
Abstract
Aging is associated with progressive tissue dysfunction, leading to frailty and mortality. Characterizing aging features, such as changes in gene expression and dynamics, shared across tissues or specific to each tissue, is crucial for understanding systemic and local factors contributing to the aging process. We performed RNA sequencing on 13 tissues at six different ages in male and female African turquoise killifish, the shortest-lived vertebrate that can be raised in captivity. This comprehensive, sex-balanced ‘atlas’ dataset revealed varying strength of sex–age interactions across killifish tissues and age-altered genes and biological pathways that are evolutionarily conserved in mice and humans. We discovered a female-biased myeloid shift with age in the killifish hematopoietic organ, developed tissue-specific ‘transcriptomic clocks’ and identified biomarkers predictive of chronological age. We showed the importance of sex-specific clocks for selected tissues, validated the tissue clocks with an independent transcriptomic dataset and used them to evaluate different lifespan interventions in the killifish. Our work provides a comprehensive resource for studying aging dynamics across tissues in the killifish, a powerful vertebrate aging model.
Similar content being viewed by others
Main
Aging is the greatest risk factor for disease and death in humans. It is a highly complex process, characterized by progressive cellular and tissue dysfunction. Such dysfunction is accompanied by shared molecular features, referred to as ‘hallmarks of aging’1, such as chronic inflammation, loss of proteostasis and dysregulated nutrient sensing. Recent work in mice and humans suggests that these aging hallmarks can differ between males and females in specific tissues2,3,4,5,6,7,8. Moreover, the amplitude and the onset age of these hallmarks can also differ among the tissues of an organism9. Currently, the extent of sex dimorphism in tissue aging, including age-altered gene pathways and aging trajectories, is not well understood. Understanding the age–sex relationship across diverse tissues will augment our understanding of sex-specific interventions to slow and even reverse aging.
We used the African turquoise killifish (Nothobranchius furzeri) as a naturally accelerated vertebrate aging model. The killifish has emerged as a new vertebrate model in aging research because it has conserved aging signatures and a short lifespan, which are attractive features for rapid lifespan and healthspan intervention testing10,11,12,13,14,15,16,17,18,19. The median lifespan of the killifish is 4–6 months (about a fifth of the mouse lifespan and a seventh of the zebrafish lifespan), with vertebrate-specific genes, tissues and systems conserved with humans10,11,12,13,14,15,16,17,18,19. Several conserved aging mechanisms and interventions have been reported in this model, such as mutants of the nutrient-sensing pathway20,21 and the germline22, dietary modifications20,21,23,24, microbiome transfer25 and administration of small-molecule treatments26,27,28,29. Many of these interventions have sex-specific effects on killifish lifespan, suggesting interesting age–sex relationships in killifish that can provide critical insights into the role of biological sex in regulating vertebrate aging.
Transcriptomic analysis (for example, RNA sequencing (RNA-seq) or single-cell RNA-seq) has been applied in the killifish to understand the aging signatures of tissues or cell types and the effects of aging interventions20,21,22,23,28,30,31,32,33,34,35,36,37,38,39. These studies have identified the crucial gene pathways and biological processes altered by tissue aging, such as elevated inflammation20,22,30,34,36,40 and loss of proteostasis30,33,41. However, publications in killifish have mostly focused on a single tissue or sex and sampled only a few time points (2–3 time points), which limits the ability to study gene dynamics across time and tissues. Because direct comparison across multiple tissues is lacking, it remains unknown how similarly tissue transcriptomes change with age, how biological sex affects the aging pathways in each tissue and across tissues, and which tissues or pathways have an early onset of gene expression changes or distinct dynamics with age. A broad characterization of killifish tissue aging will be a valuable resource to pinpoint the specific aspects of vertebrate aging that can be modeled in killifish and are suitable for intervention testing. Such characterization should also allow the development of machine-learning models (‘aging clocks’) for rapid evaluation of intervention efficacy.
In this study, we comprehensively profiled the aging transcriptomes of 13 tissues across six time points for male and female killifish. To our knowledge, this 677-sample dataset is the most comprehensive, high-quality tissue aging atlas of the killifish to date. We identified distinct age–sex relationships for each tissue, the age-correlated genes and pathways shared across multiple tissues and conserved in mice and humans, and the tissue-specific genes that may contribute to a female-biased myeloid shift with age in the head kidney, a main hematopoietic compartment of the killifish. Lastly, we developed tissue-specific aging clocks that allow us to evaluate published lifespan interventions and to uncover the importance of incorporating sex-specific features in building age-prediction models.
Results
A large-scale aging atlas spans diverse tissues, multiple ages and balanced sex representation
To understand how different tissues age in the killifish, we constructed a multi-tissue transcriptomic aging atlas consisting of 677 samples collected from two independent aging cohorts of killifish (Fig. 1a). We developed a protocol for cardiac perfusion and performed this procedure on these killifish to limit the impact of circulating immune cells on the tissue transcriptome signature, thus allowing discovery of age-dependent changes in tissue-resident cell types. Thirteen tissues (bone, brain, retina/retinal pigment epithelium (RPE), fat, gut, ovaries/testes, heart, head kidney, liver, muscle, skin, spinal cord and spleen) were analyzed across six age groups spanning a population survival of 100% to ~20%, including juveniles (47–52 days), young adults (75–78 days), middle-aged adults in two time points (102–103 days and 133–134 days), old adults (147–155 days) and geriatric animals (161–162 days; Extended Data Fig. 1a). Both males and females were sampled at a similar frequency for most tissues, and the body length of each fish was also recorded (Extended Data Fig. 1b,c). This sex-balanced feature allowed us to study the effect of biological sex during killifish aging. Using a high-sensitivity, high-throughput library preparation pipeline based on Smart-seq2 (ref. 42), we generated a high-quality dataset, with over 94% of samples sequenced to >30 million paired-end reads and over 80% of samples having over 70% of reads uniquely mapped to the killifish genome. Principal component analysis (PCA) also showed sample clustering by tissue type (Fig. 1b). Per-tissue variance partitioning and PCA showed minimal contribution of cohort and RNA extraction batch (<1%; Extended Data Fig. 1e,f, and see Supplementary Table 3 for full data), confirming the quality of our dataset.

