
Abstract
Cryogenic electron microscopy (cryo-EM) single-particle analysis (SPA) has become a powerful technique for macromolecular structure determination. However, its effectiveness is often constrained by limited particle numbers or missing views. To address these challenges, we present CoCoFold, a fine-tuned framework that integrates raw cryo-EM particle images into AlphaFold to directly guide atomic model prediction. CoCoFold adopts a memory-efficient tuning strategy by introducing a fused attention mechanism into AlphaFold’s structure module. Moreover, a differentiable network links predicted structures with cryo-EM observations, enabling end-to-end refinement against experimental data. Benchmark experiments with the escalating quantity insufficiency and view-missing of cryo-EM observations, demonstrate that CoCoFold consistently outperforms state-of-the-art methods across multiple evaluation metrics.
Similar content being viewed by others

Introduction
Proteins are of fundamental importance in biological systems, as they perform a plethora of functions within living organisms. These functions span enzyme-mediated catalytic reactions, cell-signaling pathways, and the transportation of various molecules. The three-dimensional (3D) atomic models of proteins elaborate their functions in biological processes. In the domain of experimental methods for protein structure determination, cryogenic electron microscopy (cryo-EM) has emerged as a technique of remarkable popularity1,2.
Despite the great success of cryo-EM, real-world laboratories often present situations where cryo-EM observations are severely limited. These limitations can be categorized into two main scenarios. In the first scenario, the number of obtainable particles is inadequate. For instance, when studying the high-energy states of proteins in experiments, according to Boltzmann’s law, the proportion of particles in high-energy states is low3,4,5,6. Additionally, if the protein samples are of endogenous origin, the expression level of the proteins can be low, leading to a scarcity of protein particles7,8. This low abundance restricts the number of particles that can be collected through cryo-EM9. The second scenario involves the issue of preferred orientation, which occurs when particles are adsorbed by the air-water interface10,11,12,13. In such cases, the poses of the particles are highly biased. As a result, the number of particles in non-preferred views is insufficient. Such limited cryo-EM observations result in poorly reconstructed density maps, thereby presenting substantial impediments to atomic model building.
Structure prediction, an alternative methodology for deriving the 3D coordinates of atoms within protein molecules, has undergone a revolutionary transformation driven by deep-learning models14. This transformation has catalyzed novel discoveries in the field of structural biology. AlphaFold2 (AF2), created by DeepMind, stands as a cutting-edge neural network15. It can precisely predict protein structures based on their amino acid sequences, far outperforming the results achieved by its predecessors16,17. Moreover, the recently launched AlphaFold3 (AF3)18 utilizes an architecture different from that of AF2. It employs a multiscale diffusion process to predict protein-protein interactions as well as protein-ligand complexes, which involve nucleic acids and ions, with remarkable accuracy. These, as well as other methods, have demonstrated the usefulness of deep learning techniques in the prediction of protein structures19,20,21.
Even though these deep-learning-based approaches have achieved many successes in protein structure prediction, challenges still persist. There are cases where AlphaFold finds it difficult to make accurate predictions, as recently reviewed22. For instance, when proteins adopt conformations that are different from those of the homologs in the training set, or when proteins have alternative conformations23. For the latter case, although there are various methods for predicting alternative conformations24,25,26,27,28, their success does not cover all cases of inaccurate AlphaFold predictions. Such inaccuracy of AlphaFold causes its predictions to deviate from experimental observations.
Consequently, enabling the predicted structures of AlphaFold to be in alignment with cryo-EM observations assumes considerable significance. Existing studies29,30,31,32,33,34,35, although not adopting a direct approach, fall within the same purview. These include fitting the structures predicted by AlphaFold into cryo-EM density maps29,30,31,32 or leveraging the knowledge encapsulated in AlphaFold as a prior for the model building of cryo-EM maps33,34,35. These methods have demonstrated efficacy in harmonizing the predictions with experimental findings. This congruence, though, is highly contingent upon the availability of high-resolution cryo-EM density maps. When cryo-EM observations are limited, the efficacy of these approaches is compromised. Representative methods will be utilized as control methods in this study. These include Phenix29,30 and DiffModeler32, which are used for fitting predicted structures into cryo-EM maps, and multimodal approaches like the recently developed MICA35, which integrates cryo-EM densities with AlphaFold3 predictions. DeepMainmast33 and EModelX34, which use AlphaFold as a prior for model building. Also included are other approaches for obtaining atomic models from either amino acid sequences or cryo-EM maps, such as AlphaFold-Multimer (AF-m)36, AF318, and ModelAngelo37. The performance of these methods will be assessed under conditions of limited cryo-EM observations.
Fine-tuning assumes a pivotal role within the framework of deep neural networks. It represents a procedure entailing the adjustment of pre-trained weights of a neural network to facilitate enhanced adaptation to a particular task. Through fine-tuning, the neural network can effectively exploit the knowledge amassed from large-scale datasets during the pre-training phase and subsequently optimize its performance for a more precisely defined objective. In the context of protein structure prediction, tailoring AlphaFold to predict structures that are congruent with cryo-EM observations is inherently a fine-tuning process. AlphaFold has already acquired knowledge from extensive, large-scale datasets. Meanwhile, the cryo-EM observations of a specific target protein serve as the domain-specific dataset essential for the fine-tuning operation.
