
Abstract
Somatostatin, produced by pancreatic islet δ cells, is a key intra-islet paracrine factor that regulates the secretion of the glucoregulatory hormones insulin and glucagon from β cells and α cells, respectively. Here, we show that glutamate and glucagon released by α cells cooperatively activate neighbouring δ cells through AMPA and glucagon receptors, thereby enabling spatiotemporal feedback control of glucagon secretion. Crucially, prior hypoglycaemia enhances this mechanism by sensitizing δ cells to α cell-derived factors and inducing long-lasting structural and functional changes that facilitate δ cell and α cell paracrine interaction. This culminates in somatostatin hypersecretion that impairs counter-regulatory glucagon release. These hypoglycaemia-driven effects were emulated by chemogenetic activation of α cells or high concentrations of exogenous glucagon but prevented by inhibitors of glucagon receptors or the transcription factor CREB. This plasticity represents a key component of the islet’s ‘metabolic memory’, which, through impaired counter-regulatory glucagon secretion, increases the occurrence of recurrent hypoglycaemia that complicates the management of insulin-dependent diabetes.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


Data availability
All materials used are available from the authors or are commercially available. The data that support the findings of this study are available from the corresponding author upon reasonable request, including original images and videos. Representative videos are included in Supplementary Information. Source data are provided with this paper.
References
Göke, B. Islet cell function: α and β cells—partners towards normoglycaemia. Int. J. Clin. Pract. 62, 2–7 (2008).
Rorsman, P. & Ashcroft, F. M. Pancreatic β-cell electrical activity and insulin secretion: of mice and men. Physiol. Rev. 98, 117–214 (2018).
Gao, R. et al. α-Cell electrophysiology and the regulation of glucagon secretion. J. Endocrinol. 258, e220295 (2023).
Weitz, J., Menegaz, D. & Caicedo, A. Deciphering the complex communication networks that orchestrate pancreatic islet function. Diabetes 70, 17–26 (2020).
Kahn, S. E., Hull, R. L. & Utzschneider, K. M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–846 (2006).
Hermansen, K. Characterisation of the abnormal pancreatic D and A cell function in streptozotocin diabetic dogs: studies with D-glyceraldehyde, dihydroxyacetone, D-mannoheptulose, D-glucose, and L-arginine. Diabetologia 21, 489–494 (1981).
Unger, R. H. & Cherrington, A. D. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J. Clin. Investig. 122, 4–12 (2012).
Cryer, P. E. Hypoglycemia in type 1 diabetes mellitus. Endocrinol. Metab. Clin. North Am. 39, 641–654 (2010).
Heller, S. R., Peyrot, M., Oates, S. K. & Taylor, A. D. Hypoglycemia in patient with type 2 diabetes treated with insulin: it can happen. BMJ Open Diabetes Res. Care 8, e001194 (2020).
Huo, L., Harding, J. L., Peeters, A., Shaw, J. E. & Magliano, D. J. Life expectancy of type 1 diabetic patients during 1997–2010: a national Australian registry-based cohort study. Diabetologia 59, 1177–1185 (2016).
Skrivarhaug, T. et al. Long-term mortality in a nationwide cohort of childhood-onset type 1 diabetic patients in Norway. Diabetologia 49, 298–305 (2006).
Karimian, N. et al. Somatostatin receptor type 2 antagonism improves glucagon counterregulation in biobreeding diabetic rats. Diabetes 62, 2968–2977 (2013).
Yue, J. T. Y. et al. Somatostatin receptor type 2 antagonism improves glucagon and corticosterone counterregulatory responses to hypoglycemia in streptozotocin-induced diabetic rats. Diabetes 61, 197–207 (2012).
Brereton, M. F., Vergari, E., Zhang, Q. & Clark, A. Alpha-, delta- and PP-cells: are they the architectural cornerstones of islet structure and co-ordination? J. Histochem. Cytochem. 63, 575–591 (2015).
Huang, J. L. et al. Paracrine signalling by pancreatic δ cells determines the glycaemic set point in mice. Nat. Metab. 6, 61–77 (2024).
Strowski, M. Z., Parmar, R. M., Blake, A. D. & Schaeffer, J. M. Somatostatin inhibits insulin and glucagon secretion via two receptor subtypes: an in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinology 141, 111–117 (2000).
Gromada, J., Høy, M., Buschard, K., Salehi, A. & Rorsman, P. Somatostatin inhibits exocytosis in rat pancreatic α-cells by Gi2-dependent activation of calcineurin and depriming of secretory granules. J. Physiol. 535, 519–532 (2001).
Kailey, B. et al. SSTR2 is the functionally dominant somatostatin receptor in human pancreatic β- and α-cells. Am. J. Physiol. Endocrinol. Metab. 303, E1107–E1116 (2012).
Alford, F. P. et al. Glucagon control of fasting glucose in man. Lancet 304, 974–977 (1974).
Walker, J. N. et al. Regulation of glucagon secretion by glucose: Paracrine, intrinsic or both? Diabetes Obes. Metab. 13, 95–105 (2011).
Denwood, G. et al. Glucose stimulates somatostatin secretion in pancreatic δ-cells by cAMP-dependent intracellular Ca2+ release. J. Gen. Physiol. 151, 1094–1115 (2019).
Vergari, E. et al. Somatostatin secretion by Na+-dependent Ca2+-induced Ca2+ release in pancreatic delta-cells. Nat. Metab. 2, 32–40 (2020).
Unger, R. H. & Orci, L. Possible roles of the pancreatic D-cell in the normal and diabetic states. Diabetes 26, 241–244 (1977).
Hara, M., Patton, G. & Gerich, J. Increased somatostatin release from pancreases of alloxan diabetic rats perfused in vitro. Life Sci. 24, 625–628 (1979).
Abdel-Halim, S. M., Guenifi, A., Efendic, S. & Ostenson, C. G. Both somatostatin and insulin responses to glucose are impaired in the perfused pancreas of the spontaneously noninsulin-dependent diabetic GK (Goto-Kakizaki) rats. Acta Physiol. Scand. 148, 219–226 (1993).