a, Schematic for the killifish transcriptomic aging atlas. Thirteen tissues from males and females were collected for RNA-seq at the indicated time points (the animal numbers sampled are listed as ‘n’) from two independent cohorts of the GRZ killifish strain. Testes and ovaries are the male and female gonads, respectively. b, PCA for the 677 samples reveals clear clustering by tissue identity. Symbol shape indicates biological sex (F, female; M, male). Symbol color indicates tissue type. c, Tissues have varying numbers of age-correlated genes, as shown by the Spearman’s rank correlation (ρ) distribution for all the post-filtered genes in each tissue. Each dot is one gene. Tissues are ordered by the total number of age-correlated genes, in descending order from the top (the total number of age-correlated genes for each tissue are as follows: retina/RPE, 2,222 genes; muscle, 2,112; skin, 1,766; spinal cord, 1,688; brain, 1,451; fat, 1,241; heart, 873; kidney, 516; spleen, 505; gut, 392; liver, 319; gonad, 210; and bone, 198). Male and female samples are analyzed together for each tissue and time point in c to e. Box plots show the median and 25th (Q1) and 75th (Q3) percentiles; whiskers extend to Q1 − 1.5 × (Q3 − Q1) and Q3 + 1.5 × (Q3 − Q1). d, Each tissue has a distinct proportion of age-correlated genes in its transcriptome. Upregulated with age, Spearman’s rank correlation ρ > 0.5. Downregulated with age, ρ < −0.5. e, Proportion of DEGs between males and females (sex-dimorphic genes) for each tissue, at each binned age level. A break in the y axis is denoted by double slashed lines. The ‘agebins’ (x axis) correspond to the time points in a. f, Male versus female GSEA identifying pathways significantly enriched among genes upregulated or downregulated with age in each tissue. GSEA was performed using a permutation-based enrichment test implemented in clusterProfiler, using GO annotations. Enrichment is reported as the normalized enrichment score (NES). Statistical significance was assessed using permutation-derived P values and adjusted for multiple hypothesis testing using the Benjamini–Hochberg method; pathways with false discovery rate (FDR) < 0.05 were considered significant. Dot size represents −log10(FDR). g, Heat map of select GO terms, plotting the male and female Spearman’s rank correlations of the genes that drive each GO term.
To gain insights from the comprehensive dataset (‘killifish aging atlas’), we followed a systematic approach. First, we quantified the global variance associated with age, sex and age–sex interaction for each tissue. Next, we identified the age-related gene expression changes that were either sex-specific or shared across tissues and analyzed these genes’ evolutionary conservation in mouse and humans. We then examined gene expression dynamics through trajectory analysis. Finally, we constructed sex-specific and sex-combined transcriptomic clocks based on the atlas data and used them to predict the efficacy of lifespan interventions.
The impact of aging on transcriptomes varies among different tissues
To characterize the gene expression trends across age in each tissue, we leveraged the time-series nature of our dataset and used Spearman’s rank correlation to describe the monotonicity of a gene’s expression change with age (that is, expression consistently increasing or decreasing). Tissues such as muscle, skin and the retina/RPE had genes with the strongest age association, with genes achieving a Spearman’s rank correlation ρ > 0.8 (upregulated with age) or ρ < −0.8 (downregulated with age; Fig. 1c). Next, we defined age-correlated genes to have an absolute Spearman’s rank correlation greater than 0.5. We observed that among all the tissues, the muscle had the highest proportion (14.38%) of age-correlated genes in its transcriptome (Fig. 1d). Other tissues (retina/RPE, skin, spinal cord, fat, brain and heart) had an intermediate level of age-correlated genes at around 6–13%. Among the tissues with a low proportion (~2.5%) were spleen, head kidney, liver, gut, gonad and bone (note that there were technical challenges in extracting RNA from bone samples, which may affect the identification of age-correlated genes; Methods). These tissue-level differences were also observed using variance partition analysis (Methods and Extended Data Fig. 1d; ‘Age’), highlighting the varying degree to which aging affects the transcriptomes of different tissues.
Age-altered pathways are mostly shared between sexes, but sex-divergent ones exist
Tissue-specific changes with age can stem from the distinctive physiology and functions of each tissue, pointing to the unique aging mechanisms in specific tissue contexts and revealing potential nodes for targeted intervention against aging in each tissue. The tissue context can depend on the biological sex of the animal from which the tissue is derived, given that the different tissue transcriptomes had varying proportions of genes differing in expression between males and females (Fig. 1e). For example, the gonads had on average ~95% of genes differentially expressed by sex across all age groups (this high degree of sex dimorphism is expected), liver had ~25%, skin had ~15%, head kidney had ~14%, and fat had ~6% with a peak at 147–155 days of life. Consistently, variance partition analysis showed that sex accounted for a noticeable fraction of transcriptional variance in the gonad (68.70%), skin (3.55%), fat (3.49%) and head kidney (1.51%; Extended Data Fig. 1d; ‘Sex’), and the age–sex interaction (that is, genes changed with age differently in males versus females) accounted for a high fraction of variance in the liver (19.97%; Extended Data Fig. 1d; ‘Sex:age’). Prominent sex effects on tissue transcriptomes have also been observed in similar tissues in mice (for example, gonadal adipose tissue, subcutaneous adipose tissue, liver and kidney)9 and in humans (for example, visceral and subcutaneous adipose tissue, and skin)43.
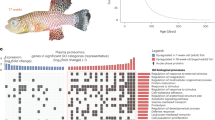
To juxtapose male versus female differences in the aging transcriptome of each tissue, we separated our datasets by tissue and sex and then calculated the Spearman’s rank correlation for each gene, followed by gene-set enrichment analysis (GSEA) or hypergeometric Gene Ontology (GO) enrichment analysis to identify the pathways altered by age for each tissue and each sex (‘sex-split’ analysis). Generally, in a given tissue type, we found that the significantly enriched GO terms changed with age in the same direction (either upregulated or downregulated) for both sexes, regardless of how sexually dimorphic the tissue transcriptome was (for example, see terms for the brain, a weakly sex-dimorphic organ, and the liver, a strongly sex-dimorphic organ; Fig. 1f and Extended Data Figs. 2 and 3a,b). The genes underlying these pathways were mostly similar between males and females, although there were differences (for example, the genes driving the ‘mitotic sister chromatid segregation’ term in the liver were somewhat different between the sexes; Fig. 1g), suggesting that aging alters many pathways similarly in male and female tissues, although the exact genes altered by age can be distinct.
Interestingly, there were also GO terms showing opposite signs of upregulation or downregulation with age in the two sexes, and often the change with age was statistically significant in only one sex (‘sex-divergent’) (Extended Data Fig. 4 and Supplementary Figs. 4 and 5). By GSEA, depending on the tissue type, the sex-divergent GO terms were upregulated with age in either male or female. These GO terms were related to proteostasis in the gut (for example, ‘protein quality control for misfolded or incompletely synthesized proteins,’ ‘response to unfolded protein’); intercellular and intracellular transport in the heart and spleen (for example, ‘peptide hormone secretion,’ ‘amino acid transport’ and ‘potassium ion transport’); and the ribosome in the spinal cord (for example, ‘ribosome biogenesis’ and ‘rRNA processing’). Two of the sex-divergent GO terms, autophagy (for example, ‘autophagosome’ and ‘lysosomal membrane’) and myeloid cell regulation (for example, ‘neutrophil activation’ and ‘granulocyte activation’), were present in various tissues such as fat, retina/RPE, gonad, head kidney, spinal cord and spleen. These results indicate that while aging can alter similar pathways in male and female tissues, the direction of these changes can diverge by sex, reflecting the distinct ways in which males and females age at the transcriptome level.
