
Abstract
Optically pure phosphonates featuring an α-functional motif serve as core structures of versatile pharmaceuticals. Pudovik reaction employing P-stereogenic H-phosphonates derived from recyclable chiral auxiliary (CA) is one of the most practical and efficient asymmetric synthetic strategies. Although several centre and axial CAs have been investigated in this field, the development of novel chiral H-phosphonates has remained largely unexplored over the past 25 years. Here, we developed a kind of H-phosphonate (CAMDOL-PHO) derived from camphor-derived 2,3-diol (CAMDOL). Stereoselective Pudovik reactions of CAMDOL-PHO with diverse aldehydes, aldimines, and nitroalkenes were well developed, affording a library of molecules of α-hydroxyl, α-amino, and β-nitro phosphonates respectively with high dr values (up to 99:1) and yields (up to 97%) as well as broad scopes. Comparative DFT and experimental studies demonstrated that CAMDOL-PHO possessed significant superiority to install the P-adjacent C-stereogenic centre, mainly benefiting from its unique diphenyl-substituted camphor skeleton with an angular methyl group.

Similar content being viewed by others
Introduction
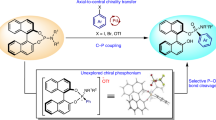
Organophosphonates are important structural units frequently found in biologically active compounds, including natural products, active pharmaceutical ingredients, and pesticides1,2,3,4,5,6,7,8,9,10. In particular, optically pure α-hydroxyl, α-amino, and β-amino phosphonic acids, and the related derivatives serve as core motifs of versatile pharmaceuticals11,12,13,14,15,16,17, such as Fosfazinomycin A18,19, Fosfomycin20,21, Alafosfalin22,23, and Phosphotyrosine24,25 (Fig. 1A). Accordingly, numerous synthetic approaches have been developed to meet the increasing need for those enantiopure organophosphorus compounds26,27,28,29,30. Pudovik reaction, known as the addition of dialkyl phosphites or the relevant P(O)H compounds to unsaturated chemical bonds (C=C, C≡C, C=O, C=N, etc.) to generate P–C functionalized phosphonates, is one of the most powerful and direct methods for the induction of phosphorus groups31,32,33,34,35. In this context, three types of asymmetric hydrophosphonylation methodologies were mainly put forward: chiral Lewis acid complex catalysis, organocatalysis, and chiral auxiliary (CA) induction36,37,38,39. More or less, the practicability of the catalytic asymmetric methods is generally limited by the use of special reagents and relatively narrow substrate scopes (always to be prepared with much effort)40. In comparison, the recyclable CA induced stereoselective Pudovik reaction employing P-stereogenic H-phosphonates has been substantiated by significant progress in the evolution of different chiral skeletons41, arising from the pioneering studies of Mislow’s menthyl-PHO40,42, Spilling’s CHDA-PHO43, Kee’s BINOL-PHO44, and Enders’s TADDOL-PHO45 (Fig. 1B). These P-chiral CA-PHOs could readily react with versatile partners to provide structurally diverse P-adjacent C-stereogenic molecules. What’s more, diastereomeric pairs in the products from CA-PHO could be unambiguously distinguished by NMR, thereby enabling the easy real-time monitoring of the stereochemical process of these reactions by 31P/1H NMR. More importantly, the CA-induction methods can generate the P–C bond and the desired C–X (X=C, O, N, etc.) motifs concurrently and generally avoid the inconvenient pre-synthesis of the relevant P-substrates. However, to some extent, further applications of these CA-PHO are much limited as a result of the relatively harsh preparation conditions, low diastereoselectivities, or confined scopes.

A Selected pharmaceutical compounds containing P-adjacent C-stereogenic center. B Previous reports for the development of CA-derived P-chiral H-phosphonates. C Our work about the design and application of center-chiral CAMDOL-derived H-phosphonate (abbrev. CAMDOL-PHO) for stereoselective Pudovik reaction.
Therefore, a critical objective for advancing stereoselective Pudovik reactions lies in developing novel induction strategies to enable chirality installation at carbon centers, particularly through rationally designed chiral auxiliaries with enhanced efficiency. However, despite the utility of the four classic CA-PHOs, almost no newly CA-PHOs have been reported over the past 25 years.
Very recently, we established a novel pattern of CA platform—camphor-derived 2,3-diols (CAMDOL)—which demonstrates exceptional utility in diastereoselective reactions, including synthesis of diverse chiral P(III)/P(V)-compounds46 and asymmetric α-functionalization of phosphonates47. In this study, we further advance CAMDOL to synthesize a centrally chiral P-chiral H-phosphonate through a streamlined one-pot protocol. This strategy enables subsequent asymmetric Pudovik additions with well-characterized aldehydes, aldimines, and nitroalkenes, thereby achieving diastereoselective construction of high-value α-hydroxyl-, α-amino-, and β-amino-phosphonates (Fig. 1C).
