
Abstract
Temperature, photosynthetically active radiation, and nutrient availability are pivotal in determining the global distribution of marine cyanobacteria. This study demonstrates that the distinct latitudinal niches of Synechococcus and Prochlorococcus are modulated by their temperature-dependent, mixotrophic nitrogen utilization strategies. Comparative genomic analysis reveals that Prochlorococcus ecotypes differ in their nitrogen transporter repertoire, with low-light ecotypes harboring ~12 transporters, while high-light ecotypes possess ~5 transporters. Conversely, Synechococcus displays greater genomic flexibility, with 17 nitrogen transporters. Reanalysis of Tara Oceans metatranscriptomic data identifies ~15 °C as a key thermal threshold for cyanobacterial distribution. In warmer regions (15-30°C, 35˚S-40˚N), Prochlorococcus optimizes nitrogen transporter functions across their ecotypes, whereas Synechococcus, leveraging its broad nitrogen transporters, exhibits functional plasticity, utilizing inorganic nitrogen in warmer waters and organic nitrogen in cooler waters ranging from -2°C to 15 °C beyond 35˚S/40˚N. Our findings underscore the critical role of temperature-driven mixotrophic nitrogen utilization in shaping the biogeographical patterns of marine cyanobacteria.

Similar content being viewed by others


Introduction
Marine cyanobacteria, Prochlorococcus and Synechococcus, have shaped Earth’s atmosphere and ecosystems for over two billion years, playing a crucial role in oxygen production during the early stages of atmospheric evolution1. Today, these cyanobacteria are the most abundant photoautotrophic microorganisms in the sunlit ocean, contributing roughly 25% of the ocean’s net primary production2,3. These metabolic processes are central to the global cycling of carbon (C) and nitrogen (N), with profound implications for regulating Earth’s climate4.
Despite both genera being widespread in oceanic waters, Prochlorococcus and Synechococcus exhibit distinct biogeographical patterns. Prochlorococcus thrives primarily in the warmer waters between 40°N and 40°S, with peak concentrations near the equator, reflecting its adaptation to tropical and subtropical regions. In contrast, Synechococcus extends its range beyond the tropics, occupying higher latitudes, with a secondary peak at ~40° (Supplementary Fig. 1)2. Although previous studies using temperature and photosynthetically active radiation (PAR) as primary drivers to construct an empirical model to successfully project these cyanobacteria’s distribution2,5, the specific mechanisms underlying their contrasting latitudinal niches remain unclear.
Nutrient availability, particularly nitrogen and phosphate, is a fundamental driver of phytoplankton growth, including that of Prochlorococcus and Synechococcus2,6. Both genera are well-adapted to nutrient-limited environments, benefiting from their small cell size, which reduces nutrient demands and increases their surface area for efficient nutrient uptake7. This evolutionary adaptation is further reflected in their streamlined genomes, with reductions in protein length and mRNA requirements8,9, enabling them to thrive in oligotrophic waters with minimal nitrogen resources7,10,11.
Recent genomic studies have revealed surprising metabolic flexibility in Prochlorococcus7,9,12, similarly, Synechococcus are also capable to uptake organic compounds in the ocean13,14,15, challenging the traditional view of these cyanobacteria as strictly photoautotrophic. Genes involved in the transport and metabolism of organic compounds suggest a potential for mixotrophic growth, wherein these organisms can exploit both organic and inorganic resources14. Mixotrophy provides significant ecological advantages, particularly in environments where nutrient availability fluctuates, although it imposes additional energetic demands on cellular processes16. Despite these challenges, mixotrophic microorganisms, including cyanobacteria, have often achieved ecological success in nutrient-variable ecosystems17,18. Given the ancient origins of marine picocyanobacteria and their evolutionary adaptations to specific ecological niches19,20,21, understanding their mixotrophic nitrogen utilization strategies is essential for predicting their responses to environmental change.
To investigate whether the distinct latitudinal distribution of Prochlorococcus and Synechococcus are shaped by mixotrophic N behavior, we conducted a comprehensive analysis of available cyanobacterial genomes, including isolates and single-cell sequences from the JGI ProPortal22,23. Our study specifically examined their capabilities for organic and inorganic nitrogen utilization, focusing on substrate-specific transport systems and ecotype-specific gene repertoires in low-light (LL) and high-light (HL) adapted Prochlorococcus strains. In parallel, we reanalyzed metagenomic and metatranscriptomic data from the Tara Ocean Expedition24 to assess how these cyanobacteria’s nitrogen utilization preferences correspond to global temperature patterns. Our findings reveal a tight linkage between mixotrophic nitrogen behavior and the biogeographical distribution of marine cyanobacteria, offering new insights into their ecological success across latitudinal gradients.
Results
Marine cyanobacteria inorganic and organic N transporters at the genomic level
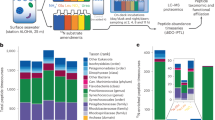
To assess the capability of Prochlorococcus and Synechococcus to utilize organic and inorganic N, we collected and analyzed all available genomes from ProPortal database (as of December 2022), with a focus on genes involved in N transport (Supplementary Note 1). A stable genome-genome phylogenetic tree25 was constructed using the GTDB-Tk toolkit to classify ecotypes based on phylogenetic relationships. The presence or absence of genes related to organic and inorganic N assimilation was visualized at the tips of the phylogenetic tree, with different colors representing gene occurrence patterns. Our comparative analysis revealed that Synechococcus exhibited a broader capacity for nitrogen mixotrophy, assimilating a wider variety of nitrogen forms (n = 17) compared to all ecotypes of Prochlorococcus (Fig. 1). Among the Prochlorococcus ecotypes, low-light (LL) adapted strains harbored a greater number of N transport systems, with the ecotypes ranked as follows: LLI (n = 12) > LLII/III (n = 11) > LLIV (n = 10). Conversely, the high-light (HL) ecotypes exhibited fewer N transporters, with HLI and HLII each possessing only five organic N transporters. The LL ecotypes also demonstrated greater diversification in inorganic nitrogen transport systems, including nitrate/nitrite uptake pathways, which likely represents an adaptation to warm, low-light environments (Supplementary Note 1).