Some age-altered pathways are unique to each tissue
Several pathways were altered with age in only one or a few tissues. For example, GSEA showed that in the muscle, some age-downregulated terms were related to angiogenesis (for example, ‘blood vessel development’) and ossification (Fig. 1f,g; Muscle). In the gut, metabolism-related pathways were altered with age, such as ‘regulation of gluconeogenesis’ (Fig. 1f,g; Gut). Even though most GO terms were consistently upregulated or downregulated with age across tissues (Extended Data Fig. 2 and Extended Data Fig. 3a,b), there were also pathways with strong tissue-dependent changes with age by GSEA (and somewhat by hypergeometric GO analysis). For example, for both sexes by GSEA, ribosome-related terms (for example, ‘ribosome’ and ‘rRNA processing’) were upregulated with age in skin and the brain, but downregulated with age in spleen, fat and the retina/RPE. In females, the terms related to the extracellular matrix (ECM; for example, ‘extracellular structure organization’ and ‘ECM organization’) were upregulated in the liver, fat, retina/RPE and ovary, but downregulated in skin, muscle and bone. How ribosome-related and extracellular matrix-related processes are modulated by aging may be tuned to the different demands of ribosome activity and extracellular organization and function in different tissues.
Immune and ECM genes change with age across multiple tissues
What pathways are commonly altered with age across multiple tissues? The shared changes could indicate systemic factors that regulate aging or shared cross-tissue consequences of the aging process. We identified several pathways that were commonly altered with age in at least six tissues (Extended Data Fig. 2). For both sexes, these pathways included upregulation of ‘immune response’ and downregulation of cell cycle (for example, ‘DNA replication’) and mitochondria terms (for example, ‘mitochondrial matrix’ and ‘mitochondrial gene expression’). Specifically for males, ECM-related terms (for example, ‘ECM organization’ and ‘extracellular structure organization’) were shared across tissues (Extended Data Fig. 2). These pathways have been reported to be changed with age in a subset of killifish tissues previously20,21,30,33,34,36,40 and are reminiscent of key hallmarks of aging, including upregulation of ‘chronic inflammaging’ and ‘cellular senescence’ and altered ‘mitochondrial functions’ and ‘intercellular communication’1.
Complementarily, we analyzed male and female samples together and identified 47 age-correlated genes shared across at least six tissues, including 22 upregulated with age (Spearman’s rank correlation ρ > 0.5) and 25 downregulated genes (ρ < −0.5; Fig. 2a). Of these 47 genes, 14 of the upregulated and 25 of the downregulated genes with age have human orthologs (annotated with human gene names using capital letters in Fig. 2a). Consistently, the pathways enriched for the cross-tissue age-correlated genes included immune response (upregulated) and ECM organization (downregulated) terms (Fig. 2b).

a, Spearman’s rank correlation (ρ) heat maps for the genes upregulated (left) or downregulated (right) with age shared across at least six tissues. Gray box indicates that Spearman’s correlation was not calculated because the expression level of a particular gene was lower than the expression threshold (transcripts per million (TPM) > 0.5 in over 80% of samples). Killifish-specific genes are named after their gene locus numbers (for example, LOC107378024) or are designated in lowercase letters. When a killifish gene has a human ortholog, the human gene name is used and shown in uppercase letters. b, Hypergeometric GO enrichment results for the genes upregulated (top) or downregulated (bottom) with age that are shared across at least five tissues. Enrichment significance was assessed using a hypergeometric test implemented in GOstats, with the background (‘universe’) defined as all genes with non-NA (not available) FDR-adjusted P values for the corresponding comparison. P values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method, and GO terms with FDR < 0.05 were considered significant. Dot size represents −log10 of the FDR-adjusted P value (that is, FDR after multiple hypothesis testing). c, Representative maximum z-projected hybridization chain reaction (HCR; RNA in situ) images for ncRNA-3777 mRNAs in male and female guts, at young (57–60 days) and old (120–130 days) ages. Scale bar, 5 µm. d, Quantification of HCR images as the average number of ncRNA-3777 transcripts per cell. Here and after, one individual fish represented an independent biological replicate. Each dot is a fish; n = 4 fish per condition. In-graph statistical comparisons were performed using a two-sided Mann–Whitney U-test between the indicated groups. Below-graph statistics were assessed using a two-way analysis of variance (ANOVA) including age, sex and the age–sex interaction as factors. e, Normalized RNA-seq expression values (DESeq2-normalized counts) in the gut for the ncRNA-3777 gene are shown for individual animals across age bins and sex. Dots represent expression values from individual fish. Bars indicate the mean expression for each sex and age bin, and error bars denote ± s.e.m., calculated across biological replicates within each group. Age bins correspond to defined ranges of days after hatching and are color coded as indicated. f, Representative maximum z-projected HCR (RNA in situ) images for IGF2BP3 with conditions as described in c. g,h, Quantification and statistics were performed as in d and e, respectively, for IGF2BP3. i, z-scaled locally estimated scatterplot smoothing (LOESS) regression fits of the gene expression trajectories across age for the genes ncRNA-3777 and IGF2BP3. j, Age-associated gene expression change (log2-fold change) in mouse and killifish brain for the genes in the ‘Common Aging Score’ gene set, which is a published brain-wide gene signature of aging defined in Hahn et al.57. Differential expression was performed using DESeq2, comparing 133–134 days and 47–52 days in killifish (corresponding to ~30% and ~100% colony survival, respectively) or between 27 months and 3 months in mice (corresponding to 50–75% and 100% colony survival, respectively). Statistical significance was assessed using the two-sided Wald test implemented in DESeq2, and P values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method. The 13 of 82 mouse genes with significant age-associated change in the killifish are shown (FDR-adjusted P value < 0.05). Green denotes a mouse; pink denotes a killifish. The mouse data are from Schaum et al.9.
We used RNA in situ hybridization to validate the age-altered expression of two of the top shared age-correlated genes in the gut, the tissue with the highest absolute Spearman’s rank correlation for these genes. We found that the transcript of the killifish gene LOC107373777 (hereafter referred to as ncRNA-3777; new locus name LOC129165246 in the 2024 NfuGRZ-RIMD1 reference genome) was mostly localized to the nucleus, and its level increased with age (Fig. 2c–e,i). This gene was annotated in the reference genome to encode a long noncoding RNA (lncRNA) of unknown function. We used the noncoding RNA sequence database RNAcentral44 to identify 83 RNA sequences in humans, 8 sequences in mice and 4 sequences in zebrafish that shared similarity to ncRNA-3777 (Extended Data Fig. 5a). Almost all these sequences were predicted to be lncRNAs, and most had unknown functions. One of the interesting sequences was the human lncRNA (URS00025BE4AF_9606), which shared the highest query coverage (37.19%) and sequence identity (43.46%) with the killifish ncRNA-3777. This human lncRNA is part of the PVT1 noncoding RNA locus, which has been identified as a candidate oncogene in several tumor types (for example, breast45,46,47, ovarian48,49,50,51 and non-small cell lung cancers52), possibly indicating a physiological role for ncRNA-3777 in the killifish.
In contrast to ncRNA-3777, the transcript of the IGF2BP3 gene (killifish gene name: LOC107383282) was both nuclear and cytoplasmic, and its level decreased with age (Fig. 2f–i). The human ortholog of the IGF2BP3 gene encodes an RNA-binding protein that promotes insulin growth factor 2 (IGF2) protein translation53. The amino acid sequence (581 residues) of the killifish IGF2BP3 protein was 75.91% identical to the human protein by alignment. AlphaFold54,55,56 predicted the killifish IGF2BP3 protein to have six protein domains with a relatively high average predicted local distance difference test of 75. The predicted structure largely overlapped with the human ortholog protein (Extended Data Fig. 5b). Therefore, the killifish IGF2BP3 gene encodes a protein highly conserved in mammals, and its downregulation with age may suggest dampening of IGF2-dependent signaling with age in the killifish.