Results
Preparation of P-chiral CAMDOL-PHO
Inspired by Ender’s creative work about TADDOL-PHO synthesis45, we first attempted to prepare the enantiomeric CAMDOL-PHO 1 in the same manner (Fig. 2). As expected, CAMDOL could be efficiently converted to CAMDOL-PCl in nearly quantitative yield by treatment with PCl3/Et3N. Moreover, CAMDOL-PCl was comparatively stable for several months at room temperature, and could even be easily purified by silica gel column chromatography. However, the subsequent selective hydrolysis of exocyclic P–Cl bond of CAMDOL-PCl proved to be a formidable challenge. Even under severe conditions including usage of high temperature and concentrated hydrochloric acid, no P–Cl bond cleavage reaction was observed, which is much different from the behaviours of reported TADDOL-PCl or BINOL-PCl48. According to our recent work about SiO2-promoted efficient hydrolysis of phosphonites49, we rationalized that CAMDOL-POR, as an alternative substrate, might obey the P–OR detaching rule to give the desired CAMDOL-PHO. To our delight, CAMDOL-POMe, derived from CAMDOL-PCl and NaOMe did transform into CAMDOL-PHO completely when managing it by routine column isolation directly. Moreover, this two-step synthesis could be conducted readily by a one-pot process without isolation of the intermediates CAMDOL-PCl and CAMDOL-POMe, affording the CAMDOL-PHO 1 in 85% isolated yield at a decagram scale. The absolute configuration of the P-chirality center for CAMDOL-PHO 1 was assigned as R-configuration based on its X-ray crystallography.

a CAMDOL (30 mmol), PCl3 (1.05 equiv.), Et3N (3 equiv.), THF, 0 °C, 1 h, >99% conversion; b H2O (10 equiv.), Et3N (10 equiv.), THF, 0 °C, 1 h; c NaOMe (2 equiv.), 0 °C, 1 h, >99% conversion; then quenched with saturated NH4Cl aqueous, and purified by flash column chromatography to give 1 in 85% isolated yield and >99:1 dr.
Pudovik reaction of aldehydes with CAMDOL-PHO
At the outset, to evaluate the validity of this center-chiral CAMDOL-PHO 1 in stereoselective Pudovik addition with aldehydes, benzaldehyde (PhCHO) 2a was selected as the model substrate to optimize the reaction conditions (Table 1). Initially, no desired α-hydroxyphsphonate 3a was observed when employing either inorganic base (K2CO3) or organic base (Et3N, pyridine, DBU, or BTMG) at 0 °C (entries 1–5). Further evaluation of other stronger bases (LiHMDS, NaHMDS, nBuLi, and LDA) at a relatively lower temperature (−40 °C) revealed that LDA exhibited superior conversion and selectivity (entries 6–9). Lowering the temperature from −40 °C to −78 °C with LDA as the base, the diastereomeric ratio of 3a increased from 93:7 to 97:3 without any loss in yield, indicating that a lower temperature has a beneficial impact on diastereoselective induction for this reaction (entries 9–11). Other types of solvents, such as PhMe, Et2O, DCM, MeOH, and EtOAc, all resulted in a remarkable reduction in both yields and diastereoselectivities, especially for the protonic solvent (MeOH) (entries 12–16). By augmenting the dosage of LDA from 1 to 1.2 equivalent, the best yield of 85% and dr value of 99:1 of 3a were achieved (entriy 17). Nevertheless, an identical yield but a lower dr was obtained when 1.5 equivalent of LDA was used (entry 18).