In this study, we introduce CoCoFold, an approach designed to fine-tune AlphaFold, enabling it to better align with limited cryo-EM observations. Inspired by cross-domain fine-tuning techniques in natural language processing38 and multimodal integration, CoCoFold harnesses raw particle images to fine-tune AlphaFold. This paper is organized as follows. First, we demonstrate that as the quantity of obtainable particles gradually decreases (the first scenario of limited cryo-EM observations), CoCoFold exhibits remarkable resilience and outperforms other state-of-the-art algorithms. Subsequently, we show that as the problem of missing view becomes increasingly severe (the second scenario of limited cryo-EM observations), CoCoFold remains robust, once again surpassing competing algorithms.
Results
The design of CoCoFold
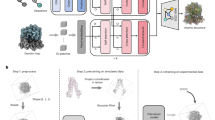
CoCoFold is a fine-tuned structure prediction framework designed to integrate cryo-EM particle images and amino acid sequences for the prediction of atomic models. The architectural design of the framework is built upon AF-m and incorporates a tunable refinement of information flow (Fig. 1). Leveraging this integrated information flow, the framework refines the predictive outputs generated by AF-m, with the specific aim of adjusting these computational predictions to achieve alignment with experimental observations derived from cryo-EM. The pipeline starts with a standard AF-m forward information flow, in which the input sequence is processed through 48 Evoformer blocks and 8 structure modules with multiple recycling iterations15, yielding high-quality sequence and pairwise representations (Fig. 1a). To improve training efficiency with cryo-EM data, we freeze the Evoformer and fine-tune only the structure module of AlphaFold (Fig. 1a). Input features are extracted from the final recycling iteration, where representations reach their highest degree of refinement, to initialize the fine-tuning process.

a CoCoFold consists of three parts: a fixed part (frozen AF-m weights), a fine-tuning part (trainable weights initialized from AF-m), and a training part. The gradient descent employed for fine-tuning the fine-tuning part is backpropagated from the training part, which constitutes a Gaussian mixture Molmap module. This module generates a density map of the predicted structure using a Gaussian Mixture Model (GMM), e.g., MolMap. Projections are then computed under specified poses and CTFs and subsequently compared with raw particle images. The resulting FRC loss is backpropagated through the Gaussian mixture Molmap module, while the weights within this module undergo training simultaneously. b The fine-tuning part operates as follows: The Evoformer generates fixed pair representations and MSA representations, adhering to the original information flow of AF-m. A parallel refinement branch incorporates a lightweight attention mechanism and linear transformation to adapt MSA representations based on image-derived constraints. These updated features are integrated with the IPA module and backbone frames module to produce refined structures. Here, “f” and “fimage” denote features derived from the original information flow and the image-constrained information flow, respectively. c The training component operates as follows: First, the predicted atomic model is aligned to the experimental coordinate system using a fixed affine transformation. This transformation is derived by aligning the model to the initial map reconstructed from particle images. Notably, the map serves solely to provide the affine transformation matrix and does not participate in gradient updates. Subsequently, a simulated density based on GMM is generated, which is then utilized to produce 2D projections for iterative fine-tuning.
To incorporate the information (or say, structural constraints) from cryo-EM observations and prevent the model parameters from drifting excessively from the pre-trained physical priors, we introduce a lightweight tuning information flow (Fig. 1b), where multiple sequence alignment (MSA) representations are refined through an attention module and a linear transformation before being merged with the AF-m information flow (Supplementary Fig. 1). This design allows CoCoFold to effectively reconcile sequence-derived features with image-based structural information and improve stability during fine-tuning. The updated representations are passed through the invariant point attention (IPA)15 and backbone frame generation modules, yielding an initial atomic model (Fig. 1b).
To establish a consistent coordinate system between the prediction and experimental data, we applied existing cryo-EM software to process the particle images and obtain a coarse map. The initial predicted structures are then subject to a rigid-body alignment to the cryo-EM density map. Importantly, this reconstruction map is used solely for this initial spatial positioning (Supplementary Fig. 2). Subsequently, the aligned atomic coordinates are then converted into a simulated density map using a Gaussian mixture MolMap module (Fig. 1c), in which each atom is represented by a 3D Gaussian distribution with learnable amplitude and variance39,40. To capture local heterogeneity due to noise or resolution variation, these parameters are made spatially adaptive. Simulated 2D projections are generated under the estimated poses and modulated by contrast transfer functions (CTFs). These are compared to experimental particle images in Fourier space using a Fourier ring correlation (FRC) loss, which is backpropagated to refine the structure module and Gaussian parameters.
Residues with low confidence (pLDDT15 < 30) are removed, and the model is relaxed using ISOLDE41 to ensure structural plausibility. Through this cross-modal fine-tuning, CoCoFold generates atomic models that are both physically reasonable and consistent with cryo-EM observations.
Performance under scarce-particle conditions
The performance of CoCoFold under conditions of particle scarcity was first assessed. For this purpose, ten datasets (five experimental datasets and five simulated datasets) were employed (Supplementary Table 1). Particle numbers in the experimental datasets were systematically reduced through random selection, which yielded a total of 19 test cases. For simulated datasets, the simulated data tool in CryoSPARC was used to generate 10 cases. The resolution of the density maps reconstructed from these particle-reduced cases is provided in Supplementary Table 2. CoCoFold was benchmarked against five state-of-the-art approaches: DiffModeler32, DeepMainmast33, EModelX34, MICA35, and ModelAngelo37. All competing methods use amino acid sequences and reconstructed cryo-EM maps as inputs, with DiffModeler, DeepMainmast and MICA further incorporating AlphaFold predictions. To ensure a fair comparison, for each such test case, density maps were reconstructed from the particles of the respective test cases, thereby ensuring consistent input conditions across all methods (Supplementary Fig. 3).
