
Abstract
Multiple lines of evidence support permanent oxygenation of the Earth’s atmosphere during the early Paleoproterozoic (~2.5–2.1 billion years ago). Here, we report geochemical data, along with sulfide textures, for >2.31-bilion-year-old sedimentary rocks from the upper Espanola and Serpent formations that underlie the third glacial diamictite of the Huronian Supergroup, Canada. Redox-sensitive element contents and the isotopic compositions of molybdenum and sulfur are comparable to upper continental crust value. Subangular to rounded pyrite grains of detrital origin are observed in sandstones of the Serpent Formation. Our data indicate limited oxidative weathering of crustal sulfide prior to the third glaciation of the Huronian succession, consistent with low atmospheric oxygen levels (<5×10−4 of the present atmospheric level). We suggest that such low atmospheric oxygen levels persisted for long periods prior to the final glaciation of the Huronian succession.
Similar content being viewed by others


Introduction
A major increase in Earth’s oxygen levels (pO2), commonly known as the Great Oxidation Event (GOE), occurred during the early Paleoproterozoic (2.5–2.1 Ga)1,2. Although transient episodes of water column oxygenation have been reported for the Neoarchean3,4,5, the persistence of mass independent S isotope fractionations (MIF-S) suggests that pO2 was persistently below 10−6 of the present atmospheric level (PAL)6,7,8. By contrast, pO2 increased to much higher levels during the GOE1,2. The disappearance of detrital uraninite and pyrite, along with the loss of MIF-S signals and the appearance of red beds, indicate that pO2 increased to above 10−6–10−4 PAL by ~2.3 Ga6,9,10,11. Oxygen levels may have become even higher between ~2.3 and 2.1 Ga, due to a large increase in organic carbon burial represented by highly positive carbon isotope values (δ13C) during the Lomagundi-Jatuli Event12,13,14.
While it is generally agreed that the GOE represents one of the most prominent redox shifts in Earth’s history, the details of this transition are still debated. Recent quadruple sulfur isotope data (Δ33S and Δ36S) obtained at high stratigraphic resolution allow for conflicting interpretations of atmospheric oxygenation during the early stages of the GOE (between 2.5 and 2.3 Ga)15,16,17,18,19,20,21,22. The disappearance of the MIF-S signal has been documented for the Polisarka Formation (2.50‒2.43 Ga) on the Fennoscandian Shield, northwest Russia19. This record is considered to represent the first disappearance of the MIF-S signal, and suggests the onset of the GOE prior to the first of multiple glaciations that occurred in association with rising oxygen levels during the early Paleoproterozoic.
Clear MIF-S signals have, however, been reported from younger sedimentary rocks, including ~2.3 Ga sedimentary rocks from the Rooihoogte and lower Timeball Hill formations of the Transvaal Supergroup, South Africa16,20,21,22, and from the Kazput and Kungarra formations in the Turee Creek Group, Australia17,18 (Fig. 1). The pronounced MIF-S signals (Δ33S values up to ~9‰) from the Rooihoogte and lower Timeball Hill formations have been interpreted to reflect photochemical reactions in a functionally anoxic atmosphere16,20,21,22. Viewed this way, oscillations in pO2 levels across the MIF-S threshold of 10−6 PAL7,8 between 2.5 and 2.3 Ga have been inferred20,23,24. By contrast, MIF-S signals from the Kazput and Kungarra formations have been attributed to weathering of old crustal sulfides possessing MIF-S signals (the crustal memory effect)17,18,22. In the latter case, a permanent increase in pO2 above the MIF-S threshold has been implied immediately after the initial disappearance of the MIF-S signal between 2.50 and 2.43 Ga19.

a Location of the Huronian Supergroup32 and core Kerr-McGee150-4 (white star). b Stratigraphic correlation between the Huronian Supergroup in Canada and the Transvaal Supergroup of the Transvaal Basin in South Africa24,27. Published radiometric ages24,25,26,27,28,40,85 and redox indicators (Quadruple sulfur isotope records15,16,20,21,29 and redox-sensitive detrital minerals9,30,32,33,34) are also shown. MIF-S and MDF-S refer to mass-independent and mass-dependent sulfur isotope fractionation, respectively. The quadruple sulfur isotope data from the Huronian Supergroup may have been affected by post-depositional alteration31.
To understand the trajectory of pO2 during the early stages of the GOE, we focus on the redox history recorded in the Huronian Supergroup, Canada, which is dominated by clastic sedimentary rocks deposited between 2.45 and 2.31 Ga (Fig. 1)25,26,27,28. Quadruple sulfur isotope data have also been reported for authigenic sulfides preserved in the Huronian Supergroup29,30,31. Small MIF-S signals (Δ33S values up to 1‰) reported from the McKim and Pecors formations in the lower part of the Huronian Supergroup may reflect low pO2 conditions (<10−6 PAL) during the early stages of the GOE (Fig. 1b)29. However, subsequent studies have suggested that the authigenic sulfide minerals, particularly pyrrhotite and chalcopyrite, may have formed during regional metamorphism and may record the isotopic composition of sulfur remobilized from detrital sulfides31.
The sedimentary rocks of the Huronian Supergroup have also been used to investigate paleo-weathering conditions based on redox-sensitive minerals9,30,32,33,34. Detrital pyrite and uraninite, which are stable under reducing environments, have been reported from fluvial sandstones of the Livingstone Creek, Matinenda and Missisagi formations in the lower part of the Huronian Supergroup (Fig. 1b). Slight increases in redox-sensitive element abundances, including Mo, Re and Os, in the Pecors and Espanola formations in the lower part of the Huronian Supergroup have been interpreted to reflect transient episodes of oxidative sulfide weathering35,36. The Serpent Formation, which underlies the third Huronian glacial diamictite in the Gowganda Formation (Fig. 1b), may also preserve detrital pyrite32, but no petrological evidence has been reported32,34. Hematitic red beds, which generally form under oxygenated conditions, are commonly found above the Gowganda Formation9,33,34. Enrichments in Mn in the Gowganda Formation also support highly oxidizing environments in the upper part of the Huronian Supergroup37. These studies indicate that the Huronian Supergroup spans an interval of dynamic shifts in atmospheric redox state, providing an opportunity to trace the detailed trajectory of pO2 during the early stages of the GOE.
Here, we document sulfide mineral textures and report major and trace element concentrations, along with isotopic compositions of Mo (δ98/95Mo), S (δ34S) and organic carbon (δ13Corg), for the upper part of the Espanola Formation (upper Espanola) and the overlying Serpent Formation (Supplementary Tables 1, 2). Some of the sulfides were further analyzed for in situ trace element concentrations (Supplementary Table 3). We also provide bulk quadruple sulfur isotope data for the entire Huronian Supergroup to complement existing data29,30,31 (Supplementary Table 4). Based on these data, we evaluate the role of oxidative processes in chemical weathering and sediment transport during deposition of the upper Espanola and Serpent formations.