With the optimized conditions in hand, we then performed a series of experiments to define the scope of functional groups and electronic constraints that are readily tolerated on the aldehyde component. As shown in Fig. 3, numerous aromatic aldehydes can be readily employed with CAMDOL-PHO to produce the target chiral α-hydroxyphosphonates without any detectable loss in diastereopurity (3a–3y). An ortho-fluoro substituent of benzaldehyde, as well as an ortho-methoxyl group, brought about a little decrease in the yield and dr (3b and 3i). In comparison, those meta- or para-substituents (such as F, Cl, Br, OMe, Me, CF3, CN, and NO2) in benzaldehyde’s ring can be incorporated without significant impact on yield or selectivity, and all of these substrates could give nearly stereospecific products (3c–3h, 3j–3p) with 99:1 dr values. Surprisingly, nitrobenzaldehyde featuring other types of groups provided different results that ortho-nitrobenzaldehyde featuring para-sulfide substituent gave 3q in 65% yield and 99:1 dr, whereas meta-nitrobenzaldehyde featuring para-ether group gave 3r in only 55% yield and 90:10 dr. It should be mentioned that aromatic aldehydes bearing a reactive Br or Cl group do not undergo lithiation at the halogenated site or interfere with the reaction efficiency, allowing all the desired addition products in good yields and up to 99:1 dr values (3e–3h). Heteroaromatic aldehydes with an indole, thiophene, or furan ring, all occurred smoothly to deliver α-hydroxyphosphonates with good yields and high levels of enantiocontrol (3t–3v). The conjugated aromatic vinyl or acetylenyl aldehyde reacted selectively at the carbonyl site to generate the corresponding α-hydroxyphosphonates with good yields and excellent stereoseletivities (3w–3x). The fused aromatic aldehyde, 2-naphthaldehyde, allowed the formation of 3y in 88% yield and 99:1 dr. As for aliphatic (long chain alkyl, α-branched alkyl, cycloalkyl, and conjugated vinyl) aldehyde, generally a slight decrease in diastereopurity was observed due to their well-recognized less steric hindrance (3za–3zc, 3zd–3zg). The absolute configuration of the new chiral center for α-hydroxyphosphonate 3a was assigned as R-configuration based on its X-ray crystallography.

A Scope in aromatic aldehyde. B Scope in aliphatic aldehyde.
To demonstrate the preparative utility of our reaction, a 10 mmol scale-up synthesis was performed, and 3a was afforded in 78% yield and 99:1 dr (Fig. 4A). One of the most advantages of CA induction methods is the recoverability, so did the CAMDOL. Hydrolysis of 3a with conc. HCl afforded the α-hydroxyphosphonic acid 4a in 90% yield. In the meanwhile, more than 95% CAMDOL was recovered in this process. Methylation of crude 4a with diazomethane gave the dimethyl α-hydroxyphosphonate 4b in 98.5:1.5 dr with an almost quantitative conversion (Fig. 4B). The acidic hydrolysis of 3r and 3 s offered the CD-45 tyrosin phosphatase inhibitor 4c50 and myo-inositol tyrosine phosphatase inhibitor 4d51,52 both in 90% yields without loss of the C-chirality, as further confirmed by diazomethylation with TMSCHN2 (Fig. 4C).

A Gram scale-up experiment. B CAMDOL removal and O-methylation experiments. C Synthetic utility experiments.
Pudovik reaction of aldimines with CAMDOL-PHO
Based on the above investigations, our diastereoselective Pudovik reaction of aldimines was first evaluated by using N-benzylidene-p-toluenesulfonamide 5a with 1 and a series of bases (Table 2). A survey of the base in THF revealed that BTMG provided the highest yield and level of enantiocontrol for α-amino phosphonate 6a at −40 °C (entries 1–7). As the temperature of the reaction decreased, a dramatic increase in disastereoselectivity and reaction efficiency was achieved using BTMG as the base (entries 4, 8–11). Solvent screening experiments displayed that THF was the optimal solvent, indicating that reaction media indeed influence this stereoselective process remarkably (entries 11–16). By augmenting the dosage of BTMG from 1 to 1.2 equivalent, a yield of 95% and a dr of 99:1 of 6a were obtained for this transformation (entry 17), whereas a slight decrease in yield and diastereoselectivity was monitored when 1.5 equivalent LDA was used (entry 18).
Having identified optimal conditions of this diastereoselective Pudovik reaction for the synthesis of α-aminophosphonates, we aimed to define the scope of the aldimine precursors. As revealed in Fig. 5, a series of differently substituted aromatic and aliphatic aldimines were well tolerated with CAMDOL-PHO 1. Surprisingly, aromatic aldimines bearing a variety of electronic and steric groups, such as halo, alkyl, alkoxyl, and nitro, can be incorporated without significant impact on yield or selectivity (6a–6r). The fused aromatic aldimine, for example, 2-naphthaldimine, enabled the synthesis of 6 s in 95% yield and 99:1 dr. Heteroaromatic aldimines with a furan, thiophene, pyrrole, or indole ring, all occurred successfully to provide α-aminophosphonates with excellent yields and high levels of enantiocontrol (6t–6w). Notably, a strong electron-deficient pyridine ring on the aldimine component is readily accommodated to furnish 6x in a yield of 90% and a dr of 95:5. Differing from the aforementioned aldehyde-based reaction, aliphatic substituents were not detrimental to this process with good diastereoselectivites (up to 93:7 dr), illustrating the high-value of CAMDOL-PHO-induced diastereocontrol for Pudovik reaction. Specifically, linear, branched, and cyclic aliphatic carbon skeletons were all well tolerated (6ya–6yf). In comparison, several N-protecting groups were explored and the results showed that the Ns group (6zb) had a comparable effect on this reaction, whereas other groups (Boc, tBuSO, and PhPO) were all not beneficial for the conversion (6za, 6zc–6zd). The absolute configuration of the new chiral center for α-aminophosphonate of 6a was assigned as S-configuration by X-ray crystallography.