Analysis of age-associated gene pathways reveals conservation across vertebrates
To evaluate conservation at the gene and pathway levels, we first compared the age-associated differentially expressed genes (DEGs) in killifish tissues to those in mouse and human counterparts. Skeletal muscle, skin and adipose tissues showed many shared upregulated and downregulated aging DEGs between killifish and both mammals (Extended Data Fig. 5c), and most tissues had at least a few statistically significant DEGs sharing the same direction of change with age between killifish and either mouse or human (albeit with differences in the magnitude of change). Next, we compared the killifish brain age-DEGs to the mouse brain ‘common aging score’, which consists of 82 genes that define a brain-wide mouse brain aging signature57. We found that the killifish brain differentially expressed 13 common aging score genes with age (Fig. 2j), and 11 of these genes changed in the same direction as in mice, including genes related to the complement cascade (for example, B2m, C1qb, C1qc and C4b), microglial activation (for example, C1qb, C1qc, C4b, Ctss, Ctsz, Pld4 and Plek) and lysosomal pathways (for example, Ctss and Ctsz). These genes exemplified the conserved transcriptomic features of the vertebrate aging brain.
Lastly, we assessed conservation at the pathway level by GSEA. We found that heart, visceral adipose tissue, brain and the immune system (for example, killifish kidney versus human blood) displayed the largest overlap of age-altered pathways between killifish and humans (Extended Data Fig. 6a). In comparison, between killifish and mice, adipose tissue (subcutaneous and mesenteric), heart, and the liver exhibited the largest pathway-level overlap (Extended Data Fig. 6b). Specific age-related transcriptional programs are broadly conserved across the three vertebrate species, including immune regulation and metabolic remodeling (Extended Data Fig. 6c). For example, immune-related pathways were upregulated with age (for example, ‘B cell activation’ in adipose tissue, ‘regulation of lymphocyte proliferation’ in the heart, and ‘T cell activation’ and ‘T cell migration’ in the brain). Specific metabolic pathways were downregulated with age (for example, ‘lipid oxidation’ and ‘regulation of lipid localization’ in adipose tissue and ‘aerobic respiration’ in heart). The convergence of these aging-associated molecular signatures across fish, rodents and humans underscored the evolutionarily conserved features exhibited by vertebrate aging.
Trajectory analysis reveals different classes of gene expression behaviors
While uncovering monotonic changes with age is informative, Spearman’s rank correlation cannot distinguish linear from nonlinear changes, or genes with stable age trajectories from those with complex dynamics (for example, U-shape). Previous studies revealed that age-related gene expression changes can be non-monotonic9,28. To explore these age-related dynamics, we performed hierarchical clustering of gene expression trajectories in each tissue, dividing the genes into ten clusters, which was the optimal cluster number determined by balancing the cluster robustness (based on silhouette score) with the gene-set size for functional enrichment analysis (Supplementary Fig. 6 and Methods). We observed that the expression trajectory clusters had unique dynamics. For example, in the brain, while clusters 1, 2 and 3 all declined with age, their trajectories had distinct shapes (Fig. 3a). Cluster 1 showed a logarithmic pattern, decreasing at early age then flattening in the remaining ages. This cluster was mainly enriched in cell cycle (for example, ‘mitotic cell cycle’ and ‘cell cycle’) and nervous system development (Fig. 3b; cluster 1) terms. Cluster 2 followed a linear pattern and was enriched in pathways related to nervous system development (for example, ‘neuron projection guidance’ and ‘neuron differentiation’; Fig. 3b; cluster 2). Lastly, cluster 3 showed a complex behavior of declining at early age, remaining flat at middle age and then declining further at old age. This cluster was enriched in mRNA regulation terms (for example, ‘mRNA processing’ and ‘mRNA splicing via spliceosome’; Fig. 3b; cluster 3). The distinct expression dynamics of these pathways may indicate different regulatory networks or the underlying reasons for the decline with age. For instance, the cluster 1 (cell cycle) pattern in the brain may result from the cessation of the killifish’s rapid growth from adolescence to adulthood (Extended Data Fig. 1c). Consistently, other tissues had clusters with a similar logarithmic shape (an inflection point at ~80 days) and were enriched in cell cycle pathways (for example, cluster 8 in gut and cluster 7 in muscle; Extended Data Fig. 7). Given that the neurogenesis terms were present in both cluster 1 and cluster 2 in the brain, it may suggest some processes related to the reduced neurogenesis as killifish age are decoupled from reduced cell division in middle-aged and old brains. Lastly, the cluster 3 (mRNA regulation) pattern may reflect distinct regulatory inputs between the two phases of decline or regulation to sustain expression at middle age. Therefore, by studying gene dynamics, we can gain insights into which biological processes may be co-regulated (or not) during aging.

a, Hierarchical clustering of the gene expression trajectories for the brain. Hierarchical clustering was performed on the LOESS regression aging trajectory of the gene expression in the brain for the 10,847 genes expressed in all tissues, resulting in ten clusters of gene expression behavior over time. Clustering and trajectory fitting were descriptive and did not involve statistical hypothesis testing. Each line represents the trajectory of an individual gene, and the average trajectory for the cluster is depicted by the black line. The most significant GO term from hypergeometric GO enrichment (lowest FDR-adjusted P value among terms related to biological processes) for each cluster is listed. The number of genes in each cluster is indicated by ‘n’. b, Hypergeometric GO enrichment (terms related to biological processes) for the genes in each cluster. Enrichment significance was assessed using a hypergeometric test implemented in GOstats, with the background (‘universe’) defined as all genes with non-NA FDR-adjusted P values used in the clustering analysis. P values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method. Select GO terms significantly enriched (FDR-adjusted P value < 0.05) for each cluster are plotted. The dot color represents the enrichment score of each GO term, with the maximum value of the scale adjusted to 20 to enhance the color resolution of GO terms with lower enrichment. Dot size indicates –log10 of the FDR-adjusted P value (that is, FDR after multiple hypothesis testing). Cluster 10 does not have any significant GO terms, so the lowest P value terms are plotted.
Cell-type composition changes with age in the killifish kidney marrow
Given the strong systemic immune signatures, we sought to better understand how the primary hematopoietic compartment, the head kidney, of the killifish changes with age. As in other teleost fish, killifish kidneys consist of two parts. The head kidney is the anterior portion of the kidney, composed of two bilateral lobes containing hematopoietic tissue, which we sampled in our atlas58. The trunk kidney is located posteriorly along the dorsal body wall and mainly contains exocrine tissue58. PCA analysis of the head kidney transcriptomic samples showed strong separation by age along principal component 1 and by sex along principal component 2 (Fig. 4a). We identified 516 genes with absolute Spearman’s rank correlation values of greater than 0.5 in the kidney samples. Several genes primarily expressed in T cells, B cells and lymphoid progenitors were negatively correlated with age (ρ < 0.5), while those primarily expressed in macrophages, neutrophils and other myeloid cells were positively correlated with age (ρ > 0.5; Fig. 4b and Extended Data Fig. 9a)59. These differences were stronger in female head kidneys than in male head kidneys (Fig. 4b), with higher absolute Spearman’s rank correlations and greater statistical significance. At a pathway level, ‘B cell receptor signaling pathway’ and ‘DNA recombination’ terms were downregulated with age (Fig. 4c). These observations are reminiscent of the ‘myeloid bias’ phenomenon in mice and zebrafish, where the cell-type composition of the hematopoietic lineage changes with age, with an increase in the ratio of myeloid lineage cells to lymphoid cells in old age60,61,62,63.

