/6J mice136. Collectively, current evidence shows that depression involves locus-specific and cell type-specific alterations in DNA methylation across the HPA-axis, neurotrophic and glutamatergic pathways. Clarifying the causal relevance of these signatures will require longitudinal human studies and targeted epigenetic manipulation in defined neural circuits.
Histone modifications and depression
Histone crotonylation and depression
Histone crotonylation (Kcr) is the introduction of crotonyl groups to the amino acid residues of histone proteins. Crotonylation modification has been confirmed to promote gene expression. After the introduction of negatively charged crotonyl group, the positive charge of histone decreases, and binds to negatively charged DNA more loosely, which is conducive to the binding of transcription factors137.
Chromosome domain Y-like protein (CDYL) (a transcriptional co-suppressor and histone H3K9/27 methyllysine) acts as crotonyl-CoA hydrase and negatively regulates histone Kcr. In the CSDS model, histone Kcr in the anterior limbic cortex region of stress-sensitive mice was significantly decreased, while the expression of CDYL protein continued to increase. CDYL converts crotonyl-CoA to β-hydroxybutyryl-CoA through C-terminal hydrase, reduces crotonylation donors, and thereby inhibits Kcr. The N-terminal chromodomain of CDYL recruits H3K27 trimethyl transferase EZH2, which cooperates to inhibit the transcription of the target gene nerve growth factor inducible (VGF). The downregulation of VGF reduces the formation of dendritic spines, damages synaptic plasticity, and ultimately leads to depressive-like behavior138. Together, these findings highlight histone crotonylation as a stress-sensitive epigenetic mechanism that regulates synaptic plasticity and depressive-like behaviors, primarily via CDYL–EZH2–VGF signaling. Nonetheless, crotonylation studies remain limited, and its region specificity, behavioral causality, and interaction with other histone marks require further clarification.
Histone acetylation and depression
The first evidence of histone acetylation’s role in depression came from studies showing that HDAC inhibitors (HDACi), used alone or with antidepressants, enhanced antidepressant responses in animal models139,140,141. The study found that histone acetylation (H3K14ac) in NAc was temporarily reduced after chronic social defeat stress and then continued to increase with a decrease in HDAC2 levels, as was also observed in postmortem brain tissue NAC of depressed patients. The injection of MS275 (a specific class I HDACi) into NAc was found to have a powerful antidepressant effect, suggesting that histone acetylation plays an adaptive role in the stress response140. HDAC5, a class II HDAC, may play a similar elastic role in NAc. Mice affected by chronic social failure stress showed decreased HDAC5 expression, while imipramine treatment increased HDAC5 expression. Mice lacking HDAC5 exhibit more depression-like behavior after prolonged social failure stress92. Neuronal plasticity and cognitive function are associated with transcriptional changes mediated by HDAC-mediated epigenetic modifications129,142,143. Overexpression of HDAC2 in neurons induces a decrease in dendritic spine density, synaptic number, synaptic plasticity, and memory formation in mice. Long-term use of HDAC inhibitors can improve the number of synapses and learning disabilities. Virus-mediated knockdown of HDAC2 can also restore structural and synaptic plasticity, ultimately improving memory loss associated with neurodegenerative diseases144,145. Together, these data position histone acetylation as a dynamic regulator of stress adaptation and antidepressant responsiveness across multiple brain circuits. However, acetylation patterns vary by cell type, histone residue, and stress paradigm, and the therapeutic specificity of HDAC inhibitors remains an important challenge for clinical translation.
RNA modifications and depression
RNA molecules can create a dynamic and reversible regulatory network through covalent modifications, influencing gene expression by affecting RNA stability, translation efficiency, and post-transcriptional regulation. Over 100 types of RNA modifications have been identified, including N6-methyladenine (m6A) and N1-methyladenine, primarily in rRNA and tRNA146. Recent studies highlight the significant role of RNA modifications, particularly m6A, in the onset and progression of nervous system diseases, with m6A homeostasis being crucial for maintaining brain functional balance147,148,149. Its methylation levels are carefully controlled by a ternary “writing-erasing-reading” regulatory system: writers, also known as methyltransferases, include Wilms tumor 1 associated protein (WTAP), methyltransferase-like 3 (METTL3), and methyltransferase-like 14 (METTL14), which are responsible for installing the m6A modification. The eraser includes several demethylases, such as AlkB homologate 5 (ALKBH5) and fat mass and obesity associated protein (FTO)150. It is noteworthy that this regulatory system has a cross-dialogue with epigenetic mechanisms—m6A modification can target the regulation of mRNA metabolism of genes associated with histone modification, suggesting that there is a collaborative regulatory network between the epigenome and the epigenome151,152.
In patients with MDD and in depressive models, the mRNA levels of FTO and m6A methyltransferases (METTL3, METTL14, and WTAP) are significantly down-regulated. Experiments showed that knocking down FTO in adult hippocampal neurons induced depression-like behavior, while overexpressing FTO reversed such behavior in response to unpredictable chronic mild stress. MeRIP-Seq analysis revealed that FTO knockout increased hippocampal m6A methylation, whereas overexpression decreased it. FTO enhances the stability of ADRB2 mRNA by demethylating its 3’ UTR. Moreover, knocking down YTHDF2, an m6A reading protein, elevated ADRB2 expression. Activation of ADRB2 counteracted the behavioral and synaptic deficits in FTO-deficient mice, with the ADRB2 agonist formoterol alleviating depression-like behavior and restoring dendritic spine density, while an antagonist blocked these effects. ADRB2 activation also promoted SIRT1 expression via up-regulation of c-MYC, influencing hippocampal synaptic plasticity and depression-like symptoms149. mRNA levels of ALKBH5 were elevated in the blood and PFC of MDD patients as well as in mouse models of depression, and selective knocking out ALKBH5 in astrocytes produced antidepressant like behavior. ALKBH5 reduces glutamate uptake by astrocytes by reducing m6A modification of glutamate transporter GLT-1, inhibiting its translation and protein expression. Glutamate homeostasis and synaptic dysfunction ultimately lead to depression-like behavior153. The study found that the plasma circHECW2 level in MDD patients was significantly increased and was positively correlated with depression severity and anxiety symptoms. CircHECW2 regulates Gng4 mRNA stability through WTAP-mediated m⁶A methylation, and GNG4 deletion leads to astrocyte dysfunction, which further affects astrocyte function and depressive phenotype152. CircSTAG1 expression was significantly down-regulated in plasma and whole blood of MDD patients and in hippocampus and peripheral blood of mice with chronic unpredictable stress. And its expression was negatively correlated with the depression scale score of MDD patients. Fatty acid amide hydrolase (FAAH) is an endocannabinoid-degrading enzyme, and its overexpression is related to depression. CircSTAG1 inhibits its nuclear translocation by binding to the m6A demethylase ALKBH5, thereby reducing the m6A demethylation of the target gene FAAH mRNA, and the increase of m6A methylation of FAAH mRNA leads to the decrease of its stability. Ultimately, reduced expression of FAAH protein induces astrocyte damage, leading to depression-like behavior150. In summary, the close relationship between RNA modification and depression not only provides a new perspective on our understanding of the complexity of depression but also opens new paths for future treatment strategies. However, the directionality, cell specificity, and functional hierarchy among different RNA modifications remain incompletely defined, and future studies are needed to determine which modifications are causal drivers versus secondary stress responses.
Noncoding RNAs and depression
Studies have shown that after death, the expression level of miR-1202 in the brain tissues and blood samples of MDD patients decreased significantly154. The expression levels of miR-15, miR-182 and miR-206 were higher than those in healthy people and regulated the BDNF gene155. However, miR-204-5p is downregulated in the ventral prefrontal cortex, activating the JAK2/STAT3 signaling pathway, causing neuronal damage and leading to depressive and anxiety-like behaviors156. In addition, the expression of miR-16-2 in the brains of MDD patients was also significantly decreased157. A study analyzed the correlation between miR-30e ss178077483 and MDD patients and control subjects, and found that miR-30e ss178077483 was significantly positively correlated with MDD158. miRNAs, such as miR-34a, -34b, -34c, -137, and -140-3p, -21-3p, -30d-5p, -330-5p, -378a-5p, -134, -19b-3p have potential roles in the diagnosis of patients with depression159. All the above indicate that multiple types of miRNAs are involved in the pathological process of depression and affect the occurrence and development of the disease.
LncRNA can be involved in the pathogenesis of MDD by regulating neurotrophic factors, affecting the development and function of the CNS, as well as synaptic function160. LncRNA BDNF-AS is associated with supporting neuronal survival and synaptic plasticity161. Magnetic resonance imaging was used to measure the hippocampal volume. The differences in ncRNA BASP-AS1 between MDD patients and healthy controls were analyzed, and it was found that the expression of BASP1-AS1 was higher in MDD patients162. LncRNA XR_351665 can up-regulate the expression of DNMT1 by sponging miR-152-3p, thereby promoting the development of depression induced by chronic pain163. LncRNA NONHSAG045500 activates the cAMP-PKA-CREB pathway, improves the level of 5-HT, reduces the expression of 5-HT transporter, and exerts an inhibitory effect on depression-like behaviors164. The studies indicate that lncRNA is linked to synaptic plasticity and may trigger depressive symptoms by regulating miRNA, making it a potential biomarker for depression.