Genes encoding inorganic N transporters, amino acid transporters, and small organic molecules transporters are depicted (A); A bubble matrix summarizes the presence of nitrogen assimilation-related genes across Prochlorococcus and Synechococcus members, expressed as percentages of total genomes (B). The genome-genome phylogenetic tree is constructed using GTDB-tk, with selected members of Synechococcus and Prochlorococcus ecotypes highlighted as pie slices. The solid gray circles represent isolates and empty circles for single-cell genomes. Bars adjacent to the phylogenetic branches indicate gene presence (colored segments) or absence (gaps). The bubble matrix shows nitrogen transporter gene prevalence of Synechococcus and Prochlorococcus: bubble size corresponds to the percentage of genomes containing these genes, while color indicates relative abundance (red: ~5%, yellow: ~50%, dark blue: ~100%). This dual representation allows for comparative insights into nitrogen assimilation-related gene distributions between the two genera.
We further investigated amino acid uptake mechanisms by mapping the genomes to the KEGG ABC-transporters pathway (https://www.kegg.jp/pathway/map02010)26, identifying 14 genes encoding amino acid transporters (Fig. 1A, B). These transporters were categorized into two groups: commonly distributed transporters (present in >40% of ecotypes) and less common transporters (present in <40% of ecotypes). The commonly distributed amino acid transporters include aapJ (general L-amino acids), glnQ (glutamine), livF and lifH (branched-chain amino acids), metn2 (methionine), gltJ (glutamate/aspartate), and tcyN (L-cystine). A significant proportion of Synechococcus, Prochlorococcus LLI, and LLIV ecotypes shared these transporters, with aapJ (85-95%), glnQ (85–90%), gltJ (82–95%), livF (86–95%), livH (82–95%) and metn2 (57–95%) being notably prevalent (Fig. 1A, B). The LLII/III ecotypes of Prochlorococcus displayed similar transporter profiles but in fewer members (10–60%). In contrast, the HL ecotypes (HLI and HLII) showed a reduced repertoire of transporters, with only four amino acid transporters: livF (69–83%), livH (67–81%), metN2 (14–62%), and tcyN (16–45%). These patterns of amino acid transporter distribution align with the observed trends in inorganic nitrogen transporter distribution, wherein Synechococcus consistently exhibited a broader spectrum of nitrogen transporters compared to Prochlorococcus, and LL ecotypes displayed greater diversification in nitrogen source acquisition compared to HL ecotypes.
The urea transporter gene urtA, commonly found in marine cyanobacteria7, was detected in over 71% of the studied genomes, with the exception of Prochlorococcus HLVI (12%) and LLII/III (13%). Additionally, the ability to assimilate cyanate, an organic nitrogen compound, was assessed through the presence of the cynA gene, which encodes a cyanate ABC-type transporter. Cyanate assimilation was found to be rare, with only 8-13% of Synechococcus, Prochlorococcus HLI, and HLII strains possessing this gene (Fig. 1A, B), suggesting that cyanate utilization is an uncommon trait in these ecotypes despite its documented importance in other cyanobacterial studies27. This underscores that compared to urea and amino acids, cyanate is a less preferred organic N source for Synechococcus and Prochlorococcus populations.
Mixotrophic N acquisitions and temperature-driven ecological boundary for cyanobacteria
To explore the spatial variations in N-assimilation-related genes within the Prochlorococcus and Synechococcus population, and to assess their coordinated behavior in response to niche adaptation, we analyzed N-assimilation protein sequences derived from genomic data using metagenomic datasets from the Tara Oceans project. These datasets corresponded to two distinct water layers: surface waters (5 m, SF) and the deep chlorophyll maximum (DCM). The N-assimilation genes were classified into four functional categories (Supplementary Note 2, Supplementary Table 1), with their abundance normalized to 100%. Our analysis indicated that organic N-assimilation genes were distributed in conjunction with inorganic nitrogen assimilation genes across different marine environments (Supplementary Note 2, Fig. 2). In the non-polar regions, amino acid transporter genes (n = 13) accounted for ~50% of the total N-assimilatory genes in both the DCM and SF layers. Genes associated with nitrate assimilation (n = 5), ammonium transport (n = 1), and small organic molecule transport (n = 2) each contributed about 15% to the N-assimilation gene pool. In contrast, in polar regions, amino acid transporters were overwhelmingly predominant, constituting over 90% of the N-assimilatory gene repertoire.

The pie map shows the four major nitrogen uptake genes categories (Supplementary Table 1) from the surface (SF) (A) and deep chlorophyll maximum (DCM) (C) samples, where different colors represent specific gene categories, blue for amino acids transporters (n = 13), yellow for the ammonium transporters (n = 1), red for the nitrate assimilation-related genes (n = 4) and green for small organic molecule transporters (n = 2). The extended bar plots compare the proportions (%) of these gene categories between polar and non-polar regions for SF (B) and DCM (D) samples, with the blue and yellow bars indicate the polar and non-polar samples, respectively.