Feniuk, W., Jarvie, E., Luo, J. & Humphrey, P. P. Selective somatostatin sst2 receptor blockade with the novel cyclic octapeptide, CYN-154806. Neuropharmacology 39, 1443–1450 (2000).
Alexander, G. M. et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39 (2009).
Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G. & Deisseroth, K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268 (2005).
Bonaventura, J. et al. High-potency ligands for DREADD imaging and activation in rodents and monkeys. Nat. Commun. 10, 4627 (2019).
Charpantier, E., Cancela, J. & Meda, P. Beta cells preferentially exchange cationic molecules via connexin 36 gap junction channels. Diabetologia 50, 2332–2341 (2007).
Serre-Beinier, V. et al. Cx36 preferentially connects beta-cells within pancreatic islets. Diabetes 49, 727–734 (2000).
DiGruccio, M. R. et al. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol. Metab. 5, 449–458 (2016).
Svendsen, B. et al. Insulin secretion depends on intra-islet glucagon signaling. Cell Rep. 25, 1127–1134.e2 (2018).
Richards, P. et al. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes 63, 1224–1233 (2014).
Rouille, Y., Westermark, G., Martin, S. K. & Steiner, D. F. Proglucagon is processed to glucagon by prohormone convertase PC2 in alpha TC1-6 cells. Proc. Natl Acad. Sci. USA 91, 3242–3246 (1994).
Holst, J. J. The physiology of glucagon-like peptide 1. Physiol. Rev. 87, 1409–1439 (2007).
van der Meulen, T. et al. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 21, 769–776 (2015).
Cabrera, O. et al. Glutamate is a positive autocrine signal for glucagon release. Cell Metab. 7, 545–554 (2008).
Muroyama, A. et al. A novel variant of ionotropic glutamate receptor regulates somatostatin secretion from delta-cells of islets of Langerhans. Diabetes 53, 1743–1753 (2004).
Ogita, K. et al. Inhibition of vesicular glutamate storage and exocytotic release by Rose Bengal. J. Neurochem. 77, 34–42 (2001).
Qiao, A. et al. Structural basis of Gs and Gi recognition by the human glucagon receptor. Science 367, 1346–1352 (2020).
Xu, Y. & Xie, X. Glucagon receptor mediates calcium signaling by coupling to Gαq/11 and Gαi/o in HEK293 cells. J. Recept. Signal. Transduct. Res. 29, 318–325 (2009).
Hamilton, A. et al. Adrenaline stimulates glucagon secretion by Tpc2-dependent Ca2+ mobilization from acidic stores in pancreatic α-cells. Diabetes 67, 1128–1139 (2018).
Acreman, S. et al. The endoplasmic reticulum plays a key role in α-cell intracellular Ca2+ dynamics and glucose-regulated glucagon secretion in mouse islets. iScience 27, 109665 (2024).
Zhang, Q. et al. R-type Ca2+-channel-evoked CICR regulates glucose-induced somatostatin secretion. Nat. Cell Biol. 9, 453–460 (2007).
Kim, A. et al. Arginine-vasopressin mediates counter-regulatory glucagon release and is diminished in type 1 diabetes. eLife 10, e72919 (2021).
Oduori, O. S. et al. Gs/Gq signaling switch in β cells defines incretin effectiveness in diabetes. J. Clin. Investig. 130, 6639–6655 (2020).
Heller, S. R. & Cryer, P. E. Reduced neuroendocrine and symptomatic responses to subsequent hypoglycemia after 1 episode of hypoglycemia in nondiabetic humans. Diabetes 40, 223–226 (1991).
Bisgaard Bengtsen, M. & Møller, N. Experimentally induced hypoglycemia-associated autonomic failure in humans: determinants, designs, and drawbacks. J. Endocr. Soc. 6, bvac123 (2022).
Cryer, P. E. in Endocrine Emergencies: Recognition and Treatment (eds Loriaux, L. & Vanek, C.) 27–35 (Springer, 2021).
Arrojo, E. D. R. et al. Structural basis for delta cell paracrine regulation in pancreatic islets. Nat. Commun. 10, 3700 (2019).
Koo, S.-H. et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437, 1109–1114 (2005).
Deisseroth, K., Bito, H. & Tsien, R. W. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron 16, 89–101 (1996).
Kaestner, K. H., Powers, A. C., Naji, A., HPAP Consortium & Atkinson, M. A. NIH initiative to improve understanding of the pancreas, islet, and autoimmunity in type 1 diabetes: the Human Pancreas Analysis Program (HPAP). Diabetes 68, 1394–1402 (2019).
Cryer, P. E., Davis, S. N. & Shamoon, H. Hypoglycemia in diabetes. Diabetes Care 26, 1902–1912 (2003).
Lai, B. K. et al. Somatostatin is only partly required for the glucagonostatic effect of glucose but is necessary for the glucagonostatic effect of KATP channel blockers. Diabetes 67, 2239–2253 (2018).
Singh, B., Khattab, F. & Gilon, P. Glucose inhibits glucagon secretion by decreasing [Ca2+]c and by reducing the efficacy of Ca2+ on exocytosis via somatostatin-dependent and independent mechanisms. Mol. Metab. 61, 101495 (2022).
Zhang, Q. et al. Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 18, 871–882 (2013).
Vieira, E., Salehi, A. & Gylfe, E. Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic alpha cells. Diabetologia 50, 370–379 (2007).
Göpel, S. et al. Capacitance measurements of exocytosis in mouse pancreatic α-, β- and δ-cells within intact islets of Langerhans. J. Physiol. 556, 711–726 (2004).
Gandasi, N. R. et al. GLP-1 metabolite GLP-1(9–36) is a systemic inhibitor of mouse and human pancreatic islet glucagon secretion. Diabetologia 67, 528–546 (2024).
Finan, B., Capozzi, M. E. & Campbell, J. E. Repositioning glucagon action in the physiology and pharmacology of diabetes. Diabetes 69, 532–541 (2019).
Ionescu-Tirgoviste, C. et al. A 3D map of the islet routes throughout the healthy human pancreas. Sci. Rep. 5, 14634 (2015).