a, Principal component (PC) analysis of all head kidney transcriptomes coded by age (in days) and sex. b, Dot plot of the select cell-type marker genes for lymphoid and myeloid lineage cells. Either the zebrafish ortholog (lowercase) or the human ortholog (uppercase) is written before the ‘/’ symbol, and the killifish gene name is written after the ‘/’ symbol. Spearman’s rank correlation coefficients (ρ) were calculated separately for each sex across biological replicate samples to assess age-associated expression changes for each gene. Statistical significance was assessed using a two-sided Spearman’s rank correlation test, and P values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method. Dot size represents the –log10 of the FDR-adjusted P value, and dot color corresponds to the Spearman’s rank correlation ρ value calculated separately for each sex. Each dot represents one gene. The cell-type specificity of each gene’s expression was based on a published killifish kidney single-cell RNA-seq dataset59 (Extended Data Fig. 9). c, Hypergeometric GO enrichment analysis (biological process terms) for genes upregulated (right) or downregulated (left) with age in the head kidney when both sexes were analyzed together. Enrichment significance was assessed using a hypergeometric test implemented in GOstats, with the background (‘universe’) defined as all genes with non-NA FDR-adjusted P values included in the analysis. P values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method, and GO terms with FDR-adjusted P value < 0.05 were considered significant. Dot color represents the enrichment score of each GO term. Dot size indicates –log10 of the FDR-adjusted P value (that is, FDR after multiple hypothesis testing). d, Schematic of the flow cytometry assay to quantify different immune cell lineages in the killifish. Dissected head kidney tissue was dissociated into a single-cell suspension and analyzed by FACS. e, Representative forward-scatter versus side-scatter flow cytometry plots from male and female killifish. Myeloid and lymphoid gates are depicted as the percentage of total live cells. f, Quantification of myeloid: lymphoid ratio (total myeloid events: total lymphoid events) from flow cytometry data. Each dot is a fish, and 12 males and 6–8 females at each time point were analyzed for e and f. Statistical significance was assessed using a two-sided Mann–Whitney U-test. g, Scatterplot of the counts normalized by DESeq2 for irf4a (LOC107383908), with each dot representing the expression of irf4a in an individual head kidney sample in the atlas dataset with n as reported in Extended Data Fig. 1b. Red denotes female; blue denotes male. h, Representative maximum z-projected HCR images of male and female kidney sections at young or old ages. The sections were stained with DAPI (blue) and the HCR probes against irf4a (red) and ptprc (white) mRNAs. Scale bars, 5 µm. i, Quantification of the HCR images in h. The average number of irf4a mRNA transcripts per cell is shown; only interstitial regions were quantified. Each dot is a fish. n = 4 animals per sex and age group. In-graph statistical comparisons were performed using a two-sided Mann–Whitney U-test. Below-graph statistics were assessed using two-way ANOVA including age, sex and the age–sex interaction as factors.
Do cell-type compositional changes contribute to the age-related alteration in gene expression in the killifish head kidney? We first performed single-cell deconvolution of the kidney transcriptomes using a published kidney single-cell RNA-seq dataset59 (Extended Data Fig. 8a). For females, two deconvolution models (dtangle and svr) showed a higher proportion of myeloid cells (a summation of macrophages, neutrophils, mast cells and thrombocytes) at older ages (Extended Data Fig. 8b; ‘Myeloid cells’) mostly due to elevation of macrophages or neutrophils (Extended Data Fig. 8c), but the proportion of lymphoid cells (a summation of natural killer/T cells and B cells) did not change with age significantly (Extended Data Fig. 8b; ‘Lymphoid cells’). For males, one deconvolution model (dtangle) showed a significantly lower proportion of lymphoid cells (Extended Data Fig. 8b), particularly B cells (Extended Data Fig. 8c), at older ages, and most myeloid cells except neutrophils did not change with age significantly (Extended Data Fig. 8b,c). With some limitations (Methods), the deconvolution analysis supported an alteration in cell-type composition with age in the killifish kidney.
Next, we orthogonally measured the different immune cell populations. We optimized a head kidney dissociation protocol followed by fluorescence-activated cell sorting (FACS; Fig. 4d and Extended Data Fig. 9b). We validated a FACS gating strategy developed for zebrafish (based on forward scatter and side scatter64) by performing RNA-seq on the FACS-sorted cells and found enrichment for either lymphoid or myeloid cell-type-specific expression in the expected cell populations (Extended Data Fig. 9c,d). Using this strategy, we observed that females (and less so for males) exhibited age-related cell-type compositional changes (Fig. 4e,f and Extended Data Fig. 9e). There was a significant increase in the ratio of putative myeloid to putative lymphoid cells in old females (133–137 days old) compared to young females (59–61 days old; P = 0.0080), whereas such increase was milder and did not reach statistical significance in males of the same chronological age (136–143 days versus 55–62 days old; P = 0.3095) or in males that survived to a similar percentile of the colony (151–179 days versus 51–59 days old; P = 0.2778). This more pronounced cell-type compositional change in females was consistent with the stronger age correlation observed in gene expression for females (Fig. 4b). Such sex differences may occur because the females in our cohorts were shorter-lived than males (Extended Data Fig. 1a) and likely aged more rapidly than males.
Interestingly, among the most strongly downregulated genes were the two orthologs of the lymphoid transcription factor IRF4 gene in mammals65,66,67 and zebrafish68 (Fig. 4b). These two killifish paralogs of IRF4, irf4a (killifish name: LOC107383908) and irf4b (killifish name: irf4), had differing expression levels and patterns (Fig. 4g and Extended Data Fig. 9f–h)59, with irf4a more strongly downregulated with age (Fig. 4b). We validated the irf4a transcript levels by RNA in situ hybridization, showing that irf4a mRNA could be coexpressed with ptprc mRNA (CD45, a pan-leukocyte marker) in cells of the hematopoietic tissue-enriched interstitial regions of the killifish head kidney (Extended Data Fig. 9i) and decreased with age (two-way ANOVA, P = 0.0549 for the ‘age’ variable; Fig. 4h,i). While we could not validate Irf4a protein expression (no fish-specific Irf4a antibody exists currently), our results raise an interesting possibility that irf4a downregulation with age may reduce lymphoid cell differentiation, leading to increased relative abundance of myeloid cells.