CircRNAs are ideal biomarkers for the diagnosis of depression165,166. Compared with patients with type 2 diabetes mellitus (T2DM) and depressive T2DM, the study found that 183 his-circRNAs were significantly upregulated and 64 were downregulated in patients with T2DM. Differentially expressed circRNA can serve as an important clue for studying the pathogenesis of depression in patients167. CircRNA is linked to microglial apoptosis. In LPS-induced mouse models, circDYM was found to inhibit microglial apoptosis via the CEBPB/ZC3H4 axis, improving depressive-like behaviors168. Studies have shown that extracellular vesicle-mediated delivery of circDYM can treat depression by inhibiting microglial activation and binding transcription factor TATA-box binding protein-related factor 1 to suppress neuroinflammation169. CircRNAs, as endogenous miR-9 sponges, can target miR-9 to participate in the ubiquitination of HSP90, thereby regulating microglial polarization and reducing depressive-like behaviors170. In conclusion, circRNAs can affect the occurrence of depressive symptoms by influencing the molecular activities of body cells and miRNAs, etc.
Yet ncRNA signatures remain heterogeneous across brain regions, patient populations, and experimental models, and only a minority have been validated functionally, highlighting the need for more mechanistic and clinically oriented studies (Table 4).
Shared epigenetic mechanisms in depression-drug addiction comorbidity
To better integrate these findings, we introduce a unifying three-axis framework, stress-response regulation, reward-circuit remodeling, and synaptic plasticity, which provides a coherent structure for understanding how epigenetic alterations converge across depression, drug addiction, and drug addiction-induced depression. This framework positions the mechanisms reviewed above not as isolated pathways, but as interconnected processes that jointly shape vulnerability and behavioral outcomes (Fig. 6).

a miRNAs. a1 Cocaine-induced upregulation of miR-212/132 in the striatum modulates MeCP2 and BDNF signaling, constituting a shared regulatory node implicated in both drug reinforcement and depressive vulnerability. a2 Cocaine also alters miR-16, increasing SERT expression and 5-HT reuptake, a pathway linked to serotonergic dysfunction in addiction and depression; b LncRNAs. b1 Malat1 is involved in both drug exposure and stress-related phenotypes and regulates transcriptional programs affecting neuronal excitability and synaptic plasticity. b2 NEAT1 is downregulated by METH but upregulated by heroin; through competitive interaction with miR-320-3p, it increases CRHR1 expression and contributes to hippocampal impairment and depressive-like outcomes; c circRNAs. c1 circHIPK2 is elevated following METH exposure and CUS. Acting as a sponge for miR-124-2HG, circHIPK2 influence astrocyte activation and behavioral sensitivity, whereas knockdown of circHIPK2 or microbiome-mediated suppression alleviates both addiction-related and depressive phenotypes. c2 Across both addictive drug exposure and depression, dysregulated circRNAs converge on m⁶A-dependent pathways, suggesting a shared circRNA–m⁶A regulatory axis that may represent a unifying mechanism and a promising future therapeutic target. Metastasis-associated lung adenocarcinoma transcript 1 (Malat1), nuclear paraspeckle assembly transcript 1 (NEAT1), circular RNA homeodomain-interacting protein kinase 2 (circHIPK2), methyl-CpG-binding protein 2 (MeCP2), brain-derived neurotrophic factor (BDNF), serotonin transporter(SERT), 5-Hydroxytryptamine (5-HT),corticotropin-releasing hormone receptor 1 (CRHR1), chronic unpredictable stress (CUS), N6-methyladenosine(m⁶A). (Figure was created by Adobe Illustrator 2023).
Multiple lines of evidence indicate that chronic stress and drug addiction converge on common epigenetic targets, particularly stress-axis regulators and neuroplasticity genes. Additionally, in MDD, aberrant DNA methylation at stress-related loci, including the glucocorticoid receptor gene NR3C1 and FKBP5 and neurotrophic genes (such as BDNF) are well documented9. Remarkably, addictive drugs similarly remodel these same loci171. For example, early postnatal alcohol exposure leads to persistent hypermethylation of the FKBP5 promoter in the hippocampus, likely disrupting HPA axis feedback regulation and enhancing susceptibility to depression-like phenotypes through epigenetically mediated stress-response dysregulation172. Similarly, experimental studies have shown that cocaine administration markedly increased FKBP5 expression in stress-related brain regions, such as the paraventricular nucleus and bed nucleus of the stria terminalis during early withdrawal, implicating a convergent role for FKBP5 dysregulation across substances in stress-related mood vulnerability74. Moreover, increased NR3C1 promoter methylation-particularly at the exon 1H region, has been observed in the PFC of individuals with alcohol use disorder, accompanied by reduced NR3C1 mRNA and protein expression171. Given NR3C1’s central role in HPA axis regulation and its known involvement in depression, such epigenetic dysregulation may represent a shared vulnerability mechanism across addiction and mood disorders. It is important to note that, despite these convergent findings, direct mechanistic evidence demonstrating that addictive substances induce depressive phenotypes via specific epigenetic pathways remains extremely limited, with only a few studies providing clear causal links.
Neurotrophic and synaptic plasticity pathways likewise show overlapping epigenetic regulation. BDNF, a key mediator of neuronal survival and plasticity, is epigenetically modulated in both disorders. Depressed subjects and stress models reduced BDNF levels, and chronic drug exposure reprogramed its expression173,174. For instance, repeated cocaine in rodents enhanced histone H3 acetylation at the BDNF promoter in PFC, boosting BDNF transcription175. By contrast, alcohol exposure tended to impose repressive marks on BDNF. Early-onset alcohol use increased H3K27 trimethylation at amygdalar BDNF and Arc loci, correlating with decreased gene expression42. Noncoding RNAs also contribute: the antisense lncRNA BDNF-AS was upregulated in individuals with alcohol use disorder and suppressed BDNF expression. Increasing prefrontal BDNF experimentally reduced cocaine-seeking behavior, underscoring BDNF as a shared epigenetic target linking mood regulation and drug reward42,176.
Critically, these shared targets are embedded in complex epigenetic networks and feedback loops. DNA methylation, histone modifications and ncRNAs often act in concert at the same loci. Chronic stress or drug use can simultaneously alter promoter methylation and histone marks to stably repress or activate synaptic genes177,178,179. Moreover, several miRNAs form homeostatic loops that span both stress and reward pathways. For instance, cocaine upregulates miR-212/132 in the striatum, which feeds back via CREB, MeCP2 and BDNF to limit cocaine reinforcement180. Conversely, acute stimulant exposure downregulates miR-495, relieving its suppression of BDNF and calcium/calmodulin-dependent protein kinase II alpha in NAc115. Such interactions create crosstalk between epigenetic layers: MeCP2, a methyl-DNA binding protein, can recruit histone modifiers, while histone acetylation status influences DNA methyltransferase access to chromatin181,182. Together, this multi-layer remodeling across the PFC, NAc, amygdala, and hippocampus provides a mechanistic bridge linking depressive and addictive pathology (Fig. 7).

The figure depicts how shared and substance-specific epigenetic mechanisms converge on BDNF in the context of drug addiction and depression. a Alcohol use disorder is associated with increased levels of the lncRNA BDNF-AS. Alcohol exposure imposes repressive H3K27me3 marks on BDNF and Arc loci in the amygdala, thereby suppressing BDNF transcription; b cocaine enhances miR-212/132 expression in the striatum, reducing MeCP2 levels and consequently decreasing BDNF. MeCP2, a methyl-DNA–binding protein, can recruit histone-modifying enzymes, and histone acetylation states further influence DNA methyltransferase access to chromatin; c prenatal cocaine exposure induces persistent histone deacetylation at the BDNF and NR3C1 promoters, increasing long-term vulnerability to depression-like phenotypes; d in contrast, repeated cocaine administration increases H3 acetylation at the BDNF promoter in the prefrontal cortex, leading to elevated BDNF expression. Acute stimulant exposure downregulates miR-495 in the nucleus accumbens, which increases BDNF and Camk2a expression. Together, these shared and substance-specific epigenetic pathways highlight BDNF as a central molecular node linking addiction-related neuroadaptations with stress-related depressive phenotypes. Brain-derived neurotrophic factor (BDNF), BDNF antisense RNA (BDNF-AS), activity-regulated (Arc), methyl-CpG binding protein 2 (MeCP2), nuclear receptor subfamily 3 group C member 1 (NR3C1), calcium/calmodulin-dependent protein kinase II alpha (Camk2a). (Figure was created by Adobe Illustrator 2023).
Collectively, these findings show that stress-axis genes, neurotrophic pathways and multi-layer epigenetic networks converge across depression and addiction, providing a unified mechanistic basis for their frequent comorbidity (Table 5).
Concluding remarks and future directions
Researches on the epigenetic mechanisms underlying depression and addiction have uncovered diverse alterations in DNA methylation, histone modifications and noncoding RNA pathways across stress- and reward-related circuits77,179. These findings outline a molecular framework, which link mood dysregulation, addictive behaviors and highlight how depression and drug exposure lead to enduring changes in gene regulation and neural function. However, many important questions remain unresolved.
Although many studies have reported changes in DNA methylation or histone states alongside depressive or addiction-related phenotypes, it is still unclear whether these changes exert causal influence or simply arise as consequences of the disease process. This uncertainty is due in part to the historical lack of tools capable of manipulating specific epigenetic marks and evaluating their behavioral effects. Only with the recent development of locus-targeted tools, such as clustered regularly interspaced short palindromic repeats (CRISPR-Cas9) or transcription activator-like effector nucleases-based epigenetic editors, has it become feasible to directly test how defined chromatin or methylation changes influence neural and behavioral outcomes9,183,184.