Motté, E. et al. Composition and function of macroencapsulated human embryonic stem cell-derived implants: comparison with clinical human islet cell grafts. Am. J. Physiol. Endocrinol. Metab. 307, E838–E846 (2014).
Barg, S., Galvanovskis, J., Göpel, S. O., Rorsman, P. & Eliasson, L. Tight coupling between electrical activity and exocytosis in mouse glucagon-secreting alpha-cells. Diabetes 49, 1500–1510 (2000).
Dean, P. M. Ultrastructural morphometry of the pancreatic β-cell. Diabetologia 9, 115–119 (1973).
Tong, J. C. L. et al. Localized GLP1 receptor pre-internalization directs pancreatic alpha cell to beta cell communication. Cell Metab. 37, 1698–1714.e5 (2025).
Hoffman, E. G., D’Souza, N. C., Liggins, R. T. & Riddell, M. C. Pharmacologic inhibition of somatostatin receptor 2 to restore glucagon counterregulation in diabetes. Front. Pharm. 14, 1295639 (2023).
Patel, Y. C. Somatostatin and its receptor family. Front. Neuroendocrinol. 20, 157–198 (1999).
Hill, T. G. et al. Loss of electrical β-cell to δ-cell coupling underlies impaired hypoglycaemia-induced glucagon secretion in type-1 diabetes. Nat. Metab. 6, 2070–2081 (2024).
Abitbol, A. et al. 219-OR: glucagon counterregulation in a hypoglycemic clamp in type 1 diabetes is increased by ZT-01, a novel somatostatin receptor 2 antagonist—a phase 1b study. Diabetes 72, 219-OR (2023).
Citri, A. & Malenka, R. C. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33, 18–41 (2008).
Han, M. H. et al. Role of cAMP response element-binding protein in the rat locus ceruleus: regulation of neuronal activity and opiate withdrawal behaviors. J. Neurosci. 26, 4624–4629 (2006).
Cox, L. J., Hengst, U., Gurskaya, N. G., Lukyanov, K. A. & Jaffrey, S. R. Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat. Cell Biol. 10, 149–159 (2008).
Viosca, J., Lopez de Armentia, M., Jancic, D. & Barco, A. Enhanced CREB-dependent gene expression increases the excitability of neurons in the basal amygdala and primes the consolidation of contextual and cued fear memory. Learn. Mem. 16, 193–197 (2009).
Garg, S. K. et al. Hypoglycemia begets hypoglycemia: the order effect in the ASPIRE in-clinic study. Diabetes Technol. Therapeutics 16, 125–130 (2014).
Schroeder, E. B. et al. Predicting the 6-month risk of severe hypoglycemia among adults with diabetes: development and external validation of a prediction model. J. Diabetes Complications 31, 1158–1163 (2017).
Cariou, B. et al. Frequency and predictors of confirmed hypoglycaemia in type 1 and insulin-treated type 2 diabetes mellitus patients in a real-life setting: results from the DIALOG study. Diabetes Metab. 41, 116–125 (2015).
Gerich, J. E., Langlois, M., Noacco, C., Karam, J. H. & Forsham, P. H. Lack of glucagon response to hypoglycemia in diabetes: evidence for an intrinsic pancreatic alpha cell defect. Science 182, 171–173 (1973).
Segel, S. A., Paramore, D. S. & Cryer, P. E. Hypoglycemia-associated autonomic failure in advanced type 2 diabetes. Diabetes 51, 724–733 (2002).
Fanelli, C. G. et al. Meticulous prevention of hypoglycemia normalizes the glycemic thresholds and magnitude of most of neuroendocrine responses to, symptoms of, and cognitive function during hypoglycemia in intensively treated patients with short-term IDDM. Diabetes 42, 1683–1689 (1993).
Adriaenssens, A. et al. A transcriptome-led exploration of molecular mechanisms regulating somatostatin-producing D-cells in the gastric epithelium. Endocrinology 156, 3924–3936 (2015).
Briant, L. J. et al. Functional identification of islet cell types by electrophysiological fingerprinting. J. R. Soc. Interface 14, 20160999 (2017).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Benito, E., Valor, L. M., Jimenez-Minchan, M., Huber, W. & Barco, A. cAMP response element-binding protein is a primary hub of activity-driven neuronal gene expression. J. Neurosci. 31, 18237–18250 (2011).
Acknowledgements
This study was supported by a Diabetes UK RD Lawrence Fellowship (Q. Zhang, 14/0005128) and project grants by the EFSD (‘New Targets for Diabetes or Obesity-related Metabolic Diseases’ educational research grant from MSD; to Q. Zhang), the Royal Society (International Exchanges 2017 Cost Share (China) grant to Q. Zhang, IEC\NSFC\170394), national funds via FCT under project UIDP/04539/2020 (Q. Zhang), the FEDER within the scope of COMPETE 2030 and by national funds via FCT (MPr-2023-12-SACCCT-Projetos de IC&DT Programme, COMPETE2030-FEDER-00698600, https://doi.org/10.54499/2023.16678.ICDT; Q. Zhang), the RCUK Medical Research Council (P.R., MR/VO11979/1), the Leona M. and Harry B. Helmsley Charitable Trust (G-1912-03553, G-2305-06047 and 2012-04188 to P.R.), the National Natural Science Foundation of China (82200887 to R.G. (Oxford)) and SSMF (Svenska sällskapet för medicinsk forskning to C.M.). M.G.-T. is supported by a European Union Marie Skłodowska-Curie Fellowship (grant 01108327, T2D-Omics). J.C.-S. is supported by the Knut and Alice Wallenberg Foundation (Wallenberg Molecular Medicine Fellow), the Swedish Research Council (grant 2021-05109) and the Erling Persson Foundation. Human islet work at Oxford was supported by the NIHR Oxford Biomedical Research Centre. Human islets for research from Alberta were provided by the Alberta Diabetes Institute IsletCore at the University of Alberta in Edmonton (http://www.bcell.org/adi-isletcore.html) with the assistance of the Human Organ Procurement and Exchange (HOPE) programme, Trillium Gift of Life Network (TGLN) and other Canadian organ procurement organizations. All donors’ families gave informed consent for the use of pancreatic tissue in research. We are grateful to the donors and their families. We thank F. Reimann and F. Gribble (University of Cambridge) for the Sst-iCre mouse line and REMD Biotherapeutics for REMD-477 and P. E. MacDonald (University of Alberta), D. Hodson (University of Oxford) and E. Coutinho (University of Coimbra) for valuable discussions throughout the study.