Sex-specific transcriptomic aging clocks can outperform sex-combined clocks in specific cases
Our comprehensive transcriptomic aging atlas allows us to develop age-prediction models for each tissue, known as ‘aging clocks’69,70,71,72. Using molecular features from large datasets (for example, DNA methylation73,74,75,76, transcriptomes75,77,78, proteomes79), these machine-learning models first learn patterns from samples of known chronological ages (‘training’) and then compare the molecular pattern of a query sample (which is not used in the training set) with the learned patterns to find the age best matched by the query, the ‘predicted age.’ Development of these clocks has accelerated evaluation of genetic, pharmacological and lifestyle aging interventions. For example, the epigenetic aging clocks trained on chronological age predict animals and humans to have ‘younger’ age when they are subjected to beneficial health interventions such as diet and exercise74,80,81,82 and lifespan-extending genetic manipulations76,83,84.
To build tissue-specific transcriptomic aging clocks, we used three machine-learning modeling strategies (Fig. 5a), including the nonlinear Bayesian pipeline BayesAge 2.0 (ref. 85), Elastic Net regression (a hybrid model of Lasso and Ridge regression) and principal component-based regression86 (PC-R; Methods). Applied to our dataset, these models had different prediction precision and residual behaviors, which measures whether a model’s predictions underestimate or overestimate the true values (see the brain as an example in Supplementary Fig. 7), and thus we reported the results of all three.

a, Workflow of the three machine-learning models used to construct the transcriptomic clocks, including BayesAge 2.0, Elastic Net and PC-R. b, Bar plots of performance metrics for sex-combined and sex-split (‘M-specific’ and ‘F-specific’) clocks built using BayesAge 2.0, Elastic Net and PC-R. Performance is shown as the coefficient of determination (R²) between chronological and predicted age. Tissues were grouped by whether one or both sex-split clocks outperformed the sex-combined clock (‘Higher than sex-combined’). ‘Neither’ indicates that the sex-combined clock performed better than both sex-split clocks. Testis and ovary were excluded from sex-combined models due to a high degree of sex dimorphism. Male-specific bone and retina models were not plotted due to poor performance. S. cord, spinal cord. c, Distribution of feature importance scores for the brain sex-combined clock built with Elastic Net and PC-R. A gray dashed line marks the 90th percentile threshold. A green dashed line marks the top BayesAge 2.0 brain clock gene (H2AFV/LOC107386217), where the ranking was based on absolute Spearman rank correlation. d, Percentage of optimal BayesAge 2.0 tissue clock genes that also fall within the top decile of feature importance scores in both Elastic Net and PC-R clocks. e, Scatterplot of transcriptomic age (tAge) versus chronological age for the optimal BayesAge 2.0 sex-combined brain clock, defined as the model with the highest concordance between predicted and chronological age across all gene numbers tested. Each dot is one fish; sample sizes for brain samples are as reported in Extended Data Fig. 1b. The solid line indicates the best-fit linear regression (ordinary least squares), and the shaded band shows the 95% confidence interval for the fitted regression. Below, gene frequency scatterplots show the top ten age-correlated genes in the sex-combined brain clock. LOESS fits (black) are shown for visualization only and were not used for statistical inference. f, Scatterplot of tAge versus chronological age for the optimal BayesAge 2.0 sex-combined gut clock, defined as above. Each dot is one fish; sample sizes for gut samples are as reported in Extended Data Fig. 1b. The solid line indicates the best-fit linear regression (ordinary least squares), and the shaded band shows the 95% confidence interval for the fitted regression. Below, gene frequency scatterplots show the top ten age-correlated genes in the sex-combined gut clock, with LOESS fits shown in pink for visualization only. MAE, mean absolute error.
Because our dataset was relatively sex-balanced, for each tissue, we compared the performance of the aging clocks developed using each sex’s transcriptome (‘sex-split’) with those built from sex-combined transcriptomes. We observed that some sex-specific clocks performed better than the sex-combined clocks, and this feature varied by tissue type and machine-learning model used to generate the clocks (Fig. 5b). For example, for the gut, the sex-combined clock performed better than the sex-split clocks in BayesAge 2.0 and the Elastic Net model, but the performance was similar between the sex-combined and the male-specific clock in PC-R (Fig. 5b; ‘Gut’). For the heart, the male-specific clock had higher performance than the sex-combined clock in all three machine-learning models (Fig. 5b; ‘Heart’), whereas for the brain, the female-specific clock had higher performance (Fig. 5b; ‘Brain’).
The three machine-learning models differed in how generally splitting the analysis by sex could improve clock performance. BayesAge 2.0 benefited the most when sex-split datasets were used: for 11 of the 12 tissues tested, the sex-split clocks (either or both sexes) had higher performance than the sex-combined clocks (Fig. 5b). In contrast, for Elastic Net and PC-R, about half of the tissues achieved an improvement in clock performance when sex-split datasets were used (Fig. 5b). The feature selection strategy of BayesAge 2.0 may render it more sensitive to the age changes unique to either sex (see the Methods for discussion). For example, the liver sex-specific clocks performed better than the sex-combined clock only in BayesAge 2.0 (Fig. 5b). Interestingly, most of the genes underlying the sex-specific liver clocks were distinct from those for the sex-combined clocks (Supplementary Fig. 8a,b). Given that the liver transcriptomes had a high proportion of sex-specific aging features (that is, high ‘sex:age interaction’ in Extended Data Fig. 1d), the improved performance of sex-split liver clocks may suggest that the sex heterogeneities in liver aging between males and females were masked when combined. Therefore, while sex-split clocks do not always improve the clock performance, they can in specific cases and should be tested for developing better age-prediction models.
The three machine-learning models identify a common set of aging biomarker genes in each tissue
We identified the aging biomarker genes shared across the three sex-combined machine-learning models. By comparing genes contributing to the optimal BayesAge 2.0 clock with the top 10% of the genes from the Elastic Net and PC-R clocks (see an example of one gene shown in Fig. 5c), we found that brain and gut tissues exhibited the highest gene overlap (92% and 70%, respectively), while the spleen had the lowest (8%; Fig. 5d). The human orthologs of these genes appeared functionally related, possibly reflecting key functional changes related to aging. For example, several key genes in the brain, such as CENPF, SMC4 and RCC2, have been linked to cell division regulation (Fig. 5e), and DLL1 has been implicated in adult neural stem cell maintenance87, consistent with reduced neurogenic capacity of the aged killifish brain37. In the gut, the key genes were associated with nutrient sensing, including the neuroendocrine peptide PTHLH and the G-protein signaling regulator RGS3 (Fig. 5f). Collectively, the genes underlying the aging clocks share related functions, pointing to key regulators of tissue aging dynamics.
Transcriptomic aging clocks can predict age in an independent aging dataset and rejuvenation interventions in killifish
To cross-validate our transcriptomic aging clocks, we applied the clocks to a published transcriptomic dataset36, which consists of four tissues (brain, heart, muscle and spleen) and compares young (6 weeks old) versus old (16 weeks old) samples derived from both male and female killifish (Extended Data Fig. 10a). Due to the strong batch effects between the atlas and query datasets, when we directly applied the atlas clocks to predict sample age, the predicted ages of the young and old query samples failed to robustly separate (Extended Data Fig. 10b). Thus, we developed ‘transfer calibration’ strategies to improve the age-prediction performance of each machine-learning model (Extended Data Fig. 10c and Methods). All three sex-combined machine-learning models (BayesAge 2.0, Elastic Net and PC-R) predicted the old samples to be significantly ‘older’ than the young samples for all four tissues (Extended Data Fig. 10d–g), validating that the calibrated atlas clocks can be used to predict age. The sex-specific clocks also separated young and old samples, although the post-calibration sample size was insufficient to achieve adequate statistical power.