Results and discussion
Samples and geological setting
The Huronian Supergroup outcrops along the north shore of Lake Huron and is divided into four groups in ascending stratigraphic order: the Elliot Lake Group, the Hough Lake Group, the Quirke Lake Group and the Cobalt Group (Fig. 1). The lithofacies of the upper three groups change cyclically from glacial deposits to deltaic mudstones or carbonates, and then to fluvial sandstones38,39 (Fig. 1b). Volcanic rocks in the Thessalon Formation have been dated at 2453 ± 6 Ma based on U-Pb analyses of zircon grains25,26 (Fig. 1b). U-Pb zircon ages of 2310 ± 5 and 2318 ± 8 Ma are reported from tuffs in the Gordon Lake Formation27,28 (Fig. 1b). These dates constrain the depositional ages of the Espanola and Serpent formations to between 2.45 and 2.31 Ga. The entire Huronian Supergroup was cut by the Nipissing diabase intrusion at 2219 ± 4 Ma40 (Fig. 1b) and deformed and metamorphosed to sub-greenschist to greenschist facies during the Penokean orogeny at ~1.85 Ga39. Felsic plutonism along the north shore of Lake Huron may have resulted in metasomatic addition of K and Na to Huronian sedimentary rocks at ~1.7 Ga41,42.
The lower part of the Huronian Supergroup (below the Gowganda Formation) is considered to have been deposited in a continental rift basin38,39. The Quirke Lake Group consists of the Bruce Formation, the Espanola Formation and the Serpent Formation, in ascending stratigraphic order (Fig. 1b). The Espanola Formation is the only widespread carbonate unit in the Huronian Supergroup and directly overlies the second glacial diamictite in the Huronian Supergroup (i.e., the Bruce Formation)38,39. The sedimentary rocks of the postglacial Espanola Formation can be subdivided into three parts: a lower carbonate member, a middle siltstone member and an upper dolomitic mudstone member39,43,44. A local fourth member is reported from the southern part of the Huronian Supergroup43,44. The presence of fine, rippled laminations suggests that the lower three members were deposited in an environment dominated by low energy conditions, either in a shallow marine or lacustrine environment39,43,44. The presence of graded beds containing carbonate grains suggests that some of the carbonate grains in these members may be detrital in origin43,44. The fourth sandstone member either represents a shallow-marine deposit dominated by storm and/or tidal processes, or a fluvial deposit, as indicated by abundant large-scale cross beds in this member43,44.
The Serpent Formation mainly consists of fine- to medium-grained feldspathic sandstones with abundant albite32,41,45. The preservation of albite-rich detritus likely reflects weak to moderate chemical weathering during deposition of the Serpent Formation32,45. Mudstones and pebbly sandstones with local development of granular conglomerates are minor constituents of the formation32. Large-scale crossbedding developed in the sandstones, and the presence of desiccation features in the mudstones indicate that the Serpent Formation is a dominantly fluvial (braided stream) deposit32. The Serpent Formation is overlain by the Gowganda Formation, the uppermost glacial diamictite unit of the Huronian Supergroup, with an erosional contact32. This diamictite has been correlated with the diamictite at the base of the Rooihoogte Formation of the Transvaal Supergroup27 (Fig. 1b).
The samples analyzed were selected from a 1700 m long drill core (Kerr-McGee 150-4) drilled near Elliot Lake (46°28'16“N, 82°41'54“W; Fig. 1a). The same core has been analyzed by previous studies for quadruple sulfur isotopes in sulfide grains throughout the Huronian Supergroup, although the Serpent Formation has not previously been targeted29,31. The core spans the Matinenda Formation to the Lorrain Formation, and was cut by Nipissing diabase intrusions in the Mississagi Formation, the boundary between the Bruce and Espanola formations, and the Lorrain Formation. Samples covering the upper Espanola Formation and the entire Serpent Formation were analyzed for redox-sensitive elements and Mo, S and organic carbon isotopic compositions (see Methods; Fig. 2). We also measured bulk quadruple sulfur isotopes on 23 samples from other formations through core 150-4, specifically from the Matinenda Formation to the Lorrain Formation (see Methods; Supplementary Fig. 1). These samples were selected because they contain sufficient authigenic sulfide for isotopic analysis.

Polished sections of a laminated mudstone in the upper Espanola Formation at 860.62 m, b poorly sorted sandstone in the Serpent Formation at 795.93 m, and c granule-rich sandstone in the Serpent Formation at 762.23 m. Reflected light photomicrographs of d disseminated pyrrhotite at 860.62 m, e disseminated pyrite at 817.96 m, f subangular to rounded pyrite grains interspersed in sandstones at 795.93 m, g, h subangular to rounded pyrite grains concentrated in discrete layers at 762.23 m, and i a euhedral pyrite grain at 690.98 m. Scanning electron microscope images showing subangular to rounded pyrite grains at j, k 795.93 m, l, m 762.23 m, n, o 690.98 m. Rounded zircon and apatite grains are also shown in panels (l), (m) and (o).
Samples from the upper Espanola Formation are laminated dark gray to black mudstones (Fig. 2a). Some of the samples contain abundant calcite and/or dolomite, although quartz and plagioclase are also present. Samples from the Serpent Formation consist mainly of light gray to dark gray, poorly sorted, fine- to medium-grained sandstones (Fig. 2b). Pebbly sandstones are also collected from the core (Fig. 2c). The sandstone samples are dominated by quartz, K-feldspar and plagioclase. Small amounts of carbonate (mostly calcite) are also present in the lower part of the Serpent Formation. Samples from the upper Espanola and Serpent formations often contain sulfide minerals (described below).
Sulfides in the upper Espanola and the Serpent formations
Sulfides with a variety of morphologies commonly occur in samples from the upper Espanola and Serpent formations (Fig. 2). As described in previous studies29,31, euhedral pyrite grains and disseminated sulfides (pyrite and pyrrhotite) are present in the studied core (Fig. 2d, e and i). These authigenic sulfides would have formed during diagenesis, metamorphism and/or hydrothermal alteration29,30,31. Subangular to rounded pyrite grains also occur in sandstone samples of the Serpent Formation (Fig. 2f–h, j–o). It has been proposed that subangular to rounded pyrite grains can form through the replacement of Fe-Ti oxides, such as titaniferous magnetite and ilmenite46. Indeed, we found an aggregate consisting of pyrite and Ti oxide that may reflect the pyritization of Ti-bearing phases (Fig. 2m). The pyrite in this aggregate has a high Ti content (~1.5 wt%; Supplementary Table 3). However, in situ analyses of trace elements indicate that all of the analyzed subangular to rounded pyrite grains have low Ti concentrations (<120 μg/g; Supplementary Table 3). Hence, the observed subangular to rounded pyrite grains are unlikely to have formed by replacing pre-existing detrital Fe-Ti oxides. In addition, subangular to rounded grains are more common than authigenic sulfides in sandstone samples of the Serpent Formation (Supplementary Fig. 2). An opposite trend would be expected if the samples had been strongly affected by post-depositional pyritization.