To further elucidate the influence of environmental factors on mixotrophic nitrogen utilization, we conducted a beta diversity analysis using the STAMP software suite28. This analysis was based on the abundance matrix of organic and inorganic N-assimilation genes across Synechococcus and six ecotypes of Prochlorococcus (Supplementary Table 2). Principal component analysis (PCA) revealed that temperature was the strongest predictor of variation in organic and inorganic N-assimilation patterns (Fig. 3A–G). To quantify the impact of seawater temperature on nitrogen assimilation strategies in marine cyanobacteria, we calculated both the Simpson and Shannon diversity indices using metagenomic data. The Simpson diversity index, which provides an overview of gene diversity without considering abundance29, ranged from ~0.4 at -2°C, increased to ~0.9 at 12.5°C, and stabilized around 15°C (Fig. 3H). The Shannon diversity index30, which reflects community evenness, indicated that communities in the DCM exhibited significantly greater evenness compared to those in the SF (p < 0.05) (Fig. 3I). This difference in evenness occurred despite the SF primarily harboring Prochlorococcus HL ecotypes and Synechococcus, while the DCM was dominated by Prochlorococcus LL ecotypes.

The abundance of 100 individual genes from Synechococcus and six ecotypes of Prochlorococcus (both HLs and LLs) is generated and their diversity is analyzed. The figure incorporates principal component analysis (PCA), diversity indices, and metadata associations to explore patterns in gene diversity and abundance. The PCA is generated using STAMP clustering each sample based on their N assimilation-related genes characteristic and grouping the samples based on their in-situ metadata of temperature with interval 5°C (A), sampling layers of surface (SF) and deep chlorophyll maximum (DCM) (B), regions (Ocean) where the samples are collected (C), NO2- + NO3- concentrations (µmol L-1) (D), ammonium concentrations (µmol L-1) at 5 m depth (E), Iron (Fe) concentration (µmol L-1) (F), and phosphate concentrations (µmol L-1) (G). The Simpson diversity distribution across temperature gradients (H), with black squares representing Simpson index values for individual stations, connected by lines for continuity. The Shannon diversity index of samples from the SF and DCM layers at temperature warmer than 15°C only (I), the solid boxes indicate the interquartile range (Q1 to Q3), the whisker indicates the data quartiles, the middle line indicates the median value and the star symbol indicates the average diversity.
Mixotrophy N utilization as a response to seawater temperature
To elucidate the interplay between mixotrophy and marine cyanobacteria adaptation to ocean temperature, we analyzed global ocean metatranscriptomic data. Our findings aligned with the metagenomic analyses, where temperature is the main driver of mixotrophic nitrogen (N) utilization (Supplementary Note 3). Variations in nutrient concentrations, including ammonium, nitrate + nitrite, iron, and phosphate, were observed across different latitudes and temperature gradients. However, no clear pattern emerged linking N-transport-related gene expression to these nutrient gradients (Supplementary Note 3). Although earlier studies suggested that Prochlorococcus could not thrive in polar regions due to growth inhibition at low temperatures2, more recent research has documented the presence of Prochlorococcus in colder environments, down to ~7°C, and even in polar regions31,32. Therefore, a comprehensive understanding of marine cyanobacteria’s mixotrophy utilization of N sources across varying temperatures and latitudes, irrespective of their absolute abundance, is essential. Our results revealed that mixotrophic activity is widespread in both Prochlorococcus and Synechococcus (Fig. 4A, B), but no general pattern was observed between SF and DCM populations. Pearson correlation analysis revealed a significant positive relationship between temperature gradients and ammonium transport, while a negative correlation was observed with amino acid transport (Fig. 4C).

The transcript of the N assimilation-related genes distributed in the Tara Ocean metatranscriptomic data, the pie map represents the categories nitrogen uptake genes from the surface (SF) (A) and DCM (B) samples, where different colors of the pie slices represent different categories, blue for amino acids transporters, yellow for the ammonium transporters, red for the nitrate assimilation-related and green for small organic molecules. The correlation analysis of the N assimilation-related genes and environmental factors (C). The contribution of N assimilation-related genes based on sample temperature (D) and latitude (E). The colors represent different N-sources, blue for the amino acids, yellow for the ammonium, red for nitrate, and green for the small organic molecules (urea and cyanate). The transcript distribution on different temperatures as the specific HL and LL ecotypes of Prochlorococcus are displayed separately (F), the different colors and shapes indicate assimilatory genes involved in different nitrogen uptake-related specific to Synechococcus, HL, and LL of Prochlorococcus. The gradient of sample temperature metadata across different latitudes (G), and the different colors indicate the epipelagic layer of SF and DCM.
In agreement with our metagenomic findings, which suggested a diversity threshold around 15°C (Fig. 3H), the metatranscriptomic analysis also identified ~15°C as a critical point where a distinct shift in the expression of amino acid versus ammonium transporters occurred (Fig. 4D). This shift was observed between ~40°N and 35°S (Fig. 4E), underscoring the role of temperature in shaping nitrogen acquisition strategies. In regions warmer than 15 °C, ammonium transporters accounted for 70-85% of transcripts (Fig. 4F), predominantly expressed by Prochlorococcus. In contrast, in regions beyond 35˚S/40˚N colder than 15°C, Synechococcus relied heavily on amino acid uptake genes (~90%). Notably, the glnQ gene, which encodes a glutamine transporter, constituted the majority of Synechococcus amino acid transporters (Supplementary Note 3). The glnQ transcript correlates negatively with most other N-transport-related genes (p < 0.01) (Supplementary Note 3), underscoring the cruciality of glutamine for Synechococcus in the colder regions.
