
Abstract
A challenge in gel science is the construction of hydrogels with high stiffness, particularly at ultralow solid contents. In this study, we are inspired by the biosynthesis of plant cell walls. In plants, cellulose chains synthesized by enzymes crystallize in situ into nanofibers, which coassemble with cellulose-binding polysaccharides to form cell walls with remarkable mechanical properties. Here, we enzymatically synthesize low-molecular-weight (LMW) cellulose in vitro in the presence of cellulose-binding polysaccharides. Various cellulose-binding polysaccharides facilitate the formation of hydrogels, with carboxymethyl cellulose (CMC) yielding the stiffest hydrogels. Remarkably, the hydrogels formed with 0.5% (w/v) CMC exhibit Young’s modulus of 386 kPa at a low solid content of 1.34% (w/v). The hydrogels are composed of ribbon-shaped nanofibers that are lamellar crystals of LMW cellulose, with CMC adsorbed on their surfaces. Analyses suggest that higher concentrations of CMC increase the crosslinking point density and enhance the uniformity at the micrometer and larger scales of the nanofibrillar networks, yielding highly stiff hydrogels at a low solid content.

Similar content being viewed by others
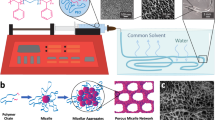


Introduction
Hydrogels are soft materials that contain a large amount of water and are necessary in healthcare, medicine, food, and other fields. One of the major challenges in gel science is the development of hydrogels with excellent mechanical properties1,2,3. Advances in the design principle of polymer hydrogels in this century have allowed for the production of stiff, strong, or tough hydrogels. Examples include tetra- and tri-branched gels4,5, slide-ring gels6,7, double network gels8,9, and highly entangled gels10,11. Nevertheless, although gel stiffness is an important mechanical property for biomedical and other applications12,13, constructing hydrogels with high stiffness, especially at ultralow solid contents, remains challenging. This is because the stiffness of polymer gels is the sum of the positive entropic elasticity, which is proportional to the polymer chain density, and the negative energy elasticity, which originates from attractive polymer–solvent interactions14. In other words, polymer gel stiffness is determined by the polymer chain density, polymer species, and solvent species. Heterogeneous self-assembled networks can form hydrogels that exhibit much higher stiffness15,16,17,18,19. For example, ultrastiff and water-rich “hydrospongels” composed of self-assembled cyano-p-aramid nanofibers were recently reported to exhibit a compressive modulus of 610 kPa at a solid content of 1 wt%19.
To construct stiff hydrogels with a low solid content, this study was inspired by the biosynthesis of plant cell walls, which are composed of a crystalline polysaccharide cellulose and other polysaccharide species20,21,22,23. In nature, cellulose synthases, which are embedded in the plasma membrane, catalyze the polymerization reaction of uridine 5′-diphosphate glucose (UDP-glucose) monomers to produce cellulose chains (Fig. 1a). The nascent cellulose chains crystallize in situ into nanofibers, which immediately coassemble with cellulose-binding polysaccharides (e.g., hemicellulose). The resultant plant cell walls with heterogeneous, hierarchically assembled network structures exhibit excellent mechanical properties and play the role of skeletal functions in plants. Despite this inspirational self-assembly pathway, the biosynthesis of plant cell walls has received little attention so far for creating stiff hydrogels.

Schematic illustration of a the biosynthesis of plant cell walls and b this study. In both systems, cellulose chains are synthesized by enzymes and self-assemble in situ into nanofibers, which coassemble with cellulose-binding polysaccharides into higher-order structures.
Herein, we report the construction of hydrogels with Young’s modulus of 386 kPa at a low solid content of 1.34% (w/v) via enzymatic synthesis of low-molecular-weight (LMW) cellulose in the presence of cellulose-binding polysaccharides in fully aqueous media under mild conditions (Fig. 1b). This study exploited the cellodextrin phosphorylase (CDP)-catalyzed oligomerization reaction for cellulose synthesis24,25. In this reaction, when nascent oligosaccharide chains have become sufficiently long to be water-insoluble, the LMW cellulose chains self-assemble in situ into crystalline nanostructures. The degree of polymerization (DP) of LMW cellulose synthesized by this reaction is typically ~10. In this study, this reaction for LMW cellulose synthesis was first performed in aqueous solutions of various water-soluble polysaccharides, including hemicelluloses, pectins, cellulose derivatives, and dextrans. Among the water-soluble polysaccharides tested, hemicelluloses, pectins, and cellulose derivatives, which can interact attractively with cellulose, induced hydrogel formation. The hydrogels formed with carboxymethyl cellulose (CMC) exhibited the highest modulus. Then, the CMC concentration during LMW cellulose synthesis was varied from 0.05 to 0.5% (w/v). The mechanical and structural properties of the resulting LMW cellulose–CMC hydrogels were characterized.
Results and discussion
Screening of polysaccharides
We screened various polysaccharide candidates to identify the polysaccharide species that facilitate the formation of higher-order structures with LMW cellulose. It is noted that the CDP-catalyzed oligomerization reaction in the absence of polysaccharides yields nanosheet-shaped lamellar crystals of LMW cellulose as sedimentated precipitates26,27. Commercially available hemicelluloses, pectins, cellulose derivatives, and dextrans were tested in this study. Hemicellulose and pectin are matrix polysaccharides that constitute, together with cellulose, the growing cell wall20. Hemicellulose is a representative group of cellulose-binding polysaccharides. Xylan and mannan were used in this study. Pectin can also interact attractively with cellulose depending on its sugar side chain structures28,29. Apple- and citrus-derived pectins were used in the present study. CMC30, methyl cellulose31, and hydroxyethyl cellulose32 are semisynthetic polymers derived from cellulose and attractively interact with cellulose because of their structural similarity. Moreover, dextran, having weak interactions with cellulose32, and its carboxymethylated derivative (carboxymethyl dextran) were also used in this study.
