
Abstract
Palladium-catalyzed switchable β-carbon elimination to extrude norbornene for selective synthesis of different heterocyclic skeletons is a long-standing challenge. Herein, we describe the switchable chemoselective synthesis of biologically active C4-carbamoylated indoles and C10-aminophenanthridinones by controlling β-carbon elimination, using chlorocarbamates as C–H carbamoylation reagents. Among them, C4-carbamoylated indoles are used for the synthesis of M2 receptor antagonists and p38α inhibitors. Mechanistic studies show that when β-carbon elimination is inhibited, the catalytic cycle goes through C–N bond coupling and retro-Diels-Alder reaction to generate C4-carbamoylated indoles. Alternatively, when β-carbon elimination to extrude norbornene occurs, an intramolecular C–H arylation cyclization reaction generates C10-aminophenanthridinones.

Similar content being viewed by others
Introduction
Indoles and phenanthridinones feature unique biological activities and are key structural motifs in small molecule drugs and natural products1,2,3,4,5,6,7. Among them, carbamoylated indole8,9 and amino phenanthridinone10,11 are important drug building blocks for pharmaceuticals. For example, C4-carbonyl piperidine indoles have been evaluated as M2 receptor antagonists for the treatment of Alzheimer’s disease12, while their difluoro derivatives are considered antagonists of orexin receptors for the treatment of insomnia (Scheme 1)13. C4-Carbocyclic amide indoles are labeled as monoacylglycerol lipase modulators for the treatment of pain, psychiatric disorders, and neurological disorders14. Specifically, a C4-carbonyl piperazinyl indole has been identified as highly active p38α inhibitors (IC50 = 1.54 μM)15. On the other hand, amino phenanthridinone is a potent inhibitor of poly(ADP-ribose) polymerases PARP1 and PARP2 and exhibits anti-cancer, cardioprotective, and neuroprotective properties10,11. Therefore, the efficient and rapid assembly of C4-carbamoylated indoles and C10-amino phenanthridinones is of continued strong importance for drug development.

A Bioactive molecules. B Classic Pd/NBE chemistry. C The “ortho constraint” group is an amino group. D Overcome the Pd–N coordination preference. E Switchable β-carbon elimination. F Synthesis of C4-carbamoylated indoles and C10-aminophenanthridinones via β-carbon elimination switch.
Palladium and norbornene (Pd/NBE) cooperative catalysis16,17,18,19,20,21,22,23,24,25,26, ortho-C–H functionalization of iodobenzene, has attracted widespread attention and was first pioneered by Catellani27. Likewise, Lautens disclosed innovative palladium and norbornene-catalyzed domino cyclization reactions, which have been applied in the chemical synthesis of natural products28,29,30,31,32,33,34. Among them, β-carbon elimination to extrude norbornene is a key step in the Pd/NBE-catalyzed cycle and has attracted considerable attention. In 2018, the Dong group made a breakthrough discovery, exploiting the steric effect of C1-modified norbornenes to simulate “ortho constraint” and achieved the first example of single C–H functionalization of iodobenzene without a ortho substituent35,36,37. Recently, they further utilized this strategy to accelerate the β-carbon elimination, suppressing the formation of nitrene cyclization products, and successfully achieved ortho C–H secondary amination38. Interestingly, the Jiao group employed heterocyclic olefin ligands to achieve a β-carbon elimination similar to norbornene39,40. In particular, when iodobenzene has an amino group at the ortho position, Lautens found that the strong coordination effect of the amino group induce the reaction to proceed towards C–N bond coupling41. In 2019, we effectively altered the direction of the reaction by modulating the groups on o-iodoaniline, steering the reaction towards C–H bond functionalization and subsequently enabling C–N bond coupling cyclization42,43,44,45. The rapid C–N coupling precluded the subsequent β-carbon elimination to extrude norbornene derivatives. Therefore, controllable β-carbon elimination to extrude norbornene for the selective synthesis of two different scaffolds remains elusive. Herein, we describe a strategy for the chemo-divergent synthesis of bioactive C4-carbamoylated indoles or C10-amino phenanthridinone through the judicious choice of the reaction conditions for selective β-carbon elimination.