In addition, most of what we know about epigenetic regulation in depression and addiction comes from rodent models. Paradigms, such as CUMS or CSDS, are useful for probing mechanisms, but they capture only fragments of the human condition and cannot fully reproduce the complexity of real depressive or addictive states185. As a result, it is still uncertain to what extent epigenetic findings from mouse models can be applied to humans. A related limitation is that many experiments rely on whole-brain or large tissue samples, which mix together signals from neurons, glia and other cell types. These cells carry distinct chromatin and DNA methylation patterns, so important cell-specific changes can easily be obscured186,187. Newer methods, such as Fluorescence-Activated Cell Sorting-based nuclear isolation and single-cell or single-nucleus epigenomic profiling, are only now allowing researchers to look past these averaged signals and observe epigenetic changes at true cellular resolution. However, such high-resolution datasets are still limited, leaving important gaps in our understanding188. Together, these challenges highlight how far the field still is from fully connecting epigenetic findings in animal models and bulk tissues to the complex biology of human depression and addiction.
Moreover, most studies examine patients and controls at only one time point, so it is still difficult to tell how epigenetic marks change during the illness or whether they reflect causes or consequences189. Common factors, such as smoking, medications, and metabolic conditions can also leave strong background signals190,191, and differences in how studies define behavioral or clinical phenotypes further reduce comparability. In addition, future research must pay closer attention to biological moderators, such as sex, age, genetic background, and early-life stress. Evidence has indicated that males and females exhibited markedly different epigenetic responses to stress, and early adversity can leaft persistent marks on the epigenome thereby influencing later vulnerability to depression or addiction192,193. Together, these issues make it challenging to determine which epigenetic alterations have real biological significance in depression or addiction.
Future progress in this field will require advances on several fronts. First, methodological improvements remain essential. As single-cell and multi-omics technologies continue to mature, researchers will increasingly be able to profile DNA methylation, histone modifications, chromatin accessibility, and gene expression within the same cell, thereby generating a more accurate picture of epigenetic changes associated with depression and addiction186,187. Recent developments in CRISPR-cas9-based epigenome editing tools, DNA methylation probes, and programmable systems for modifying histone marks at specific loci have opened new possibilities for testing whether particular epigenetic signatures exert causal effects194,195. However, the application of these tools in the nervous system is still in its early stages, especially with respect to behavioral validation.
Second, future therapeutic exploration will require greater precision. Conventional epigenetic drugs, such as HDAC inhibitors or DNMTi, broadly alter chromatin states and often exhibit substantial off-target effects, limiting their suitability for treating depression or addiction in a targeted manner196. Emerging approaches that modulate noncoding RNAs, including miRNA mimics or inhibitors and antisense oligonucleotides targeting lncRNAs, have shown some promise in animal models, but further validation is still needed197,198,199. Clarifying how addiction and depression intersect at the level of epigenetic regulation will lay essential groundwork for translating these mechanistic insights into more precise and effective therapeutic strategies.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
De Sa Nogueira, D., Merienne, K. & Befort, K. Neuroepigenetics and addictive behaviors: where do we stand? Neurosci. Biobehav. Rev. 106, 58–72 (2019).
Korpi, E. R. et al. Mechanisms of action and persistent neuroplasticity by drugs of abuse. Pharmacol. Rev. 67, 872–1004 (2015).
Nestler, E. J. & Lüscher, C. The molecular basis of drug addiction: linking epigenetic to synaptic and circuit mechanisms. Neuron 102, 48–59 (2019).
Zhao, Y. et al. MicroRNAs in drug addiction: current status and future perspectives. Pharmacol. Ther. 236, 108215 (2022).
Stringaris, A. Editorial: what is depression? J. Child Psychol. Psychiatry 58, 1287–1289 (2017).
Nestler, E. J. The biology of addiction. Sci. Signal. 18, eadq0031 (2025).
Hu, H. Reward and aversion. Annu. Rev. Neurosci. 39, 297–324 (2016).
Abdoli, N. et al. The global prevalence of major depressive disorder (MDD) among the elderly: a systematic review and meta-analysis. Neurosci. Biobehav. Rev. 132, 1067–1073 (2022).
Yuan, M. et al. Epigenetic regulation in major depression and other stress-related disorders: molecular mechanisms, clinical relevance and therapeutic potential. Signal Transduct. Target. Ther. 8, 309 (2023).
Meng, Y. et al. The changes of blood and CSF ion levels in depressed patients: a systematic review and meta-analysis. Mol. Neurobiol. 61, 5369–5403 (2024).
Penner-Goeke, S. & Binder, E. B. Epigenetics and depression. Dialogues Clin. Neurosci. 21, 397–405 (2019).
Miller, L. & Campo, J. V. Depression in adolescents. N. Engl. J. Med. 385, 445–449 (2021).
Sales, A. J. & Joca, S. R. L. Antidepressant administration modulates stress-induced DNA methylation and DNA methyltransferase expression in rat prefrontal cortex and hippocampus. Behav. Brain Res. 343, 8–15 (2018).
Li, Q. et al. Microglia sing the prelude of neuroinflammation-associated depression. Mol. Neurobiol. 62, 5311–5332 (2025).
Wong, C. C. Y., Mill, J. & Fernandes, C. Drugs and addiction: an introduction to epigenetics: an introduction to epigenetics in addiction. Addiction 106, 480–489 (2011).
Descalzi, G. et al. Epigenetic mechanisms of chronic pain. Trends Neurosci. 38, 237–246 (2015).
Aljabali, A. A. A. et al. Deciphering depression: epigenetic mechanisms and treatment strategies. Biology 13, 638 (2024).
Rivier, C. A. et al. Bidirectional relationship between epigenetic age and stroke, dementia, and late-life depression. Nat. Commun. 16, 1261 (2025).
Wang, H., Dong, X., Awan, M. U. N. & Bai, J. Epigenetic mechanisms involved in methamphetamine addiction. Front. Pharmacol. 13, 984997 (2022).
Leung, J. et al. Methamphetamine exposure and depression—a systematic review and meta-analysis. Drug Alcohol Rev. https://doi.org/10.1111/dar.13670 (2023).
Costa, G. & Gołembiowska, K. Neurotoxicity of MDMA: main effects and mechanisms. Exp. Neurol. 347, 113894 (2022).
Huang, C.-L., Tsai, I.-J. & Lee, C. W.-S. Risk of psychosis in illicit amphetamine users: a 10 year retrospective cohort study. Evid. Based Ment. Health 25, 163–168 (2022).
Zweben, J. E. et al. Psychiatric symptoms in methamphetamine users. Am. J. Addctn. 13, 181–190 (2004).
Larkin, H. D. First amphetamine transdermal patch approved for ADHD. JAMA 327, 1642 (2022).
Arroyo-García, L. E. et al. Amphetamine sensitization alters hippocampal neuronal morphology and memory and learning behaviors. Mol. Psychiatry 26, 4784–4794 (2021).
Hui, R. et al. Betaine improves METH-induced depressive-like behavior and cognitive impairment by alleviating neuroinflammation via NLRP3 inflammasome inhibition. Prog. Neuropsychopharmacol. Biol. Psychiatry 135, 111093 (2024).
Tan, Y. et al. The role and mechanism of TRIM13 regulation of TRAF6 ubiquitination in the synergy of inflammatory responses and neurotoxicity induced by METH and HIV- 1 tat protein in astrocytes. Neurotox. Res. 43, 21 (2025).
Cox, B. M. et al. Behavioral and neurochemical effects of repeated MDMA administration during late adolescence in the rat. Prog. Neuro Psychopharmacol. Biol. Psychiatry 48, 229–235 (2014).
Biezonski, D. K. & Meyer, J. S. Effects of 3,4-methylenedioxymethamphetamine (MDMA) on serotonin transporter and vesicular monoamine transporter 2 protein and gene expression in rats: implications for MDMA neurotoxicity. J. Neurochem. 112, 951–962 (2010).
Schweppe, C. A. et al. Neurochemical and behavioral comparisons of contingent and non-contingent methamphetamine exposure following binge or yoked long-access self-administration paradigms. Psychopharmacology 237, 1989–2005 (2020).
Støier, J. F., Konomi-Pilkati, A., Apuschkin, M., Herborg, F. & Gether, U. Amphetamine-induced reverse transport of dopamine does not require cytosolic Ca2. J. Biol. Chem. 299, 105063 (2023).
Pang, L. & Wang, Y. Overview of blood-brain barrier dysfunction in methamphetamine abuse. Biomed. Pharmacother. 161, 114478 (2023).
Ali, T. et al. Melatonin prevents neuroinflammation and relieves depression by attenuating autophagy impairment through FOXO3a regulation. J. Pineal Res. 69, e12667 (2020).
Zhang, K. et al. Hyperactive neuronal autophagy depletes BDNF and impairs adult hippocampal neurogenesis in a corticosterone-induced mouse model of depression. Theranostics 13, 1059–1075 (2023).
Miao, Z.-F., Cho, C. J., Wang, Z.-N. & Mills, J. C. Autophagy repurposes cells during paligenosis. Autophagy 17, 588–589 (2021).