The CDP-catalyzed synthesis of LMW cellulose was performed in the presence of a polysaccharide at 0.05% (w/v). Notably, this polysaccharide concentration is much lower than the polymer concentrations typically used to induce macromolecular crowding in vitro33. The polysaccharides apparently dissolved under the conditions for LMW cellulose synthesis. Xylan, mannan, apple- and citrus-derived pectins, CMC, methyl cellulose, and hydroxyethyl cellulose, which potentially interact with cellulose, induced the formation of turbid hydrogels (Fig. 2a). Especially, the hydrogels formed with CMC had higher uniformity than the other hydrogels, as revealed by optical microscopy observations (Supplementary Fig. 1). It seems that the polysaccharides potentially having attractive interactions with cellulose coassemble with LMW cellulose to form network structures, inducing hydrogel formation.

a Photographs of the products. b Averaged initial slope values of the force–stroke curves obtained by indentation tests for the hydrogels, which correspond to relative moduli of the hydrogels. “N.A.” denotes not applicable because of a non-gel state of the sample. The error bars represent the standard deviation of three individual trials.
In contrast, dextran and carboxymethyl dextran did not induce hydrogel formation, and LMW cellulose was obtained as sedimentated precipitates (Fig. 2a). This observation is similar to that observed for LMW cellulose synthesis in the absence of polysaccharides26,27. It is noted that we previously reported that concentrated dextran at 10% (w/v) facilitated the self-assembly of LMW cellulose into nanofibrous networks via crowding effects, such as the decrease in diffusion rate and the depletion stabilization, where adsorption of dextran onto the LMW cellulose nanofibers was negligible34. The concentration of dextran in this study (0.05% (w/v)) was insufficient to create crowding conditions. This implies that the hydrogel formation with hemicelluloses, pectins, and cellulose derivatives was caused by attractive interactions between those polysaccharides and LMW cellulose, rather than crowding effects.
When the polysaccharide concentration was increased from 0.05 to 0.5% (w/v), all of the tested polysaccharides induced hydrogel formation (Supplementary Fig. 2). Nevertheless, the hydrogels formed with CMC exhibited greater uniformity than those formed with the other polysaccharides (Supplementary Fig. 3).
The hydrogels formed with polysaccharides at 0.05% (w/v) were subjected to indentation tests to compare their stiffnesses (Supplementary Fig. 4). The averaged initial slope values of the force–stroke curves, which are proportional to the moduli of the material, are shown in Fig. 2b. The hydrogel with CMC was shown to have the highest stiffness; its modulus was 6–12 times higher than those of the other hydrogels. This high modulus can be attributed to the high apparent uniformity of hydrogels with CMC. Among the polysaccharides that induced hydrogel formation, CMC is highly anionic at the neutral pH for LMW cellulose synthesis (i.e., pH 7.5). The negatively charged polysaccharide chains seemed to prevent their coassemblies with LMW cellulose from aggregating, producing hydrogels with a relatively high uniformity. It is mentioned that the order of the moduli of the gels did not exactly correspond to the order of their uniformity observed under optical microscopy (Supplementary Fig. 1), suggesting that gel network structures at the micrometer scale or below also played an important role in determining gel stiffness.
Mechanical properties of the hydrogels with CMC
We focused on CMC for further studies. The CMC concentration was varied in a range of 0.05–0.5% (w/v). In this CMC concentration range, hydrogels were produced via enzymatic synthesis of LMW cellulose (Fig. 3a). Notably, hydrogels produced with higher concentrations of CMC were more translucent, suggesting a higher uniformity of the gel network structures (see the structural analyses results shown below). The hydrogels were self-standing without apparent changes in their shape, irrespective of CMC concentrations. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry confirmed the successful synthesis of LMW cellulose, even in the presence of CMC (Supplementary Fig. 5). The average DP and molar-mass dispersity (ÐM = \({\bar{M}}_{{{{\rm{w}}}}}/\,{\bar{M}}_{{{{\rm{n}}}}}\)) values were calculated to be 8.5–8.8 and 1.02–1.04, respectively (Table 1). The values hardly varied irrespective of the presence or concentration of CMC.

a Photographs of the hydrogels in vials and the cylindrical hydrogels used for compression tests. b Stress–strain curves obtained by compression tests and c Young’s moduli of the hydrogels. d Total solid content (LMW cellulose + CMC) and LMW cellulose content of the hydrogels. The error bars represent the standard deviation of three individual trials.
Compression tests were performed on the self-standing gels (Fig. 3b). It was found that the Young’s moduli monotonically increased with the CMC concentration (Fig. 3c and Table 1). At 0.5% (w/v) of CMC, the hydrogels exhibited a Young’s modulus of 386 kPa. Through compression tests, water was squeezed out from the hydrogels, indicating a porous, sponge-like structure of the LMW cellulose–CMC coassembled networks (Supplementary Fig. 6).
Dynamic viscoelastic measurements of the disc-shaped hydrogels (~1 mm in thickness) with 0.5% (w/v) CMC revealed storage moduli (G’) more than one order higher than loss moduli (G”), with little frequency dependence in an angular frequency range of 10–1–102 rad s–1 (Supplementary Fig. 7). It is noted that G’ was measured to be ~5.4 kPa, which gives, with Young’s modulus value, an apparent Poisson’s ratio larger than the unity, according to the linear elastic theory. A possible reason for this unusual value is that the gel samples with a thickness of ~1 mm used for the measurements might have mechanical heterogeneity within the material. Although further study is required to quantitatively discuss the shear modulus and Poisson’s ratio, dynamic viscoelastic measurements demonstrated that the system was in a gel state.