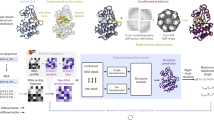
Using the PDB deposition model as the ground truth, the accuracy of the atomic models output by all methods was evaluated via MM-align42, which reported four metrics: coverage (fraction of aligned PDB residues), root-mean-square deviation (RMSD), template modeling score (TM-score), and sequence identity. Both CoCoFold and DiffModeler achieved near-complete average coverage (>96%) (Fig. 2a). CoCoFold produced significantly more accurate structures, with a lower mean RMSD of 1.82 Å, compared to higher variability in DiffModeler (Fig. 2b). This discrepancy became more pronounced under severe particle depletion, where conventional methods exhibited marked drops in coverage (<75%) and decreased TM-score (<0.75), likely due to resolution loss of reconstructed maps. Also, CoCoFold exhibited tightly clustered RMSD values (0.48–3.29 Å), while DiffModeler showed broader dispersion (0.94–4.10 Å), highlighting CoCoFold’s robustness under scarce-particle conditions (Fig. 2b). TM-scores and sequence identities further confirmed this advantage (Fig. 2c, d). To further characterize model degradation trends against reducing particles, we plotted coverage, RMSD, TM-score and sequence identity against the number of particles for three experimental datasets (Fig. 3, left three columns). CoCoFold (Fig. 3, blue line) consistently maintained high TM-scores and low RMSDs, even at minimal particle counts (e.g., 103), while DeepMainmast, EModelX and MICA exhibited sharp declines.

a–d correspond to the coverage, RMSD, TM-score, and sequence identity of CoCoFold and other methods, respectively. Beyond RMSD and TM-scores, coverage is defined as the proportion of residues in the PDB structure that are successfully aligned to the built model via MM-align. Meanwhile, sequence identity refers to the percentage of paired residues in the alignment that exhibit identical amino acid sequences. e, f Comparison of predicted structures by CoCoFold, AF-m, and DiffModeler across three test datasets under the scenario of the fewest particles. e For each dataset, the predicted models were superposed onto the ground-truth structures (displayed as transparent tubes). Six regions of interest (ROIs) are outlined in red boxes, while black arrows highlight the major mismatches or deviations observed in the predictions of AF-m and DiffModeler. f Zoomed-in views of selected regions. Each panel corresponds to the boxed ROIs in (e), and it compares the local atomic details between the model predicted by CoCoFold (shown in purple) and the ground truth (shown in cyan and transparent).

The left three columns correspond to conditions with escalating scarce-particle, whereas the right three columns correspond to conditions with escalating missing-view. The four rows correspond to coverage, RMSD, TM-score, and sequence identity, respectively.
We next conducted a visual comparison of structures predicted or built from the most limited particle subsets of three experimental datasets. Only CoCoFold, AF-m, and DiffModeler produced usable atomic models under these conditions, as DeepMainmast, EModelX, ModelAngelo, and MICA either failed or generated poor-quality predictions. The ground truth is depicted as a semi-transparent light steel blue model, which is superimposed on all three models in Fig. 2e. Across all three viable models, CoCoFold’s predictions closely align with the ground truth (Fig. 2e, left column), whereas AF-m and DiffModeler exhibit shifts or mispositioned helices (Fig. 2e, right two columns, emphasized by black arrows). Zoomed-in views of the representative segments of helices (Fig. 2e, f, dark magenta) that are mispositioned by AF-m and DiffModeler but correctly placed by CoCoFold are depicted in Fig. 2f.
Performance under missing-view conditions
We then evaluated the performance of CoCoFold under conditions of missing views, where a certain range of cryo-EM observation poses is absent. To simulate these conditions, we progressively expanded the missing cone across ten benchmark datasets. As exemplified by Cav1.2 in Fig. 4a, each sphere shows available projection directions (blue and red, where red indicates a more concentrated angular distribution) and increasing angular gaps (dashed line). 26 test cases were constructed for the same ten datasets, consistent with the aforementioned study (Supplementary Table 3). For each protein, two or four levels of missing views were established, with the missing cone ranging from none to ± 80°. The missing cone ranges, the conical Fourier shell correlation area ratio (cFAR) and the sampling compensation factor (SCF) of these datasets are provided in Supplementary Table 3. The corresponding density maps are shown in Supplementary Fig. 4.

a The visualization of progressively expanded missing-view artifacts is presented using the Cav1.2 dataset as an example, where each sphere represents the available projection directions (blue: dispersed distribution; red: concentrated distribution) and angular gaps (denoted by dashed lines). b–e correspond to the coverage, RMSD, TM-score, and sequence identity of CoCoFold and other methods, respectively.