Moore, J., Megaly, M., MacNeil, A. J., Klentrou, P. & Tsiani, E. Rosemary extract reduces Akt/mTOR/p70S6K activation and inhibits proliferation and survival of A549 human lung cancer cells. Biomed. Pharmacother. 83, 725–732 (2016).
Yang, G. et al. Protective effect of gastrodin against methamphetamine-induced autophagy in human dopaminergic neuroblastoma SH-SY5Y cells via the AKT/mTOR signaling pathway. Neurosci. Lett. 707, 134287 (2019).
Li, B. et al. Effects of DDIT4 in methamphetamine-induced autophagy and apoptosis in dopaminergic neurons. Mol. Neurobiol. 54, 1642–1660 (2017).
Koob, G. F. A role for brain stress systems in addiction. Neuron 59, 11–34 (2008).
Armario, A. Activation of the hypothalamic–pituitary–adrenal axis by addictive drugs: different pathways, common outcome. Trends Pharmacol. Sci. 31, 318–325 (2010).
Chen, Y., Chen, J., Xing, Z., Peng, C. & Li, D. Autophagy in neuroinflammation: a focus on epigenetic regulation. Aging Dis. 15, 739–754 (2024).
Bohnsack, J. P., Teppen, T., Kyzar, E. J., Dzitoyeva, S. & Pandey, S. C. The lncRNA BDNF-AS is an epigenetic regulator in the human amygdala in early onset alcohol use disorders. Transl. Psychiatry 9, 34 (2019).
Daziani, G. et al. Synthetic cathinones and neurotoxicity risks: a systematic review. Int. J. Mol. Sci. 24, 6230 (2023).
Ricci, V. & Maina, G. Clinical and public health challenge of handling synthetic cathinone and cannabinoid abuse in pediatric care: a narrative review. Pediatr. Rep. 17, 19 (2025).
Riley, A. L. et al. Abuse potential and toxicity of the synthetic cathinones (i.e., “bath salts). Neurosci. Biobehav. Rev. 110, 150–173 (2020).
Espinosa-Velasco, M. et al. Behavioural and neurochemical effects after repeated administration of N-ethylpentylone (ephylone) in mice. J. Neurochem. 160, 218–233 (2022).
Hadlock, G. C. et al. 4-methylmethcathinone (mephedrone): neuropharmacological effects of a designer stimulant of abuse. J. Pharm. Exp. Ther. 339, 530–536 (2011).
López-Arnau, R. et al. Neuronal changes and oxidative stress in adolescent rats after repeated exposure to mephedrone. Toxicol. Appl. Pharmacol. 286, 27–35 (2015).
Correia, B., Fernandes, J., Botica, M. J., Ferreira, C. & Quintas, A. Novel psychoactive substances: the razor’s edge between therapeutical potential and psychoactive recreational misuse. Medicines 9, 19 (2022).
Siedlecka-Kroplewska, K., Szczerba, A., Lipinska, A., Slebioda, T. & Kmiec, Z. 3-fluoromethcathinone, a structural analog of mephedrone, inhibits growth and induces cell cycle arrest in HT22 mouse hippocampal cells. J. Physiol. Pharm. 65, 241–246 (2014).
Ashton, C. H. Adverse effects of cannabis and cannabinoids. Br. J. Anaesth. 83, 637–649 (1999).
Gobbi, G. et al. Association of cannabis use in adolescence and risk of depression, anxiety, and suicidality in young adulthood: a systematic review and meta-analysis. JAMA psychiatry 76, 426–434 (2019).
Bambico, F. R., Nguyen, N.-T., Katz, N. & Gobbi, G. Chronic exposure to cannabinoids during adolescence but not during adulthood impairs emotional behaviour and monoaminergic neurotransmission. Neurobiol. Dis. 37, 641–655 (2010).
Pistis, M. et al. Adolescent exposure to cannabinoids induces long-lasting changes in the response to drugs of abuse of rat midbrain dopamine neurons. Biol. Psychiatry 56, 86–94 (2004).
Jefsen, O. H., Erlangsen, A., Nordentoft, M. & Hjorthøj, C. Cannabis use disorder and subsequent risk of psychotic and nonpsychotic unipolar depression and bipolar disorder. JAMA Psychiatry 80, 803–810 (2023).
Breijyeh, Z., Jubeh, B., Bufo, S. A., Karaman, R. & Scrano, L. Cannabis: a toxin-producing plant with potential therapeutic uses. Toxins 13, 117 (2021).
Guldager, M. B., Biojone, C., da Silva, N. R., Godoy, L. D. & Joca, S. New insights into the involvement of serotonin and BDNF-TrkB signalling in cannabidiol’s antidepressant effect. Prog. Neuropsychopharmacol. Biol. Psychiatry 133, 111029 (2024).
Vowles, K. E. et al. Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain 156, 569–576 (2015).
Pfeiffer, A., Brantl, V., Herz, A. & Emrich, H. M. Psychotomimesis mediated by κ opiate receptors. Science 233, 774–776 (1986).
Ballantyne, J. C. & Mao, J. Opioid therapy for chronic pain. N. Engl. J. Med. 349, 1943–1953 (2003).
Davis, M. A., Lin, L. A., Liu, H. & Sites, B. D. Prescription opioid use among adults with mental health disorders in the United States. J. Am. Board Fam. Med. 30, 407–417 (2017).
Rosoff, D. B., Smith, G. D. & Lohoff, F. W. Prescription opioid use and risk for major depressive disorder and anxiety and stress-related disorders: a multivariable Mendelian randomization analysis. JAMA Psychiatry 78, 151–160 (2021).
Faria, J. et al. Comparative study of the neurotoxicological effects of tramadol and tapentadol in SH-SY5Y cells. Toxicology 359–360, 1–10 (2016).
Lin, X. et al. Chronic high-dose morphine treatment promotes SH-SY5Y cell apoptosis via c-jun N-terminal kinase-mediated activation of mitochondria-dependent pathway. FEBS J. 276, 2022–2036 (2009).
Taghizadehghalehjoughi, A. et al. Effect of fentanyl and remifentanil on neuron damage and oxidative stress during induction neurotoxicity. J. Cell. Mol. Med. 28, e18118 (2024).
Mehranpour, M., Moghaddam, M. H., Abdollahifar, M.-A., Salehi, M. & Aliaghaei, A. Tramadol induces apoptosis, inflammation, and oxidative stress in rat choroid plexus. Metab. Brain Dis. 38, 2679–2690 (2023).
Roque Bravo, R. et al. Cocaine: an updated overview on chemistry, detection, biokinetics, and pharmacotoxicological aspects including abuse pattern. Toxins 14, 278 (2022).
Goeders, N. E. The HPA axis and cocaine reinforcement. Psychoneuroendocrinology 27, 13–33 (2002).
Mantsch, J. R. et al. Daily cocaine self-administration under long-access conditions augments restraint-induced increases in plasma corticosterone and impairs glucocorticoid receptor-mediated negative feedback in rats. Brain Res. 1167, 101–111 (2007).
Leventhal, A. M. et al. Agitated depression in substance dependence. Drug Alcohol Depend. 116, 163–169 (2011).
Pomara, C. et al. Data available on the extent of cocaine use and dependence: biochemistry, pharmacologic effects and global burden of disease of cocaine abusers. Curr. Med. Chem. 19, 5647–5657 (2012).
Perrine, S. A., Sheikh, I. S., Nwaneshiudu, C. A., Schroeder, J. A. & Unterwald, E. M. Withdrawal from chronic administration of cocaine decreases delta opioid receptor signaling and increases anxiety- and depression-like behaviors in the rat. Neuropharmacology 54, 355–364 (2008).
Ma, D.-D. et al. Ephedrine and cocaine cause developmental neurotoxicity and abnormal behavior in zebrafish. Aquat. Toxicol. 265, 106765 (2023).
Connelly, K. L. & Unterwald, E. M. Chronic cocaine administration upregulates FKBP5 in the extended amygdala of male and female rats. Drug Alcohol Depend. 199, 101–105 (2019).
Hamilton, P. J. & Nestler, E. J. Epigenetics and addiction. Curr. Opin. Neurobiol. 59, 128–136 (2019).
Stewart, A. F., Fulton, S. L. & Maze, I. Epigenetics of drug addiction. Cold Spring Harb. Perspect. Med. 11, a040253 (2021).
Cadet, J. L. & Jayanthi, S. Epigenetics of addiction. Neurochem. Int. 147, 105069 (2021).
Paoli, C., Misztak, P., Mazzini, G. & Musazzi, L. DNA methylation in depression and depressive-like phenotype: biomarker or target of pharmacological intervention? Curr. Neuropharmacol. 20, 2267–2291 (2022).
Mattei, A. L., Bailly, N. & Meissner, A. DNA methylation: a historical perspective. Trends Genet. 38, 676–707 (2022).
Cadet, J. L. & Jayanthi, S. Epigenetic landscape of methamphetamine use disorder. Curr. Neuropharmacol. 19, 2060–2066 (2021).
Barrès, R. et al. Non-CpG methylation of the PGC-1α promoter through DNMT3B controls mitochondrial density. Cell Metab. 10, 189–198 (2009).
Cecil, C. A. M. et al. DNA methylation and substance-use risk: a prospective, genome-wide study spanning gestation to adolescence. Transl. Psychiatry 6, e976–e976 (2016).