A Scope in aromatic aldimine. B Scope in aliphatic aldimine. C Scope in N-group of aldimine.
In order to examine the preparative utility of aldimine-based Pudovik reaction, the addition of CAMDOL-PHO 1 to 5a was performed on a 10 mmol scale with BTMG as the base to afford 6a in 90% yield and 99:1 dr (Fig. 6A). Enantiopure α-amino phenylphosphonic acid 7a was obtained in 95% yield from 6a using aqueous HCl in toluene at 110 °C, along with the recovery of CAMDOL in high yield (>95%). The CAMDOL skeleton of 6a could be selectively removed with HCl at a lower temperature (70 °C), followed by diazomethylation with TMSCHN2 to give 7aa without loss of the C-chirality (Fig. 6B). Hydrolysis of another α-amino phosphonate 6yc afforded the bioactive phospholeuine 7b, a LAP inhibitor53,54, whose er was determined via a manipulation similar to 7aa, yielding 7ba with an er of 99:1 (Fig. 6C).

A Gram scale-up experiment. B CAMDOL removal experiment. C Synthetic utility experiment.
Pudovik reaction of nitroalkenes with CAMDOL-PHO
Investigations towards the development of nitroalkene-based Pudovik reaction began by reacting CAMDOL-PHO with trans-β-nitrostyrene 8a under the above similar conditions (Table 3). We were pleased to find that K2CO3 promoted the reaction in a yield of 60% and a dr of 55:45, recommending the feasibility of the asymmetric synthesis of β-nitrophosphonates 9a (entry 1). Then, base screening experiments disclosed that NaHMDS could provide the high yield and level of enantiocontrol for 9a in yield of 75% and dr of 83:17 (entries 1–9). Decreasing the reaction temperature resulted in an increase of both conversion and efficiency (−78 °C, 90% yield, 93:7 dr; entries 10–11). The impact of reaction media on diastereoselectivity was probed with several aprotic solvents, and MeTHF was proved as the optimal solvent with a superior diastereomeric ratio of 95:5 (entries 11–16). Defining examples of the conversion were found that the optimization of the dosage of NaHMDS of 0.8 equivalent afforded a yield of 95% and a dr of 95:5, whereas 1.1 equivalent led to a slight decrease in both yield and diastereoselectivity of 9a (entries 17–18).
Having developed the optimal reaction conditions for diastereoselective Pudovik reaction for the synthesis of β-aminophosphonates, we next turned our attention to the nitroalkene substrate scope (Fig. 7). As disclosed in Fig. 7, a diverse range of nitroalkenes can be readily tolerated to furnish the desired enantioenriched β-nitrophosphonates. In most cases of the aromatic nitroalkenes, excellent levels of asymmetric induction and good conversion were obtained with either a meta- or a para-substituent (F, Cl, Br, CF3, CN, Me, iPr, and OMe) of 8a (9c–9d, 9f, 9h–9l, 9n–9p). Comparatively, an ortho-group of 8a, such as F, Br, and OMe, gave the products in relatively low yield and dr, suggesting that a neighboring group of 8a is adverse to this transformation (9b, 9g, and 9m). Nitroalkene incorporated with a naphthyl or phenanthryl ring was readily tolerated with even better diastereoselectivity (9q–9r). Especially for a more rigid phenanthryl ring, a dr of 99:1 was observed (9r). Except pyrrole (9s), other heteroaromatic nitroalkenes with a furan, thiophene, or pyridine ring, occurred successfully to provide β-nitrophosphonates with excellent yields and high levels of enantiocontrol (9t–9v). As for aliphatic nitroalkenes, straight-chain alkyl (9wa–9wb), branched alkyl (9wc), and cycloalkyl (9xa–9xc) all delivered the corresponding products with good yield and stereocontrol. The absolute configuration of the new chiral center for β-nitrophosphonate of 9a was assigned as R-configuration by X-ray crystallography.

A Scope in aromatic nitroalkene. B Scope in aliphatic nitroalkene.
A demonstration of the preparative utility of nitroalkene-based Pudovik reaction was presented by the addition of CAMDOL-PHO 1–8a, which was performed on a 10 mmol scale with NaHMDS as the base to afford 9a in 88% yield and 95:5 dr (Fig. 8A). After recrystallization, the dr value of 9a could be improved to 99:1. Exposure of 9a by catalytic NiCl2 with NH3·BH3, followed by removal of CAMDOL using HCl, provided the corresponding β-amino phenylphosphonic acid 10a—a potential GABAB receptor antagonist55,56—in 95% yield. Simultaneously, >95% of CAMDOL was recovered. The er of 10a was further confirmed by CAMDOL cleavage, followed by diazomethylation with TMSCHN2, to yield 10aa with 99:1 er (Fig. 8B).