A key functionality of the aging clocks is to evaluate the beneficial or detrimental effects of interventions using transcriptomic age. To explore this application, we made age predictions on four published transcriptomic datasets of lifespan-extending interventions. We calculated the ∆tAge score, where a negative ∆tAge indicated a sample had a ‘younger’ age compared to the control samples’ median predicted age. The first intervention is dietary restriction, which we reported to extend male lifespan in killifish by 16–22% but has no effect on female lifespan23 (Fig. 6a). Our sex-split liver clocks of all three models revealed that for males, dietary restriction decreased the predicted age of the liver transcriptomes (∆tAge) in comparison to the ad libitum paradigm (P = 0.0286 by BayesAge 2.0 and PC-R and P = 0.1143 for Elastic Net; Mann–Whitney U-test; Fig. 6a). In contrast, for females, dietary restriction did not significantly decrease the predicted age of the liver transcriptome (∆tAge) in comparison to the ad libitum paradigm (P = 0.4857 for Elastic Net and P = 0.6857 by BayesAge 2.0 and PC-R, Mann–Whitney U-test; Fig. 6a). This finding is consistent with the observation that this dietary-restriction paradigm does not extend female lifespan23.

a, Predicted ages for liver samples from male and female killifish fed on ad libitum (AL) or dietary restricted (DR) diets using sex-specific liver clocks (data from a published dataset23). Age prediction was performed using three different modeling strategies: BayesAge 2.0, Elastic Net regression and PC-R. Each dot represents the predicted ΔtAge (tAge scaled by the median of AL samples) for the liver transcriptome of one individual fish (n = 4 fish per condition). Statistical significance between AL and DR conditions for each model was assessed using a two-sided Mann–Whitney U-test. No adjustment for multiple comparisons was performed. Calibration was not applied for BayesAge 2.0 and PC-R due to minimal batch effects. b, Predicted ages for fat samples from male killifish wild-type (WT) and a constitutive AMPKγ1 mutant (UBI: γ1(R70Q)), either fed or fasted, using fat clocks (data from a published dataset21). Predicted tAge values were scaled by the median of young WT samples. Calibration was applied. Statistical testing was performed as described in panel a using a two-sided Mann–Whitney U-test without multiple-comparison adjustment. Sample sizes are reported in Supplementary Table 23. c, Predicted ages for liver samples from male killifish WT and a constitutive AMP biosynthesis mutant (APRT+/-), either fed or fasted, using liver clocks (data from a published dataset20). Predicted tAge values were scaled, calibration was applied, and statistical testing was performed as described in panel b using a two-sided Mann–Whitney U-test without multiple-comparison adjustment. P values were not calculated when only a single sample was available for comparison in the testing data. NA, not available. Sample sizes are reported in Supplementary Table 23. d, Predicted ages for gut samples from male killifish WT at young and old ages. WT males received young (6 weeks old) gut microbiome transfer at 9.5 weeks old (Ymt), and WT males received middle-aged gut microbiome transfer at 9.5 weeks old (Omt). The transcriptomes were generated when the fish were 16 weeks old (~7 weeks after treatment). Data were from a published dataset25. Predicted tAge values were scaled, calibration was applied, and statistical testing was performed as described in b using a two-sided Mann–Whitney U-test without multiple-comparison adjustment. Sample sizes are reported in Supplementary Table 23.
We tested two transcriptomic datasets from genetic mutants, including a constitutive AMPKγ1 mutant (UBI:γ1(R70Q))21 and an AMP biosynthesis mutant (APRT+/−)20. Both mutants extend the lifespan of male killifish. For both transcriptomic datasets, all three machine-learning models successfully separated the young and old tissue samples, particularly when the fish were under the ‘fed’ condition (Fig. 6b,c). The sex-combined Elastic Net and PC-R models predicted that the UBI:γ1(R70Q) mutant had a ‘younger’ transcriptomic age (∆tAge) compared to the wild type (P = 0.0952 for Elastic Net and P = 0.0357 for PC-R, Mann–Whitney U-test), specifically at the old age and under the ‘fed’ condition (Fig. 6b), consistent with the longer lifespan of UBI:γ1(R70Q) males21. For the AMP biosynthesis mutant, we did not have sufficient statistical power to predict age for the APRT+/− mutant versus wild type (Fig. 6c) because the sample size of this dataset was relatively limited.
The last intervention evaluated was gut microbiome ‘transfer.’ It has been reported that exposing the gut microbiome of 6-week-old males (donors) to 9.5-week-old males (recipients; known as ‘Ymt’) extends the lifespan of the recipients, but no lifespan extension is observed when 9.5-week-old male donors are used (known as ‘Omt’)25. All three machine-learning models successfully separated young and old samples (Fig. 6d). The male-specific gut Elastic Net clock predicted that Ymt trended toward a ‘younger’ transcriptomic age (∆tAge) compared to the wild-type control (P = 0.200, Mann–Whitney U-test), which is consistent with the lifespan extension effect of Ymt. Interestingly, Omt was predicted to have a ‘younger’ age than the wild-type control (P = 0.0667 for BayesAge 2.0, P = 0.1143 for Elastic Net, and P = 0.1333 for PC-R, Mann–Whitney U-test), even though no lifespan extension was observed for Omt25. The original publication reported that the Omt treatment reduced expression of inflammation-related genes (for example, lyz, marco and plek) compared to the Ymt treatment25, which might explain the lower predicted age of Omt. Alternatively, the Omt treatment might have ‘rejuvenated’ specific aspects of the gut transcriptome, but these changes were insufficient to extend lifespan.
Altogether, we conclude that for at least a subset of published killifish intervention datasets, the transcriptomic clocks can make age predictions on unseen data (such as those in interventions) that are consistent with biological contexts and provide insights into biological age.
Discussion
Here we present a comprehensive aging transcriptome atlas of 13 tissues for male and female killifish, which we made accessible through an open-access online portal (Methods). Our analyses reveal varying age–sex relationships for each tissue, identifying several sex-dimorphic tissues (for example, gonads, liver, gut, head kidney) that benefit from analyzing each sex separately. Time-series correlation and gene expression trajectory analyses have identified age-correlated genes and pathways common across multiple tissues, including several ‘hallmarks of aging’ and consistent with the findings in mammals (for example, Tabula Muris Senis9), highlighting evolutionary conservation between killifish and mammals.