Cadet, J. L. et al. Genome-wide DNA hydroxymethylation identifies potassium channels in the nucleus accumbens as discriminators of methamphetamine addiction and abstinence. Mol. Psychiatry 22, 1196–1204 (2017).
Numachi, Y. et al. Methamphetamine alters expression of DNA methyltransferase 1 mRNA in rat brain. Neurosci. Lett. 414, 213–217 (2007).
Biagioni, F. et al. Methamphetamine persistently increases alpha-synuclein and suppresses gene promoter methylation within striatal neurons. Brain Res. 1719, 157–175 (2019).
LaPlant, Q. et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 13, 1137–1143 (2010).
Ploense, K. L. et al. Prolonged-access to cocaine induces distinct Homer2 DNA methylation, hydroxymethylation, and transcriptional profiles in the dorsomedial prefrontal cortex of male Sprague-Dawley rats. Neuropharmacology 143, 299–305 (2018).
Bohnsack, J. P. & Pandey, S. C. Histone modifications, DNA methylation, and the epigenetic code of alcohol use disorder. Int. Rev. Neurobiol. 156, 1–62 (2021).
Browne, C. J., Godino, A., Salery, M. & Nestler, E. J. Epigenetic mechanisms of opioid addiction. Biol. Psychiatry 87, 22–33 (2020).
Shvedunova, M. & Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 23, 329–349 (2022).
Malvaez, M., Mhillaj, E., Matheos, D. P., Palmery, M. & Wood, M. A. CBP in the nucleus accumbens regulates cocaine-induced histone acetylation and is critical for cocaine-associated behaviors. J. Neurosci. 31, 16941–16948 (2011).
Renthal, W. et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 56, 517–529 (2007).
Pandey, S. C., Ugale, R., Zhang, H., Tang, L. & Prakash, A. Brain chromatin remodeling: a novel mechanism of alcoholism. J. Neurosci. 28, 3729–3737 (2008).
Ferguson, D. et al. Essential role of SIRT1 signaling in the nucleus accumbens in cocaine and morphine action. J. Neurosci. 33, 16088–16098 (2013).
Ferguson, D. et al. SIRT1-FOXO3a regulate cocaine actions in the nucleus accumbens. J. Neurosci. 35, 3100–3111 (2015).
Sheng, J., Lv, Z. G., Wang, L., Zhou, Y. & Hui, B. Histone H3 phosphoacetylation is critical for heroin-induced place preference. NeuroReport 22, 575–580 (2011).
Wang, Z., Yan, P., Hui, T. & Zhang, J. Epigenetic upregulation of PSD-95 contributes to the rewarding behavior by morphine conditioning. Eur. J. Pharmacol. 732, 123–129 (2014).
Egervari, G. et al. Striatal H3K27 acetylation linked to glutamatergic gene dysregulation in human heroin abusers holds promise as therapeutic target. Biol. Psychiatry 81, 585–594 (2017).
Jalali Mashayekhi, F. et al. Expression levels of the BDNF gene and histone modifications around its promoters in the ventral tegmental area and locus ceruleus of rats during forced abstinence from morphine. Neurochem. Res. 37, 1517–1523 (2012).
Wei, L. et al. Microinjection of histone deacetylase inhibitor into the ventrolateral orbital cortex potentiates morphine induced behavioral sensitization. Brain Res. 1646, 418–425 (2016).
Wang, Y. et al. Inhibition of histone deacetylase in the basolateral amygdala facilitates morphine context-associated memory formation in rats. J. Mol. Neurosci. 55, 269–278 (2015).
Chen, W.-S. et al. Effects of histone deacetylase inhibitor sodium butyrate on heroin seeking behavior in the nucleus accumbens in rats. Brain Res. 1652, 151–157 (2016).
Torres, O. V. et al. CAMKII-conditional deletion of histone deacetylase 2 potentiates acute methamphetamine-induced expression of immediate early genes in the mouse nucleus accumbens. Sci. Rep. 5, 13396 (2015).
Torres, O. V. et al. An acute methamphetamine injection downregulates the expression of several histone deacetylases (HDACs) in the mouse nucleus accumbens: potential regulatory role of HDAC2 expression. Neurotox. Res. 30, 32–40 (2016).
Martin, T. A. et al. Methamphetamine causes differential alterations in gene expression and patterns of histone acetylation/hypoacetylation in the rat nucleus accumbens. PLoS ONE 7, e34236 (2012).
Lepack, A. E. et al. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 368, 197–201 (2020).
Stewart, A. F., Lepack, A. E., Fulton, S. L., Safovich, P. & Maze, I. Histone H3 dopaminylation in nucleus accumbens, but not medial prefrontal cortex, contributes to cocaine-seeking following prolonged abstinence. Mol. Cell. Neurosci. 125, 103824 (2023).
Fulton, S. L. et al. Histone H3 dopaminylation in ventral tegmental area underlies heroin-induced transcriptional and behavioral plasticity in male rats. Neuropsychopharmacology 47, 1776–1783 (2022).
Zhu, L. et al. A novel microRNA, novel-m009C, regulates methamphetamine rewarding effects. Mol. Psychiatry 27, 3885–3897 (2022).
Oliver, R. J. & Mandyam, C. D. Regulation of adult neurogenesis by non-coding RNAs: implications for substance use disorders. Front. Neurosci. 12, 849 (2018).
Zhao, Y. et al. Decreased expression of plasma microRNA in patients with methamphetamine (MA) use disorder. J. Neuroimmune Pharm. 11, 542–548 (2016).
Zhang, K. et al. MiR-181a is a negative regulator of GRIA2 in methamphetamine-use disorder. Sci. Rep. 6, 35691 (2016).
Bosch, P. J., Benton, M. C., Macartney-Coxson, D. & Kivell, B. M. MRNA and microRNA analysis reveals modulation of biochemical pathways related to addiction in the ventral tegmental area of methamphetamine self-administering rats. BMC Neurosci. 16, 43 (2015).
Im, H.-I., Hollander, J. A., Bali, P. & Kenny, P. J. MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat. Neurosci. 13, 1120–1127 (2010).
Bastle, R. M. et al. In silico identification and in vivo validation of miR-495 as a novel regulator of motivation for cocaine that targets multiple addiction-related networks in the nucleus accumbens. Mol. Psychiatry 23, 434–443 (2018).
Sauvageau, M. et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. eLife 2, e01749 (2013).
Ferrè, F., Colantoni, A. & Helmer-Citterich, M. Revealing protein–lncRNA interaction. Brief. Bioinform. 17, 106–116 (2016).
Zhu, J., Fu, H., Wu, Y. & Zheng, X. Function of lncRNAs and approaches to lncRNA-protein interactions. Sci. China Life Sci. 56, 876–885 (2013).
Gu, Y.-J., Chen, L., Cheng, L., Zhou, M.-Y. & Wang, Y. Non-coding RNA: insights into the mechanism of methamphetamine neurotoxicity. Mol. Cell. Biochem. 476, 3319–3328 (2021).
Zhu, L. et al. Methamphetamine induces alterations in the long non-coding RNAs expression profile in the nucleus accumbens of the mouse. BMC Neurosci. 16, 18 (2015).
Chen, Y. et al. Identification of functional CircRNA-miRNA-mRNA regulatory network in dorsolateral prefrontal cortex neurons of patients with cocaine use disorder. Front. Mol. Neurosci. 15, 839233 (2022).
Deng, B., Tang, X. & Wang, Y. Regulation and bioinformatic analysis of circ_0015891/miR-129-1-3p axis in methamphetamine-induced dopaminergic apoptosis. Front. Endocrinol. 13, 999211 (2022).
Huang, R. et al. Circular RNA HIPK2 regulates astrocyte activation via cooperation of autophagy and ER stress by targeting MIR124-2HG. Autophagy 13, 1722–1741 (2017).
Yu, H. et al. The role of circTmeff-1 in incubation of context-induced morphine craving. Pharm. Res. 170, 105722 (2021).
Li, J. et al. Profiling circular RNA in methamphetamine-treated primary cortical neurons identified novel circRNAs related to methamphetamine addiction. Neurosci. Lett. 701, 146–153 (2019).
Malhi, G. S. & Mann, J. J. Depression. Lancet 392, 2299–2312 (2018).
Maurer, D. M., Raymond, T. J. & Davis, B. N. Depression: screening and diagnosis. Am. Fam. Physician 98, 508–515 (2018).
Ménard, C., Hodes, G. E. & Russo, S. J. Pathogenesis of depression: insights from human and rodent studies. Neuroscience 321, 138–162 (2016).
Uchida, S., Yamagata, H., Seki, T. & Watanabe, Y. Epigenetic mechanisms of major depression: targeting neuronal plasticity. Psychiatry Clin. Neurosci. 72, 212–227 (2018).
Park, C. et al. Stress, epigenetics and depression: a systematic review. Neurosci. Biobehav. Rev. 102, 139–152 (2019).
Zhu, J.-H., Bo, H.-H., Liu, B.-P. & Jia, C.-X. The associations between DNA methylation and depression: a systematic review and meta-analysis. J. Affect. Disord. 327, 439–450 (2023).
Sales, A. J., Maciel, I. S., Suavinha, A. C. D. R. & Joca, S. R. L. Modulation of DNA methylation and gene expression in rodent cortical neuroplasticity pathways exerts rapid antidepressant-like effects. Mol. Neurobiol. 58, 777–794 (2021).