A Gram scale-up experiment. B Synthetic utility experiment.
Mechanistic insights
Control experiments using Menthyl-PHO, TADDOL-PHO, and BINOL-PHO as representative chiral H-phosphonates were conducted for asymmetric Pudovik reaction with aldehyde 2a, aldimine 5a, and nitroalkene 8a, respectively. The results demonstrated that TADDOL-PHO exhibited higher conversion and diastereomeric ratio than Menthyl-PHO and BINOL-PHO, though still lower than CAMDOL-PHO (Fig. 9A). Notably, BINOL-PHO failed to react with any substrate under our optimal conditions. These findings highlight the significantly superior stereoinductive capability of centrally chiral CAMDOL-PHO over other CA-derived H-phosphonates in Pudovik reaction.

A Control experiments of the representative chiral H-phosphonates for Pudovik reaction under our optimal conditions. The dr of Menthyl-PHO, BINOL-PHO, TADDOL-PHO are all >99:1. B DFT calculations. i Energy profile for the P–C formation; ii Calculated structures of the transition states.
The results showed that both the conversion and diastereomeric ratio of TADDOL-PHO were superior to Menthyl-PHO and BINOL-PHO, albeit inferior to those of CAMDOL-PHO (Fig. 9A). Notably, BINOL-PHO did not react with any tested substrate under the corresponding optimal conditions. Obviously, the center-chiral CAMDOL-PHO has a very superior stereoinduction ability for Pudovik reaction than other types of CA derived H-phosphonates.
Density functional theory (DFT) calculations were carried out to further investigate the origin of the stereoselectivity in the Pudovik reaction of 2a with CAMDOL-PHO (Fig. 9B). Deprotonation of the phosphoric acid with LDA is barrierless and irreversible, yielding Li-phosphonate (Supplementary Fig. S349†). Coordination of 2a to the Li+ facilitates the formation of a stable complex I, which is prone to the hydride transfer process. Driven by alkali metal ion activation, the nucleophilic attack of phosphonate on 2a requires only a barrier of 1.7 kcal/mol to form the P–C bond. The work-up procedure with NH4Cl is performed to convert compound II into the final product 3a (Fig. 9B-i).
The privileged scaffold of CAMDOL induces an asymmetric reaction pocket (Fig. 9B-ii). The methyl group on the chiral carbon causes the adjacent phenyl group (Ph1) to adopt a vertical orientation, while the other phenyl group (Ph2) protrudes from the plane. This geometry divides the pocket into two distinct sections: a large area and a small area, allowing for the differentiation of substituents on the substrates. Upon examining the enantiomeric transition states for P–C bond formation, it was observed that significant steric hindrance occurs between the phenyl group of 2a and the extended Ph2 of CAMDOL-PHO (Fig. 9B-ii, TS-S). On the contrary, the orientation of Ph1 reserves ample space for the bulky substituent of the substrate, resulting in a lower energy transition state TS-R that favors the production of the R-configured product II. The energy barrier difference between the two transition states is 2.5 kcal/mol, corresponding to a calculated ee value of 97%, which closely aligns with the experimental value of 98%.
Discussion
In summary, we have developed CAMDOL-PHO, a novel centrally chiral H-phosphonate, for diastereoselective Pudovik reaction, allowing direct access to carbon-phosphorus stereogenicity in a wide variety of structural contexts. Based on CAMDOL-PHO, three subtypes of Pudovik reaction, involving aldehydes, aldimines, and nitroalkenes were established, displaying broad functional group tolerance, operational simplicity, scalable practicability, and diverse applicability for the preparation of bioactive molecules. Furthermore, these hydrophosphorylations of unsaturated bonds demonstrate atom economy, as all key starting materials are incorporated into the products, coupled with facile recovery of the CA for late-stage diversification. We anticipate that this P-chiral H-phosphonate will complement existing CA-PHOs in asymmetric induction and emerge as a privileged scaffold for constructing high-value molecular architectures.