In some cases, subangular to rounded pyrite grains are concentrated in discrete layers (Fig. 2g, h) and are associated with other heavy minerals, such as zircon and apatite (Fig. 2l, m, o). The subangular to rounded pyrite grains are typically ~50–100 μm in size and are generally smaller than the surrounding silicate minerals (Fig. 2j–o). These characteristics of the pyrite grains are similar to those described for detrital pyrite grains in the ~2.42 Ga Naragas and Heynskop formations, Transvaal Supergroup in the Griqualand West Basin, South Africa11, and the ~2.45 Ga Kungarra Formation, Turee Creek Group, Australia47.
Chemostratigraphic trends through the upper Espanola and Serpent formations
Geochemical profiles through the study section are shown in Fig. 3 (see Supplementary Tables 1, 2). Magnesium and Ca concentrations decrease at the base of the Serpent Formation, reflecting changes in mineralogical composition. Chemical Index of Alteration (CIA) values range from 43 to 74, and are in line with those reported for the Serpent Formation in previous studies41,45,48,49. When plotted on an Al2O3-CaO+Na2O-K2O (A-CN-K) diagram, the data deviate from the predicted weathering trend of feldspar dissolution, which should be parallel to the A-CN boundary (Fig. 4). The measured data are more consistent with a trend previously reported for Serpent mudstone samples affected by K-metasomatism41,45. Hence, the CIA values of the analyzed samples are likely to be affected by K-metasomatism and provide minimum estimates of the original values. When corrected for the effects of K-metasomatism (see Fig. 4), the analyzed samples have higher CIA values, ranging from ~45 to ~85.

The CIA values are not corrected for K-metasomatism. The solid line in the CIA profile shows the average chemical composition of the source rocks48. The dashed lines denote average UCC compositions50. The gray area in the δ98/95Mo profile indicates the average Archean UCC value52. The hatched area in the δ34S profile represents the isotopic composition of the mantle53. The errors for δ98/95Mo and δ34S are in 2 SD and 1 SD, respectively, of the sample reproducibility.

Data from the present study (filled circles) and the previous study45 (open circles) are shown. The white star denotes the chemical composition of unweathered source rocks estimated by the previous study45. The weathering trend is expected to be parallel to the A-CN boundary and to join the source rock compositions. To correct for the effects of K-metasomatism, the raw data are projected back onto the expected weathering trend from the K apex. Filled and open squares represent the corrected data from the present and previous45 studies, respectively.
Redox-sensitive element contents are normalized to Al to examine the degree of authigenic enrichment (Fig. 3), giving values that are generally comparable to average values for upper continental crust (UCC)50. Similar patterns hold for V, Cr, Mo and U concentrations when they are compared with other detrital tracers51,52 (Fig. 5). In situ measurements indicate that these elements are not necessarily concentrated in sulfide minerals (Supplementary Table 3). The lack of strong authigenic enrichment of redox-sensitive elements occurs alongside low TOC and TS contents (<0.04 wt% and <0.7 wt%, respectively; Supplementary Table 2).

The δ98/95Mo values of the studied interval range from −0.06 to 0.79‰ (Fig. 3). Except for two samples in the upper Espanola Formation with higher δ98/95Mo values (>0.5‰), most of the samples have δ98/95Mo values close to 0‰ (Fig. 3). For example, the samples in the Serpent Formation have an average δ98/95Mo of +0.14 ± 0.26‰ (2 SD). This value is identical, within uncertainty, to the average Archean UCC value (+0.03 ± 0.18‰, 2 SD) estimated from Archean and early Paleoproterozoic glacial diamictites52. The two higher values in the upper Espanola Formation may have been affected by authigenic incorporation of Mo into carbonate minerals, as these samples have high Ca and Mg contents (Fig. 3). The δ34S values range from −1.6 to 5.1‰ (Fig. 3), close to the isotopic composition of the mantle (0 ± 2‰)53. The narrow range in δ34S from the upper Espanola and Serpent formations contrasts with previously reported large δ34S variations, ranging from −5 to 35‰, in pyrite grains from the lower part of the Espanola and the Gordon Lake formations29.
Quadruple sulfur isotopes through the Huronian Supergroup
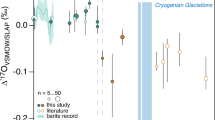
When considered in terms of Δ33S and Δ36S, most of the quadruple sulfur isotope analyses performed through the Huronian Supergroup plot close to the array reflecting isotopic fractionation due to microbial sulfate reduction utilizing a mass-dependent sulfur source (Δ36S/Δ33S = −7; Fig. 6)54. However, most samples from the Serpent and Mississagi formations have Δ36S/Δ33S ratios that are close to, but slightly lower than, the Δ36S/Δ33S Archean reference array (Δ36S/Δ33S = −0.9)54 that reflects photochemically-induced isotopic fractionations in an anoxic atmosphere. Similar low Δ36S/Δ33S values are also reported from pre-GOE pyritic black shales of the Abitibi greenstone belt in the Superior Province (Fig. 6), which is one of the major provenances of the Huronian Supergroup55. Consistent with the Δ36S/Δ33S ratios, the samples from the Serpent Formation exhibit MIF-S signals (i.e., Δ33S = 1.2 to 1.3‰, Δ36S = −1.5 to −2.0‰), although the magnitude of Δ33S (~0.4 ‰) and Δ36S (−0.5 to −1.2‰) values in the Mississagi Formation is relatively small (Supplementary Table 4).

The black dotted line (Δ36S/Δ33S = −0.9) represents photochemically induced MIF-S anomalies reported from Archean sulfides, whereas the gray dashed line (Δ36S/Δ33S = −7) shows the variation caused by mass-dependent fractionation processes including microbial sulfate reduction54. The sulfur isotopic compositions of 2.7-Ga pyritic black shales from the Superior Province are also plotted55. The errors for the Δ33S and Δ36S values are ±0.05‰ and ±0.3‰, respectively.
Effects of post-depositional alteration
To discuss the trajectory of pO2 during the early stages of the GOE using data from the upper Espanola and Serpent formations, we evaluate the effects of post-depositional alteration on the abundance and isotopic compositions of redox-sensitive elements. The Huronian Supergroup experienced multiple tectonic events, such as the emplacement of the Nipissing diabase at 2.22 Ga, deformation during the Penokean orogeny at ~1.9–1.8 Ga, and the intrusion of granitic plutons at ~1.75 Ga39,41,42. These events likely caused metasomatic addition of K and Na to Huronian sedimentary rocks and disturbed the U-Pb isotopic system, although the Sm-Nd isotopic system was not affected41,42. The A-CN-K diagram indicates that the samples analyzed in the present study are also highly likely to have been affected by the post-depositional addition of K (Fig. 4). To assess the effects of post-depositional alteration on the redox-sensitive element data, we focus on the Mo isotope data because both oxidative and reductive alteration can cause large isotopic fractionations56,57,58,59,60.