The solid content of the hydrogels was quantified as follows. The total solid content values, which were the sum of the LMW cellulose and CMC amounts constituting the gel networks, were measured by weighing the insoluble products after drying. The LMW cellulose content values were estimated with the assumption that monomers consumed by the CDP-catalyzed reaction were completely converted into solid-state LMW cellulose. The experimental details are described in the Methods.
It was found that the solid content values were very low (Fig. 3d and Table 1). In particular, the hydrogels formed with 0.5% (w/v) CMC had a total solid content of 1.34% (w/v). Despite this low solid content, the hydrogels exhibited a high Young’s modulus of 386 kPa. This mechanical performance closely approaches that of a recently reported, ultrastiff and water-rich hydrospongel with a compressive modulus of 610 kPa at a solid content of 1 wt%19. The hydrospongel was produced via the synthesis of poly(2-cyano-p-phenylene terephthalamide) in organic solvents under N2, dissolution of the polymer into potassium hydroxide–dimethyl sulfoxide for several days, and self-assembly of the polymer during dialysis against water. On the other hand, the biomolecular hydrogels developed in this study were produced in one step in fully aqueous media under mild conditions. Moreover, the Young’s modulus of 386 kPa at 1.34% (w/v) is a remarkable value for a low solid content gel, as shown in Supplementary Fig. 8, which compares the mechanical performance of our hydrogels with previously reported low solid content hydrogels, including peptide-based and nanocomposite hydrogels15,19,35,36,37,38,39,40,41,42,43,44,45,46,47. The plant cell wall-inspired strategy allows the creation of stiff hydrogels with an ultralow solid content through a sustainable route.
The LMW cellulose content values were roughly constant irrespective of CMC concentration, albeit with a slight decrease in the value at 0.5% (w/v) CMC (Fig. 3d and Table 1). This indicates that CMC had little effect on the CDP-catalyzed oligomerization reactions for LMW cellulose synthesis. The differences in total solid content and LMW cellulose content correspond to the amounts of CMC constituting the gel networks and were, in a CMC concentration range of 0–0.4% (w/v), almost the same as the CMC concentrations in the reaction mixtures. At 0.5% (w/v) CMC, the amount of CMC constituting the gel networks was calculated to be 0.36 ± 0.12% (w/v), suggesting that a small amount of CMC remained dissolved within the gel networks. Nevertheless, CMC mostly coassembled quantitatively with LMW cellulose to form gel networks. In fact, Fourier transform infrared (FTIR) absorption spectroscopy indicated increasing amounts of carboxylate groups of CMC in the products (Supplementary Fig. 9)48,49,50.
The hydrogels exhibited non-swelling behavior. When the hydrogels were immersed in water for one week, no apparent change in their shape was observed (Supplementary Fig. 10a). Additionally, their Young’s moduli hardly decreased by immersion in water, indicating the high stability of the coassembled state of water-soluble CMC and LMW cellulose (Supplementary Fig. 10b, c). Furthermore, the hydrogels hardly swelled even in an aqueous buffer solution at pH 4 (Supplementary Fig. 11a). Despite the protonation of the carboxylate groups, Young’s moduli of the hydrogels were mostly maintained (Supplementary Fig. 11b, c). This maintenance of the mechanical performance despite the significant structural change in CMC implies that crystalline LMW cellulose was responsible for the high stiffness of the hybrid gels, as discussed in more detail below.
Structure of the hydrogels with CMC
The hydrogels formed with CMC were structurally characterized. Optical microscopy observation of the hydrogel at 0.05% (w/v) CMC revealed a non-uniform optical density at the submillimeter scale (Fig. 4a). Increases in CMC concentration to 0.1 and 0.2% (w/v) decreased the scale where the non-uniformity lay to tens of micrometers. At 0.5% CMC, the hydrogels were uniform, as observed using optical microscopy. Given that the total solid content and LMW cellulose content were roughly constant irrespective of CMC concentration, the higher uniformity at higher CMC concentrations was considered to significantly contribute to the higher gel stiffnesses.

a Optical microscopy images of the hydrogels after binary processing. b SEM images of nanofibrous networks of the hydrogels. c AFM images of the mechanically crushed nanofibers. d XRD profiles of solid components of the hydrogels.
The gel network structures were observed by scanning electron microscopy (SEM) after freeze-drying of the hydrogels and subsequent osmium coating. The images revealed that the gel networks formed with 0.05% (w/v) CMC were composed of ribbon-shaped nanofibers (Fig. 4b, top left). The thickness of the nanoribbons was measured using atomic force microscopy (AFM) after mechanically crushing the gels to be ~6 nm (Fig. 4c, top left; Table 1). Additionally, X-ray diffraction (XRD) analysis showed three major peaks at 2θ = 12.2, 20.0, and 22.1° for \(1\bar{1}0\), 110, and 020 of cellulose II allomorph26,51 (Fig. 4d, second from the top). The nanoribbon thickness was comparable to the molecular chain length of LMW cellulose with a DP of 9 in cellulose II allomorph (i.e., 4.7 nm)34, suggesting that the ribbon-shaped nanofibers were lamellar crystals of LMW cellulose (Supplementary Fig. 12). Such assembled structures are common among in vitro LMW cellulose assemblies24,25. CMC seemed to be adsorbed on nanofiber surfaces, given that objects other than the ribbon-shaped nanofibers were absent in SEM images (Fig. 4b, top left) and AFM images (Fig. 4c, top left). The lamellar crystals of LMW cellulose appeared to be prevented from aggregation by CMC chains adsorbed on their surfaces through excluded volume effect and electrostatic repulsion, producing hydrogels with relatively high uniformity (Fig. 2a).