Results and discussion
Reaction conditions and chemo-divergent synthesis
We initiated our studies with o-iodoanilines with different protecting groups on the amine (1a, 1aa-1ae) as substrates, phenylcarbamic chloride as the C–H carbamoylation reagent, and norbornene as the transient mediator to optimize the reaction conditions. To our delight, when we used o-iodoaniline 1a as the substrate and P(m-MeO-C6H4)3 as the ligand, we obtained the C10-amino phenanthridinone product 4a with a yield of 24%. The main side product was the intramolecular C–N bond coupling cyclization product 5, which resulted from the failure of norbornene extrusion (Table 1). Subsequently, we probed the reaction temperature and ligands. When the temperature was raised to 120 °C and P(m-MeO-C6H4)3 was used as the ligand, the yield increased to 35%. Then, different bases were tested, and we found that potassium carbonate could significantly increase the yield of the target product to 51%. Finally, we evaluated different additives. Notably, the addition of a catalytic amount of pivalic acid further increased the yield to 88%, while the formation of byproduct 5 was completely suppressed.
Due to the formation of byproduct 5, we attempted to replace norbornene (NBE) with norbornadiene (NBD), speculating that byproduct 5 would undergo a retro-Diels-Alder reaction to generate the C4-carbamoylated indole product 3a. After systematic optimization, we were pleased to find that when using P-(m-F-C6H4)3 as the ligand, cesium carbonate as the base, and cesium fluoride as the additive, the desired product 3a could be obtained in a yield of 32% after stirring at 100 °C for 12 h, followed by a reaction at 140 °C for another 12 h. Subsequently, we further probed various palladium sources and found that palladium trifluoroacetate (Pd(TFA)2) provided the highest yield of 53%. Additionally, we screened norbornadiene derivatives (N1 and N2), but no improvement catalytic efficiency was observed. Under these conditions, we further tested different N-groups on the nitrogen of o-iodoaniline, albeit with the original aniline 1a providing optimal results. Based on the by-products we detected, we speculate that the steric hindrance of the tert-butyl group plays two key roles: first, it effectively suppresses direct C−N coupling prior to ortho C−H functionalization; second, it inhibits the nucleophilic substitution between carbamic chloride and the amino group. Finally, we obtained the optimal reaction conditions for selectively generating the C4-carbamoylated indole product (3a, Condition A) and the C10-amino phenanthridinone product (4a, Condition B).
Substrate scope and product derivatization
With the optimal conditions in hand (for detailed procedures, see “Methods” section), we explored the robustness towards decorated C4-carbamoylated indoles (Scheme 2, Nuclear magnetic spectrums are provided in the NMR spectra file). First, the scope of the C−H carbamoylation reagent was investigated. Both electron-donating (−Me, −OMe) and electron-withdrawing (−F, −CO2Me) substituted phenylcarbamic chlorides were found to yield the desired products in moderate to good yields. It is worth noting that the highly reactive N-allyl-carbamoyl chloride reagent also smoothly provided the C4-carbamoylated indole product 3h with a yield of 34%. Subsequently, we attempted to synthesize various C4 non-cyclic carbamoyl-substituted indoles, such as those containing dimethylamine, diethylamine, and dibenzylamine, and obtained the corresponding products in good to excellent yields (57–81%). Additionally, various C4-cyclic carbamoyl-substituted indoles could be synthesized with excellent yields, regardless of whether the cyclic amides were five-membered, six-membered, or seven-membered rings. Particularly, acid-labile acetals and piperazines, which are very common in drug building blocks, are also compatible with this reaction. The corresponding indole products 3o and 3q are precursors to M2 receptor antagonists and p38α inhibitors, respectively, highlighting the practical value of this method in medicinal chemistry. It is noteworthy that reactions of the same substrate with phenylcarbamoyl chloride under two distinct conditions proceed with high chemoselectivity: Condition A affords carbamoylated indoles (>20:1 selectivity), while Condition B also exhibits high selectivity, yielding amino phenanthridinones.