Author information
Authors and Affiliations
Contributions
Q. Zhang and P.R. conceived the study. All the authors performed the experiments and analysed the data. Q. Zhang and R.G. (Oxford) designed the experiments and interpreted the results. M.G.-T. and J.C.-S. performed the transcriptomic analysis. R.G. (Oxford), S.A., P.R. and Q. Zhang wrote the paper. Q. Zhang, R.G. (Oxford) and P.R. secured funding. All authors read and approved the final version of the paper. Q. Zhang is the guarantor of this work, and as such, has full access to all the data in the study and takes responsibility for the integrity of the data and accuracy of the analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Metabolism thanks Alejandro Caicedo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Revati Dewal, in collaboration with the Nature Metabolism team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Crosstalk between α- and δ-cells is not mediated by electrical coupling.
a-b, Glucagon (a) and SST (b) secretion at 1 or 6 mM glucose in the absence or presence of 1 μM J60 in Gcg-hM3Dq− islets (6 experiments/group from 4 mice). **p = 0.002, ***p = 0.0002 vs. 1 mM glucose alone and **p = 0.002, ***p = 0.0004 vs. 1 mM glucose with J60; two-sided unpaired t-test. c, Experimental setup of the patch-imaging technique. Left: schematic diagram of the ‘patch-imaging’ experimental paradigm. An α-cell within an intact Sst-GCaMP6 islet is voltage-clamped (‘patched’) and electrically stimulated (with a depolarisation, electrical stimulus) using a patch-clamp amplifier. A fluorescent tracer (red) is infused through the patch pipette to mark the ‘patched’ α-cell. Ca2+ response in an adjacent δ-cell is monitored as changes in GCaMP6f fluorescence (green line). Green=δ-cell (light green indicates increase in fluorescence), orange=non-patched α-cell, blue=β-cell. Right: Image captured during the patch-imaging experiment. Sst-GCaMP6 islet is outlined by the green dashed line. Red shows an α-cell infused with sulforhodamine B (SRB) through an electrode (arrow, white dashed line). The adjacent δ-cell is shown in green (GCaMP6f fluorescence). Scale bar:10 μm. d, Representative traces of capacitance measurements (ΔCm; red traces) showing an α-cell without exocytosis in response to progressively-longer depolarisations (10 to 200-ms, from -70 to 0 mV; top), and its adjacent δ-cell was without [Ca2+]i responses (ΔF/F0; green). The dashed vertical lines indicate the onset of the depolarisations with indicated durations in the α-cell. Representative of 11 cell pairs in 11 islets from 8 mice. e, Assessment of dye diffusion from an α-cell to its neighbouring cells. Top: an α-cell is patch-clamped and injected with propidium iodide (PI, a connexin 36-permeable dye) through an electrode (white dashed lines, indicated by arrow). The adjacent δ-cells are shown in green (GCaMP6f fluorescence). Bottom: same image as the top panel but without green channel. PI is clearly confined within the patched α-cell and does not diffuse into its neighbouring cells. Scale bar=10 μm. Representative of 4 cells in 4 islets from 2 mice. f, Exocytosis (ΔCm; red, middle) triggered by a train of pulses (10 200-ms depolarizations from -70 to 0 mV; top) of the α-cell shown in (E) and [Ca2+]i oscillations (ΔF/F0; green, bottom) of the adjacent δ-cell recorded simultaneously. Vertical dashed line indicates onset of the depolarization of the α-cell and horizontal dashed lines mark baselines. Representative of 4 cells in 4 islets from 2 mice. In dot plots, rectangles and error bars behind data points represent mean values ± S.E.M.
Extended Data Fig. 2 α-cell-activity-dependent activation of δ-cells involves glucagon, glutamate, not GLP-1 or GRPP.
a, SST secretion at 1 mM glucose in response to REMD-477 (REMD) in the presence and absence of GLP-1 receptor blocker exendin-(9-39) as indicated (5 experiments using islets from 3 mice per group). **p = 0.004 (REMD alone) and **p = 0.003 (REMD with exendin-(9-39)) vs. control. b, as in (a) but the effects of REMD and exendin-(9-39) were tested at 1 mM glucose in the presence of 1 µM glucagon in indicated groups (5 experiments using islets from 3 mice per group). ***p = 0.0003 vs. control (1 mM glucose alone); #p = 0.01, ####p < 0.0001 vs. 1 mM glucose with 1 µM glucagon; §§p = 0.009 vs. 1 mM glucose with 1 µM glucagon and 20 nM REMD. c, SST secretion at 1 mM glucose in response to GRPP and GLP-1 receptor blocker exendin-(9-39) (n = 9 experiments/group for control and n = 4 experiments/group for exendin-(9-39) and GRPP, 8 mice; concentrations as indicated). d-e, Representative δ-cell [Ca2+]i oscillations (ΔF/F0) in Sst-GCaMP6 islets at 1 mM glucose in response to 1 μM exendin-(9-39) (d). The δ-cell Ca2+ spike frequency is summarized in (f) (123 cells from 7 islets, 2 mice). f-g, Representative δ-cell [Ca2+]i oscillations (ΔF/F0) in Sst-GCaMP6 islets at 1 mM glucose in response to 1 μM GRPP (f). The δ-cell Ca2+ spike frequency is summarized in (g) (105 cells from 4 islets, 2 mice). h, Insulin secretion measured at 1 mM glucose in response to 1 µM glucagon in the presence and absence of corticotropin-releasing hormone receptor antagonist Astressin2B (5 experiments using islets isolated from 4 mice). i, As in (h) but the effect on SST secretion was measured (5 experiments using islets from 4 mice). ***p = 0.0002 (glucagon alone) and ***p = 0.0001 (glucagon with Astressin2B) vs. control. j, Immunofluorescent staining of GluR3 (top, green) or GluR4 (bottom, green) and SST (red). Areas in the yellow quadrangles are displayed on expanded scales. Scale bar=20 μm. Representative of 14 islets from 2 mice for GluR3 and 11 islets from 2 mice for GluR4. In dot plots, rectangles and error bars behind data points represent mean values ± S.E.M. and violin plots the median with quartiles. In (a-c, i), one-way ANOVA with Tukey post hoc test was used.