Xiu, J. et al. Elevated BICD2 DNA methylation in blood of major depressive disorder patients and reduction of depressive-like behaviors in hippocampal BICD2-knockdown mice. Proc. Natl. Acad. Sci. USA 119, e2201967119 (2022).
Nagy, C. et al. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol. Psychiatry 20, 320–328 (2015).
Poulter, M. O. et al. GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biol. Psychiatry 64, 645–652 (2008).
Uchida, S. et al. Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 69, 359–372 (2011).
Chan, J. C. & Maze, I. Histone crotonylation makes its mark in depression research. Biol. Psychiatry 85, 616–618 (2019).
Liu, Y. et al. Chromodomain Y-like protein–mediated histone crotonylation regulates stress-induced depressive behaviors. Biol. Psychiatry 85, 635–649 (2019).
Sun, H., Kennedy, P. J. & Nestler, E. J. Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology 38, 124–137 (2013).
Covington, H. E. et al. Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 29, 11451–11460 (2009).
Covington, H. E. et al. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron 71, 656–670 (2011).
Guan, Z. et al. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell 111, 483–493 (2002).
Levenson, J. M. & Sweatt, J. D. Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 6, 108–118 (2005).
Guan, J.-S. et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60 (2009).
Gräff, J. et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226 (2012).
Zhao, B. S., Roundtree, I. A. & He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42 (2017).
Wilkinson, E., Cui, Y.-H. & He, Y.-Y. Context-dependent roles of RNA modifications in stress responses and diseases. IJMS 22, 1949 (2021).
Jung, Y. & Goldman, D. Role of RNA modifications in brain and behavior. Genes Brain Behav. 17, e12444 (2018).
Liu, S. et al. Fat mass and obesity-associated protein regulates RNA methylation associated with depression-like behavior in mice. Nat. Commun. 12, 6937 (2021).
Huang, R. et al. N6-methyladenosine modification of fatty acid amide hydrolase messenger RNA in circular RNA STAG1–regulated astrocyte dysfunction and depressive-like behaviors. Biol. Psychiatry 88, 392–404 (2020).
Kan, R. L., Chen, J. & Sallam, T. Crosstalk between epitranscriptomic and epigenetic mechanisms in gene regulation. Trends Genet. 38, 182–193 (2022).
Bai, Y. et al. Engagement of N6-methyladenisine methylation of Gng4 mRNA in astrocyte dysfunction regulated by CircHECW2. Acta Pharm. Sin. B 14, 1644–1660 (2024).
Guo, F. et al. Astrocytic ALKBH5 in stress response contributes to depressive-like behaviors in mice. Nat. Commun. 15, 4347 (2024).
Lopez, J. P. et al. miR-1202 is a primate-specific and brain-enriched microRNA involved in major depression and antidepressant treatment. Nat. Med. 20, 764–768 (2014).
Mert, A. et al. miRNAs in major depression: possible association of miR-17 and miR-92 with childhood traumas. Clin. Psychopharmacol. Neurosci. 23, 133–143 (2025).
Chen, X. et al. MicroRNA-204-5p deficiency within the vmPFC region contributes to neuroinflammation and behavioral disorders via the JAK2/STAT3 signaling pathway in rats. Adv. Sci. 12, 2403080 (2025).
Wang, Y. et al. Down-regulated miR-16-2 in peripheral blood is positively correlated with decreased bilateral insula volume in patients with major depressive disorder. J. Affect. Disord. 338, 137–143 (2023).
Xu, Y. et al. A polymorphism in the microRNA-30e precursor associated with major depressive disorder risk and P300 waveform. J. Affect. Disord. 127, 332–336 (2010).
Martinez, B. & Peplow, P. V. MicroRNAs as potential diagnostic biomarkers for bipolar disorder. Neural Regen. Res. 20, 1681–1695 (2025).
Zhong, X.-L., Du, Y., Chen, L. & Cheng, Y. The emerging role of long noncoding RNA in depression and its implications in diagnostics and therapeutic responses. J. Psychiatr. Res. 164, 251–258 (2023).
Huang, X., Luo, Y., Mao, Y. & Ji, J. The link between long noncoding RNAs and depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 73, 73–78 (2017).
Yrondi, A. et al. Association between the expression of lncRNA BASP-AS1 and volume of right hippocampal tail moderated by episode duration in major depressive disorder: a CAN-BIND 1 report. Transl. Psychiatry 11, 469 (2021).
Ding, X. et al. LncRNA XR_351665 contributes to chronic pain-induced depression by upregulating DNMT1 via sponging miR-152-3p. J. Pain 24, 449–462 (2023).
Cui, X., Xu, Y., Zhu, H., Wang, L. & Zhou, J. Long noncoding RNA NONHSAG045500 regulates serotonin transporter to ameliorate depressive-like behavior via the cAMP-PKA-CREB signaling pathway in a model of perinatal depression. J. Matern. Fetal Neonatal Med. 36, 2183468 (2023).
Cui, X. et al. hsa_circRNA_103636: potential novel diagnostic and therapeutic biomarker in major depressive disorder. Biomark. Med. 10, 943–952 (2016).
Gan, H. et al. Circular RNAs in depression: biogenesis, function, expression, and therapeutic potential. Biomed. Pharmacother. 137, 111244 (2021).
Jiang, G. et al. Relationships of circular RNA with diabetes and depression. Sci. Rep. 7, 7285 (2017).
Zhou, Z. et al. CircDYM attenuates microglial apoptosis via CEBPB/ZC3H4 axis in LPS-induced mouse model of depression. Int. J. Biol. Macromol. 254, 127922 (2024).
Yu, X. et al. Extracellular vesicle-mediated delivery of circDYM alleviates CUS-induced depressive-like behaviours. J. Extracell. Vesicles 11, e12185 (2022).
Zhang, Y. et al. CircDYM ameliorates depressive-like behavior by targeting miR-9 to regulate microglial activation via HSP90 ubiquitination. Mol. Psychiatry 25, 1175–1190 (2020).
Gatta, E. et al. Genome-wide methylation in alcohol use disorder subjects: implications for an epigenetic regulation of the cortico-limbic glucocorticoid receptors (NR3C1). Mol. Psychiatry 26, 1029–1041 (2021).
Dursun, I. et al. Exploring epigenetic modification of the stress-related FKBP5 gene in mice exposed to alcohol during early postnatal development. Alcohol 123, 11–17 (2025).
Castrén, E. & Monteggia, L. M. Brain-derived neurotrophic factor signaling in depression and antidepressant action. Biol. Psychiatry 90, 128–136 (2021).
Ornell, F. et al. Serum BDNF levels increase during early drug withdrawal in alcohol and crack cocaine addiction. Alcohol 111, 1–7 (2023).
Schmidt, H. D. et al. Increased brain-derived neurotrophic factor (BDNF) expression in the ventral tegmental area during cocaine abstinence is associated with increased histone acetylation at BDNF exon I-containing promoters. J. Neurochem. 120, 202–209 (2012).
Drake, J. et al. Assessing the role of long noncoding RNA in nucleus accumbens in subjects with alcohol dependence. Alcohol Clin. Exp. Res. 44, 2468–2480 (2020).
Qureshi, I. A. & Mehler, M. F. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci. 13, 528–541 (2012).
Robison, A. J. & Nestler, E. J. Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 12, 623–637 (2011).
Nestler, E. J. Epigenetic mechanisms of depression. JAMA Psychiatry 71, 454–456 (2014).
Hollander, J. A. et al. Striatal microRNA controls cocaine intake through CREB signalling. Nature 466, 197–202 (2010).
Jones, P. L. et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191 (1998).
Fuks, F. et al. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 278, 4035–4040 (2003).
Zhang, X. et al. A programmable CRISPR/dCas9-based epigenetic editing system enabling loci-targeted histone citrullination and precise transcription regulation. J. Genet. Genom. 51, 1485–1493 (2024).
Bohnsack, J. P. et al. Targeted epigenomic editing ameliorates adult anxiety and excessive drinking after adolescent alcohol exposure. Sci. Adv. 8, eabn2748 (2022).
Duan, Z. & Lu, J. DNA methyltransferases in depression: an update. Front. Psychiatry 11, 538683 (2020).
Peterson, V. M. et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 35, 936–939 (2017).
Clark, S. J. et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat. Commun. 9, 781 (2018).
Clark, S. J., Lee, H. J., Smallwood, S. A., Kelsey, G. & Reik, W. Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol. 17, 72 (2016).
Januar, V., Saffery, R. & Ryan, J. Epigenetics and depressive disorders: a review of current progress and future directions. Int. J. Epidemiol. 44, 1364–1387 (2015).
Engelmann, J. et al. Epigenetic signatures in antidepressant treatment response: a methylome-wide association study in the EMC trial. Transl. Psychiatry 12, 268 (2022).
Keller, M. et al. Genome-wide DNA promoter methylation and transcriptome analysis in human adipose tissue unravels novel candidate genes for obesity. Mol. Metab. 6, 86–100 (2017).
Cheng, Z., Su, J., Zhang, K., Jiang, H. & Li, B. Epigenetic mechanism of early life stress-induced depression: focus on the neurotransmitter systems. Front. Cell Dev. Biol. 10, 929732 (2022).
Jaric, I., Rocks, D., Cham, H., Herchek, A. & Kundakovic, M. Sex and estrous cycle effects on anxiety- and depression-related phenotypes in a two-hit developmental stress model. Front. Mol. Neurosci. 12, 74 (2019).