The δ98/95Mo values of the upper Espanola and Serpent formations show a narrow variation, with a mean value close to the average UCC value, except for the samples rich in carbonate minerals (Fig. 3). If the samples were affected by modern oxidative weathering, they would have δ98/95Mo values and Mo concentrations lower than the UCC values. This is because, (1) Mo would be effectively released from the primary minerals during modern weathering, and (2) part of the dissolved Mo would be trapped in weathered products (e.g., clay minerals and iron oxides) that preferentially adsorb isotopically light Mo56,57. In addition, hydrothermal alteration would likely have resulted in large variations in δ98/95Mo due to local isotopic fractionation, possibly caused by Rayleigh distillation58, whereby δ98/95Mo values of sulfides from a single terrestrial hydrothermal deposit have been shown to vary by up to 3.0‰59,60. Hence, the δ98/95Mo values obtained in the present study are not consistent with intense post-depositional alteration. The lack of strong enrichment in other redox-sensitive elements, coupled with the absence of clear Ce anomalies, also suggests limited addition of these elements by secondary processes (Figs. 3 and 5, Supplementary Fig. 3). The analyzed samples contain euhedral and disseminated sulfides that may have formed during post-depositional alteration (Fig. 2d, e, i). However, in situ measurements indicate that these sulfides are not the major host of redox-sensitive elements (Supplementary Table 3). Thus, although our samples are likely affected by K and Na metasomatism (Fig. 4), these processes appear to have had little influence on the redox-sensitive element data.
Atmospheric oxygen levels between the Bruce and Gowganda glaciations
The subangular to rounded pyrite grains found in sandstones of the Serpent Formation (Fig. 2j–o) are similar to those reported from the Transvaal Supergroup11 and Turee Creek Group47. Rounded pyrite can be supplied through submarine erosion of sulfides hosted in underlying sediments (i.e., reworked pyrite61) and/or erosion of continental sulfides (i.e., detrital pyrite11). However, sulfide minerals are not abundant in the underlying Espanola Formation, which would be inconsistent with the submarine erosion of sulfides. In addition, sulfides eroded from the Espanola Formation would exhibit large variations in δ34S values if the previously reported δ34S values, ranging from +2 to 31‰, represent the isotopic characteristics of sulfides in the Espanola Formation29. Hence, we suggest that the subangular to rounded pyrite grains in the Serpent Formation represent detrital pyrite grains, as proposed previously32. Detrital pyrite has also been reported from other Paleoproterozoic sedimentary rocks11,47. However, most of the detrital pyrite is found in sedimentary rocks deposited before the first Paleoproterozoic glaciation that likely occurred after the initial loss of the MIF-S signal between 2.50 and 2.43 Ga11,19,46,47. Our findings from the Serpent Formation are much younger—by tens of millions of years—than those reported in previous studies.
The positive Δ33S values in the Serpent Formation may have been strongly affected by the presence of detrital sulfides possessing Archean MIF-S signals (Fig. 6). The Δ36S/Δ33S ratios also support this interpretation, given that sulfides in the Abitibi greenstone belt of the 2.7-Ga Superior Province could have been supplied to the Serpent Formation45,55 (Fig. 6). Although samples containing authigenic (i.e., euhedral and disseminated) sulfides were selected for quadruple sulfur isotope analysis (Supplementary Fig. 1), some of the euhedral and disseminated sulfides in the Serpent Formation may have formed by recrystallization of detrital sulfides associated with post-depositional processes31. Therefore, the MIF-S signals in the Serpent Formation do not provide constraints on the isotopic composition of dissolved sulfate in seawater at the time of deposition.
The presence of detrital pyrite in the Serpent Formation is consistent with the abundance and isotopic composition of redox-sensitive elements in the Serpent Formation (Fig. 3). Molybdenum hosted in sulfide would be oxidized and mobilized during oxidative sulfide weathering3,5. In addition, intense acidity generated by sulfide weathering can solubilize Cr in reduced form62. Sediments affected by oxidative sulfide weathering would thus be expected to be depleted in these elements compared to UCC values63. Alternatively, accumulation of these elements in seawater due to oxidative sulfide weathering would then result in enrichment in the sediments deposited under anoxic conditions3,5. However, the Al-normalized values of Cr and Mo in the Serpent Formation are comparable to those reported for average UCC50,51,52 (Fig. 3). In addition, the lack of enrichment in V/Al and U/Al also suggests limited oxidative V and U cycling at the time of deposition, although sulfide may not be the major source of these elements64,65. The δ98/95Mo values in the Serpent Formation, which are close to crustal values, also indicate minimal influence from oxidative weathering56,57 and/or limited authigenic enrichment of Mo from contemporaneous seawater4,5. Most of the Mo in the samples may be hosted in detrital minerals, such as Ti-oxides and pyrite, rather than in authigenic minerals formed during deposition or early diagenesis.
Since the source rocks of the Serpent Formation underwent moderate chemical weathering prior to deposition45 (Fig. 4), ineffective oxidative weathering would have been due to low pO2 at the time of deposition. A model that considers both chemical and physical erosion processes shows that the preservation of detrital pyrite grains also depends on total travel distance and initial grain size11. However, assuming a large initial grain size, the model predicts that all pyrite would be destroyed at pO2 levels above 5 × 10−4 PAL in a large sediment transport system (~1000 km). The detrital zircon ages of the Huronian Supergroup suggest that the sediment was likely transported ~1000 km by a long fluvial system from the source areas in the Superior Province66. Hence, our results from the Serpent Formation suggest an upper limit on pO2 of ~5 × 10−4 PAL at the time of deposition. Given the presence of detrital pyrite grains in the Mississagi, Matinenda and Livingstone Creek formations (Fig. 1b)9,30,34, intervals of low pO2 (<~5 × 10−4 PAL) appear to have been common prior to the Gowganda glaciation (Fig. 1b), although transient accumulation of O2 may have occurred given that fluctuations in pO2 between ~10−7 and ~10−3 PAL were likely possible on geologically short timescales67. As described above, there is disagreement concerning the trajectory of atmospheric oxygenation in the early stages of the GOE based on the multiple S isotope record17,18,20,21,22. Our findings support a scenario whereby pO2 did not remain at a substantially elevated level after 2.50‒2.43 Ga, and instead, pO2 may have fluctuated considerably above and below the 10−6 PAL threshold, at least until the Gowganda glaciation.