To further validate the temperature effect and the preference for amino acids below 15°C and inorganic nitrogen above 15°C, we calculated the ratio of ecotype-specific glutamine-metabolism-related genes to the corresponding recA gene (DNA recombinant A) (Fig. 5, Supplementary Fig. 8), serving as a proxy for transcript abundance per cell33,34. The ratio of glnA (glutamine synthetase, GS) to recA demonstrated temperature sensitivity across all ecotypes, with the highest ratios observed in high-light (HL) ecotypes, showing a two-fold increase in the glnA/recA ratio at 15°C, and a four-fold increase at 30°C. This highlights the temperature dependence of GS activity and glutamine transport.

The schematic framework illustrates different N sources assimilate through permeases, transporters and metabolisms (A). The potential active metabolic pathways in cold regions (<15°C) are highlighted with red-orange dash lines (--) and boxes. The key pathways include: ammonium is transported via ammonium transporter (amt1); nitrate/nitrite is transported by nrtP and focA, then nitrate is reduced to nitrite by nitrate reductase (narB), and nitrite is further reduced to ammonium by nitrite reductase (nirA); The urea and cyanate are transported via urtA and cynA and converted to ammonium by urease and cyanase. *The GS (glutamine synthetase glnA) converts ammonium and glutamate to glutamine, and glutamate is regenerated by GOGAT (glutamate synthase) by incorporating 2-OG into glutamine the carbon backbone. **The two main sources of 2-OG and isocitrate dehydrogenase (icd) participate in generating 2-OG from the tricarboxylic acid cycle (TCA), and RuBisCO (cbbL ribulose bisphosphate carboxylase large chain) catalyzes the fixation of CO2, which is subsequently converted to 2-OG via the Calvin cycle. ***The other amino acids are metabolized by converting them into organic molecules or biodegrading them to ammonium. The functional gene ratios in Synechococcus, LLII/III and LLIV Prochlorococcus ecotypes normalized to recA across different water temperature intervals (5 °C bins): glnA/recA (B), glnQ/recA (C), icd/recA (D), and cbbL/recA (E). The color-filled boxes represent interquartile ranges (Q1 to Q3), the middle lines indicate the medians, the stars indicate the average scores, and the whiskers indicate the data quartiles.
To delve deeper into Synechococcus metabolic pathway in regions below 15°C, we examined the ratio of glnA, glnQ, cbbL (ribulose bisphosphate carboxylase large chain), and icd (isocitrate dehydrogenase gene) to recA (Fig. 5A). These genes are key components of the glutamine synthetase/glutamate synthase (GS/GOGAT) cycle, which maintains intracellular N balance35. In warmer regions (>15°C), the glnQ/recA ratio decreased significantly (Fig. 5B, C), inversely correlating with the glnA/recA ratio, highlighting the negative relationship between glutamine conversion and its uptake pathway. The synthesis of glutamine is highly dependent on 2-OG (2-Oxoglutarate), derived from both the tricarboxylic acid (TCA) and Calvin cycles7. Thus, we also analyzed the ratios of cbbL and icd to recA. The expression of icd gene, a marker for respiratory activity, showed that temperature significantly affects Synechococcus respiration (Fig. 5D), while cbbL expression varied with temperature, indicating temperature-driven differences in photosynthetic activity (Fig. 5E).
Further analysis of SF and DCM samples revealed that for regions below 15°C, photosynthetically active radiation (PAR) had no significant effect on the respiratory (icd) and photosynthetic system (cbbL) of Synechococcus and Prochlococcus LL ecotypes (Supplementary Note 4). However, in regions warmer than 15°C, PAR significantly impacts cbbL expression but not respiratory activity in Synechococcus (Supplementary Note 4), PAR plays a crucial role in regulating marine cyanobacterial activity in warmer environments, which in turn affects inorganic nitrogen uptake. Additional analysis of Synechococcus adaptation in colder regions (<15°C) revealed that glutamine and glutamate metabolism pathway facilitates the conversion of glutamine to glutamate without energy expenditure. This process is followed by glutamate oxidation to 2-OG, producing NADPH, a key survival strategy in these cold environments (Supplementary Note 4).
Discussions
Our findings confirm that both Prochlorococcus and Synechococcus possess the capability to utilize organic and inorganic nitrogen sources (Fig. 2, Supplementary Note 5), aligning with previous research on the ecological success of these cyanobacteria17,18. Despite the inherent challenges of a mixotrophic lifestyle16, their success is attributable to functional redundancy within planktonic prokaryotes36,37, driven by interactions between the pangenome and environmental factors38,39,40, which define their ecological roles41,42. The evolutionary optimization of N transport systems in these cyanobacteria has led to efficient mechanisms for nutrient uptake, shaped by environmental pressures and redundancy11.
Our analysis reveals that Synechococcus exhibits a broader range of N source utilization compared to Prochlorococcus, highlighting its versatility in nitrogen acquisition (Fig. 1, Supplementary Note 6). In particular, Prochlorococcus exhibits genome reduction8, optimizing its metabolic pathways to reduce cellular N requirements. However, significant variation in genome size43 and N transport capacity exists between different ecotypes. Low-light (LL) ecotypes of Prochlorococcus show a higher abundance of N transporters, likely an adaptation to the diverse N availability in deeper water layers (Supplementary Note 5). This adaptation contrasts with high-light (HL) ecotypes, which have fewer N transport systems11. Synechococcus, on the other hand, competes across a wider range of temperatures and nitrogen sources due to its larger genome and broader physiological versatility.