CoCoFold was again benchmarked against the five aforementioned competing methods. As demonstrated by the coverage metric (Fig. 4b), the atomic models output by CoCoFold and DiffModeler can cover the majority of the input density maps, whereas other methods cannot. Specifically, CoCoFold achieves a mean coverage of 0.99, while that of DiffModeler stands at 0.83 and others’ mean coverages are smaller than 0.72. With regard to the modeled regions, CoCoFold exhibits superior accuracy (Fig. 4c–e). Quantitatively, CoCoFold attains a mean RMSD of 1.86 Å, in contrast to 2.72 Å for DiffModeler. CoCoFold also achieved a mean TM-score of 0.97, well above DiffModeler (0.79). These results underscore CoCoFold’s advantage not only in residue coverage but also in the atomic-level accuracy of the modeled regions. Across all four evaluation metrics—coverage, RMSD, TM-score, and sequence identity, CoCoFold not only achieves the best mean values but also exhibits concentrated distributions with narrow interquartile ranges. This indicates that its predictions are both accurate and consistently stable across diverse test cases, in contrast to the broader and more variable distributions observed for competing methods.
To further investigate performance degradation trends under increasingly severe missing view conditions, we plotted coverage, RMSD, TM-score and sequence identity across a range of missing cones for three proteins (Fig. 3, right three columns). CoCoFold (blue line) demonstrated remarkable stability, with TM-scores consistently exceeding 0.92, even in extreme scenarios such as ±80° missing cones. In contrast, other methods exhibited a steep decline in performance once the missing region surpassed ±30°, underscoring their limited tolerance to missing views.
Discussion
This study aimed to solve the key challenge of integrating deep learning-based protein structure prediction with limited cryo-EM observations. Results show CoCoFold, an AlphaFold fine-tuning framework, effectively addresses both sparse particles3,4,5,6,7,8,9 and severe missing views10,11,12,13, the two main scenarios of limited cryo-EM observations. In scarce-particle scenarios (caused by low high-energy state abundance or low endogenous protein expression), CoCoFold maintained high accuracy by using raw cryo-EM particles for fine-tuning, leveraging AlphaFold’s pre-trained global folding knowledge while integrating experimental cues. For missing views, it remained robust: fine-tuning AlphaFold directly on raw particles, which retain angular information (which will be lost in map reconstruction), enabled adjustments to predictions and strong performance in RMSD, TM-score, and sequence identity.
Compared with five existing approaches DiffModeler32, DeepMainmast33, EModelX34, MICA35, and ModelAngelo37, CoCoFold differs in its treatment of experimental data. Most prior approaches rely on reconstructed density maps as input, inevitably subject to information loss during map refinement, especially in high-frequency regions critical for atomic accuracy. In contrast, CoCoFold does not use the density map as an optimization target. While a coarse reconstruction is requisite for the initial rigid-body alignment of the coordinate systems, the subsequent fine-tuning process relies directly on particle images, thereby avoiding information losses inherent to map reconstruction and maintaining access to raw experimental information. This design makes CoCoFold robust in challenging conditions with few particles or highly anisotropic data.
Performance of CoCoFold also derives from cross-domain fine-tuning (inspired by NLP38) and multimodal integration. CoCoFold proactively aligns its predictions with experimental observations. Additionally, leveraging the pre-trained weights of AlphaFold—derived from a large-scale dataset PDB, addresses a critical limitation of AlphaFold15,18,36 itself, namely its difficulty in predicting structures of non-homologous proteins or proteins with alternative conformations22,23. Fine-tuning on target-specific cryo-EM data further enables the adaptation of AlphaFold’s globally acquired structural knowledge to the unique features of the protein under investigation. This design aligns with the broader shift toward multimodal integration in structural biology, a field increasingly dominated by cryo-EM and deep learning techniques. In this context, CoCoFold exemplifies how fine-tuning can serve as a bridge between pre-trained deep learning models and experimental data, thereby extending the utility of both cryo-EM and AlphaFold in structural biology research.
While CoCoFold demonstrates superior robustness in challenging scenarios, it is important to delineate its optimal scope of application compared to map-based approaches. In “ideal” regimes—where particle abundance and angular coverage are sufficient to reconstruct high-resolution density maps (typically <3.5 Å)—automated model building tools that rely directly on the density (e.g., MICA) perform exceptionally well. In such cases, CoCoFold yields results that are comparable to, but not necessarily superior to, these methods, as the high-fidelity experimental data render the strong structural prior from AlphaFold less critical (Supplementary Fig. 5 row 1, 3). However, the unique value of CoCoFold emerges precisely when these conditions are not met. As experimental observations degrade (e.g., scarce particles or missing views), the quality of reconstructed maps deteriorates rapidly, causing map-based methods to fail (Supplementary Fig. 5 row 2, 4). By contrast, CoCoFold’s strategy of fine-tuning against raw particles enables it to maintain structural accuracy even when a reliable density map cannot be reconstructed.
Current cryo-EM workflows often generate datasets containing millions of particles, raising concerns about training costs. However, CoCoFold does not require the exhaustive use of all available particles. Indeed, one can first select using tools like CryoSieve43 to get a representative subset of high-quality particles. To validate this, we performed an experiment on the MSP-1 dataset (75.6k particles). By filtering the dataset down to a representative subset of 3000 particles (approx. 40 min processing) and fine-tuning for only 2 epochs (~21 min), we achieved a TM-score of 0.97 and RMSD of 1.97 Å (Supplementary Fig. 6). This demonstrates that CoCoFold is highly practical even for massive datasets, provided that a high-quality particle subset is used.