Methods
Synthesis of CAMDOL-PHO (1)
A 500 mL three-necked flask equipped with a stirrer was charged with CAMDOL (6.44 g, 20 mmol). The flask was sealed with a gas-tight septum and subjected to evacuation, followed by three cycles of backfilling with N₂. Subsequently, anhydrous THF (200 mL) was added, followed by the addition of Et3N (8.4 mL, 3 equiv.) using a syringe. The reaction mixture was stirred at 0 °C for 0.5 h. Thereafter, PCl3 (19.2 mL, 1 M in THF, 1.1 equiv.) was introduced dropwise via syringe at 0 °C and stirred for an additional 2 h. During this period, 31P NMR was employed to confirm the complete conversion of the reaction. Then, NaOMe (7.5 mL, 5.4 M, 3 equiv.) was added at 0 °C for another 2 h. After the full conversion confirmed by 31P NMR, the reaction mixture was quenched with saturated aqueous NH4Cl solution and allowed to stir until no further effervescence occurred. The phases were then separated, the aqueous layer underwent extraction twice with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. Finally, purification of the crude product was achieved through flash column chromatography.
General procedure for Pudovik reaction
As for the aldehyde-based Pudovik reaction, a 25 mL Schlenk tube equipped with a stirrer was charged with CAMDOL-PHO (1, 0.4 mmol, 1 equiv.). When a solid aldehyde was employed, it was added at this stage. The vial was sealed with a gas-tight septum and subjected to evacuation followed by three cycles of backfilling with N2. Subsequently, dry THF (4 mL) was introduced, followed by the addition of aldehyde (2, 1.2 equiv.) using a microsyringe. The reaction mixture was stirred at −78 °C for 0.5 h. Thereafter, LDA (0.48 mL, 1 M in THF, 1.2 equiv.) was administered dropwise via syringe at −78 °C and stirred for an additional 24 h. During this period, 31P NMR was utilized to verify the complete conversion of the reaction. Next, the reaction was quenched with saturated aqueous NH4Cl solution and allowed to stir until no further effervescence was observed. The phases were then separated, and the aqueous layer underwent extraction twice with EtOAc. The combined organic layers were dried over Na2SO4, filtered, concentrated, and finally purified by flash column chromatography.
As for the aldimine-based Pudovik reaction, aldehyde was replaced with aldimine (5, 1.2 equiv.), and LDA was substituted with BTMG (1.2 equiv.) on the basis of above conditions. As for the nitroalkene-based Pudovik reaction, THF was substituted with 2-MeTHF, aldehyde was replaced with nitroalkene (8, 1.2 equiv.), and LDA was replaced with LiHMDS (0.8 equiv.).
Data availability
General information, experimental details, and analytical data: NMR spectra, HRMS data and computational details can be found in the Supplementary Information. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2402270 (1), 2330640 (3a), 2330642 (6a), and 2330643 (9a), respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif and are enclosed as Supplementary Data 1–4. Computational chemistry details are available in Supplementary Data 5–6. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Vel’tishchev, Y. E., Yur’eva, ÉA., Kudrin, A. N., Korytnyi, A. M. & Varsanovich, E. A. Biologically-active phosphonic acids and their rerivatives (review). Pharm. Chem. J. 17, 175–182 (1983).
Tusek–Bozic, L. Aminophosphonate metal complexes of biomedical potential. Curr. Med. Chem. 20, 2096–2117 (2013).
Demkowicz, S., Rachon, J., Daśko, M. & Kozak, W. Selected organophosphorus compounds with biological activity. Appl. Med. RSC Adv. 6, 7101–7112 (2016).
Horsman, G. P. & Zechel, D. L. Phosphonate biochemistry. Chem. Rev. 117, 5704–5783 (2017).
Lorke, D. E., Stegmeier-Petroianu, A. & Petroianu, G. A. Biologic activity of cyclic and caged phosphates: a review. J. Appl. Toxicol. 37, 13–22 (2017).
Dembitsky, V. M., Gloriozova, T. A. & Savidov, N. Steroid phosphate esters and phosphonosteroids and their biological activities. Appl. Microbiol. Biotechnol. 102, 7679–7692 (2018).
Yu, H., Yang, H., Shi, E. & Tang, W. Development and clinical application of phosphorus-containing drugs. Med. Drug Discov. 8, 100063 (2020).
Manghi, M. C. et al. The use of phosphonates in agriculture. Chemical, biological properties and legislative issues. Chemosphere 283, 131187 (2021).
Kolodiazhna, A. O. & Kolodiazhnyi, O. I. Chiral organophosphorus pharmaceuticals: properties and application. Symmetry 15, 1550 (2023).
Ung, S. P. M. & Li, C.-J. From rocks to bioactive compounds: a journey through the global P(v) organophosphorus industry and its sustainability. RSC Sustain. 1, 11–37 (2023).
Matoba, K., Yonemoto, H., Fukui, M. & Yamazaki, T. Structural modification of bioactive compounds. II. Syntheses of aminophosphonoic acids. Chem. Pharm. Bull. 32, 3918–3925 (1984).