An increase in the CMC concentration narrowed the ribbon-shaped nanofibers. SEM images showed that ribbon-shaped nanofibers were narrower at higher CMC concentrations up to 0.2% (w/v) (Fig. 4b, top). Although at higher CMC concentrations, nanofibers observed by SEM were further narrower and thus apparently had rounded shapes due to osmium coating layers with thicknesses of several nanometers (Fig. 4b, bottom), AFM observations for samples without coating layers clearly revealed ribbon shapes of the nanofibers with narrower widths (Fig. 4c and Table 1). Their thicknesses were ~ 6 nm (Table 1), indicating that the ribbon-shaped nanofibers were lamellar crystals of LMW cellulose irrespective of the CMC concentration. Narrower nanofibers, while maintaining similar solid content, correspond to a greater total combined length of the nanofibers. The greater total combined length of nanofibers at higher CMC concentrations could lead to denser networks, contributing to higher stiffness of the hydrogels.
XRD analyses showed that CMC reduced the crystallite size of LMW cellulose more in the width direction than in the length direction of the nanofibers. The anisotropic shape of the nanofibers would be due to the crystal habit of cellulose II; it was previously reported that lamellar single crystals of LMW cellulose grew preferentially in the direction parallel to the (\(1\bar{1}0\)) plane, yielding rectangular sheet-shaped crystals26,52. This crystal habit of cellulose II was considered to yield the highly anisotropic ribbon-shaped nanofibers in this study. Specifically, the (\(1\bar{1}0\)) plane seemed to be perpendicular to the width direction of the nanofibers (Supplementary Fig. 12). The crystallite size in the direction perpendicular to each lattice plane (\({D}_{{hkl}}\)) of the LMW cellulose was evaluated from the XRD profiles using the Scherrer equation (Supplementary Fig. 13). The calculated crystallite sizes were 10–30 nm (Table 1) and hereafter used only qualitatively. \({D}_{1\bar{1}0}\) is the crystallite size in the width direction of the nanofibers, whereas \({D}_{110}\) and \({D}_{020}\) are those in the length direction. Notably, \({D}_{1\bar{1}0}\) of LMW cellulose crystals decreased with increasing CMC concentrations, while decreases in \({D}_{110}\) and \({D}_{020}\) were relatively small (Table 1). This means that CMC reduced the crystallite size of LMW cellulose more in the width direction than in the length direction of the nanofibers (Supplementary Fig. 12). This more decreased crystallite size in the width direction may correlate with the narrower nanofibers at higher CMC concentrations.
While decreasing crystallite size, CMC enhanced the crystallinity of the LMW cellulose fraction. The degree of crystallinity (\({\chi }_{{{{\rm{c}}}}}\)) values, which were calculated based on the total solid content including both LMW cellulose and CMC from the XRD profiles, hardly decreased with increasing CMC concentrations, despite the increasing noncrystalline CMC fraction (Fig. 4d, Supplementary Fig. 13, and Table 1). This means that CMC increased the crystallinity of the LMW cellulose fraction. The maintenance of the high crystallinity of nanofibers should be important for the high stiffnesses of the hydrogels.
SEM observations suggested that an increase in the CMC concentration caused an increase in the number of physical crosslinking points among the nanofibers (Fig. 4b). At 0.05% (w/v) CMC, numerous segments along the nanofiber length without physical crosslinks extending over >1 µm were observed (Fig. 4b, top left). Such nanofiber segments became fewer with increasing CMC concentration. At 0.5% (w/v) CMC, nanofiber lengths between physical crosslinking points were mostly a few hundred nanometers (Fig. 4b, bottom right). Higher concentrations of CMC might effectively act as an adhesive to physically crosslink the nanofibers. The plausible increase in the number of physical crosslinking points at higher CMC concentrations could lead to the higher stiffnesses of the hydrogels. Nevertheless, the possibility of artifacts made during the SEM sample preparation is noted.
Considering all the results, the hydrogel formation process is proposed as follows: The enzymatically synthesized LMW cellulose crystallized in situ into lamellar crystals, which adsorbed CMC. The adsorption of CMC stabilized the nanocrystals in a dispersed state for further growth into ribbon-shaped nanofibers. Additionally, CMC might be preferentially adsorbed onto the long side rather than the short side of the nanoribbons (Supplementary Fig. 12), thereby narrowing the ribbon-shaped nanofibers while reducing the crystallite size of the LMW cellulose in the nanoribbon width direction and maintaining the high crystallinity of the nanoribbons. Moreover, CMC probably acted as an adhesive to physically crosslink the nanofibers. As a result, hydrogels with higher uniformity at the micrometer and larger scales and greater numbers of nanofibers and physical crosslinking points were produced, which exhibited high moduli despite their ultralow solid contents.
Conclusions
In summary, we demonstrated a plant cell wall-inspired strategy for creating hydrogels with high moduli at ultralow solid contents. First, LMW cellulose was enzymatically synthesized in the presence of various polysaccharides. Cellulose-binding polysaccharides generally facilitated the formation of hydrogels, with CMC yielding hydrogels with the highest modulus. It was found that hydrogels formed with 0.5% (w/v) CMC exhibited Young’s modulus of 386 kPa at a solid content of 1.34% (w/v). Structural characterizations revealed that the hydrogels were composed of ribbon-shaped nanofibers that were lamellar crystals of LMW cellulose, with CMC adsorbed on their surfaces. Detailed analyses suggested that the adsorption of CMC promoted the formation of stiff hydrogels in several ways: it prevented aggregation of the nanofibers, enhancing the uniformity of hydrogels at the micrometer and larger scales. It also narrowed the nanoribbons to increase the number of nanofibers, while maintaining their crystallinity. Additionally, it likely acted as an adhesive to physically crosslink the nanofibers. Consequently, the resulting hydrogels exhibited high uniformity and high crosslinking point density, making them highly stiff despite their ultralow solid contents. Collectively, this study presents a biological pathway in plants as a source of inspiration for creating soft materials with excellent mechanical properties via a sustainable route.