Reaction conditions: substrate 1 (0.2 mmol), 2 (0.4 mmol, 2.0 equiv), Pd(TFA)2 (10 mol%), P-(m-F-C6H4)3 (20 mol%), norbornadiene (0.5 mmol, 2.5 equiv), Cs2CO3 (0.8 mmol, 4.0 equiv), CsF (0.05 mmol, 0.25 equiv), toluene (2.0 mL), 100 °C 12 h, and then 140 °C, 12 h. Isolated yields. a1.0 mmol scale.
Next, we explored the functional group tolerance of o-iodoanilines. Electron-donating groups such as methyl (-Me) and methoxy (-OMe), as well as electron-withdrawing groups, including ester (-CO₂Me), trifluoromethyl (-CF₃), cyano(-CN), and especially the strongly electron-withdrawing nitro (-NO₂) group, were all well-tolerated, providing the desired products in good to excellent yields. Halogen groups (-F, -Cl) that can alter the lipophilicity of drugs are also compatible with this transformation. It is worth noting that this reaction can also be applied to the synthesis of C4-carbamoylated azaindole (3ri). Furthermore, N-adamantyl indole (3rj) could be obtained using this method.
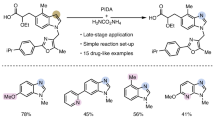
We carried out studies on the deprotection and derivatization of the thus-obtained products 3 to enhance the application value of the reaction (Scheme 3, for detailed procedures, see Section 3.1–3.3 in the Supplementary Information). Firstly, the C4-carbamoylated indoles can be easily deprotected using hydrochloric acid to remove the tert-butyl protecting group, and an aldehyde group can be introduced at the C3 position using N,N-dimethylformamide (DMF). In addition, the Boc protecting group on the piperazine nitrogen of compound 3q can be efficiently removed by the addition of silica gel and heating, furnishing product 3q’ as precursor for the synthesis of p38α inhibitors.

A Formylation at the C3-position of indole and protecting group removal. B Synthesis of a p38α inhibitor precursor by Boc deprotection of indole 3q.
After exploring the substrate scope of the C4-carbamoylated indoles, we immediately proceeded to investigate the functional group tolerance for the synthesis of C10-amino phenanthridinones (Scheme 4, for detailed procedures, see “Methods” section). Electron-donating groups (-Me) and electron-withdrawing groups (-CO2Me, -NO2 and -CF3), as well as halogens (-F, -Cl and -Br) on o-iodoanilines 1, were all well-tolerated and provide the desired products in excellent yields. Notably, the synthesis of aza-C10-amino phenanthridinone was also achievable via our strategy with a yield of 67%. Subsequently, the functional group tolerance of various phenylcarbamic chlorides was also tested, and it was found that methoxy, ester, and fluoro groups could all be tolerated to afford the desired products 4 in excellent yields. Interestingly, we also attempted the reaction using allyl carbamoyl chloride, and the olefin coupling cyclization product was selectively furnished under otherwise identical conditions.

Reaction conditions: substrate 1 (0.2 mmol), 2 (0.4 mmol, 2.0 equiv), Pd(OAc)2 (10 mol%), P(m-MeO-C6H4)3 (20 mol%), NBE (0.2 mmol, 1.0 equiv), K2CO3 (0.8 mmol, 4.0 equiv), PivOH (0.1 mmol, 0.5 equiv), toluene (2.0 mL), 120 °C, Ar. Isolated yields.
Mechanistic investigation by density functional theory (DFT) calculations
Based on the optimized conditions, we can infer that substrate 1a initially undergoes an oxidative addition reaction onto a palladium(0) species to form intermediate I. It then undergoes migratory insertion with norbornadiene, accompanied by C–H bond activation and cyclization, leading to the formation of the five-membered-ring palladium intermediate II. Subsequently, oxidative addition with chloroformamide occurs, forming intermediate III, which then undergoes reductive elimination to generate intermediate IV, featuring ortho-carbamoylation of iodobenzene. At this stage, two pathways emerge, leading respectively to the desired C4-carbamoylated indoles and C10-amino phenanthridin-6(5H)-ones.