The limitation of CoCoFold lies in its continued constraint by the amino acid sequence length capacity of AlphaFold. Specifically, for large protein supercomplexes whose sequence lengths exceed the processing capability of AlphaFold, CoCoFold fails to maintain its efficacy. How to leverage the fine-tuning functionality of CoCoFold to enable it to surpass AlphaFold’s inherent amino acid sequence length limitation remains an important question that requires further investigation.
Additionally, as with all refinement approaches that rely on projection matching, CoCoFold relies on the accuracy of upstream pose and CTF estimates. In practice, however, this assumption is generally reliable in the resolution regime where CoCoFold is intended to operate. Once a dataset has been refined to produce a 4–7 Å reconstruction, the pose dispersion is inherently constrained to within a few degrees—significant pose inaccuracies would smear secondary-structure features and prevent the map from reaching this resolution. Even in missing-view scenarios, overestimated FSC resolution typically arises from directional anisotropy rather than large per-particle pose errors; poses for the observed directions remain accurate after CryoSPARC/Relion refinement. Finally, because the differentiable projection operator provides explicit gradients with respect to pose parameters, extending CoCoFold to perform joint pose refinement is mathematically feasible and represents a promising direction for future development.
Methods
Overview of CoCoFold
CoCoFold consists of the following two steps (Supplementary Fig. 2): (1) Initialization: we first applied existing cryo-EM software to process the particle images and obtain a coarse map. Meanwhile, we used AlphaFold to generate a predicted structure. The predicted structure was then aligned to the coarse map through an affine transformation, establishing a consistent coordinate frame. (2) Fine-tuning: based on the obtained affine transformation, we constructed an end-to-end differentiable architecture to fine-tune AlphaFold such that its output better fits the experimental particle images after training. It is worth noting that this affine transformation is fixed during fine-tuning, and therefore, no map information is required in this step.
Fine-tuned structure module
CoCoFold builds upon the OpenFold PyTorch implementation, using parameters from AlphaFold-Multimer v2.3 to initialize the pre-trained model. For each input sequence, a MSA is first constructed, followed by a standard forward pass through AlphaFold-Multimer. The structural module parameters from the final recycling iteration are retained for fine-tuning, while the Evoformer is kept frozen throughout training. This design enables efficient gradient updates while preserving the pretrained sequence representation backbone.
To enable interaction between particle images and sequence features and to improve stability during fine-tuning with extremely noisy particle data, we introduce a lightweight attention adapter inserted within the structure module (Supplementary Fig. 1). This adapter is analogous to regularization techniques used in large-model fine-tuning, where small trainable parameters constrain updates and prevent the model from drifting too far from the pretrained AF-m parameters. Ablation studies confirm that removing this module leads to training instability and gradient explosion (Supplementary Table 4). Specifically, three learnable projection matrices—Wq, Wk, and Wv—are initialized from a normal distribution and updated solely via gradients from the particle projection loss. These matrices operate on the MSA representation s to produce query, key, and value vectors:
The resulting attention output follows the same procedure as the IPA mechanism used in AlphaFold’s Structure module.
To ensure numerical stability, we enforce that the output of this modified attention block matches the original IPA output in both mean and variance. This constraint prevents gradient explosion and supports smoother optimization. Additionally, the inclusion of this cross-attention information flow regularizes the network, reducing the risk of overfitting during training on small cryo-EM datasets.
Gaussian mixture Molmap module
To refine the atomic model with particle images, we represent a density map of the model by Gaussian mixture, which is a sum of Gaussian distributions (Supplementary Fig. 7). The formulation is:
where Aj is the amplitude, N represents the total number of atoms, c = [cx, cy, cz] denotes the atom center and \({{{{\boldsymbol{\sigma }}}}}^{2}=[{\sigma }_{x}^{2},{\sigma }_{y}^{2},{\sigma }_{z}^{2}]\) are the widths in three axes in the real space. Aj is initialized with its atomic number and σx, σy, σz are initialized with \(\frac{3}{\sqrt{2}\pi }\). The projection under given poses (rotation R and translation t) is:
where \({{{{\bf{c}}}}}^{{\prime} }\) takes the first and second rows of Rc, bi-cubic interpolation is used for calculating the translation. Since the orientation of the atomic model predicted by AlphaFold may differ from that of the particle images, the rotation matrix R is actually the composition of two rotation matrices: one that rotates the atomic model into the density map and another that rotates it by a specific angle to project onto the particle. To determine the initial affine transformation, we employ a two-step protocol to ensure reproducibility. First, a coarse manual alignment is performed to orient the model globally. Second, the “Fit in Map” tool in ChimeraX is utilized to mathematically refine the fit. This step serves solely to align the coordinate systems of the prediction and the experimental data. The reconstruction map is not utilized subsequently for gradient calculation or structural fine-tuning. The fitting procedure can also be performed by patches. We fitted HSL-Dimer (PDB 8ZVQ) and RhlR-PqsE complex (PDB 8DQ0) into the density map separately by chains, while the other data were fitted to the density map as a whole.
To verify the resilience of this alignment workflow against typical residual errors, we performed a sensitivity check on the MSP-1 dataset (1.1k particles case). We introduced rotational perturbations to the aligned model and observed that the fine-tuning optimization reliably recovers the native structure even with initial deviations of up to 10°. This confirms that the pipeline possesses a sufficient capture radius to accommodate the minor variances expected from automated rigid-body fitting tools.