As indicated by the presence of detrital sulfide grains in the present study, oxidative sulfide weathering during the early stages of the GOE (e.g., 2.5–2.3 Ga) was limited by the supply of O2 to the weathering environment (i.e., O2-limited regime5). In the subsequent period (up to ~2.1 Ga), sulfide weathering rates likely increased in response to rising pO213,14. As pO2 levels were probably higher than ~10−4–10−3 PAL13, sulfide weathering during this period may have been limited by the supply of sulfide minerals rather than by O2 supply (i.e., sulfide-limited regime5). Increased sulfide weathering rates likely led to transient growth of the marine sulfate reservoir14. It has been proposed that oxidative sulfide weathering coupled with carbonate dissolution can act as a transient source of CO2 to the atmosphere68,69. This suggests that CO2 emissions associated with sulfide weathering were negligible during the early stages of the GOE (e.g., 2.5–2.3 Ga) and thus likely had little impact on climate. By contrast, during the subsequent period, active sulfide weathering may have enhanced CO2 release to the atmosphere, possibly causing a climatic shift to a greenhouse state. As the shift in the sulfide weathering regime broadly coincides with the end of the Paleoproterozoic glacial interval at ~2.3–2.2 Ga (Fig. 1)27,28, this shift may have contributed to the termination of one of the most intense and prolonged intervals of climatic fluctuation in Earth history.
Methods
Sample preparation
Thin sections and powdered materials from the upper Espanola and Serpent formations were prepared for geochemical analyses. Thin sections of representative samples were used to study sulfide grains. Their textures were first examined by reflected light microscopy. The thin sections were then carbon coated for observation using a scanning electron microscope (SEM; HITACHI SU3500) equipped with an energy dispersive X-ray spectrometer (EDS; Oxford Instruments X-max70) at the Geological Survey of Japan (GSJ). Compositional data obtained from the EDS analysis were used to identify heavy minerals in the thin sections. Some of the sulfides were further analyzed using a laser ablation inductively coupled plasma mass spectrometer (LA-ICP-MS; Agilent 8900) equipped with a femtosecond laser ablation system (Laser Blender Raijin, Seishin Trading Co., Ltd.) at GSJ. Trace metal concentrations of the sulfide minerals were measured with a laser spot size of 20 µm and a repetition rate of 20 Hz. BCR-2g (USGS) and synthetic pyrite (Johnson Matthey-Alfa) were used as external standards71. Powdered samples were used to determine major and trace element contents, and Mo, S and organic carbon isotope measurements. The surface layers of drill core samples were first removed using a diamond cutter and polished with corundum powder to avoid contamination from the cutter. The polished samples were cleaned in an ultrasonic bath with distilled water and dried in an oven at 110 °C. The samples were then crushed and pulverized in an agate mortar.
Major and trace element concentrations
Major element concentrations of the bulk rock samples were analyzed at GSJ. Powdered samples were ignited at 1000 °C for 4 h to remove volatile components. An accurately weighed (~0.5 g) aliquot of the ignited sample was mixed with 5 g of lithium tetraborate and fused to prepare glass beads. The glass beads were analyzed using an X-ray fluorescence spectrometer (PANalytical AXIOS). Major element concentrations were used to calculate chemical index of alteration (CIA)48 values, which have been developed to quantify the degree of chemical weathering of silicate materials. As some of the samples contain carbonate minerals, we followed the approach of ref. 72 and corrected for Ca hosted in carbonate minerals: i.e., the molar fraction of CaO was assumed to be equal to that of Na2O if the molar fraction of CaO was greater than that of Na2O. This assumption is based on the observation that calcium plagioclase is less stable than sodium plagioclase during chemical weathering.
Trace element concentrations (Mo, Cr, V and U) were determined by ICP-Q-MS (Agilent 7500cx) at GSJ73. About 0.1 g of powdered sample was digested first with concentrated HNO3 and then with HClO4–HF solution. The samples were completely dissolved with 6 M HNO3 and diluted with 2% HNO3 for measurement. Trace element concentrations in geological reference materials (GSJ JB-2 and USGS SCo-1) were also measured and were within the range of previously published values (Supplementary Table 1).
Mo isotope analysis
Mo isotope analysis was performed using previously described methods74. Powdered samples containing ~300 ng of Mo were completely dissolved using HNO3–HClO4–HF, and mixed with 97Mo-100Mo enriched double spike. Molybdenum was separated from the matrix using anion and cation exchange resins (Biorad AG1-X8 and AG 50W-X8, respectively). The Mo fraction was digested with H2O2–HNO3 to destroy organic matter leached from the resins. Molybdenum isotopic compositions were measured using a multi-collector ICP-MS (Thermo NEPTUNE) at GSJ73. Purified samples dissolved in 3% HNO3 were introduced via an ESI Apex-Ω desolvating nebulizer. Each sample was measured three times, and the mean values and two standard deviations were reported. The isotopic compositions of Mo were expressed using conventional delta notation relative to NIST SRM 3134 (i.e., δ98/95MoNIST = 0‰). The accuracy and precision of the analysis were monitored by processing geochemical reference materials (GSJ JB-2 and USGS SCo-1) along with the samples (Supplementary Table 1).
Total sulfur analysis
For total sulfur (TS) analysis, powdered samples of 2–5 mg were loaded into tin capsules with ~3 mg of V2O5 powder. The tin capsules were then introduced into an elemental analyzer/isotope ratio mass spectrometer (EA/IRMS) system (Isoprime-EA, Isoprime Ltd.) at the University of Tsukuba75. The TS contents were determined by calibration of ion currents at m/z 64 in the IRMS. IAEA-S-1 (δ34S = −0.3‰) and IAEA-S-2 (δ34S = +21.8‰) were used for calibration of sulfur concentrations and isotopic compositions76. The isotopic compositions of sulfur were expressed using δ notation with respect to the VCDT standard: δ34S (‰) = (34Rsample/34Rstandard − 1) × 1000, where 34R is the 34S/32S ratio. Measurements were performed four times for each sample to determine average TS and δ34S values. Analytical uncertainties were expressed as one standard deviation of the repeated measurements (Supplementary Table 2).
Organic carbon analysis
A modified version of the Finnigan Delta Plus XP isotope-ratio mass spectrometer coupled to a Flash EA1112 elemental analyzer through a ConFlo III interface at JASMTEC was used to measure the concentrations and isotopic compositions of total organic carbon77,78,79. Whole rock powders (~0.3 g) were treated with 6 M HCl for 2 days to decarbonate. The samples were then washed several times with distilled water and dried in an oven. The dried samples of 30 mg were loaded into tin capsules and then introduced into the mass spectrometer system. The TOC content of the samples was determined by calibrated ion currents at m/z 44, 45 and 46 in the mass spectrometer. The carbon isotopic composition was expressed in δ notation with respect to the VPDB standard: δ13C (‰) = (13Rsample/13Rstandard − 1) × 1000, where 13R is the 13C/12C ratio. The results for TOC and δ13Corg are summarized in Supplementary Table 2.