Temperature emerged as a critical driver of nitrogen assimilation dynamics in both cyanobacteria. A distinct threshold at 15 °C marks a shift in nitrogen source utilization preferences (Fig. 3A), with Prochlorococcus favoring ammonium transport in warmer regions spanning 35˚S to 40˚N with temperature higher than 15°C, while Synechococcus relies on amino acid uptake, particularly glutamine, in colder environments beyond 35˚S/40˚N from freezing -2°C to 15°C (Fig. 4F), highlighting the role of cyanobacteria as modulators of marine nitrogen cycling. Glutamine is crucial for cellular nitrogen regulation and precursor for N compounds44, the transition at 15°C coincides with shifts in both genomic expression and physiological responses, as evidenced by the glnA/recA and glnQ/recA ratios (Fig. 5B, C), which indicate high activity of glutamine uptake and utilization in the colder regions (<15°C) and high glutamine synthetase in the warmer regions (15−30°C). This temperature-driven specialization is consistent with previous observations of microbial substrate affinity changes at lower temperatures45 and aligns with findings regarding cyanobacterial preferences for inorganic nitrogen46,47. This temperature and nitrogen source interplay may also play a role in the increasing harmful cyanobacteria blooms, as evident in the coastal waters48 and in the wintertime of boreal lakes49. The occurrence of these harmful cyanobacteria in the open ocean waters under the global change scenario needs further explorations, particularly in the aspect of nitrogen utilization strategies, due to the increasing anthropogenic nitrogen input into global ocean.
Synechococcus’s reliance on amino acid uptake in colder waters (<15°C) in the (sub)polar regions is further supported by the glutamine/2-OG ratio, which regulates key nitrogen regulatory genes like glnA and ntcA7. This mechanism enables Synechococcus to manage nitrogen balance through the glutamine-glutamate system, which also facilitates the production of NADPH and 2-OG through the gdhA gene50. The adaptability of Synechococcus in colder environments, especially in regions below 15°C, highlights the physiological advantages conferred by its larger genome and its ability to shift from inorganic to organic nitrogen utilization.
The observed differences between Prochlorococcus and Synechococcus suggest a temperature-dependent partitioning of ecological niches. While Prochlorococcus favors inorganic nitrogen in warmer waters and exhibits niche diversification through HL and LL ecotypes across the water column, Synechococcus demonstrates a preference for organic nitrogen, especially amino acids, in colder regions beyond 35˚S/40˚N with temperature <15°C. This temperature-driven shift in nitrogen source utilization reflects an evolutionary response to environmental gradients, enabling both cyanobacteria to maintain their ecological success across diverse marine habitats. This adaptation highlights the influence of temperature over PAR as the driver of Synechococcus latitudinal distribution, in regions warmer and colder than 15°C (Fig. 5D, E). Furthermore, our analysis of metatranscriptomic data supports the notion that temperature, more than nutrient concentrations such as ammonium, nitrate, iron, or phosphate, plays a dominant role in shaping nitrogen assimilation strategies (Supplementary Note 3).
This latitudinal distribution patterns of nitrogen utilization and their linkage to temperature variation provide critical insights into the responses of marine cyanobacteria to climate change. As global sea surface temperatures continue to rise, the ecological niches of Prochlorococcus and Synechococcus are likely to shift, with potential consequences for marine primary productivity and nutrient cycling. For instance, the expansion of Prochlorococcus into higher latitudes could lead to an increased reliance on inorganic nitrogen sources in regions traditionally dominated by Synechococcus. This shift may alter the balance of carbon and nitrogen fluxes since N constrains on carbon uptake and influences on global carbon-climate feedback51.
Understanding the interplay between temperature and nutrient assimilation also informs efforts to model and predict the impacts of climate change on oceanic nutrient dynamics. Incorporating the physiological responses of cyanobacteria to temperature gradients into Earth system models will enhance our ability to forecast changes in primary productivity, nutrient cycling, and carbon sequestration. Moreover, the adaptability of Synechococcus to utilize organic nitrogen in colder regions emphasizes the need to account for mixotrophy in nutrient and ecosystem models, as this trait may become increasingly relevant under future climate scenarios.
In general, our findings highlight the critical role of Prochlorococcus and Synechococcus in regulating marine biogeochemical cycles through their temperature-dependent nitrogen source utilization strategies. By linking nutrient dynamics to broader environmental gradients, this work provides a foundation for understanding how climate change may reshape the ecological roles and global distributions of these cyanobacteria, with cascading effects on marine ecosystems and global biogeochemical cycles. In the future, studies should conduct specific manipulative experiments to separate the regulatory effect of temperature from nutrients on cyanobacteria distribution, and to explore their mixtrophy effect on the interactions among carbon, nitrogen, and phosphorus cycles to elucidate their collective impact on cyanobacteria behavior and climate regulation.
Methods
Isolates and single-cell genomes collection
As of December 2022, all the available genomes of Prochlorococcus and Synechococcus from JGI ProPortal (https://img.jgi.doe.gov/cgi-bin/proportal/main.cgi) were downloaded22. In total, 773 Prochlorococcus and 136 Synechococcus genomes were collected, including 489 and 50 genomes of Prochlorococcus and Synechococcus single-cell genomes published by Berube in 201823. The sequence data formats-related work was performed using BBmap tools52.
Genomes quality control
The genome quality control parameters were quantified using CheckM (version v1.2.2)53, and the genome checking was performed using the checkm lineage_wf workflow with the default parameters (https://github.com/Ecogenomics/CheckM/wiki). The genome quality and completeness results from the workflow were used to filter genomes with low quality. The filtering was performed to reduce the usage of incomplete genomes, and at the same time, preserve large portions of genomes to construct genome-genome phylogenetic trees and gene content analysis. The standard used for this study was the same as GTDB25, which filtered genomes with estimated quality lower than score 50, where the estimated quality is defined as genome completeness – 5x contamination. After quality filtering, 322 and 76 genomes of Prochlorococcus and Synechococcus remained for further analysis (Supplementary Data 1).