We use the average of the correlation coefficients over Fourier rings as our reconstruction loss:
where \({{{\mathcal{P}}}},{{{\mathcal{I}}}}\) are Fourier transform of the projections and particle images, \({{{\mathcal{C}}}}\) where P, I are Fourier transform of the projections and particle images, C denotes the CTF, b is the boxsize and k, θ are fast Fourier transform polar coordinates. We also add some penalty to the widths and amplitude to prevent unphysical expansion of Gaussian widths (over-smoothing), as visually demonstrated in Supplementary Fig. 8 and quantified in Supplementary Table 4.:
ISOLDE relaxation
Following fine-tuning, predicted models are relaxed using ISOLDE. This step does not modify the global fold: TM-score and backbone RMSD before and after relaxation basically remain unchanged (Supplementary Table 4). Instead, ISOLDE primarily resolves local stereochemical inconsistencies. Clash analysis in ChimeraX shows that unrelaxed models exhibit tens to hundreds of clashes, whereas relaxed models consistently show 0–10 clashes (Supplementary Fig. 9). Thus, ISOLDE serves as a geometric cleanup step improving physical plausibility without affecting the accuracy of the predicted conformation.
Input data requirements and use of orientations/CTF parameters
CoCoFold assumes that particle orientations (Euler angles and in-plane shifts) and CTF parameters have been estimated through a standard upstream cryo-EM workflow. These parameters are treated as fixed inputs to the differentiable projection operator and are not updated during fine-tuning. For each predicted atomic model, we compute its 2D projections at the known orientations and apply the corresponding per-particle CTF before computing the FRC loss. No pose refinement or CTF re-estimation is performed inside CoCoFold.
CoCoFold relies on the accuracy of upstream pose and CTF estimates. To rigorously assess this dependency, we performed static perturbation experiments by introducing synthetic Gaussian noise to the input poses (Supplementary Fig. 10). Our results reveal a dual behavior: at noise levels typical of high-quality consensus refinement (σ = 1°, mean deviation ~1. 6°), the model demonstrates robust stability, with the TM-score decreasing only marginally from 0.970 to 0.960. However, significant performance degradation occurs at larger perturbations (σ ≥ 3°, mean deviation > 4.8°), where the RMSD deteriorates from 1.82 Å to 3.66 Å. This confirms that CoCoFold is robust to minor residual errors but remains physically constrained by the experimental data, preventing the hallucination of high-resolution features when geometric consistency is lost.
Training hyperparameters setting
We use ADAM44 optimizer to train the neural network. The learning rates for Gaussian mixtures’ amplitudes, widths, and structure module are 0.01, 0.005, and 0.0001, respectively. Training batch size is 32, and gradient accumulation is used to achieve this. Training epoch is set to 10. The particles are randomly sampled during training. Reconstruction loss and penalty loss have the same weight.
Computational resources and efficiency
All fine-tuning experiments were conducted on a single NVIDIA A100 (80GB) GPU. We utilized gradient accumulation to manage memory usage, with effective batch sizes of 32. As detailed in Supplementary Table 5, the fine-tuning process is computationally accessible due to the frozen Evoformer backbone. The execution times reported in the table correspond to processing a standardized volume of 20,480 particle images. Empirically, we observed that CoCoFold typically reaches convergence within this volume, requiring only 0.5 to 1.5 h for most targets, and within 4.5 h for large complexes (e.g., Cav1.2, >2500 residues). However, to ensure the utmost robustness for the benchmark results presented in this study, we employed a rigorous protocol using the full particle dataset for 10 epochs. Consequently, the total training time for these specific experiments scales linearly with the dataset size (estimated as \(\frac{Total\,Particles}{20,480}\times Reported\,Time\times Epochs\)). Peak memory usage ranges from 19 to 77 GB, depending on the box size and sequence length. The inference time after training is up to a few seconds and can be negligible. Note that the reported execution times correspond to the fine-tuning phase specifically; MSA generation and initial feature extraction are standard pre-processing steps inherent to the AlphaFold pipeline and are excluded from these metrics.
Comparison between direct 2D image supervision and 3D map supervision
To evaluate the benefit of fine-tuning directly against raw particle images, we compared CoCoFold with a map-based approach that replaces 2D image supervision with projections of a reconstructed 3D density map. This analysis highlights an intrinsic limitation of map-based optimization: the irreversible loss of high-frequency information during 3D reconstruction. Because reconstruction is an averaging procedure, it behaves effectively as a low-pass filter. Under scarce-particle or missing-view conditions, this leads to pronounced resolution degradation and anisotropic blurring, diminishing the fidelity of the supervisory signal provided by the map.
We performed a controlled experiment on the MSP-1 dataset (PDB 6ZBH), using only 1.1 k particles to simulate a low-resolution reconstruction scenario. Two fine-tuning strategies were examined:
-
Direct 2D supervision (CoCoFold) Fine-tuning the AlphaFold model parameters using raw particle images.
-
3D map supervision. Fine-tuning using projections of the reconstructed 3D map obtained from the same limited particle set.