Methods
Materials
α--Glucose 1-phosphate (αG1P) disodium salt n-hydrate was purchased from Nacalai Tesque (Kyoto, Japan) or FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Apple- and citrus-derived pectins, and dextran (molecular weight ~150,000) were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Carboxymethyl dextran sodium salt (molecular weight ~40,000), xylan from corn core, and trifluoroacetic acid aqueous solution were purchased from Tokyo Chemical Industry (Tokyo, Japan). Sodium dodecyl sulfate, super-DHB, ProteoMass MALDI-MS Standards (Bradykinin fragment 1–7, P14R, and ACTH fragment 18–39), and acetonitrile were purchased from Sigma–Aldrich (Missouri, USA). All other reagents were purchased from Nacalai Tesque (Kyoto, Japan). Ultrapure water (>18.2 MΩ·cm) was obtained from Milli-Q Advantage A-10 (Merck Millipore, Massachusetts, USA) and used throughout the study.
Enzymatic synthesis of LMW cellulose
The enzymatic synthesis of LMW cellulose was conducted using CDP in the presence of CMC or other water-soluble polysaccharides, namely, xylan, mannan, apple- and citrus-derived pectins, methyl cellulose, hydroxyethyl cellulose, dextran, and carboxymethyl dextran. CDP derived from Acetivibrio thermocellus DSM 1313 was used for the oligomerization reaction53. Typically, αG1P monomer, D-glucose primer, CDP, and water-soluble polysaccharides were mixed at 0.2 M, 0.05 M, 1 U mL–1, and 0.05% (w/v), respectively, in 0.5 M 4-(2-hydroxyethyl)−1-piperazineethanesulfonic acid buffer solution (pH 7.5) containing 50 µM ethylenediaminetetraacetic acid. The mixtures were incubated at 60 °C for 3 days for the enzymatic synthesis of LMW cellulose.
For SEM, the gelled products (1 mL) were soaked in ultrapure water at 4 °C for at least one week for purification. The ultrapure water was exchanged daily. For other characterizations, the gelled products (0.3 mL) after the reaction were mechanically collapsed by pipetting to obtain product dispersions. The dispersions were purified with ultrapure water by performing at least five centrifugation (20,400 × g)–redispersion cycles to remove more than 99.999 % of the soluble fraction of reaction mixtures. The purified products were stored in a dispersion state at 4 °C or after freeze-drying until use. For quantification of the total solid content, a volume of the product dispersion was dried at 105 °C for 24 h, and the resultant residues were weighed.
The conversion of αG1P monomers through the enzymatic LMW cellulose synthesis was measured by quantifying the liberated phosphate ions. Phosphate ions were quantified as previously described54. Briefly, the product solutions were 10-fold diluted with a 3-morpholinopropane-1-sulfonic acid buffer solution (50 mM, pH 7.5) before centrifugation at 2040 × g at 4 °C for 10 min. The supernatants were adequately diluted with the buffer solution and mixed with molybdenum reagent and sodium ascorbate. After incubation at 30 °C for 15 min, the solutions were mixed with sodium dodecyl sulfate and subjected to absorbance measurement at 850 nm. A standard curve was used to quantify the phosphate ions.
Characterizations of products
For MALDI-TOF mass spectrometry, the never-dried, purified products were used. Their dispersion at 0.01% (w/v) in an acetonitrile–water mixture (1:1, v/v) containing 2 mg mL–1 super-DHB and 0.5 mM trifluoroacetic acid was mounted onto a sample target plate five times with 1 μL each. Spectra were obtained using a MALDI-8030 instrument (Shimadzu, Kyoto, Japan) with pulsed ion extraction in the linear positive mode at room temperature and calibrated with Bradykinin fragment 1–7 (757.3997 Da), P14R (1,533.8582 Da), and ACTH fragment 18–39 (2,465.1989 Da). The average DP and ÐM were calculated using the following equations:

where \({\bar{M}}_{{{{\rm{n}}}}}\) is the number-average molecular weight, \({N}_{{{{\rm{i}}}}}\) is the peak area of i-mer species, \({M}_{{{{\rm{i}}}}}\) is the molar mass of that species, and \({\bar{M}}_{{{{\rm{w}}}}}\) is the weight-average molecular weight. The peak areas of the sodium and potassium ion adducts of an \(i\)-mer species were summed for the calculations.
For attenuated total reflection-FTIR absorption spectroscopy, the freeze-dried products were mounted onto an FT/IR-4100 instrument (JASCO, Tokyo, Japan). The measurements were conducted at a cumulative measurement number of 100 and a resolution of 2.0 cm−1 under ambient conditions.