Adden, N. et al. Phosphonic acid monolayers for binding of bioactive molecules to titanium surfaces. Langmuir 22, 8197–8204 (2006).
Kukhar, V. P., Sorochinsky, A. E. & Soloshonok, V. A. Practical synthesis of fluorine-containing α- and β-amino acids: recipes from Kiev, Ukraine. Future Med. Chem. 1, 793–819 (2009).
Bansal, R. K. (ed.) Phosphorous Heterocycles I (Springer Berlin, 2009).
Naydenova, E. D., Todorov, P. T. & Troev, K. D. Recent synthesis of aminophosphonic acids as potential biological importance. Amino Acids 38, 23–30 (2010).
Villemin, D. & Didi, M. A. Aminomethylenephosphonic acids syntheses and applications (a review). Orient. J. Chem. 31, 1–12 (2015).
Kaboudin, B., Daliri, P., Faghih, S. & Esfandiari, H. Hydroxy- and amino-phosphonates and -bisphosphonates: synthetic methods and their biological applications. Front. Chem. 10, 890696 (2022).
Huang, Z., Wang, K.-K. A. & van der Donk, W. A. New insights into the biosynthesis of fosfazinomycin. Chem. Sci. 7, 5219–5223 (2016).
Takeuchi, M. et al. Fosfonochlorin, a new antibiotic with spheroplast forming activity. J. Antibiot. 42, 198–205 (1989).
Falagas, M. E., Vouloumanou, E. K., Samonis, G. & Vardakas, K. Z. Fosfomycin. Clin. Microbiol. Rev. 29, 321–347 (2016).
Dijkmans, A. C. et al. Fosfomycin: pharmacological, clinical and future perspectives. Antibiotics 6, 24 (2017).
Atherton, F. R. et al. Phosphonopeptide antibacterial agents related to alafosfalin: design, synthesis, and structure-activity relationships. Antimicrob. Agents Chemother. 18, 897–905 (1980).
Lejczak, B., Kafarski, P., Sztajer, H. & Mastalerz, P. Antibacterial activity of phosphono dipeptides related to alafosfalin. J. Med. Chem. 29, 2212–2217 (1986).
Yaffe, M. B. Phosphotyrosine-binding domains in signal transduction. Nat. Rev. Mol. Cell Biol. 3, 177–186 (2002).
Vogel, W., Lammers, R., Huang, J. & Ullrich, A. Activation of a phosphotyrosine phosphatase by tyrosine phosphorylation. Science 259, 1611–1614 (1993).
Kolodiazhnyi, O. I. Asymmetric synthesis of hydroxyphosphonates. Tetrahedron Asymmetry 16, 3295–3340 (2005).
Cao, H.-Q., Li, J.-K., Zhang, F.-G., Cahard, D. & Ma, J.-A. Asymmetric synthesis of chiral amino carboxylic-phosphonic acid derivatives. Adv. Synth. Catal. 363, 688–729 (2021).
Merino, P., Marqués-López, E. & Herrera, R. P. Catalytic enantioselective hydrophosphonylation of aldehydes and imines. Adv. Synth. Catal. 350, 1195–1208 (2008).
Zhao, D. & Wang, R. Recent developments in metal catalyzed asymmetric addition of phosphorus nucleophiles. Chem. Soc. Rev. 41, 2095–2108 (2012).
Kolodiazhnyi, O. I., Kukhar, V. P. & Kolodiazhna, A. O. Asymmetric catalysis as a method for the synthesis of chiral organophosphorus compounds. Tetrahedron Asymmetry 25, 865–922 (2014).
Semenzin, D., Etemad-Moghadam, G., Albouy, D., Diallo, O. & Koenig, M. Dual radical/polar Pudovik reaction: application field of new activation methods. J. Org. Chem. 62, 2414–2422 (1997).
Salin, A. V. et al. The Pudovik reaction catalyzed by tertiary phosphines. Curr. Org. Synth. 13, 132–141 (2016).
Fener, B. E. et al. Scope and limitations of the s-block metal-mediated Pudovik reaction. Chem. Eur. J. 26, 7235–7243 (2020).
Abell, J. P. & Yamamoto, H. Catalytic enantioselective Pudovik reaction of aldehydes and aldimines with tethered bis(8-quinolinato) (TBOx) aluminum complex. J. Am. Chem. Soc. 130, 10521–10523 (2008).
Yokomatsu, T., Yamagishi, T. & Shibuya, S. Enantioselective synthesis of α-hydroxyphosphonates through asymmetric Pudovik reactions with chiral lanthanoid and titanium alkoxides. J. Chem. Soc. Perkin Trans. 1, 1527–1534 (1997).
Gnas, Y. & Glorius, F. Chiral auxiliaries-principles and recent applications. Synthesis 2006, 1899–1930 (2006).