Quadruple sulfur isotope analysis
For quadruple sulfur isotope analysis, sulfide extraction was performed using a method modified from refs. 70,80. Whole rock powders (2–10 g) were leached with 15 mL of 5 M HCl to extract acid volatile sulfur in a glass bottle with an alkaline zinc (Zn) trap (a mixture of 0.2 M Zn acetate and 2 M NaOH solution at the mixing ratio of 3:5). The leached samples were reacted with 20 mL of Cr (II) solution with HCl for more than 1 day to convert pyrite sulfur to ZnS in the same alkaline Zn trap81. The ZnS precipitates were extracted from the alkaline Zn trap and reacted with 0.1 M AgNO3 solution to convert them to Ag2S. The Ag2S was converted to SF6 by reaction with ~300 torr of elemental fluorine at 300 °C overnight in a nickel bomb apparatus. The SF6 was first purified by gas chromatography and then introduced into a Thermo-Fisher MAT 252 mass spectrometer at Institute of Science Tokyo to measure multiple sulfur isotopes82. The capital delta values of sulfur isotopes were defined as follows: Δ33S (‰) = 1000 × ln (δ33S/1000 + 1) − 0.515 × 1000 × ln (δ34S/1000 + 1) and Δ36S (‰) = 1000 × ln (δ36S/1000 + 1) − 1.90 × 1000 × ln (δ34S/1000 + 1). These values are normalized to the VCDT scale by analyzing IAEA-S183,84. Typical analytical precision of δ34S, Δ33S and Δ36S values are ±0.6‰, ±0.05‰ and ±0.3‰, respectively. The results of the δ34S, Δ33S and Δ36S values are summarized in Supplementary Table 4.
Data availability
The geochemical data reported in the present study are included in Supplementary Information and are also available through Figshare (https://doi.org/10.6084/m9.figshare.30134803).
References
Holland, H. D. The oxygenation of the atmosphere and oceans. Philos. Trans. R. Soc. B 361, 903–915 (2006).
Lyons, T. W., Reinhard, C. T. & Planavsky, N. J. The rise of oxygen in Earth’s early ocean and atmosphere. Nature 506, 307–315 (2014).
Anbar, A. D. et al. A whiff of oxygen before the great oxidation event?. Science 317, 1903–1906 (2007).
Ostrander, C. M. et al. An expanded shale δ98Mo record permits recurrent shallow marine oxygenation during the Neoarchean. Chem. Geol. 532, 119391 (2020).
Johnson, A. C. et al. Reconciling evidence of oxidative weathering and atmospheric anoxia on Archean Earth. Sci. Adv. 7, eabj0108 (2021).
Farquhar, J., Bao, H. & Thiemens, M. Atmospheric influence of Earth’s earliest sulfur cycle. Science 289, 756–758 (2000).
Pavlov, A. & Kasting, J. Mass-independent fractionation of sulfur isotopes in Archean sediments: strong evidence for and anoxic Archean atmosphere. Astrobiology 2, 27–41 (2002).
Zahnle, K., Claire, M. & Catling, D. E. The loss of mass-independent fractionation in sulfur due to a Palaeoproterozoic collapse of atmospheric methane. Geobiology 4, 271–283 (2006).
Roscoe, S. M. Huronian Rocks and Uraniferous Conglomerates in the Canadian Shield, 68-40 (Geological Survey of Canada, 1969).
Chandler, F. W. Proterozoic Redbed Sequences of Canada, 311 (Geological Survey of Canada, 1980).
Johnson, J. E., Gerpheide, A., Lamb, M. P. & Fischer, W. W. O2 constraints from Paleoproterozoic detrital pyrite and uraninite. Geol. Soc. Am. Bull. 126, 813–830 (2014).
Karhu, J. A. & Holland, H. D. Carbon isotopes and the rise of atmospheric oxygen. Geology 24, 867–870 (1996).
Bekker, A. & Holland, H. D. Oxygen overshoot and recovery during the early Paleoproterozoic. Earth Planet. Sci. Lett. 317-318, 295–304 (2012).
Planavsky, N. J., Bekker, A., Hofmann, A., Owens, J. D. & Lyons, T. W. Sulfur record of rising and falling marine oxygen and sulfate levels during the Lomagundi event. Proc. Natl. Acad. Sci. USA 109, 18300–18305 (2012).
Guo, Q. et al. Reconstructing Earth’s surface oxidation across the Archean-Proterozoic transition. Geology 37, 399–402 (2009).
Luo, G. et al. Rapid oxygenation of Earth’s atmosphere 2.33 billion years ago. Sci. Adv. 2, e1600134 (2016).
Philippot, P. et al. Globally asynchronous sulphur isotope signals require re-definition of the Great Oxidation Event. Nat. Commun. 9, 2245 (2018).
Killingsworth, B. A. et al. Constraining the rise of oxygen with oxygen isotopes. Nat. Commun. 10, 4924 (2019).
Warke, M. R. et al. The Great Oxidation Event preceded a Paleoproterozoic “snowball Earth. Proc. Natl. Acad. Sci. Usa. 117, 13314–13320 (2020).
Poulton, S. W. et al. A 200-million-year delay in permanent atmospheric oxygenation. Nature 592, 232–236 (2021).
Izon, G. et al. Bulk and grain-scale minor sulfur isotope data reveal complexities in the dynamics of Earth’s oxygenation. Proc. Natl. Acad. Sci. USA 119, e2025606119 (2022).
Uveges, B. T., Izon, G., Ono, S., Beukes, N. J. & Summons, R. E. Reconciling discrepant minor sulfur isotope records of the Great Oxidation Event. Nat. Commun. 14, 279 (2023).
Gumsley, A. P. et al. Timing and tempo of the Great Oxidation Event. Proc. Natl. Acad. Sci. USA 114, 1811–1816 (2017).
Millikin, A. E. et al. A new Re-Os age constraint informs the dynamics of the Great Oxidation Event. Geology 52, 857–862 (2024).
Krogh, T. E., Davis, D. W., Corfu, F. & Pye, E. G. Precise U-Pb zircon and baddeleyite ages for the Sudbury area In The Geology and Ore Deposits of the Sudbury Structure (eds. Pye, E. G., Naldrett, A. J. & Giblin, P. E.) 431–446 (Ontario Geological Survey, 1984).
Ketchum, K. Y., Heaman, L. M., Bennett, G. & Hughes, D. J. Age, petrogenesis and tectonic setting of the Thessalon volcanic rocks, Huronian Supergroup, Canada. Precambrian Res. 233, 144–172 (2013).
Rasmussen, B., Bekker, A. & Fletcher, I. R. Correlation of Paleoproterozoic glaciations based on U–Pb zircon ages for tuff beds in the Transvaal and Huronian Supergroups. Earth Planet. Sci. Lett. 382, 173–180 (2013).
Rasmussen, B., Zi, J. W. & Bekker, A. New U-Pb zircon tuff ages and revised stratigraphic correlations in the Superior craton during the Great Oxidation Episode. Earth Planet. Sci. Lett. 640, 118779 (2024).
Papineau, D., Mojzsis, S. J. & Schmitt, A. K. Multiple sulfur isotopes from Paleoproterozoic Huronian interglacial sediments and the rise of atmospheric oxygen. Earth Planet. Sci. Lett. 255, 188–212 (2007).