Therefore, we investigated the reaction mechanism using density functional theory (DFT) calculations (see Section 5.1 in the Supplementary Information). We first calculated intermediate IV with different anions and found that intermediates A and A-I, coordinated with chloride and iodide ions respectively, have similar relative Gibbs free energies (Scheme 5B). However, their coordination with cesium carbonate releases 3.5 kcal/mol and 2.1 kcal/mol of energy, respectively. Subsequently, bidentate coordination of cesium carbonate to palladium displaces the chloride or iodide ions, releasing a significant amount of energy. Consequently, the bidentate cesium carbonate-coordinated intermediate C is the most stable and is identified as the key intermediate for the subsequent steps. The cesium carbonate of intermediate C can deprotonate the aniline to generate the dearomatized intermediate D, which subsequently undergoes palladium migration and coordinates with the amino group to form intermediate E (with an energy barrier of 23.3 kcal/mol). At this stage, the coordination between cesium carbonate and palladium in intermediate E is very weak, allowing another molecule of cesium carbonate to easily remove it, leading to the formation of intermediate F. Intermediate F undergoes a reductive elimination process to produce compound G, along with the formation of a Pd(0) species that participates in the next catalytic cycle. Finally, a retro-Diels–Alder reaction occurs at high temperature to produce the final product 3a. Intermediate C can also undergo a β-carbon elimination transition state TS-CD1 to form intermediate D1. The energy barrier for this step is 29.3 kcal/mol, so under the optimal reaction conditions, the reaction proceeds in the direction of forming the target product 3a. This process subsequently undergoes a cesium carbonate-assisted C–H bond activation and cyclization reaction, leading to the formation of product 4a.

A Possible reaction mechanisms for two competing products. B Intermediate IV with different anions. C Density functional theory (DFT) to study the reaction pathway at the PBE0-D3BJ/Def2-TZVP, SMD (toluene)//PBE0-D3BJ/Def2-SVP, 373.15 K level of theory.
Based on the mechanistic insights above and prior studies on pivalate-assisted C–H bond activation46,47, we propose a plausible reaction mechanism (Scheme 6). Initially, a Pd(0) complex undergoes oxidative addition with o-iodoaniline 1a to form intermediate I. Subsequent carbonate-assisted C–H activation generates a five-membered palladacycle II. It then engages in oxidative addition with phenylcarbamoyl chloride, followed by reductive elimination to yield o-carbamoylated intermediate IV. At this stage, the catalytic cycle diverges into two distinct pathways:

Pathway A Catalytic cycle for the synthesis of C4-carbamoylated indoles. Pathway B Mechanistic pathway for the construction of C10-aminophenanthridinones.
Pathway A: Intermediate IV undergoes C–N bond coupling followed by a retro-Diels-Alder reaction to afford indole product 3a.
Pathway B: β-Carbon elimination of intermediate IV extrudes norbornene (see Supplementary Fig. 1 in the Supplementary Information), generating a palladium species that coordinates with pivalic acid to form intermediate V2. Finally, a pivalate-assisted concerted metalation-deprotonation (CMD) process delivers the amino phenanthridinone product.
Conclusions
In summary, our findings addressed the long-standing challenge of constructing diverse heterocyclic skeletons by a chemo-divergent assembly strategy governed by β-carbon elimination pathways. With chlorocarbamates as versatile C–H carbamoylation reagents, a dual-path synthetic platform was established: (a) When β-carbon elimination is suppressed, the catalytic cycle proceeds via C–N bond coupling and retro-Diels-Alder reaction to furnish biologically significant C4-carbamoylated indoles, pivotal intermediates for M2 receptor antagonists and p38α inhibitors. (b) In sharp contrast, enabling β-carbon elimination to extrude norbornene triggers an intramolecular C–H arylation, efficiently delivering C10-amino phenanthridinone scaffolds. This work not only provides a versatile platform for constructing diverse heterocyclic skeletons but also offers valuable insights into the molecular control of β-carbon elimination processes. This offers a new concept and approach for chemoselectivity research in organic chemistry.