Extended Data Fig. 3 Glucagon and glutamate synergistically mediate α-cell activity-dependent effects on δ-cells.
a, Schematic showing the experimental setup of the ‘puff-imaging’ technique. An Sst-GCaMP6 islet is immobilized by a suction pipette (left). A series of treatment solutions can be rapidly delivered to the islet through a puffing pipette (‘puffer’, right) controlled by an oocyte injector (Nanoliter, not shown). δ-cell Ca2+ activity is reported by changes in GCaMP6f fluorescence (ΔF/F0) using an optical apparatus mounted on the microscope (not shown). Scale bar:50 μm. b, Representative δ-cell [Ca2+]i response (ΔF/F0) in Sst-GCaMP6 islet to acute applications of glucagon (concentrations as indicated) at 1 mM glucose. The vertical dashed lines indicate the times at which glucagon was applied. Puffing duration=1.5 s. c-e, Representative δ-cell [Ca2+]i oscillations (ΔF/F0) in Sst-GCaMP6 islets at 1 mM glucose pre-treated with 100 nM rose bengal or not (control) for >1 h prior to imaging (c). Rose bengal was also included in the extracellular media during perfusion. The δ-cell Ca2+ spike frequency (control, n = 79 cells from 3 islets; rose bengal, n = 88 cells from 5 islets; 4 mice; ***p = 0.0001 vs. control, two-sided Mann-Whitney test) and percentage of active δ-cell (control, n = 3 islets; rose bengal, n = 4 islets; 4 mice; **p = 0.005 vs. control, two-sided unpaired t-test) are respectively summarised in (d) and (e). f, SST secretion at 1 mM glucose in mouse islets pre-treated with 100 nM rose bengal (n = 3 experiments) or not (control, n = 4 experiments) for >1 h (islets were from 3 mice). *p = 0.01 vs. control, two-sided unpaired t-test. g-h as (c-d) but show the response to 1 μM glucagon under indicated conditions (control, n = 79 cells from 3 islets; rose bengal, n = 88 cells from 5 islets; 4 mice; ****p < 0.0001 vs. control, two-sided Mann-Whitney test). i, as (f) but shows the effect of rose bengal pre-treatment in the presence of 1 μM glucagon on SST secretion at 1 mM glucose (4 experiments/group from 4 mice). *p = 0.02, **p = 0.0023 vs. control; ##p = 0.0015 (without rose bengal) and ##p = 0.0017 (with rose bengal) vs. without glucagon in the same treatment group; two-sided unpaired t-test. j, SST secretion at 1 mM glucose in response to CNQX or CP465022 in the presence of 1 μM glucagon (5 experiments/group from 3 mice). *p < 0.05, ****p < 0.0001 vs. 1 mM glucose alone and ####p < 0.0001 vs. 1 mM glucose in the presence of 1 μM glucagon. k, shows the effects of REMD or CP465022 alone or in combination on SST secretion at 1 mM glucose (n = 6 experiments/group for control, REMD alone and CP465022 alone; n = 7 experiments for REMD with CP465022; 4 mice; concentrations as indicated). ***p < 0.001, ****p < 0.0001 vs. control; REMD + CP465022 vs. REMD alone, p = 0.24. In dot plots, rectangles and error bars behind data points represent mean values ± S.E.M. and violin plots the median with quartiles. In (j-k), one-way ANOVA with Tukey post hoc test was used.
Extended Data Fig. 4 Glucagon stimulates δ-cell Ca2+ activity via Gs and Gq pathways.
a, δ-cell [cAMP]i (cADDis signal in Sst-tdRFP islets; ΔF/F0) at 1 mM glucose in response to 1 μM glucagon and 10 μM forskolin (positive control). Data represents mean value (solid red trace) ± SEM (shaded ribbon) (41 cells from 15 islets, 2 mice). Timings and durations of the application of treatments are indicated with horizontal red bars and grey shaded areas. The inset summarizes δ-cell [cAMP]i AUC in the absence or presence of 1 μM glucagon. ****p = 2.9×10−9 vs. control, two-sided unpaired t-test. b, Representative δ-cell [Ca2+]i oscillations (ΔF/F0) in Sst-GCaMP6 islets at 1 mM glucose in the absence or presence of 1 μM glucagon in response to pre-treatment and continuous presence of control medium, ESI-05, PKI 14-22 amide, ryanodine, YM-254890 or xestospongin C. Concentrations and treatments are as indicated. c, Percentage of active δ-cell at 1 mM glucose under indicated experimental conditions as shown in (b) (control, n = 3 islets; ESI-05, n = 7 islets; PKI, n = 5 islets; ryanodine, n = 5 islets; YM-254890, n = 5 islets; Xestospongin C, n = 5 islets; 4 mice). *p < 0.05, **p < 0.01 vs. control, one-sided unpaired t-test. d, Summary of δ-cell Ca2+ spike frequency under indicated experimental conditions as shown in (b) (control, n = 79 cells in 3 islets; ESI-05, n = 133 cells in 7 islets; PKI, n = 71 cells in 5 islets; ryanodine, n = 72 cells in 5 islets; YM-254890, n = 86 cells in 5 islets; Xestospongin C, n = 102 cells in 5 islets, 4 mice). For comparison, the responses with and without glucagon but in the absence of blockers are shown as the controls (dark grey, data taken from Fig. 3h). **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. control of the respective groups, two-sided Mann-Whitney test; ###p = 0.0001 vs. without glucagon, two-sided Wilcoxon matched-pairs signed rank test. e-f, SST secretion at 1 mM glucose with and without 1 μM glucagon in mouse (e) (n = 4 experiments for control, n = 3 experiments/group for all other groups, 4 mice) and human (f) islets (n = 11 experiments for control, n = 10 experiments/group for all other groups, 3 donors, different colours mark individual donors) pre-treated with conditions as indicated. *p < 0.05, **p < 0.01, ***p < 0.001, ****p = 7.4×10−5 vs. control of the respective groups; #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 vs. without glucagon in the same pre-treatment group, two-sided unpaired t-test. In (b-f), glucagon was applied acutely and all the blockers were applied during preincubation and perfusion (Ca2+ imaging) or incubation (secretion experiments). In dot plots, rectangles and error bars behind data points represent mean values ± S.E.M. and violin plots the median with quartiles.