As shown in Supplementary Fig. 11, direct 2D image supervision achieves substantially higher structural accuracy (TM-score 0.97; RMSD 1.82 Å) than the map-based approach (TM-score 0.86; RMSD 4.91 Å). These results confirm that retaining the original 2D particle information is particularly advantageous in low-data regimes, where the 3D reconstruction is unable to preserve high-resolution detail.
Data availability
Three of five experimental datasets were downloaded from EMPIAR (EMPIAR-1043745, EMPIAR-1124746, EMPIAR-12180)47, while the other two experimental datasets (HSL-dimer48 and Cav1.249) were collected in-house and are available at https://github.com/jwliaomath/CoCoFold-datasets. Five simulated datasets’ maps were downloaded from EMDB (EMD-1466950, EMD-1463650, EMD-4308951, EMD-2888852, EMD-2864053). The atomic models of all datasets, which were used as the ground truth in this study, were downloaded from the PDB (6ZBH, 8DQ0, 9C91, 8ZVQ, 8WE6, 7ZDT, 7ZD5, 8VAE, 8F6Q, 8EW2). The numerical source data underlying the graphs presented in the main figures are provided in Supplementary Data 1.
Code availability
An implementation of this work is available at: https://github.com/jwliaomath/CoCoFold.
References
Bai, X.-c., McMullan, G. & Scheres, S. H. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 40, 49–57 (2015).
Cheng, Y. Single-particle cryo-EM–how did it get here and where will it go. Science 361, 876–880 (2018).
Mäeots, M.-E. & Enchev, R. I. Structural dynamics: review of time-resolved cryo-EM. Acta Crystallogr. D Struct. Biol. 78, 927–935 (2022).
Ourmazd, A. Cryo-EM, XFELs and the structure conundrum in structural biology. Nat. Methods 16, 941–944 (2019).
Henzler-Wildman, K. & Kern, D. Dynamic personalities of proteins. Nature 450, 964–972 (2007).
Fan, X. et al. CryoTRANS: predicting high-resolution maps of rare conformations from self-supervised trajectories in cryo-EM. Commun. Biol. 7, 1058 (2024).
Mesa, P., Deniaud, A., Montoya, G. & Schaffitzel, C. Directly from the source: endogenous preparations of molecular machines. Curr. Opin. Struct. Biol. 23, 319–325 (2013).
Rogawski, R. & Sharon, M. Characterizing endogenous protein complexes with biological mass spectrometry. Chem. Rev. 122, 7386–7414 (2022).
Liang, K. et al. Conservation and specialization of the ycf2-FtsHi chloroplast protein import motor in green algae. Cell Press J. 187, 5638–5650 (2024).
Lyumkis, D. Challenges and opportunities in cryo-EM single-particle analysis. J. Biol. Chem. 294, 5181–5197 (2019).
Drulyte, I. et al. Approaches to altering particle distributions in cryo-electron microscopy sample preparation. Acta Crystallogr. D Struct. Biol. 74, 560–571 (2018).
Li, B., Zhu, D., Shi, H. & Zhang, X. Effect of charge on protein preferred orientation at the air-water interface in cryo-electron microscopy. J. Struct. Biol. 213, 107783 (2021).
Zhang, H. et al. Addressing preferred orientation in single-particle cryo-EM through AI-generated auxiliary particles. https://doi.org/10.1101/2023.09.26.559492 (2023).
Abriata, L. A. The nobel prize in chemistry: past, present, and future of AI in biology. Commun. Biol. 7, 1409 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Jumper, J. et al. Applying and improving alphafold at caps14. Proteins 89, 1711–1721 (2021).
Senior, A. W. et al. Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710 (2020).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Ahdritz, G. et al. OpenFold: retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. Nat. Methods 21, 1514–1524 (2024).
Hayes, T. et al. Simulating 500 million years of evolution with a language model https://doi.org/10.1101/2024.07.01.600583 (2024).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Chakravarty, D., Lee, M. & Porter, L. L. Proteins with alternative folds reveal blind spots in AlphaFold-based protein structure prediction. Curr. Opin. Struct. Biol. 90, 102973 (2025).
Schafer, J. W. & Porter, L. L. Alphafold2’s training set powers its predictions of fold-switched conformations https://doi.org/10.1101/2024.10.11.617857 (2024).
Monteiro Da Silva, G., Cui, J. Y., Dalgarno, D. C., Lisi, G. P. & Rubenstein, B. M. High-throughput prediction of protein conformational distributions with subsampled AlphaFold2. Nat. Commun. 15, 2464 (2024).
Vani, B. P., Aranganathan, A. & Tiwary, P. Exploring kinase asp-phe-gly (DFG) loop conformational stability with AlphaFold2-RAVE. J. Chem. Inf. Model. 64, 2789–2797 (2024).
Stein, R. A. & Mchaourab, H. S. SPEACH_af: sampling protein ensembles and conformational heterogeneity with alphafold2. PLoS Comput. Biol. 18, e1010483 (2022).
Kalakoti, Y. & Wallner, B. AFsample2: predicting multiple conformations and ensembles with AlphaFold2 https://doi.org/10.1101/2024.05.28.596195 (2024).
Del Alamo, D., Sala, D., Mchaourab, H. S. & Meiler, J. Sampling alternative conformational states of transporters and receptors with AlphaFold2. Elife 11, e75751 (2022).
Kim, D. N. et al. Cryo_fit: democratization of flexible fitting for cryo-EM. J Struct Biol. 208, 1–6 (2019).