Methods
Synthesis of C4-carbamoylated indole 3 (general procedure)
In a 20 mL tube, 1 (0.2 mmol), Pd(TFA)2 (10 mol%), P(m-F-C6H4)3 (20 mol%), Cs2CO3 (0.8 mmol, 4.0 equiv.) and CsF (0.05 mmol, 0.25 equiv.) were added and charged with argon more than three times (The tube was sealed with tipping plug). Toluene (2 mL), 2 (0.4 mmol, 2.0 equiv.) and NBD (0.5 mmol, 2.5 equiv.) were injected into the tube via plastic syringes. The resulting yellow suspension was stirred vigorously at room temperature for 15 min before being placed in a pre-heated oil bath at 100 °C stirring at 900–1200 rpm for 12 h, then rises to 140 °C for 12 h. After the reaction was completed, the residue was purified with chromatography column on silica gel or preparative TLC (PTLC) (petroleum ether/EtOAc = 1:1–4:1).
Synthesis of C10-amino phenanthridin-6(5H)-one 4 (general procedure)
In a 20 mL tube, 1 (0.2 mmol), 2 (0.4 mmol, 2.0 equiv.), Pd(OAc)2 (10 mol%), P(m-MeO-C6H4)3 (20 mol%), K2CO3 (0.8 mmol, 4.0 equiv.) and PivOH (0.1 mmol, 0.5 equiv.) were added and charged with argon more than three times (The tube was sealed with tipping plug). NBE (0.2 mmol, 1.0 equiv.) were dissolved in toluene (2 mL), and the mixture was injected into the tube via plastic syringes. The resulting yellow suspension was stirred vigorously at room temperature for 15 min before being placed in a pre-heated oil bath at 120 °C stirring at 900–1200 rpm for 12 h. After the reaction was completed, the residue was purified with chromatography column on silica gel or preparative TLC (PTLC) (petroleum ether/EtOAc = 5:1–3:1).
Data availability
Detailed experimental procedures, characterizations of new compounds and computational studies details are available in Supplementary Information. 1H and 13C NMR spectra can be found in the Supplementary Data 1. Extra data are available from the corresponding author upon request.
References
Taylor, R. D., MacCoss, M. & Lawson, A. D. G. Rings in drugs. J. Med. Chem. 57, 5845–5859 (2014).
Zhang, S., Zhang, S., Wang, Y., Zhang, Y., Liang, S., Fan, S., Chen, D. & Liu, G. Discovery of novel phenanthridone derivatives with anti-streptococcal activity. Arch. Microbiol. 205, 371 (2023).
Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 22, 111–126 (2005).
Josephitis, C. M., Nguyen, H. M. H. & McNally, A. Late-stage C–H functionalization of azines. Chem. Rev. 123, 7655–7691 (2023).
Teng, S. & Zhou, J. S. Metal-catalyzed asymmetric heteroarylation of alkenes: diverse activation mechanisms. Chem. Soc. Rev. 51, 1592–1607 (2022).
Liu, J., Xiao, X., Lai, Y. & Zhang, Z. Recent advances in transition metal-catalyzed heteroannulative difunctionalization of alkenes via C–H activation for the synthesis of heterocycles. Org. Chem. Front. 9, 2256–2279 (2022).
Mohanty, S. R., Prusty, N., Nanda, T., Mahulkar, P. S. & Ravikumar, P. C. Directing group-assisted selective C–H activation of six-membered N-heterocycles and benzo-fused N-heterocycles. Org. Chem. Front. 11, 540–575 (2024).
Duan, S.-F. et al. Research status of indole-modified natural products. RSC Med. Chem. 14, 2535–2563 (2023).
Zhu, Y. et al. Research progress of indole compounds with potential antidiabetic activity. Eur. J. Med. Chem. 223, 113665 (2021).
Schwartz, B. & Yu, Y. Pharmaceutical combination for treatment of cancer. US 2020/0155521 A1 (2020).
Fugger, K. & West, S. Treatment of HR deficient cancer. WO 2021/048235 A1 (2021).
Palani, A. et al. Isopropyl amide derivatives of potent and selective muscarinic M2 receptor antagonists. Bioorg. Med. Chem. Lett. 14, 1791–1794 (2004).
Kuduk, S., Cooke, A., Stump, C. & Williams, P. 4-Fluoropiperidine orexin receptor antagonists. WO2014113303 (2014).
Ameriks, M. K. et al. Monoacylglycerol lipase modulators. US11993601B2 (2023).