For XRD measurements, the freeze-dried products were pressed using a hand press to prepare pellets with a diameter of 4 mm. A D8 DISCOVER instrument (Bruker, Massachusetts, USA) with Cu K\(\alpha\) radiation (\(\lambda\) = 1.542 Å) was used under ambient conditions to obtain two-dimensional XRD patterns, which were then converted into one-dimensional profiles in a 2\(\theta\) range of 5–33°(\(\theta\) is the Bragg angle). The effect of air scattering was subtracted using the following equation:
where \({I}_{{{{\rm{cor}}}}}\) is the corrected intensity, \({I}_{{{{\rm{obs}}}}}\) is the observed intensity, \(t\) is the X-ray transmittance through the sample, and \({I}_{{{{\rm{blank}}}}}\) is the intensity measured without the sample. \(t\) is the ratio of the X-ray intensities with and without the sample, which was evaluated by measuring the intensity of the direct X-ray beam using a scintillation counter. The measured \(t\) values in this study were in the range of 0.5–0.7. The amorphous cellulose halo obtained previously54 was fitted to the one-dimensional profiles in 2\(\theta\) ranges adequately selected from 14 to 16.5° for each profile before peak separation. It is noted that we confirmed that amorphous CMC exhibited a halo similar to that of amorphous cellulose. \({\chi }_{{{{\rm{c}}}}}\) (per 100% solid content) was calculated using the following equation:
where \({I}_{{{{\rm{c}}}}}\left(2\theta \right)\) and \({I}_{{{{\rm{a}}}}}\left(2\theta \right)\) are the scattering intensities from the crystalline and amorphous regions, respectively. After subtracting the halo, peak fitting with Lorentzian functions was performed using the Igor Pro 9 software (WaveMetrics, Oregon, USA), as shown in Supplementary Fig. 1355. \({D}_{{hkl}}\) was evaluated using the Scherrer equation56:
where \(K\) (=1.00) is the Scherrer constant, which depends on the relative orientation of the scattering vector to the external shape of the crystallite57. \(\beta\) is the integral width (radian). \(\theta\) is the Bragg angle (radian) for the selected hkl peak and was calibrated using the following equation:
where \({\beta }_{{{{\rm{obs}}}}}\) and \({\beta }_{{{{\rm{std}}}}}\) are the integral widths of the hkl peak without calibration and the 111 peak of silicon powder, respectively.
For SEM, small pieces of the purified gels were soaked in 10, 20, 30, 40, 50, 60, 70, 80, and 90% (v/v) ethanol, ethanol, ethanol–tert-butyl alcohol (1:1, v/v), and then tert-butyl alcohol. The resultant organogels were rapidly frozen in liquid nitrogen, cut using a razor blade, and freeze-dried. Dotite (Nissin EM, Tokyo, Japan) was used to adhere the freeze-dried gels to the substrates before osmium coating using a Neoc-Pro instrument (MEIWAFOSIS, Tokyo, Japan). The fractured surfaces were observed using a JSM-7500F instrument (JEOL, Tokyo, Japan) at an accelerating voltage of 5 kV.
For AFM, 15 µL of aqueous dispersions of the purified products at 0.001% (w/v) were spin-cast onto mica substrates at 600 rpm. After drying under ambient conditions, an SPM-9700HT instrument (Shimadzu, Kyoto, Japan) was used in the tapping mode under ambient conditions. Nine objects were observed, and their thicknesses and widths were measured at three different cross-sectional positions. The average thicknesses and widths with standard deviations were calculated from all data.
For optical microscopy, ~2-mm-thick hydrogels were prepared in 96-well plates (clear, flat bottom). A ZOE Fluorescent Cell Imager (Bio-Rad, Tokyo, Japan) was used at room temperature. The images were subjected to thresholding using the ImageJ software (https://imagej.nih.gov/ij/)58 to obtain binary images.
Mechanical tests of hydrogels
For the indentation test, hydrogels were prepared in a 1 mL vial. An AGS-X instrument (Shimadzu, Kyoto, Japan) equipped with a flat-ended cylindrical indenter with a diameter of 3 mm was operated at a velocity of 1 mm min–1 at room temperature. Three individual trials were performed to obtain the average slope of the force–stroke curves in the early stage of indentation and standard deviations.
For compression test, cylindrical hydrogels with a diameter of 15 mm and a height of 7.5 mm were prepared in a polytetrafluoroethylene mold. An AGS-X instrument (Shimadzu, Kyoto, Japan) equipped with a fixed compression plate was operated at a velocity of 2 mm min–1 at room temperature. Young’s modulus was calculated from the stress-strain curve in a strain range adequately selected from 0 to 0.1 for each profile. Average values and standard deviations were obtained from three individual trials.
For dynamic viscoelastic measurement, hydrogels with thicknesses of ~1 mm were prepared in a Petri dish-shaped stainless steel dish. The measurements were performed at 25 °C and a strain of 1%, which was within the linear viscoelasticity range, using a Physica MCR 302 instrument (Anton Paar, Graz, Austria) equipped with a 25 mm parallel plate with 320-grit sandpaper on its surface.
Data availability
The data that support the findings of this study are available from the corresponding author upon request. Source data are provided with this paper.
References
Zhang, Y. S. & Khademhosseini, A. Advances in engineering hydrogels. Science 356, 500 (2017).
Creton, C. 50th anniversary perspective: networks and gels: soft but dynamic and tough. Macromolecules 50, 8297–8316 (2017).
Zhao, X. et al. Soft materials by design: unconventional polymer networks give extreme properties. Chem. Rev. 121, 4309–4372 (2021).
Sakai, T. et al. Design and fabrication of a high-strength hydrogel with ideally homogeneous network structure from tetrahedron-like macromonomers. Macromolecules 41, 5379–5384 (2008).
Fujiyabu, T. et al. Tri-branched gels: rubbery materials with the lowest branching factor approach the ideal elastic limit. Sci. Adv. 8, eabk0010 (2022).
Okumura, Y. & Ito, K. The polyrotaxane gel: a topological gel by figure-of-eight cross-links. Adv. Mater. 13, 485 (2001).