Modi, N., Guo, J., Lee, R. A., Greenstein, A. & Lee, R. S. Targeted DNA methylation using modified DNA probes: a potential therapeutic tool for depression and stress-related disorders. IJMS 26, 5643 (2025).
Sgro, A. & Blancafort, P. Epigenome engineering: new technologies for precision medicine. Nucleic Acids Res. 48, 12453–12482 (2020).
Patel, S. & Chakravarty, S. HDAC dynamics and GR signaling: neurobiological insights and antidepressant potential. J. Ment. Health Disord. 4, 57–62 (2024).
Peedicayil, J. & Kumar, A. Epigenetic drugs for mood disorders. Prog. Mol. Biol. Transl. Sci. 157, 151–174 (2018).
Albert, P. R., Le François, B. & Vahid-Ansari, F. Genetic, epigenetic and posttranscriptional mechanisms for treatment of major depression: the 5-HT1A receptor gene as a paradigm. J. Psychiatry Neurosci. 44, 164–176 (2019).
Park, H.-S., Kim, J., Ahn, S. H. & Ryu, H.-Y. Epigenetic targeting of histone deacetylases in diagnostics and treatment of depression. Int. J. Mol. Sci. 22, 5398 (2021).
Maze, I. et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327, 213–216 (2010).
Anier, K. et al. Cocaine-induced epigenetic DNA modification in mouse addiction-specific and non-specific tissues. Neuropharmacology 139, 13–25 (2018).
Cannella, N. et al. Dnmt3a2 in the nucleus accumbens shell is required for reinstatement of cocaine seeking. J. Neurosci. 38, 7516–7528 (2018).
Anier, K., Malinovskaja, K., Aonurm-Helm, A., Zharkovsky, A. & Kalda, A. DNA methylation regulates cocaine-induced behavioral sensitization in mice. Neuropsychopharmacology 35, 2450–2461 (2010).
Chen, Z.-G. et al. Ube2b-dependent degradation of DNMT3a relieves a transcriptional brake on opiate-induced synaptic and behavioral plasticity. Mol. Psychiatry 26, 1162–1177 (2021).
Jiang, F.-Z. et al. Double dissociation of inhibitory effects between the hippocampal TET1 and TET3 in the acquisition of morphine self-administration in rats. Addctn. Biol. 26, e12875 (2021).
Zhang, J.-J. et al. DNMT3a in the hippocampal CA1 is crucial in the acquisition of morphine self-administration in rats. Addctn. Biol. 25, e12730 (2020).
Zhu, J. et al. Methylation of glucocorticoid receptor gene promoter modulates morphine dependence and accompanied hypothalamus-pituitary-adrenal axis dysfunction. J. Neurosci. Res. 95, 1459–1473 (2017).
González, B. et al. Repeated methamphetamine and modafinil induce differential cognitive effects and specific histone acetylation and DNA methylation profiles in the mouse medial prefrontal cortex. Prog. Neuropsychopharmacol. Biol. Psychiatry 82, 1–11 (2018).
González, B. et al. The effects of single-dose injections of modafinil and methamphetamine on epigenetic and functional markers in the mouse medial prefrontal cortex: potential role of dopamine receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 88, 222–234 (2019).
Hong, Q. et al. Histone 3 lysine 9 acetylation of BRG1 in the medial prefrontal cortex is associated with heroin self‑administration in rats. Mol. Med. Rep. 21, 405–412 (2020).
Kennedy, P. J. et al. Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation. Nat. Neurosci. 16, 434–440 (2013).
Franco-García, A. et al. Morphine-withdrawal aversive memories and their extinction modulate H4K5 acetylation and Brd4 activation in the rat hippocampus and basolateral amygdala. Biomed. Pharmacother. 165, 115055 (2023).
Jayanthi, S. et al. Methamphetamine downregulates striatal glutamate receptors via diverse epigenetic mechanisms. Biol. Psychiatry 76, 47–56 (2014).
Campbell, R. R. et al. HDAC3 activity within the nucleus accumbens regulates cocaine-induced plasticity and behavior in a cell-type-specific manner. J. Neurosci. 41, 2814–2827 (2021).
Aguilar-Valles, A. et al. Methamphetamine-associated memory is regulated by a writer and an eraser of permissive histone methylation. Biol. Psychiatry 76, 57–65 (2014).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550 (2018).
Tomasiewicz, H. C. et al. Proenkephalin mediates the enduring effects of adolescent cannabis exposure associated with adult opiate vulnerability. Biol. Psychiatry 72, 803–810 (2012).
Sun, H. et al. Morphine epigenomically regulates behavior through alterations in histone H3 lysine 9 dimethylation in the nucleus accumbens. J. Neurosci. 32, 17454–17464 (2012).
Damez-Werno, D. M. et al. Histone arginine methylation in cocaine action in the nucleus accumbens. Proc. Natl. Acad. Sci. USA 113, 9623–9628 (2016).
Jordi, E. et al. Differential effects of cocaine on histone posttranslational modifications in identified populations of striatal neurons. Proc. Natl. Acad. Sci. USA 110, 9511–9516 (2013).
Shi, J.-J. et al. Dorsolateral striatal miR-134 modulates excessive methamphetamine intake in self-administering rats. Metab. Brain Dis. 34, 1029–1041 (2019).
Li, W. et al. Combined diagnostic value of hsa-miR-592 and hsa-miR-9-3p in plasma for methamphetamine addicts. Int. J. Mol. Sci. 25, 8952 (2024).
Chen, F. et al. Prognostic plasma exosomal microRNA biomarkers in patients with substance use disorders presenting comorbid with anxiety and depression. Sci. Rep. 11, 6271 (2021).
Jiang, M.-J. et al. Rhynchophylline inhibits methamphetamine dependence via modulating the miR-181a-5p/GABRA1 axis. J. Ethnopharmacol. 314, 116635 (2023).
Wang, Y. et al. MicroRNA-181a is involved in methamphetamine addiction through the ERAD pathway. Front. Mol. Neurosci. 14, 667725 (2021).
Li, J. et al. Regulation of miR-128 in the nucleus accumbens affects methamphetamine-induced behavioral sensitization by modulating proteins involved in neuroplasticity. Addctn. Biol. 26, e12881 (2021).
Su, H. et al. Impact of miR-29c-3p in the nucleus accumbens on methamphetamine-induced behavioral sensitization and neuroplasticity-related proteins. Int. J. Mol. Sci. 25, 942 (2024).
Liu, D. et al. Potential Ago2/miR-3068-5p cascades in the nucleus accumbens contribute to methamphetamine-induced locomotor sensitization of mice. Front Pharm. 12, 708034 (2021).
T, N. et al. The potential involvement of miR-204-3p-axon guidance network in methamphetamine-induced locomotor sensitization of mice. Neurosci. Lett. 707, 134303 (2019).
Gu, W.-J. et al. Altered serum microRNA expression profile in subjects with heroin and methamphetamine use disorder. Biomed. Pharmacother. 125, 109918 (2020).
Cabana-Domínguez, J., Arenas, C., Cormand, B. & Fernàndez-Castillo, N. MiR-9, miR-153 and miR-124 are down-regulated by acute exposure to cocaine in a dopaminergic cell model and may contribute to cocaine dependence. Transl. Psychiatry 8, 173 (2018).
Sadakierska-Chudy, A. et al. Prolonged induction of miR-212/132 and REST expression in rat striatum following cocaine self-administration. Mol. Neurobiol. 54, 2241–2254 (2017).
Wu, Y., Rong, W., Jiang, Q., Wang, R. & Huang, H. Downregulation of lncRNA GAS5 alleviates hippocampal neuronal damage in mice with depression-like behaviors via modulation of microRNA-26a/EGR1 axis. J. Stroke Cerebrovasc. Dis. 30, 105550 (2021).
Chandrasekar, V. & Dreyer, J.-L. Regulation of MiR-124, Let-7d, and MiR-181a in the accumbens affects the expression, extinction, and reinstatement of cocaine-induced conditioned place preference. Neuropsychopharmacology 36, 1149–1164 (2011).
Zanda, M. T., Floris, G. & Daws, S. E. Orbitofrontal cortex microRNAs support long-lasting heroin seeking behavior in male rats. Transl. Psychiatry 13, 117 (2023).
Yan, B. et al. MiR-218 targets MeCP2 and inhibits heroin seeking behavior. Sci. Rep. 7, 40413 (2017).
Zanda, M. T., Saikali, L., Morris, P. & Daws, S. E. MicroRNA-mediated translational pathways are regulated in the orbitofrontal cortex and peripheral blood samples during acute abstinence from heroin self-administration. Adv. Drug Alcohol Res. 3, 11668 (2023).
Xu, W. et al. Increased expression of plasma hsa-miR-181a in male patients with heroin addiction use disorder. J. Clin. Lab. Anal. 34, e23486 (2020).
H, L. et al. Increased expression of plasma miRNA-320a and let-7b-5p in heroin-dependent patients and its clinical significance. Front. Psychiatry 12, 679206 (2021).
He, J. et al. LncRNA NPTN-IT1-201 ameliorates depressive-like behavior by targeting miR-142-5p and regulating inflammation and apoptosis via BDNF. Curr. Med. Sci. https://doi.org/10.1007/s11596-024-2917-8 (2024).
Jin, J. et al. miR-17-92 cluster regulates adult hippocampal neurogenesis, anxiety, and depression. Cell Rep. 16, 1653–1663 (2016).