Genomes gene calling and annotation
First, as reference protein sequences, all possible protein sequences N-transport-related were collected from the UniProt database54 according to the KEGG55 ABC transporter map (https://www.kegg.jp/pathway/map02010) and the nitrogen metabolisms map (https://www.kegg.jp/pathway/map00910). All keywords (orthologs and gene names) were used to download protein sequences from the UniProt database. This study focused on four main N forms, (1) ammonium, the most crucial inorganic nitrogen (reduced nitrogen) in open ocean environments; (2) nitrate+nitrite, the oxidized form of inorganic nitrogen, requiring energy to utilize11; (3) amino acids, which are essential for any life in the open ocean; and (4) small organic molecules (non-amino acids organic N compounds), which consist of cyanate and urea7. The collected protein sequences include available sequences belonging to Synechococcus and Prochlorococcus. When sequences belonging to Synechococcus and Prochlorococcus were unavailable, sequences from other cyanobacteria were incorporated into the reference sequences, such as from genera Anabaena, Nostoc, and Microcystis. When the cyanobacteria sequences were unavailable, the sequences from marine-originated bacteria were used. The protein reference sequences collected from the UniProt are attached as Supplementary Data 2.
The genome assembly files were collected from JGI ProProtal and subsequently mapped and annotated using Prokka v1.14.656 (https://github.com/tseemann/prokka), with Prodigal employed for gene calling and predictions57. Initially, a small, well-characterized set of proteins was identified using the program BLAST + , followed by more sensitive and precise searches performed with HMMER3 against the HMM databases58. The resulting.faa files, containing predicted protein sequences generated by Prokka, were validated against the UniProt protein sequences using Diamond BlastP59 with a minimum similarity threshold of 60%. Furthermore, 10 sequences from each gene with the lowest protein similarity scores (>60%) were cross-validated using online BLASTP against the NCBI nr database60. All the classified genes in this study are detailed in Supplementary Table 2 and Supplementary Table 1.
Phylogenetic tree construction
The genome-genome phylogenetic tree was constructed using GTDB-TK v2.0.025. The phylogenetic tree construction started with the command gtdbtk identify where bac120 marker genes were identified in the genomes using Prodigal (table 11 as the default)57. Next was the gtdbtk align command, the identified genes were aligned based on the AR53/BAC120 marker set25. Last, the phylogenetic tree was constructed using FastTree v2.1.761 under the WAG model of protein evolution62 with gamma-distributed rate heterogeneity (+GAMMA)63, this was performed using the command gtdbtk infer. The trees were viewed using an online tool, Interactive Tree Of Life (iTOL) v5 (https://itol.embl.de/)64. All the datasets including genome type (isolate/single-cell), functional genes (binary data), and genome classifications were visualized based on methods and templates provided by iTOL v5 web interference (https://itol.embl.de/help.cgi).
Global distribution of nitrogen assimilation genes
Tara Ocean metagenomic and metatranscriptomic data24 were collected using the links generated by Phil Ewels SRA Explorer (https://sra-explorer.info/) in January 2023. The list of samples used in this analysis is listed in Supplementary Data 3. The identified protein sequences collected are attached as Supplementary Data 4 and were used as the database for exploring the metagenomic and metatranscriptomic data. The sequence extraction and format conversion were carried out using SeqTk65.
The global distribution analysis started with the creation of a protein database containing all the N transport-related sequences from the ProPortal genomes that have been translated, annotated (Prokka), and validated (Diamond+UniProt). Each sequence was extracted from the genomes, named after its Gold Analysis (GA) number in ProPortal22 and tagged with their respective genera names and clades using a Python script. The analysis of the Tara Ocean data was performed based on the created sequences database. The forward fastq files were analyzed using Diamond blastX59 against the identified Prochlorococcus and Synechococcus nitrogen uptake genes according to our codes uploading in GitHub (https://github.com/Buce-Hetharua/Cyanobacteria-Mixotrophy). The bulk run of the Tara Ocean metagenomic and metatranscriptomic data were run using two separate bash files (*.sh), with parameters including a bit score 50 (--min-score 50), 90% similarity (--id 90) and 50% sequence coverage (--query-cover 50). According to this study, genes from Synechococcus and Prochlorococcus only share ~ 90% of similarity of their protein sequences (Supplementary Fig. 2). A combination of Python and Perl scripts were used to process the Diamond blastX results, starting with the Count-seqs-for-both-metaG-metaT.sh script, followed by Merging-files, which generated a matrix table of genes and samples. The number of reads of each gene from the matrix was normalized using their respective sequence length (Supplementary Table 2), and the sequence length was calculated using Seqkit (https://bioinf.shenwei.me/seqkit/)66. The normalization of the genes allows the comparison of each gene as how many copies per 1000 bp of sequence.
Statistical analysis
The diversity indexes were constructed based on nitrogen-assimilation-related genes, where the Shannon diversity index (H) was using the calculation H = -Σ pi * ln (pi), in which pi is the proportion of the entire community made up of species i67. The Simpson (D) diversity was calculated using D = Σ ni (ni-1) / N(N-1), where ni is the number of organisms that belong to species i, and N is the total number of organisms.
The normalized matrix of the Tara Ocean metagenomic and metatranscriptomic data were processed and analyzed using STAMP v2.1.328 (https://beikolab.cs.dal.ca/software/STAMP), then, the data matrix was further analyzed by incorporating the metadata from the samples’ respective stations24. The beta diversity analysis was generated based on a 100 genes matrix (24 nitrogen-assimilation-related genes and each ecotype), and the analysis was performed for both metagenomic and metatranscriptomic data. The beta diversity was calculated using the Kruskal-Wallis H-test, with a post-hoc test performed using Tukey-Kramer.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the sequence data, modified codes, and raw results of this study are available for the public on Buce-Hetharua GitHub web page (https://github.com/Buce-Hetharua/Cyanobacteria-Mixotrophy).