Liu, C. et al. Tough hydrogels with rapid self-reinforcement. Science 372, 1078–1081 (2021).
Gong, J. P., Katsuyama, Y., Kurokawa, T. & Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 15, 1155–1158 (2003).
Zhang, H. J. et al. Tough physical double‐network hydrogels based on amphiphilic triblock copolymers. Adv. Mater. 28, 4884–4890 (2016).
Norioka, C., Inamoto, Y., Hajime, C., Kawamura, A. & Miyata, T. A universal method to easily design tough and stretchable hydrogels. NPG Asia Mater. 13, 34 (2021).
Kim, J., Zhang, G., Shi, M. & Suo, Z. Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links. Science 374, 212–216 (2021).
Guimarães, C. F., Gasperini, L., Marques, A. P. & Reis, R. L. The stiffness of living tissues and its implications for tissue engineering. Nat. Rev. Mater. 5, 351–370 (2020).
Blache, U. et al. Engineered hydrogels for mechanobiology. Nat. Rev. Methods Primers 2, 98 (2022).
Yoshikawa, Y., Sakumichi, N., Chung, U. & Sakai, T. Negative energy elasticity in a rubberlike gel. Phys. Rev. X 11, 011045 (2021).
Wang, Q. et al. High-water-content mouldable hydrogels by mixing clay and a dendritic molecular binder. Nature 463, 339–343 (2010).
De Leon Rodriguez, L. M., Hemar, Y., Cornish, J. & Brimble, M. A. Structure–mechanical property correlations of hydrogel forming β-sheet peptides. Chem. Soc. Rev. 45, 4797–4824 (2016).
Ji, D. & Kim, J. Recent strategies for strengthening and stiffening tough hydrogels. Adv. Nanobiomed. Res. 1, 2100026 (2021).
Xu, J., Zhu, X., Zhao, J., Ling, G. & Zhang, P. Biomedical applications of supramolecular hydrogels with enhanced mechanical properties. Adv. Colloid Interface Sci. 321, 103000 (2023).
Lee, M. et al. Network of cyano-p-aramid nanofibres creates ultrastiff and water-rich hydrospongels. Nat. Mater. 23, 414–423 (2024).
Cosgrove, D. J. Growth of the plant cell wall. Nat. Rev. Mol. Cell Biol. 6, 850–861 (2005).
Purushotham, P., Ho, R. & Zimmer, J. Architecture of a catalytically active homotrimeric plant cellulose synthase complex. Science 369, 1089–1094 (2020).
Zhang, Y. et al. Molecular insights into the complex mechanics of plant epidermal cell walls. Science 372, 706–711 (2021).
Addison, B. et al. Atomistic, macromolecular model of the Populus secondary cell wall informed by solid-state NMR. Sci. Adv. 10, eadi7965 (2024).
Zhong, C. & Nidetzky, B. Bottom‐up synthesized glucan materials: opportunities from applied biocatalysis. Adv. Mater. 36, 2400436 (2024).
Hata, Y. & Serizawa, T. Nanoarchitectonics of cello-oligosaccharides: a route toward artificial nanocelluloses. Adv. Colloid Interface Sci. 336, 103361 (2025).
Hiraishi, M. et al. Synthesis of highly ordered cellulose II in vitro using cellodextrin phosphorylase. Carbohydr. Res. 344, 2468–2473 (2009).
Serizawa, T., Kato, M., Okura, H., Sawada, T. & Wada, M. Hydrolytic activities of artificial nanocellulose synthesized via phosphorylase-catalyzed enzymatic reactions. Polym. J. 48, 539–544 (2016).
Zykwinska, A. W., Ralet, M.-C. J., Garnier, C. D. & Thibault, J.-F. J. Evidence for in vitro binding of pectin side chains to cellulose. Plant Physiol 139, 397–407 (2005).
Wang, T., Zabotina, O. & Hong, M. Pectin–cellulose interactions in the Arabidopsis primary cell wall from two-dimensional magic-angle-spinning solid-state nuclear magnetic resonance. Biochemistry 51, 9846–9856 (2012).
Agarwal, D., MacNaughtan, W. & Foster, T. J. Interactions between microfibrillar cellulose and carboxymethyl cellulose in an aqueous suspension. Carbohydr. Polym. 185, 112–119 (2018).
McKee, J. R. et al. Thermoresponsive nanocellulose hydrogels with tunable mechanical properties. ACS Macro Lett. 3, 266–270 (2014).
Hu, Z., Cranston, E. D., Ng, R. & Pelton, R. Tuning cellulose nanocrystal gelation with polysaccharides and surfactants. Langmuir 30, 2684–2692 (2014).
Hata, Y., Sawada, T. & Serizawa, T. Macromolecular crowding for materials-directed controlled self-assembly. J. Mater. Chem. B 6, 6344–6359 (2018).
Hata, Y. et al. Enzymatic synthesis of cellulose oligomer hydrogels composed of crystalline nanoribbon networks under macromolecular crowding conditions. ACS Macro Lett. 6, 165–170 (2017).
Maity, I., Rasale, D. B. & Das, A. K. Sonication induced peptide-appended bolaamphiphile hydrogels for in situ generation and catalytic activity of Pt nanoparticles. Soft Matter 8, 5301–5308 (2012).
Guo, H. et al. The robust hydrogel hierarchically assembled from a pH-sensitive peptide amphiphile based on silk fibroin. Biomacromolecules 14, 2733–2738 (2013).
King, P. J. S. et al. A modular self-assembly approach to functionalised β-sheet peptide hydrogel biomaterials. Soft Matter 12, 1915–1923 (2016).