Kaurani, L. et al. Baseline levels of miR-223-3p correlate with the effectiveness of electroconvulsive therapy in patients with major depression. Transl. Psychiatry 13, 294 (2023).
Fiori, L. M. et al. Investigation of miR-1202, miR-135a, and miR-16 in major depressive disorder and antidepressant response. Int. J. Neuropsychopharmacol. 20, 619–623 (2017).
Xian, X. et al. Neuron secrete exosomes containing miR-9-5p to promote polarization of M1 microglia in depression. J. Nanobiotechnol. 20, 122 (2022).
Wei, Z.-X. et al. Exosomes from patients with major depression cause depressive-like behaviors in mice with involvement of miR-139-5p-regulated neurogenesis. Neuropsychopharmacology 45, 1050–1058 (2020).
Feng, H. et al. Decreased miR-451a in cerebrospinal fluid, a marker for both cognitive impairment and depressive symptoms in Alzheimer’s disease. Theranostics 13, 3021–3040 (2023).
Li, Y. et al. Hippocampal miR-211-5p regulates neurogenesis and depression-like behaviors in the rat. Neuropharmacology 194, 108618 (2021).
Wu, X. et al. Clinical and preclinical evaluation of miR-144-5p as a key target for major depressive disorder. CNS Neurosci. Ther. 29, 3598–3611 (2023).
Fan, C. et al. MiR-134 modulates chronic stress-induced structural plasticity and depression-like behaviors via downregulation of Limk1/cofilin signaling in rats. Neuropharmacology 131, 364–376 (2018).
Wang, H. et al. Microglia in depression: an overview of microglia in the pathogenesis and treatment of depression. J. Neuroinflamm. 19, 132 (2022).
Yu, H. et al. miR-143-3p modulates depressive-like behaviors via Lasp1 in the mouse ventral hippocampus. Commun. Biol. 7, 944 (2024).
Li, D. et al. NK cell-derived exosomes carry miR-207 and alleviate depression-like symptoms in mice. J. Neuroinflamm. 17, 126 (2020).
Fiori, L. M. et al. miR-323a regulates ERBB4 and is involved in depression. Mol. Psychiatry 26, 4191–4204 (2021).
Zheng, Y.-B. & Jin, X. Evidence for the contribution of the miR-206/BDNF pathway in the pathophysiology of depression. Int. J. Neuropsychopharmacol. pyae039, https://doi.org/10.1093/ijnp/pyae039 (2024).
Suento, W. J. et al. Prefrontal cortex miR-874-3p prevents lipopolysaccharide-induced depression-like behavior through inhibition of indoleamine 2,3-dioxygenase 1 expression in mice. J. Neurochem. 157, 1963–1978 (2021).
Xu, D.-W. et al. MiR-184-3p in the paraventricular nucleus participates in the neurobiology of depression via regulation of the hypothalamus-pituitary-adrenal axis. Neuropharmacology 260, 110129 (2024).
Yang, J.-C. et al. miR-29a-5p rescues depressive-like behaviors in a CUMS-induced mouse model by facilitating microglia M2-polarization in the prefrontal cortex via TMEM33 suppression. J. Affect. Disord. 360, 188–197 (2024).
Rodrigues, B. et al. MiR-186-5p inhibition restores synaptic transmission and neuronal network activity in a model of chronic stress. Mol. Psychiatry https://doi.org/10.1038/s41380-024-02715-1 (2024).
Huang, C. et al. miR-98-5p plays a critical role in depression and antidepressant effect of ketamine. Transl. Psychiatry 11, 454 (2021).
Wan, Y.-Q. et al. Prefrontal cortex miR-29b-3p plays a key role in the antidepressant-like effect of ketamine in rats. Exp. Mol. Med. 50, 1–14 (2018).
MiR-182-5p: a novel biomarker in the treatment of depression in CSDS-induced mice | Int. J. Neuropsychopharmacol. https://academic.oup.com/ijnp/article/27/1/pyad064/7457303?login=false (2024).
Issler, O. et al. Sex-specific role for the long non-coding RNA LINC00473 in depression. Neuron 106, 912–926 (2020).
Jiao, X. et al. LncRNA-84277 is involved in chronic pain-related depressive behaviors through miR-128-3p/SIRT1 axis in central amygdala. Front. Mol. Neurosci. 15, 920216 (2022).
Huan, Z. et al. lncRNA MIR155HG alleviates depression-like behaviors in mice by regulating the miR-155/BDNF axis. Neurochem. Res. 46, 935–944 (2021).
Zhan, T. et al. Implication of lncRNA MSTRG.81401 in hippocampal pyroptosis induced by P2X7 receptor in type 2 diabetic rats with neuropathic pain combined with depression. Int. J. Mol. Sci. 25, 1186 (2024).
Chen, H.-S. et al. Long noncoding RNA Gm2694 drives depressive-like behaviors in male mice by interacting with GRP78 to disrupt endoplasmic reticulum homeostasis. Sci. Adv. 8, eabn2496 (2022).
Yu X. et al. Plasma circRNA HIPK2 as a putative biomarker for the diagnosis and prediction of therapeutic effects in major depressive disorder. Clin. Chim. Acta 552, 117694 (2024).
He, Y. et al. Hippocampal circAnk3 deficiency causes anxiety-like behaviors and social deficits by regulating the miR-7080-3p/IQGAP1 pathway in mice. Biol. Psychiatry 95, 896–908 (2024).
Bu, T. et al. Diagnostic biomarker hsa_circ_0126218 and functioning prediction in peripheral blood monocular cells of female patients with major depressive disorder. Front. Cell Dev. Biol. 9, 651803 (2021).
Ju, M. et al. Intranasal delivery of circATF7IP siRNA via lipid nanoparticles alleviates LPS-induced depressive-like behaviors. Adv. Healthc. Mater. e2402219, https://doi.org/10.1002/adhm.202402219 (2024).
Shi, Y. et al. Potential clinical value of circular RNAs as peripheral biomarkers for the diagnosis and treatment of major depressive disorder. EBioMedicine 66, 103337 (2021).
Mews, P. et al. Alcohol metabolism contributes to brain histone acetylation. Nature 574, 717–721 (2019).
Chen, W.-Y. et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) alleviates depression-like behavior and normalizes epigenetic changes in the hippocampus during ethanol withdrawal. Alcohol 78, 79–87 (2019).
Gajewski, P. A. et al. Epigenetic regulation of hippocampal FosB expression controls behavioral responses to cocaine. J. Neurosci. 39, 8305–8314 (2019).
Feng, J. et al. Chronic cocaine-regulated epigenomic changes in mouse nucleus accumbens. Genome Biol. 15, R65 (2014).
Anderson, E. M. et al. Overexpression of the histone dimethyltransferase G9a in nucleus accumbens shell increases cocaine self-administration, stress-induced reinstatement, and anxiety. J. Neurosci. 38, 803–813 (2018).
Jiang, Z. et al. H3K9me2 regulation of BDNF expression in the hippocampus and medial prefrontal cortex is involved in the depressive-like phenotype induced by maternal separation in male rats. Psychopharmacology 238, 2801–2813 (2021).
Muñoa-Hoyos, I. et al. Morphine leads to global genome changes in H3K27me3 levels via a polycomb repressive complex 2 (PRC2) self-regulatory mechanism in mESCs. Clin. Epigenet. 12, 170 (2020).
Sun, Z.-W. et al. Blood-brain barrier dysfunction mediated by the EZH2-claudin-5 axis drives stress-induced TNF-α infiltration and depression-like behaviors. Brain Behav. Immun. 115, 143–156 (2024).
Zhou, V. W., Goren, A. & Bernstein, B. E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 12, 7–18 (2011).
Bedi, Y. S. et al. Alcohol induced increases in sperm histone H3 lysine 4 trimethylation correlate with increased placental CTCF occupancy and altered developmental programming. Sci. Rep. 12, 8839 (2022).
Elliott, E. et al. Dnmt3a in the medial prefrontal cortex regulates anxiety-like behavior in adult mice. J. Neurosci. 36, 730–740 (2016).
Guo, M.-L. et al. Cocaine-mediated downregulation of microglial miR-124 expression involves promoter DNA methylation. Epigenetics 11, 819–830 (2016).
Yang, W. et al. Knockdown of miR-124 reduces depression-like behavior by targeting CREB1 and BDNF. Curr. Neurovasc. Res. 17, 196–203 (2020).
Xu, H. et al. Role of long noncoding RNA Gas5 in cocaine action. Biol. Psychiatry 88, 758–766 (2020).
Acknowledgements
This work was supported by the grant from the National Natural Science Foundation of China (82473906).
Author information
Authors and Affiliations
Contributions
Conceptualization: Feng Guo, Xu Zhu, and Ying Pan; funding acquisition: Feng Guo; writing-original draft: Wangwei Zhang, Mingyue Xu, Chong Wang, and Hongyue Liang; writing-review and editing: Feng Guo, Xu Zhu, and Ying Pan. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Yogesh Dwivedi, Ayako Kawatake-Kuno and the other anonymous reviewer(s) for their contribution to the peer review of this work. Primary handling editor: Benjamin Bessieres. [A peer review file is available.]
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, W., Xu, M., Wang, C. et al. Addictive drug abuse and depression-a focus on epigenetics. Commun Biol 9, 297 (2026). https://doi.org/10.1038/s42003-026-09705-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-026-09705-9