Mavunkel, B. J. et al. Indole-based heterocyclic inhibitors of p38α MAP kinase: designing a conformationally restricted analogue. Bioorg. Med. Chem. Lett. 13, 3087–3090 (2003).
Ye, J. & Lautens, M. Palladium-catalysed norbornene-mediated C–H functionalization of arenes. Nat. Chem. 7, 863–870 (2015).
Della Ca, N., Fontana, M., Motti, E. & Catellani, M. Pd/Norbornene: a winning combination for selective aromatic functionalization via C–H bond activation. Acc. Chem. Res. 49, 1389–1400 (2016).
Cheng, H.-G., Chen, S., Chen, R. & Zhou, Q. Palladium(II)-initiated Catellani-type reactions. Angew. Chem. Int. Ed. 58, 5832–5844 (2019).
Wang, J. & Dong, G. Palladium/norbornene cooperative catalysis. Chem. Rev. 119, 7478–7528 (2019).
Li, R. & Dong, G. Structurally modified norbornenes: a key factor to modulate reaction selectivity in the palladium/norbornene cooperative catalysis. J. Am. Chem. Soc. 142, 17859–17875 (2020).
Dong, S. & Luan, X. Catellani reaction: an enabling technology for vicinal functionalization of aryl halides by palladium(0)/norbornene cooperative catalysis. Chin. J. Chem. 39, 1690–1705 (2021).
Cheng, H.-G., Jia, S. & Zhou, Q. Benzo-fused-ring toolbox based on palladium/norbornene cooperative catalysis: methodology development and applications in natural product synthesis. Acc. Chem. Res. 56, 573–591 (2023).
Li, X., Pan, J., Song, S. & Jiao, N. Pd-catalyzed dehydrogenative annulation approach for the efficient synthesis of phenanthridinones. Chem. Sci. 7, 5384–5389 (2016).
Rasina, D. et al. Heterogeneous palladium-catalysed Catellani reaction in biomass-derived γ-valerolactone. Green. Chem. 18, 5025–5030 (2016).
Shi, H., Herron, A. N., Shao, Y., Shao, Q. & Yu, J.-Q. Enantioselective remote meta-C–H arylation and alkylation via a chiral transient mediator. Nature 558, 581–585 (2018).
Li, J.-J. et al. Atroposelective diverse remote meta-C–H functionalization via kinetic resolution. J. Am. Chem. Soc. 147, 6594–6603 (2025).
Catellani, M., Frignani, F. & Rangoni, A. A complex catalytic cycle leading to a regioselective synthesis of o,o′-disubstituted vinylarenes. Angew. Chem., Int. Ed. Engl. 36, 119–122 (1997).
Lautens, M. & Piguel, S. A new route to fused aromatic compounds by using a palladium-catalyzed alkylation–alkenylation sequence. Angew. Chem. Int. Ed. 39, 1045–1046 (2000).
Mariampillai, B., Alliot, J., Li, M. & Lautens, M. A convergent synthesis of polysubstituted aromatic nitriles via palladium-catalyzed C−H functionalization. J. Am. Chem. Soc. 129, 15372–15379 (2007).
Rudolph, A., Rackelmann, N. & Lautens, M. Stereochemical and mechanistic investigations of a palladium-catalyzed annulation of secondary alkyl iodides. Angew. Chem., Int. Ed. 46, 1485–1488 (2007).
Weinstabl, H., Suhartono, M., Qureshi, Z. & Lautens, M. Total synthesis of (+)-linoxepin by utilizing the catellani reaction. Angew. Chem., Int. Ed. 52, 5305–5308 (2013).
Zhao, P., Guo, Y. & Luan, X. Total synthesis of dalesconol A by Pd(0)/norbornene-catalyzed three-fold domino reaction and Pd(II)-catalyzed trihydroxylation. J. Am. Chem. Soc. 143, 21270–21274 (2021).
Bai, M. et al. A Modular approach for diversity-oriented synthesis of 1,3-trans-disubstituted tetrahydroisoquinolines: seven-step asymmetric synthesis of michellamines B and C. Angew. Chem. Int. Ed. 61, e202205245 (2022).