Ulrich, T., Long, D. G. F., Kamber, B. S. & Whitehouse, M. J. In situ trace element and sulfur isotope analysis of pyrite in a Paleoproterozoic gold placer deposit, Pardo and Clement Townships. Ont., Can. Econ. Geol. 106, 667–686 (2011).
Cui, H. et al. Searching for the Great Oxidation Event in North America: a reappraisal of the Huronian Supergroup by SIMS sulfur four-isotope analysis. Astrobiology 18, 519–538 (2018).
Long, D. G. The stratigraphy and sedimentology of the Huronian (lower Aphebian) Mississagi and Serpent Formations. Ph.D. thesis, University of Western Ontario (1976).
Rainbird, R. H., Nesbitt, H. W. & Donaldson, J. A. Formation and diagenesis of a sub-Huronian saprolith: comparison with a modern weathering profile. J. Geol. 98, 801–822 (1990).
Shawwa, N. A. et al. Earth’s oldest terrestrial red beds as direct evidence for the Great Oxidation Event ca. 2.3 Ga. Precambrian Res. 409, 107423 (2024).
Goto, K. T. et al. Redox conditions in the atmosphere and shallow-marine environments during the first Huronian deglaciation: Insights from Os isotopes and redox-sensitive elements. Earth Planet. Sci. Lett. 376, 145–154 (2013).
Sekine, Y. et al. Osmium evidence for synchronicity between a rise in atmospheric oxygen and Palaeoproterozoic deglaciation. Nat. Commun. 2, 502 (2011).
Sekine, Y. et al. Manganese enrichment in the Gowganda Formation of the Huronian Supergroup: A highly oxidizing shallow-marine environment after the last Huronian glaciation. Earth Planet. Sci. Lett. 307, 201–210 (2011).
Young, G. M., Long, D. G. F., Fedo, C. M. & Nesbitt, H. W. Paleoproterozoic Huronian basin: product of a Wilson cycle punctuated by glaciations and a meteorite impact. Sediment. Geol. 141-142, 233–254 (2001).
Bennett, G. The Huronian Supergroup between Sault Ste Marie and Elliot Lake. In Field Trip Guidebook, 52 (Institute on Lake Superior Geology, 2006).
Corfu, F. & Andrews, A. J. A U-Pb age for mineralized Nipissing diabase, Gowganda, Ontario. Can. J. Earth Sci. 23, 107–109 (1986).
Fedo, C. M., Young, G. M., Nesbitt, H. W. & Hanchar, J. M. Potassic and sodic metasomatism in the southern province of the Canadian Shield: evidence from the Paleoproterozoic Serpent Formation, Huronian Supergroup, Canada. Precambrian Res. 84, 17–36 (1997).
McLennan, S. M., Simonetti, A. & Goldstein, S. L. Nd and Pb isotopic evidence for provenance and post-depositional alteration of the Paleoproterozoic Huronian Supergroup, Canada. Precambrian Res. 102, 263–278 (2000).
Young, G. M. Origin of carbonate-rich Early Proterozoic Espanola Formation, Ontario, Canada. Geol. Soc. Am. Bull. 84, 135–160 (1973).
Bernstein, L. & Young, G. M. Depositional environments of the early Proterozoic Espanola formation Ontario, Canada. Can. J. Earth Sci. 27, 539–551 (1990).
Fedo, C. M., Young, G. M. & Nesbitt, H. W. Paleoclimatic control on the composition of the Paleoproterozoic Serpent Formation, Huronian Supergroup, Canada: a greenhouse to icehouse transition. Precambrian Res. 86, 201–223 (1997).
England, G. L., Rasmussen, B., Krapez, B. & Groves, D. I. Palaeoenvironmental significance of rounded pyrite in siliciclastic sequences of the Late Archaean Witwatersrand Basin: oxygen-deficient atmosphere or hydrothermal alteration?. Sedimentology 49, 1133–1156 (2002).
Williford, K. H., Van Kranendonk, M. J., Ushikubo, T., Kozdon, R. & Valley, J. W. Constraining atmospheric oxygen and seawater sulfate concentrations during Paleoproterozoic glaciation: In situ sulfur three-isotope microanalysis of pyrite from the Turee Creek Group, Western Australia. Geochim. Cosmochim. Acta 75, 5686–5705 (2011).
Nesbitt, H. W. & Young, G. M. Early Proterozoic climates and plate motions inferred from major element chemistry of lutiles. Nature 299, 715–717 (1982).
Young, G. M. & Nesbitt, H. W. Paleoclimatology and provenance of the glaciogenic Gowganda Formation (Paleoproterozoic), Ontario, Canada: a chemostratigraphic approach. Geol. Soc. Am. Bull. 111, 264–274 (1999).
Rudnick R. L. & Gao S. Composition of the continental crust. In Treatise on Geochemistry, 3 (eds. Holland, H. D. & Turekian, K. K.) 1–64, (Elsevier, 2003).
Cole, D. B., Zhang, S. & Planavsky, N. J. A new estimate of detrital redox-sensitive metal concentrations and variability in fluxes to marine sediments. Geochim. Cosmochim. Acta 215, 337–353 (2017).
Greaney, A. T. et al. Molybdenum isotope fractionation in glacial diamictites tracks the onset of oxidative weathering of the continental crust. Earth Planet. Sci. Lett. 534, 116083 (2020).
Seal, R. R. Sulfur isotope geochemistry of sulfide minerals. Rev. Mineral. Geochem. 61, 633–677 (2006).
Ono, S., Wing, B., Johnson, D., Farquhar, J. & Rumble, D. Mass-dependent fractionation of quadruple stable sulfur isotope system as a new tracer of sulfur biogeochemical cycles. Geochim. Cosmochim. Acta 70, 2238–2252 (2006).
Kurzweil, F. et al. Atmospheric sulfur rearrangement 2.7 billion years ago: Evidence for oxygenic photosynthesis. Earth Planet. Sci. Lett. 366, 17–26 (2013).
Wang, Z. et al. Fe (hydro) oxide controls Mo isotope fractionation during the weathering of granite. Geochim. Cosmochim. Acta 226, 1–17 (2018).
Liu, J. H. et al. M. L. Molybdenum isotopic behavior during intense weathering of basalt on Hainan Island, South China. Geochim. Cosmochim. Acta 287, 180–204 (2020).
Kaufmann, A. K. C., Pettke, T. & Wille, M. Molybdenum isotope fractionation at upper-crustal magmatic-hydrothermal conditions. Chem. Geol. 578, 120319 (2021).
Greber, N. D., Hofmann, B. A., Voegelin, A. R., Villa, I. M. & Nägler, T. F. Mo isotope composition in Mo-rich high-and low-T hydrothermal systems from the Swiss Alps. Geochim. Cosmochim. Acta 75, 6600–6609 (2011).
Greber, N. D., Pettke, T. & Nägler, T. F. Magmatic–hydrothermal molybdenum isotope fractionation and its relevance to the igneous crustal signature. Lithos 190, 104–110 (2014).