References
Bekker, A. et al. Beukes. Dating the rise of atmospheric oxygen. Nature 427, 117–120 (2004).
Flombaum, P. et al. Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl Acad. Sci. USA 110, 9824–9829 (2013).
Zehr, J. P. & Ward, B. B. Nitrogen cycling in the ocean: new perspectives on processes and paradigms. Appl. Environ. Microbiol. 68, 1015–1024 (2002).
Hutchins, D. A. & Capone, D. G. The marine nitrogen cycle: new developments and global change. Nat. Rev. Microbiol. 20, 401–414 (2022).
Visintini, N., Martiny, A. C. & Flombaum, P. Prochlorococcus, Synechococcus, and picoeukaryotic phytoplankton abundances in the global ocean. Limnol. Oceanography Lett. 6, 207–215 (2021).
Flombaum, P., Wang, W. L., Primeau, F. W. & Martiny, A. C. Global picophytoplankton niche partitioning predicts overall positive response to ocean warming. Nat. Geosci. 13, 116–120 (2020).
Díez, J. et al. Regulatory and metabolic adaptations in the nitrogen assimilation of marine picocyanobacteria. FEMS Microbiol. Rev. 47, fuac043 (2023).
Read, R. W. et al. Nitrogen cost minimization is promoted by structural changes in the transcriptome of N-deprived Prochlorococcus cells. The ISME . 11, 2267–2278 (2017).
Muñoz-Marín, M. D. C. et al. Glucose uptake in Prochlorococcus: diversity of kinetics and effects on the metabolism. Front. Microbiol. 8, 248847 (2017).
Grzymski, J. J. & Dussaq, A. M. The significance of nitrogen cost minimization in proteomes of marine microorganisms. ISME J. 6, 71–80 (2012).
Berube, P. M., Rasmussen, A., Braakman, R., Stepanauskas, R. & Chisholm, S. W. Emergence of trait variability through the lens of nitrogen assimilation in Prochlorococcus. Elife 8, e41043 (2019).
Muñoz-Marín, Md. C. Mixotrophy in depth. Nat. Microbiol. 7, 1949–1950 (2022).
Muñoz-Marín, M. C. et al. Mixotrophy in marine picocyanobacteria: use of organic compounds by Prochlorococcus and Synechococcus. ISME J. 14, 1065–1073 (2020).
Muñoz-Marín, Md. C., López-Lozano, A., Moreno-Cabezuelo, J. Á., Díez, J. & García-Fernández, J. M. Mixotrophy in cyanobacteria. Curr. Opin. Microbiol. 78, 102432 (2024).
Chen, J. et al. Genomic and transcriptomic evidence for the diverse adaptations of Synechococcus subclusters 5.2 and 5.3 to mesoscale eddies. N. Phytol. 233, 1828–1842 (2022).
Tittel, J. et al. Mixotrophs combine resource use to outcompete specialists: implications for aquatic food webs. Proc. Natl Acad. Sci. USA 100, 12776–12781 (2003).
Hartmann, M. et al. Mixotrophic basis of Atlantic oligotrophic ecosystems. Proc. Natl Acad. Sci. USA 109, 5756–5760 (2012).
Våge, S., Castellani, M., Giske, J. & Thingstad, T. F. Successful strategies in size structured mixotrophic food webs. Aquatic Ecol. 47, 329–347 (2013).
Kashtan, N. et al. Single-cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus. Science 344, 416–420 (2014).
Larkin, A. A. et al. Niche partitioning and biogeography of high light adapted Prochlorococcus across taxonomic ranks in the North Pacific. ISME J.10, 1555–1567 (2016).
Doré, H. et al. Evolutionary mechanisms of long-term genome diversification associated with niche partitioning in marine picocyanobacteria. Front. Microbiol. 11, 567431 (2020).
Kelly, L., Huang, K. H., Ding, H. & Chisholm, S. W. ProPortal: a resource for integrated systems biology of Prochlorococcus and its phage. Nucleic Acids Res. 40, D632–D640 (2012).
Berube, P. M. et al. Single cell genomes of Prochlorococcus, Synechococcus, and sympatric microbes from diverse marine environments. Sci. Data 5, 1–11 (2018).
Salazar, G. et al. Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell 179, 1068–1083. e1021 (2019).
Parks, D. H. et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004 (2018).
Kanehisa, M. et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484 (2007).
Widner, B., Fuchsman, C. A., Chang, B. X., Rocap, G. & Mulholland, M. R. Utilization of urea and cyanate in waters overlying and within the eastern tropical north Pacific oxygen deficient zone. FEMS Microbiol. Ecol. 94, fiy138 (2018).
Parks, D. H., Tyson, G. W., Hugenholtz, P. & Beiko, R. G. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124 (2014).
Mouillot, D. & Lepretre, A. A comparison of species diversity estimators. Res. Population Ecol. 41, 203–215 (1999).
Levine, J. M. & HilleRisLambers, J. The importance of niches for the maintenance of species diversity. Nature 461, 254–257 (2009).
Mioduchowska, M., Pawłowska, J., Mazanowski, K. & Weydmann-Zwolicka, A. Contrasting marine microbial communities of the fram strait with the first confirmed record of cyanobacteria Prochlorococcus marinus in the Arctic region. Biology 12, 1246 (2023).
Smith, A. N. et al. Comparing Prochlorococcus temperature niches in the lab and across ocean basins. Limnol. Oceanography 66, 2632–2647 (2021).
Orellana, L. H. et al. Detecting nitrous oxide reductase (nosZ) genes in soil metagenomes: method development and implications for the nitrogen cycle. MBio 5, 01193–01114 (2014).