Extended Data Fig. 5 Previous hypoglycaemia compromises counter-regulatory glucagon response.
a, Plasma glucose levels after administration of saline (control, black, n = 6 mice; control for CYN, blue, n = 7 mice) or insulin (0.75 U/kg, hypo, red, n = 5 mice; hypo for CYN, orange, n = 7 mice) on Day 0 in wildtype mice. ****p < 0.0001 vs. control. ####p < 0.0001 vs. control for CYN154806 group (CYN154806 was only used on Day 1). b-c, Plasma glucose (b) and glucagon (c) levels measured on Day 1 during ITTs with or without CYN158406 (0.5 mg/kg, injected 15 min before insulin) in control or mice that had hypoglycaemia on Day 0. Black, control without CYN, n = 6 mice; red, hypo without CYN, n = 5 mice; blue, control with CYN, n = 7 mice; orange, hypo with CYN, n = 7 mice. Data are normalized to baseline (0 min). Summaries of AUCs of each group are shown on the right. *p = 0.02, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. control; #p < 0.05, ##p = 0.002, ####p < 0.0001 vs. hypo alone. d-e, Plasma glucose (d) and glucagon (e) in control and hypoglycaemia pretreated mice measured during ITTs on Day 2 (control: black; hypo: red; 48 h later) or Day 3 (control: blue; hypo: orange; 72 h later) (5 mice/group). Severe hypoglycaemia and slow restoration of normoglycaemia were observed in Day 2 hypo group at 90 min time point of the ITTs and experiments were stopped (no 120 min data points). Right panels summarize the AUC of each group (until 90 min in e and 60 min in f). *p < 0.05, **p = 0.002 vs. control of the respective testing day. f-g, as in d-e but data are shown as relative to baseline (0 min) (5 mice/group). **p = 0.003 vs. control of the respective testing day. h-k, Plasma glucose nadir (h), glucose AUC (i), glucagon peak (j) and glucagon AUC (k) measured during ITT after previous hypoglycaemia with indicated euglycaemic intervals (x-axes). Data are from Extended Data Fig. 5b-c and 5g-h (control, n = 16 mice; 24 h interval, n = 5 mice; 48 h interval, n = 4 mice; 72 h interval, n = 5 mice). *p = 0.02, **p < 0.05 vs. hypoglycaemia naïve mice. In dot plots, rectangles and error bars behind data points represent mean values ± S.E.M. Two-sided unpaired t-test was used in (a-g); one-sided unpaired t-test was used in (h-k). In (h-k), each mouse is one biological replicate.
Extended Data Fig. 6 Low-glucose treatment induces lasting changes in δ-cell activity and morphology.
a-b, Glucagon secretion at 1 or 6 mM glucose with or without 100 nM CYN154806 in control (6 mM; black) or islets that were incubated with low glucose (2 mM, red) for 30 min (a) or for 1 h (b) ~18 h post pretreatment (4 experiments/group from 4 mice). **p = 0.006 vs. control in the same category; ##p < 0.01, ###p < 0.001, ####p = 0.00003 vs. 1 mM glucose in the same treatment group. §p < 0.05, §§p < 0.01 vs. without CYN154806 in the same treatment group. c-d, as (a-b) but SST secretion was measured (4 experiments/group from 4 mice). #p = 0.02, ##p < 0.01 vs. 1 mM glucose in the same treatment group. e-f, Glucagon (e) and SST (f) secretion measured in islets that were pre-treated with low glucose (2 mM) with for indicated durations. Data are normalised to control (without low glucose exposure) and are derived from Extended Data Fig. 6a-d and Fig. 5d-e (control, n = 14 experiments, 8 mice; 30 min, n = 4 experiments, 4 mice; 60 min, n = 4 experiments, 4 mice; 120 min, n = 6 experiments, 8 mice). **p = 0.005, ***p < 0.001 vs. islets cultured at 6 mM throughout. g-h, Immunofluorescent staining of wildtype islets with glucagon (GCG, green) and SST (red), with merged and enlarged slides to show histological proximity between α- and δ-cells. Islets were pre-treated with control (6 mM) or 2 mM glucose for 2 h (g) or 24 h (h). Scale bar: 20 μm. i, Quantification of δ-cell protrusion length for experiments as shown in (g-h) (control 2 h, n = 78 cells from 9 islets; low glucose 2 h, n = 91 cells from 44 islets; control 24 h, n = 77 cells from 9 islets; low glucose 24 h, n = 124 cells from 12 islets, 2 mice). **p = 0.007, ***p = 0.0002 vs. control in the same category. In dot plots, rectangles and error bars behind data points represent mean values ± S.E.M. and violin plots the median with quartiles. Two-sided unpaired t-test was used in (a-f, i).