Pellissier, H. Asymmetric domino reactions. Part A: reactions based on the use of chiral auxiliaries. Tetrahedron 62, 1619–1665 (2006).
Meggers, E. Chiral auxiliaries as emerging tools for the asymmetric synthesis of octahedral metal complexes. Chem. Eur. J. 16, 752–758 (2010).
Diaz-Muñoz, G., Miranda, I. L., Sartori, S. K., de Rezende, D. C. & Alves Nogueira Diaz, M. Use of chiral auxiliaries in the asymmetric synthesis of biologically active compounds: a review. Chirality 31, 776–812 (2019).
Sun, Y.-M. et al. Addition of optically pure H-phosphinate to ketones: selectivity, stereochemistry and mechanism. Org. Biomol. Chem. 12, 9457–9465 (2014).
Gbubele, J. D. & Olszewski, T. K. Asymmetric synthesis of organophosphorus compounds using H–P reagents derived from chiral alcohols. Org. Biomol. Chem. 19, 2823–2846 (2021).
Farnham, W. B., Murray, R. K. Jr. & Mislow, K. Stereospecific alkylation of menthyl phenylphosphinate. J. Am. Chem. Soc. 92, 5809–5810 (1970).
Koeller, K. J. & Spilling, C. D. The preparation and reactions of chiral phosphorous acid diamides. Tetrahedron Lett. 32, 6297–6300 (1991).
Greene, N. & Kee, T. P. Asymmetric Silylphosphite Esters: Synthesis and Reactivity of (rac-O,O-Binaphtholato)POSiR3 (R3 = Ph3, tBuMe2, Et3). Synth. Commun. 23, 1651–1657 (1993).
Enders, D., Tedeschi, L. & Bats, J. W. Asymmetric synthesis of α-substituted β-nitrophosphonic acids by phospha-analogous Michael addition to aromatic nitroalkenes. Angew. Chem. Int. Ed. 39, 4605–4607 (2000).
Zhang, Y. et al. Universal and divergent P-stereogenic building with camphor-derived 2,3-diols. Commun. Chem. 6, 133 (2023).
Huang, Y. et al. CAMDOL-enabled diastereoselective synthesis of α-substituted phosphonates. Chem. Commun. 60, 1924–1927 (2024).
Linghu, X., Potnick, J. R. & Johnson, J. S. Metallophosphites as umpolung catalysts: the enantioselective cross silyl benzoin reaction. J. Am. Chem. Soc. 126, 3070–3071 (2004).
Guo, H., Wu, Q., Wang, S., Shu, H. & Shi, E. Facile synthesis of H-phosphinates from P(OR)3 or ClP(OR)2 via SiO2-promoted hydrolysis. J. Org. Chem. 89, 8915–8923 (2024).
Lee, K. & Burke, T. R. Jr. CD45 protein-tyrosine phosphatase inhibitor development. Curr. Top. Med. Chem. 3, 797–807 (2003).
Fujimoto, S., Tsuda, J., Kawakami, N., Tanino, H. & Shimohama, S. myo-Inositol monophosphatase in the brain has zinc ion-dependent tyrosine phosphatase activity. Gen. Pharmacol. 31, 469–475 (1998).
Puhl, A. A. et al. Kinetic and structural analysis of a bacterial protein tyrosine phosphatase-like myo-inositol polyphosphatase. Protein Sci. 16, 1368–1378 (2007).
Giannousis, P. P. & Bartlett, P. A. Phosphorus amino acid analogs as inhibitors of leucine aminopeptidase. J. Med. Chem. 30, 1603–1609 (1987).
Grembecka, J., Mucha, A., Cierpicki, T. & Kafarski, P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J. Med. Chem. 46, 2641–2655 (2003).
Cates, L. A. et al. Phosphorus analogs of γ-aminobutyric acid, a new class of anticonvulsants. J. Med. Chem. 27, 654–659 (1984).
Froestl, W. et al. Phosphinic acid analogs of GABA. 1. New potent and selective GABAB agonists. J. Med. Chem. 38, 3297–3312 (1995).
Author information
Authors and Affiliations
Contributions
J.X. and E.S. conceived the project and designed the experiments. E.S. directed the project. J.L. directed the mechanistic investigations. N.L., Q.W., and Y.H. performed the reaction. N.L. and L.P. performed the mechanistic studies. E.S. and J.L. prepared the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, N., Wu, Q., Huang, Y. et al. Stereoselective Pudovik reaction of aldehydes, aldimines, and nitroalkenes with CAMDOL-derived H-phosphonate. Commun Chem 8, 349 (2025). https://doi.org/10.1038/s42004-025-01735-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-025-01735-4