Baird, G. C. & Brett, C. E. Erosion on an anaerobic seafloor: significance of reworked pyrite deposits from the Devonian of New York State. Palaeogeogr. Palaeoclimatol. Palaeoecol. 57, 157–193 (1986).
Konhauser, K. O. et al. Aerobic bacterial pyrite oxidation and acid rock drainage during the Great Oxidation Event. Nature 478, 369–373 (2011).
Gaschnig, R. M. et al. Onset of oxidative weathering of continents recorded in the geochemistry of ancient glacial diamictites. Earth Planet. Sci. Lett. 408, 87–99 (2014).
Palmer, M. R. & Edmond, J. M. Uranium in river water. Geochim. Cosmochim. Acta 57, 4947–4955 (1993).
Shiller, A. M. & Mao, L. Dissolved vanadium in rivers: effects of silicate weathering. Chem. Geol. 165, 13–22 (2000).
Craddock, J. P. et al. B. Detrital zircon geochronology and provenance of the Paleoproterozoic Huron (∼ 2.4–2.2 Ga) and Animikie (∼ 2.2–1.8 Ga) basins, southern Superior Province. J. Geol. 121, 623–644 (2013).
Wogan, N. F., Catling, D. C., Zahnle, K. J. & Claire, M. W. Rapid timescale for an oxic transition during the Great Oxidation Event and the instability of low atmospheric O2. Proc. Natl. Acad. Sci. USA 119, e2205618119 (2022).
Calmels, D., Gaillardet, J., Brenot, A. & France-Lanord, C. Sustained sulfide oxidation by physical erosion processes in the Mackenzie River basin: climatic perspectives. Geology 35, 1003–1006 (2007).
Torres, M. A., West, A. J. & Li, G. Sulphide oxidation and carbonate dissolution as a source of CO2 over geological timescales. Nature 507, 346–349 (2014).
Hsieh, Y. P. & Shieh, Y. N. Analysis of reduced inorganic sulfur by diffusion methods: improved apparatus and evaluation for sulfur isotopic studies. Chem. Geol. 137, 255–261 (1997).
Jochum, K. P., Willbold, M., Raczek, I., Stoll, B. & Herwig, K. Chemical characterisation of the USGS reference glasses GSA-1G, GSC-1G, GSD-1G, GSE-1G, BCR-2G, BHVO-2G and BIR-1G Using EPMA, ID-TIMS, ID-ICP-MS and LA-ICP-MS. Geostand. Geoanal. Res. 29, 285–302 (2005).
McLennan, S. M. Weathering and global denudation. J. Geol. 101, 295–303 (1993).
Goto, K. T. et al. Progressive ocean oxygenation at ~2.2 Ga inferred from geochemistry and molybdenum isotopes of the Nsuta Mn deposit. Ghana. Chem. Geol. 567, 120116 (2021).
Romaniello, S. J., Herrmann, A. D. & Anbar, A. D. Syndepositional diagenetic control of molybdenum isotope variations in carbonate sediments from the Bahamas. Chem. Geol. 438, 84–90 (2016).
Maruoka, T., Koeberl, C., Hancox, P. J. & Reimold, W. U. Sulfur geochemistry across a terrestrial Permian–Triassic boundary section in the Karoo Basin, South Africa. Earth Planet. Sci. Lett. 206, 101–117 (2003).
Mayer, B. & Krouse, H. R. Procedures for sulfur isotope abundance studies. In Handbook of Stable Isotope Analytical Techniques, 1 (ed. de Groot, P. A.) 538–596 (Elsevier, 2004).
Ohkouchi, N. et al. Biogeochemical processes in the saline meromictic Lake Kaiike, Japan: implications from molecular isotopic evidences of photosynthetic pigments. Environ. Microbiol. 7, 1009–1016 (2005).
Ogawa, N. O., Nagata, T., Kitazato, H. & Ohkouchi, N. Ultra sensitive elemental analyzer/isotope ratio mass spectrometer for stable nitrogen and carbon isotopic analyses. In Earth, Life and Isotopes (eds. Ohkouchi, N., Tayasu, I. & Koba, K.) 339–353 (Kyoto Univ. Press, 2010).
Sekine, Y. et al. Anomalous negative excursion of carbon isotope in organic carbon after the last Paleoproterozoic glaciation in North America. Geochem. Geophys. Geosyst. 11, Q08019 (2010).
Aoyama, S., Nishizawa, M., Takai, K. & Ueno, Y. Microbial sulfate reduction within the Iheya North subseafloor hydrothermal system constrained by quadruple sulfur isotopes. Earth Planet. Sci. Lett. 398, 113–126 (2014).
Canfield, D. E., Raiswell, R., Westrich, J. T., Reaves, C. M. & Berner, R. A. The use of chromium reduction in the analysis of reduced inorganic sulfur in sediments and shales. Chem. Geol. 54, 149–155 (1986).
Nakagawa, M. et al. Seasonal change in microbial sulfur cycling in monomictic Lake Fukami-ike, Japan. Limnol. Oceanogr. 57, 974–988 (2012).
Ding, T. et al. Calibrated sulfur isotope abundance ratios of three IAEA sulfur isotope reference materials and V-CDT with a reassessment of the atomic weight of sulfur. Geochim. Cosmochim. Acta 65, 2433–2437 (2001).
Ono, S., Shanks, W. C., Rouxel, O. J. & Rumble, D. S-33 constraints on the seawater sulfate contribution in modern seafloor hydrothermal vent sulfides. Geochim. Cosmochim. Acta 71, 1170–1182 (2007).
Nelson, D. R., Trendall, A. F. & Altermann, W. Chronological correlations between the Pilbara and Kaapvaal cratons. Precambrian Res. 97, 165–189 (1999).
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 19K14832, 21K03733, 23K13209 and 23H00136 and the Environmental Radioactivity Research Network Center through the joint research project P-24-17. S.W.P. acknowledges financial support from the World Research Hub (WRH) Program of the International Research Frontiers Initiative, Tokyo Institute of Technology. The samples used in the present study were stored at the Ontario Geological Survey.
Author information
Authors and Affiliations
Contributions
Y.S., S.T. and E.T. designed the study. Y.S., S.T., R.T., K.G., S.Y. and E.T. collected the samples. K.T.G., Y.S., U.N., K.S., R.S., N.Ya., Y.U., S.T., N.Yo., M.H., T.M., N.O.O., N.O., Y.H., Y.K. and G.S. performed geochemical analyses. K.T.G., Y.S. and S.W.P wrote the manuscript with input from all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Somaparna Ghosh. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Goto, K.T., Sekine, Y., Nakamura, U. et al. Incomplete oxidative sulfide weathering and low atmospheric oxygen levels during the Great Oxidation Event. Commun Earth Environ 6, 906 (2025). https://doi.org/10.1038/s43247-025-02841-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43247-025-02841-w