Extended Data Fig. 7 Glucagon receptor blockade prevents hypoglycaemia-associated changes in counter-regulatory glucagon responses.
a, Plasma glucose levels after administration of saline (‘control’, black), insulin (‘hypo’, red) or the combination of insulin and Gcg-amide (1 mg/kg; ‘hypo + Gcg-amide’, blue) on Day 0 (5 mice/group). **p = 0.009, ***p = 0.0005, ****p < 0.0001 vs. control; #p = 0.04, ##p = 0.005 vs hypo alone. b-c, Plasma glucose (b) and glucagon (c) levels relative to baseline (0 min) during ITTs on Day 1 in control (black), hypo (red), and hypo + Gcg-amide (blue) groups (5 mice/group). Right panels summarise the AUCs of each group. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control. #p < 0.05, ##p < 0.01 vs. hypo alone. d, Schematic of the protocol for in vitro islet function assessment following in vivo conditioning. Wildtype mice were administered with saline (‘control’), insulin (‘hypo’) or the combination of insulin and Gcg-amide (1 mg/kg; ‘hypo+Gcg-amide’) on Day 0. On Day 1, 24 h later, islets were isolated and hormone secretion was tested. e, Plasma glucose levels on Day 0 during in vivo conditioning period in mice that underwent indicated treatments (2 mice/group). f, Glucagon secretion measured at 1 mM glucose in the presence of 100 nM CYN154806 in islets isolated from mice that were subjected to in vivo treatments as indicated 24 h ago (control, n = 10 experiments; hypoglycaemia alone, n = 8 experiments; hypoglycaemia with Gcg-amide, n = 11 experiments; 2 mice/condition). g-i, Plasma glucagon (g), glucose (h), and insulin (i) levels after administration of J60 (0.1 mg/kg, 0 min; arrow) on Day 0 in Gcg-hM3Dq− (black, n = 12 mice) and Gcg-hM3Dq+ (red, n = 9 mice) mice. *p < 0.05, **p < 0.01, ****p = 9.6×10−5 vs. Gcg-hM3Dq−. j, As in (d), but illustrating the protocol used with mice subjected to chemically-induced hyperglucagonemia conditioning. Gcg-hM3Dq− and Gcg-hM3Dq+ mice were administered with J60 or the combination of J60 and Gcg-amide (1 mg/kg; ‘Gcg-amide’) on Day 0. On Day 1, 24 h later, islets were isolated and hormone secretion was tested. k, Plasma glucose levels on Day 0 in in vivo conditioning period in Gcg-hM3Dq− and Gcg-hM3Dq+ mice that underwent indicated treatments (2 mice/group). l, Glucagon secretion measured at 1 mM glucose in the presence of 100 nM CYN154806 in islets isolated from Gcg-hM3Dq− and Gcg-hM3Dq+ mice 24 h after previous in vivo treatments as indicated (Gcg-hM3Dq−, n = 10 experiments; Gcg-hM3Dq+ with J60, n = 9 experiments; Gcg-hM3Dq+ with J60 and Gcg-amide, n = 7 experiments, 2 mice/condition). Symbols and error bars and rectangles and error bars behind data points represent mean values ± S.E.M. Two-sided unpaired t-test was used in (a-c, f-i, l). (d, j) were created with BioRender.com.
Extended Data Fig. 8 Activation of glucagon signalling underlies low-glucose-induced changes in glucagon and SST secretion in isolated islets.
a-b, Glucagon (a) and SST (b) secretion at 1 mM glucose in Gcg-hM3Dq+ islets pre-treated with conditions as indicated (7 experiments/group from 6 mice). **p = 0.0011, ***p = 0.0009 vs. control, #p = 0.02, ##p = 0.004 vs. with J60 alone. c-d, As in (a-b) but show glucagon (c) and SST (d) secretion measured in Gcg-hM3Dq− islets (7-8 experiments/group from 6 mice). e-f, Glucagon (e) and SST (f) secretion at 1 mM glucose in wildtype (C57BL/6) islets pre-treated with conditions as indicated (5 experiments/group, 8 mice). *p < 0.05 vs. control. #p = 0.015, ###p = 0.0004 vs. 2 mM glucose alone. g-h, SST (g) and glucagon (h) secretion at 1 mM glucose in islets that were pre-treated with different concentration of added exogenous glucagon (0, 0.01 µM, 0.1 µM and 1 µM) 18 h previously (5 experiments/group, 8 mice). **p < 0.01, ***p = 0.0002 vs. control (no added exogenous glucagon). i, Representative α-cell [Ca2+]i oscillations (ΔF/F0) in Gcg-GCaMP6 islets at 1 mM glucose in response to 100 nM CYN154806 pre-treated with conditions as indicated on the left to the traces. j, Percentage of active α-cell at 1 mM glucose of experiments shown in (i) (control, n = 4 islets; 2 mM glucose pretreatment, n = 6 islets; 2 mM glucose with Gcg-amide, n = 5 islets; 4 mice). *p = 0.04 vs. control. #p = 0.04 vs. 2 mM glucose alone. k, Summary of α-cell Ca2+ spike frequency of experiments shown in (i). Control ± CYN154806, n = 58 cells from 4 islets; 2 mM glucose pretreatment ± CYN154806, n = 104 cells from 6 islets; 2 mM glucose with Gcg-amide ± CYN154806, n = 85 cells from 5 islets; 4 mice. For comparison, the responses in islets that were continuously cultured at 6 mM glucose are shown as controls (data from Fig. 1d). ***p = 0.0002 vs. control; #p = 0.04 vs. 2 mM glucose alone (two-sided unmatched rank sum Mann-Whitney test). §p = 0.02, §§§p < 0.001 vs. without CYN154806 in the same treatment group (two-sided paired Wilcoxon test). l, Representative δ-cell [Ca2+]i oscillations (ΔF/F0) in Sst-GCaMP6 islets at 1 mM glucose pre-treated with conditions as indicated. m-n, Percentage of active δ-cell at 1 mM glucose (m) and summary of δ-cell Ca2+ spike frequency (n) of experiments shown in (l). In (m): control, n = 4 islets; 2 mM glucose pretreatment, n = 6 islets; 2 mM glucose with Gcg-amide, n = 5 islets; 4 mice. ***p = 0.0008 vs. control; ##p = 0.003 vs. 2 mM glucose alone; two-sided unpaired t-test. In (n): control, n = 46 cells from 4 islets; 2 mM glucose pretreatment, n = 65 cells from 6 islets; 2 mM glucose with Gcg-amide, n = 42 cells from 5 islets; 4 mice. *p = 0.048 vs. control; #p = 0.022 vs. 2 mM glucose alone; two-sided Mann-Whitney test. o-p, Glucagon (o) and SST (p) secretion at 1 mM glucose in wildtype islets pre-treated with conditions as indicated (4 experiments/group from 4 mice). *p < 0.05 for indicated comparisons. Exogenous glucagon, J60, Gcg-amide, glutamate or CNQX was applied during the preincubation with indicated glucose concentration. For clarity, pre-treatment conditions are noted with the prefix ‘Pre-‘. CYN154806 and adrenaline were applied acutely during perfusion (Ca2+ imaging). Hormone secretion experiments were conducted ~18 h post preincubation. Islets were kept in 6 mM glucose except for the duration of pret-reatments. Symbols and error bars and rectangles and error bars behind data points represent mean values ± S.E.M. Violin plots show the median with quartiles. Two-sided unpaired t-test was used in (a-h, j, m, o-p).