Si, Y. et al. Ultrahigh‐water‐content, superelastic, and shape‐memory nanofiber‐assembled hydrogels exhibiting pressure‐responsive conductivity. Adv. Mater. 29, 1700339 (2017).
Clarke, D. E., Parmenter, C. D. J. & Scherman, O. A. Tunable pentapeptide self‐assembled β‐sheet hydrogels. Angew. Chem. Int. Ed. 57, 7709–7713 (2018).
Jian, H. et al. Dipeptide self-assembled hydrogels with tunable mechanical properties and degradability for 3D bioprinting. ACS Appl. Mater. Interfaces 11, 46419–46426 (2019).
Gao, F. et al. Osteochondral regeneration with 3D‐printed biodegradable high‐strength supramolecular polymer reinforced‐gelatin hydrogel scaffolds. Adv. Sci. 6, 1900867 (2019).
Pugliese, R. & Gelain, F. Characterization of elastic, thermo-responsive, self-healable supramolecular hydrogel made of self-assembly peptides and guar gum. Mater. Des. 186, 108370 (2020).
Okesola, B. O. et al. De Novo design of functional coassembling organic–inorganic hydrogels for hierarchical mineralization and neovascularization. ACS Nano 15, 11202–11217 (2021).
Laishram, R. et al. Secondary nucleation-triggered physical cross-links and tunable stiffness in seeded supramolecular hydrogels. J. Am. Chem. Soc. 144, 11306–11315 (2022).
Hiew, S. H. et al. Modulation of mechanical properties of short bioinspired peptide materials by single amino-acid mutations. J. Am. Chem. Soc. 145, 3382–3393 (2023).
Yue, Y. Designing ultrahigh-water-content, tough, and crack-resistant hydrogels by balancing chemical cross-linking and physical entanglement. ACS Appl. Eng. Mater. 2, 638–648 (2024).
Zhang, Y., Kobayashi, K. & Wada, M. Comparative analysis of the structures and properties of cellulose hydrogels prepared using different solvent systems. Cellulose 32, 2337–2351 (2025).
Max, J.-J. & Chapados, C. Infrared spectroscopy of aqueous carboxylic acids: comparison between different acids and their salts. J. Phys. Chem. A 108, 3324–3337 (2004).
Cuba-Chiem, L. T., Huynh, L., Ralston, J. & Beattie, D. A. In situ particle film ATR FTIR spectroscopy of carboxymethyl cellulose adsorption on talc: binding mechanism, pH effects, and adsorption kinetics. Langmuir 24, 8036–8044 (2008).
Hishikawa, Y., Togawa, E. & Kondo, T. Characterization of individual hydrogen bonds in crystalline regenerated cellulose using resolved polarized FTIR spectra. ACS Omega 2, 1469–1476 (2017).
French, A. D. Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21, 885–896 (2014).
Helbert, W. & Sugiyama, J. High-resolution electron microscopy on cellulose II and α-chitin single crystals. Cellulose 5, 113–122 (1998).
Sugiura, K., Saito, M., Sawada, T., Tanaka, H. & Serizawa, T. Cellodextrin phosphorylase-catalyzed single-process production and superior mechanical properties of organic-inorganic hybrid hydrogels composed of surface-carboxylated synthetic nanocelluloses and hydroxyapatite. ACS Sustain. Chem. Eng. 10, 13484–13494 (2022).
Hata, Y., Sawada, T., Marubayashi, H., Nojima, S. & Serizawa, T. Temperature-directed assembly of crystalline cellulose oligomers into kinetically trapped structures during biocatalytic synthesis. Langmuir 35, 7026–7034 (2019).
Tashiro, M. et al. Modulating neutralization-induced self-assembly of cello-oligosaccharides by organic solvents and temperature for preparing gels with improved mechanical properties. Cellulose 31, 9057–9073 (2024).
Klug, H. P. & Alexander, L. E. X-ray diffraction procedures: for polycrystalline and amorphous materials, 2nd Edn (Wiley, New York, 1974).
Patterson, A. L. The Scherrer formula for X-ray particle size determination. Phys. Rev. 56, 978–982 (1939).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Acknowledgements
The authors wish to thank the Materials Analysis Division, Core Facility Center (Institute of Science Tokyo) for the XRD measurements and SEM observations. This study was financially supported in part by a Grant-in-Aid for JSPS Fellows (JP23KJ0932) from the Japan Society for the Promotion of Science (JSPS) for Y.S., the Environment Research and Technology Development Fund (JPMEERF20241RA1) of the Environmental Restoration and Conservation Agency provided by the Ministry of the Environment of Japan for Y.H. and T.K., a Grant-in-Aid for Scientific Research (B) (JP24K01548) from JSPS for T. Serizawa, and a Grant-in-Aid for Scientific Research on Innovative Areas (Aquatic Functional Materials) (JP20H05208 and JP22H04528) from the Ministry of Education, Culture, Sports, Science and Technology, Japan for T. Serizawa.
Author information
Authors and Affiliations
Contributions
T. Serizawa conceived and directed the project with the assistance of Y.H. and T. Sawada. M.T. and H.M. conducted the XRD analysis. R.F. and T.K. conducted the rheological analysis. Y.S. conducted the other experiments. Y.S. wrote the initial draft of the manuscript, which was critically revised by Y.H., T.K., H.M., T. Sawada, and T. Serizawa. All authors discussed the results, commented on the manuscript, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks Kazuhiro Shikinaka and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Youn Soo Kim and Jet-Sing Lee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sakurai, Y., Hata, Y., Tashiro, M. et al. Plant cell wall-inspired synthesis of biomolecular self-assembled stiff hydrogels. Commun Mater 6, 150 (2025). https://doi.org/10.1038/s43246-025-00874-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43246-025-00874-4