Zhang, B.-S. et al. Combined C-H amination and intermolecular alkyne insertion for a three-component cyclization. Cell Rep. Phys. Sci. 4, 101647 (2023).
Wang, J., Li, R., Dong, Z., Liu, P. & Dong, G. Complementary site-selectivity in arene functionalization enabled by overcoming the ortho constraint in palladium/norbornene catalysis. Nat. Chem. 10, 866–872 (2018).
Dong, Z., Lu, G., Wang, J., Liu, P. & Dong, G. Modular ipso/ortho difunctionalization of aryl bromides via palladium/norbornene cooperative catalysis. J. Am. Chem. Soc. 140, 8551–8562 (2018).
Catellani, M. & Motti, E. Selective aryl coupling via palladacycles: a new route to m-alkylbiphenyls or m-terphenyls. N. J. Chem. 22, 759–761 (1998).
Liu, X., Zhu, Q. & Dong, G. Beyond tertiary amines: introducing secondary amines by palladium/norbornene-catalyzed ortho amination. Angew. Chem. Int. Ed. 63, e202404042 (2024).
Wang, F.-Y., Li, Y.-X. & Jiao, L. Functionalized cycloolefin ligand as a solution to ortho-constraint in the Catellani-type reaction. J. Am. Chem. Soc. 145, 4871–4881 (2023).
Zheng, Y.-X. & Jiao, L. Hybrid cycloolefin ligands for palladium–olefin cooperative catalysis. Nat. Synth. 1, 180–187 (2022).
Thansandote, P., Hulcoop, D. G., Langer, M. & Lautens, M. Palladium-catalyzed annulation of haloanilines and halobenzamides using norbornadiene as an acetylene synthon: a route to functionalized indolines, isoquinolinones, and indoles. J. Org. Chem. 74, 1673–1678 (2009).
Zhang, B.-S. et al. Synthesis of C4-aminated indoles via a Catellani and retro-Diels–Alder strategy. J. Am. Chem. Soc. 141, 9731–9738 (2019).
An, Y. et al. Palladium-catalyzed C–H glycosylation and retro Diels–Alder tandem reaction via structurally modified norbornadienes (smNBDs). Chem. Sci. 12, 13144–13150 (2021).
Zhang, B.-S. et al. Direct synthesis of C4-acyl indoles via C–H acylation. Org. Lett. 26, 4998–5003 (2024).
Zhang, B.-S. et al. A switch strategy for the synthesis of C4-ethylamine indole and C7-aminoindoline via controllable carbon elimination. Chem. Sci. 15, 16169–16175 (2024).
Ackermann, L. Carboxylate-assisted transition-metal-catalyzed C−H bond functionalizations: mechanism and scope. Chem. Rev. 111, 1315–1345 (2011).
Zhang, B.-S. et al. Carboxylate ligand-exchanged amination/C(sp3)–H arylation reaction via Pd/norbornene cooperative catalysis. ACS Catal. 8, 11827–11833 (2018).
Acknowledgements
Financial support was received from the National Natural Science Foundation of China (22101232, B.-S.Z.; 22171114, Y.-M.L.) and the DFG (Gottfried Wilhelm Leibniz award to L.A., SPP2363).
Author information
Authors and Affiliations
Contributions
Conceptualization: B.-S.Z., L.A., and Y.-M.L.; methodology: B.-S.Z., Y.-M.W., and J.-S.Z.; investigation: Y.-M.W., J.-S.Z., X.-Y.F., T.-J.G., S.T., and X.-Y.G.; writing—original draft: B.-S.Z. and Y.-M.W.; writing—review & editing: B.-S.Z., Y.-M.W., S.T., X.-Y. G., L.A., and Y.-M.L.; funding acquisition: B.-S.Z.; resources: B.-S.Z., X.-C.W., and Z.-J.Q.; supervision: B.-S.Z., X.-C.W., and L.A.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Juntao Ye and the other, anonymous, reviewers for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, BS., Wang, YM., Zhou, JS. et al. Palladium-catalyzed selective assembly of carbamoylated indoles and aminophenanthridinones via β-carbon elimination switch. Commun Chem 8, 352 (2025). https://doi.org/10.1038/s42004-025-01740-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-025-01740-7