Extended Data Fig. 9 Transcriptomic analysis of CREB-regulated genes in human and mouse δ-cells, and exposure of islets to low-glucose or exogenous glucagon activates CREBs in δ-cells.
a, Examples of immunofluorescence staining of p-CREB (green), SST (red) and nuclei (DAPI, blue) in mouse islets pre-treated with indicated conditions. Scale bar:10 µm. b, Nuclear p-CREB positive fractions of δ-cells in islets under indicated pre-treatments (control, n = 4 experiments; 2 mM glucose alone, n = 7 experiments; 2 mM glucose with Gcg-amide, n = 5 experiments; 2 mM glucose with REMD, n = 4 experiments; control with glucagon, n = 4 experiments; control with forskolin, n = 5 experiments; 2 mice). **p = 0.002, ****p < 0.0001 vs. 6 mM glucose alone; ###p = 0.0002 and ####p1.5×10−5 vs. 2 mM glucose alone; two-sided unpaired t-test. c-d, Single-cell transcriptomic data from the Human Pancreas Analysis Project (HPAP, 7161 delta cells, 46 nondiabetic donors) (c) and bulk transcriptomics data from purified mouse δ-cells (d) showing expression of CREB1-related genes (see Methods). e, Correlation between the mean expression of human (c) and mouse (d) genes that are upstream (black) and downstream (red) to CREB1 activation. Two-sided Pearson correlation results in a significant positive correlation with ρ = 0.65 and p-value of 2e-14. Linear fit in grey solid line. f, Pie chart with proportion of CREB-regulated genes expressed in δ-cells from both the human (HPAP) and mouse datasets, only in one dataset, or in none. g, Schematic representation of cellular location and pathways with CREB-related genes expressed in δ-cells, highlighting genes that are upstream (black) and downstream (red) of CREB1 (Created with BioRender.com.). Symbols and error bars and rectangles and error bars behind data points represent mean values ± S.E.M.
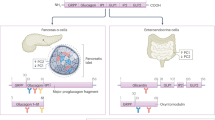
Extended Data Fig. 10 Negative feedback control of α-cells by neighbouring δ-cells and its enhancement following prior hypoglycaemia.
a, Schematic of α-/δ-cell interaction within an islet. An α-cell (left) releases glucagon (purple) and glutamate (orange) that bind to glucagon receptors (GCGR) and ionotropic glutamate receptors (iGluR), respectively, in a neighbouring δ-cell. Activation of GCGR, a class-B G protein-coupled receptor (GPCR) that is coupled to Gαs and Gαq proteins, leading to elevation of IP3 (through activation of Gαq and phospholipase C, PLC) and cAMP (via activation of Gαs and subsequent adenylyl cyclase, AC). The two second messengers potentiate or directly lead to intracellular Ca2+ release from the ER by activating IP3 receptors (IP3R, by IP3) and ryanodine receptor 3 (RyR3, by the cAMP effector Epac2). Ca2+ influx (through the iGluRs or following initiation of electrical activity) triggers Ca2+-induced Ca2+ release (CICR), from the smooth ER (sER), that strongly stimulates δ-cell exocytosis and release of somatostatin (SST, red). Secreted SST activates α-cell somatostatin receptor 2 (SSTR2), a Gi-coupled GPCR, and G-protein-coupled inwardly rectifying K+ channels (GIRK) and inhibit exocytosis and AC (which leads to reduce cytosolic cAMP and further reduction in exocytosis). These combined actions suppress glucagon secretion. b, Schematic portraying situation following prior hypoglycaemia. During prior hypoglycaemia (left), the release of abundant glucagon (purple) activates Gαs signalling in δ-cells through glucagon receptors (GCGR), leading to an increase in cAMP levels. This elevation in cAMP subsequently activates protein kinase A (PKA), which activates CREB (through phosphorylation of CREB at Ser133) in δ-cells. Activated CREB induced functional changes in δ-cells, culminating in hypersensitivity to paracrine signals from α-cell-derived glucagon (purple) and glutamate (orange) and excessive release of somatostatin (SST, red) in subsequent hypoglycaemia. In addition, δ-cell filopodia elongate to facilitate paracrine signalling in response to prior hypoglycaemia. Collectively, these heighten the somatostatin-mediated negative feedback control of α-cells by δ-cells, resulting in inappropriate suppression of glucagon secretion and compromise the counter-regulatory response to subsequent hypoglycaemia. Created with BioRender.com.
Supplementary information
Supplementary Information
Supplementary Tables 1 and 2, Figs. 1 and 2, and Videos 1–3 captions.
Supplementary Video 1
A significant proportion of δ-cells are active at 1 mM glucose within an intact islet.
Supplementary Video 2
Patch-imaging experiment shows the α-cell activity stimulates neighbouring δ-cells.
Supplementary Video 3
Local rapid application of glutamate stimulates δ-cells within an intact islet.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gao, R., Acreman, S., Dou, H. et al. Antecedent hypoglycaemia impairs glucagon secretion by enhancing somatostatin-mediated negative feedback control. Nat Metab (2026). https://doi.org/10.1038/s42255-025-01422-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42255-025-01422-7