Feike, J. et al. Measuring unbiased metatranscriptomics in suboxic waters of the central Baltic Sea using a new in situ fixation system. ISME J. 6, 461–470 (2012).
Bolay, P., Muro-Pastor, M. I., Florencio, F. J. & Klähn, S. The distinctive regulation of cyanobacterial glutamine synthetase. Life 8, 52 (2018).
Giovannoni, S. J. et al. Genome streamlining in a cosmopolitan oceanic bacterium. Genome streamlining in a cosmopolitan oceanic bacterium. Science 309, 1242–1245 (2005).
Martínez-Cano, D. J. et al. Evolution of small prokaryotic genomes. Front. Microbiol. 5, 742 (2015).
Cummins, E. A., Hall, R. J., McInerney, J. O. & McNally, A. Prokaryote pangenomes are dynamic entities. Curre. Opin. Microbiol. 66, 73–78 (2022).
Brockhurst, M. A. et al. The ecology and evolution of pangenomes. Curr. Biol. 29, R1094–R1103 (2019).
McInerney, J. O., McNally, A. & O’connell, M. J. Why prokaryotes have pangenomes. Nat. Microbiol. 2, 1–5 (2017).
Fuhrman, J. A., Cram, J. A. & Needham, D. M. Marine microbial community dynamics and their ecological interpretation. Nat. Rev. Microbiol. 13, 133–146 (2015).
Bobay, L. M. & Ochman, H. The evolution of bacterial genome architecture. Front. Genetics 8, 72 (2017).
Biller, S. J., Berube, P. M., Lindell, D. & Chisholm, S. W. Prochlorococcus: the structure and function of collective diversity. Nat. Rev. Microbiol. 13, 13–27 (2015).
Herrero, A., Muro-Pastor, A. M. & Flores, E. Nitrogen control in cyanobacteria. J. Bacteriol. 183, 411–425 (2001).
Nedwell, D. B. Effect of low temperature on microbial growth: lowered affinity for substrates limits growth at low temperature. FEMS Microbiol. Ecolo. 30, 101–111 (1999).
Reay, D. S., Nedwell, D. B., Priddle, J. & Ellis-Evans, J. C. Temperature dependence of inorganic nitrogen uptake: reduced affinity for nitrate at suboptimal temperatures in both algae and bacteria. Appl. Environ. Microbiol. 65, 2577–2584 (1999).
Sakamoto, T. & Bryant, D. A. Growth at low temperature causes nitrogen limitation in the cyanobacterium Synechococcus sp. PCC 7002. Archives Microbiol. 169, 10–19 (1997).
Telesh, I., Schubert, H. & Skarlato, S. Ecological niches of bloom-forming cyanobacteria in brackish Baltic Sea coastal waters. Estuarine Coastal Shelf Sci. 295, 108571 (2023).
Grosbois, G., Mou, T. A. & Girona, M. M. Cyanobacteria in winter: seasonal dynamics of harmful algal blooms and their driving factors in boreal lakes. Heliyon 10, e40687 (2024).
Martinez-Bilbao, M., Martinez, A., Urkijo, I., Llama, M. & Serra, J. Induction, isolation, and some properties of the NADPH-dependent glutamate dehydrogenase from the nonheterocystous cyanobacterium Phormidium laminosum. J. Bacteriol. 170, 4897–4902 (1988).
Wang, Y. P., & Houlton, B. Z. Nitrogen constraints on terrestrial carbon uptake: Implications for the global carbon-climate feedback. Geophys. Res. Lett. 36, 24 (2009).
Segata, N. et al. BBMap short-read aligner, and other bioinformatics tools. Bioinformatics 12, 17–23 (2016).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Consortium, U. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2019).
Kanehisa, M., Goto, S., Kawashima, S. & Nakaya, A. The KEGG databases at GenomeNet. Nucleic Acids Res. 30, 42–46 (2002).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 1–11 (2010).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35, D61–D65 (2007).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS ONE 5, e9490 (2010).
Whelan, S. & Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol.18, 691–699 (2001).
Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39, 306–314 (1994).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Li, H. seqtk Toolkit for processing sequences in FASTA/Q formats. GitHub 767, 69 (2012).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PloS One= 11, e0163962 (2016).
Strong, W. Biased richness and evenness relationships within Shannon–Wiener index values. Ecol. Indic. 67, 703–713 (2016).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (42106048, W2411034, 92058204, 42494885), the Hainan Provincial Natural Science Foundation of China (424RC466), and Innovational Fund for Scientific and Technological Personnel of Hainan Province (KJRC2023B04). We thank Qinglu Zeng from the Hong Kong University of Science and Technology for his quick comments on our early draft. We also thank all the reviewers and the editor for their constructive comments.
Author information
Authors and Affiliations
Contributions
B.H., M.X. and S.J.K. conceived the work and wrote the paper. B.H. collected and analyzed the published raw sequence data. B.H., M.X. and S.J.K. analyzed the data and proposed the hypothesis. M.X., B.H., S.J.K. S.S., K.Z., H.Y., and H.L. drafted the manuscript and figures. M.X. revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth and Environment thanks Vinod K. Kannaujiya, Yanyang Zhang and Irena Telesh for their contribution to the peer review of this work. Primary Handling Editors: Haihan Zhang, Alice Drinkwater. [A peer review file is available.]
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hetharua, B., Xu, M., Sun, S. et al. Temperature-driven nitrogen mixotrophy shapes marine cyanobacteria Prochlorococcus and Synechococcus latitudinal distribution pattern. Commun Earth Environ 6, 149 (2025). https://doi.org/10.1038/s43247-025-02102-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43247-025-02102-w