Adams, P. D. et al. PHENIX : a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Kidmose, R. T. et al. Namdinator - automatic molecular dynamics flexible fitting of structural models into cryo-EM and crystallography experimental maps. IUCrJ 6, 526–531 (2019).
Wang, X., Zhu, H., Terashi, G., Taluja, M. & Kihara, D. DiffModeler: large macromolecular structure modeling for cryo-EM maps using a diffusion model. Nat. Methods 21, 2307–2317 (2024).
Terashi, G., Wang, X., Prasad, D., Nakamura, T. & Kihara, D. DeepMainmast: integrated protocol of protein structure modeling for cryo-EM with deep learning and structure prediction. Nat. Methods 21, 122–131 (2024).
Chen, S. et al. Protein complex structure modeling by cross-modal alignment between cryo-em maps and protein sequences. Nat. Commun. 15, 8808 (2024).
Gyawali, R., Dhakal, A. & Cheng, J. Multimodal deep learning integration of cryo-EM and AlphaFold3 for high-accuracy protein structure determination. Commun. Chem. 8, 320 (2025).
Evans, R. et al. Protein complex prediction with AlphaFold-multimer https://doi.org/10.1101/2021.10.04.463034 (2021).
Jamali, K. et al. Automated model building and protein identification in cryo-EM maps. Nature 628, 450–457 (2024).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: Pre-training of deep bidirectional transformers for language understanding. In Proc. of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics. 4171–4186 (2019).
Chen, M. & Ludtke, S. J. Deep learning-based mixed-dimensional Gaussian mixture model for characterizing variability in cryo-EM. Nat. Methods 18, 930–936 (2021).
Chen, M., Schmid, M. F. & Chiu, W. Improving resolution and resolvability of single-particle cryoEM structures using Gaussian mixture models. Nat. Methods 21, 37–40 (2024).
Croll, T. I. ISOLDE : a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. Sect. D Struct. Biol. 74, 519–530 (2018).
Mukherjee, S. & Zhang, Y. MM-align: a quick algorithm for aligning multiple-chain protein complex structures using iterative dynamic programming. Nucleic Acids Res. 37, e83–e83 (2009).
Zhu, J. et al. A minority of final stacks yields superior amplitude in single-particle cryo-EM. Nat. Commun. 14, 7822 (2023).
Kingma, D. P. & Ba, J. Adam: A method for stochastic optimization. In Proc. 3rd International Conference on Learning Representations. (2015).
Dijkman, P. M. et al. Structure of the merozoite surface protein 1 from Plasmodium falciparum. Sci. Adv. 7, eabg0465 (2021).
Feathers, J. R., Richael, E. K., Simanek, K. A., Fromme, J. C. & Paczkowski, J. E. Structure of the RhlR-PqsE complex from Pseudomonas aeruginosa reveals mechanistic insights into quorum-sensing gene regulation. Structure 30, 1626–1636 (2022).
Ghazi Esfahani, B. et al. Structure of dimerized assimilatory NADPH-dependent sulfite reductase reveals the minimal interface for diflavin reductase binding. Nat. Commun. 16, 2955 (2025).
Peng, H. et al. Molecular determinants for the association of human hormone-sensitive lipase with lipid droplets. Nat. Commun. 16, 3497 (2025).
Gao, S. et al. Structural basis for human Cav1.2 inhibition by multiple drugs and the neurotoxin calciseptine. Cell 186, 5363–5374 (2023).
Wu, D. et al. Dissecting the conformational complexity and mechanism of a bacterial heme transporter. Nat. Chem. Biol. 19, 992–1003 (2023).
Catalano, C. et al. The CryoEM structure of human serum albumin in complex with ligands. J. Struct. Biol. 216, 108105 (2024).
Edman, N. I. et al. Modulation of FGF pathway signaling and vascular differentiation using designed oligomeric assemblies. Cell 187, 3726–3740 (2024).
Shi, H., Wu, C. & hang, X. Addressing compressive deformation of proteins embedded in crystalline ice. Structure 31, 213–220 (2023).
Acknowledgements
This work was supported by the National Key R&D Program of China (No. 2021YFA1001300) (to C.B.), the National Natural Science Foundation of China (No. 12271291) (to C.B.), National Key R&D Program of the Ministry of Science and Technology of China (2025YFA1309602 to M.H.), SMART (to M.H.), Beijing Frontier Research Center for Biological Structure (to M.H.), and the Advanced Innovation Center for Structural Biology (to M.H.). We acknowledge the support in data analysis provided by the Computing Labware for Electron-microscopy Visualization and Experimental Research (CLEVER) at Shenzhen Medical Academy of Research and Translation (SMART). The authors gratefully acknowledge Prof. Nieng Yan’s Lab for providing the Cav1.2 dataset and Prof. Qi Hu’s Lab for providing the HSL-dimer dataset.
Author information
Authors and Affiliations
Contributions
C.B. and M.H. initiated and supervised the project. J.L., D.Z., and H.Z. developed CoCoFold, carried out testing, and analyzed the data. L. Z. provided the suggestions for computing. J.L., M.H., and C.B. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Ashwin Dhakal and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liao, J., Zheng, D., Zhang, H. et al. Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95 (2026). https://doi.org/10.1038/s42004-026-01899-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-026-01899-7