In our study, most pathways altered with age show consistency between sexes, with a strong upregulation of immune response pathways in older individuals irrespective of sex across at least six tissues. This upregulation may be partly driven by increased immune cell infiltration, which has been reported in several killifish tissues21,37,38. Our integration and deconvolution of bulk RNA-seq data with single-cell datasets for killifish tissues22,37,59,88 has provided insights into cell-type composition changes, particularly in the head kidney. Interestingly, females have a stronger increase in myeloid cell proportions with age, which correlates with broader innate immune response upregulation in female organs compared to male organs, possibly suggesting a link between systemic inflammation and hematopoietic changes in head kidney. The downregulation of IRF4/irf4a, along with other factors, may contribute to this myeloid bias in females, and it will be interesting to explore the functional role of IRF4/irf4a in this process.
Another interesting class of age-altered pathways involves the ECM, which are downregulated with age in most tissues in males and some tissues in females. The ECM, which plays a central role in tissue structural maintenance and cell-cell signaling, is impacted by aging in mice, primates and humans9,75,89,90. ECM disruption can accelerate aging in mice91,92, and longevity interventions promote ECM homeostasis in Caenorhabditis elegans93. While lifespan extension has not been shown by modulating the ECM in vertebrates, our study, along with others, highlights growing evidence for the ECM regulating aging in animals.
We also identified 39 killifish age-altered genes that have human orthologs and show consistent expression changes across tissues. A prominent example is the IGF2BP3 gene, whose downregulation is a main driver of the gut aging clock and correlates with an age-dependent decline in cell division genes across tissues. The killifish IGF2BP3 gene encodes a protein highly conserved with the human IGF2BP3, which promotes IGF2 mRNA translation53 and IGF2 signaling to support cell proliferation during development94,95,96,97, cancers98,99 and adult stem cell renewal in mice and humans100. Future studies on IGF2BP3 would clarify its role in regulating killifish tissue aging and growth.
In addition to the aging pathways shared between sexes, some pathways diverge in their directions of change. Interestingly, male and female killifish often differ in their responses to lifespan interventions, including dietary restriction and intermittent fasting20,23, genetic mutations in the AMPK pathway20,21 and the germline22 and metformin treatment29. The sex-divergent pathways may contribute to the sex-specific responses to lifespan interventions. It would be interesting to screen interventions in a sex-specific manner, such as testing small molecules that specifically target female pathways.
The sex-balanced nature of our atlas dataset has enabled comparison between sex-specific and sex-combined clocks. We have shown that sex-split clocks can improve the clock performance, although this improvement varies by tissue types and machine-learning models. When applied to the independent datasets, both sex-specific and combined models generally separate young and old samples. However, the ability of these clocks to record longevity is more mixed. Future application of these clocks to additional datasets will test their effectiveness in capturing different aging interventions. As more transcriptomic datasets become available, integrating them will facilitate the generation of generalized tissue-specific clocks, for accelerating evaluation of intervention efficacy.
We acknowledge several limitations of our study, including reduced time-point coverage for certain tissues due to sample dropout and technical challenges in RNA isolation (for example, ovary, retina/RPE and bone). Future studies should also include larger sample sizes and denser time points to determine the aging rates across tissues. Furthermore, it will be important to determine the contribution of the inherent biological differences between male and female killifish (for example, body size, growth rate and metabolic rate) to the aging transcriptome. Lastly, our oldest time point corresponded to ~20% of the surviving colony, prompting questions about the changes due to a survivor phenotype versus natural decline. Future studies that monitor fish health (for example, by behavior tracking) could provide crucial insights into this question.
Within the bounds of limitations, we envision that the killifish aging atlas and the associated aging clocks will catalyze future research into the drivers and biomarkers of tissue aging, enabling the rapid evaluation of interventions in the killifish, a powerful short-lived vertebrate model for aging research. These resources will also facilitate the identification of shared aging pathways across species.
Methods
African turquoise killifish husbandry
All experiments used the GRZ strain of the African turquoise killifish species Nothobranchius furzeri. Fish were housed in a 26 °C circulating water system maintained at a conductivity of between 3,500 μS cm−1 and 4,500 μS cm−1 and a pH of between 6.5 and 7.5, with a daily water exchange of 10% using reverse-osmosis-treated water. All animals were kept on a 12-h–12-h day–night cycle. Feeding and husbandry details are described below. All fish were housed within the Stanford Research Animal Facility under protocols approved by the Stanford Administrative Panel on Laboratory Animal Care (Institutional Animal Care and Use Committee protocols 31727 and 13645).
Atlas cohorts
All fish were raised from embryos collected from group breeding tanks (one male paired with at least three females in 9.8-l tanks, and the breeders were generally 2–4 months old). Breeder tanks were fed ~18 mg Otohime fish pellets per fish (Reed Mariculture, Otohime C1) twice a day and bred with sand trays in the tanks for embryo collection. After 4–8 h, the sand trays were collected, and embryos were separated from the sand by sieving. To reduce contamination, we rinsed the embryos with 0.2% mild iodine (diluted from povidone-iodine solution (10% wt/vol, 1% wt/vol available iodine, RICCA, 3955-16) in Ringer’s solution (Sigma-Aldrich, 96724)). Decontaminated embryos were incubated in Ringer’s solution supplemented with 0.01% methylene blue (Kordon, 37344) at 28 °C in 60-mm × 15-mm Petri dishes (E and K Scientific, EK-36161) at a density between 10 and 50 embryos per plate for ~2 weeks and then placed on moist coconut fiber substrate (Amazon, B00167VVP4) at 26 °C. After ~2 weeks on coconut fiber, fish were hatched in ~1-cm-deep chilled (4 °C) 1 g l−1 humic acid solution (Sigma-Aldrich, 53680) and incubated at room temperature overnight. For the next 4 days, the hatched fish were housed at room temperature. During this period, system water was added to the hatching containers, and fish were fed 2–3 drops of live brine shrimp (hatched from premium-grade brine shrimp eggs (Brine Shrimp Direct, 12-pound carton); see published protocols for details101) once daily using plastic pipettes (Globe Scientific, 138090). Fish were housed at a density of four fish per 0.8-l tank for the following 2 weeks, then two fish per 0.8-l tank for one week, and then one fish per 0.8-l tank for one week. Fish were fed with brine shrimp twice daily. At the 5th week after hatching, each fish was transferred to a 2.8-l tank and sexed by caudal fin color: males exhibit vivid colors, but females do not. Fish with severe gill defects, curved spines and an inability to float (‘belly sliders’) were excluded. A random subset of individuals from each cohort was designated as ‘Lifespan’ animals, and these animals were not selected for harvest. Any other unharvested animals that died from natural causes were also plotted in the lifespan analysis. Cohort 1 fish were enrolled in two batches, 2 weeks apart (see Supplementary Table 1 for enrollment details). Cohort 2 fish were enrolled as an independent cohort, 6 months after cohort 1. All fish from each cohort were randomly assigned to tank locations using the ‘Randomize Range’ function in Google Sheets. Cohort 1 (first enrollment) fish were fed using an automated feeder23 under the ad libitum regimen (5 mg per feeding and fed seven times a day for a total of 35 mg of Otohime fish pellets). Cohort 1 (second enrollment) and cohort 2 were fed using a custom-made manual feeder twice a day, 18 mg per feeding, for a total of 36 mg of Otohime fish pellets. The core design of the custom feeder has the same acrylic-cut feeding disc as the automated feeder; thus, it has the same precision as the automated feeder.
Validation cohort for RNA in situ staining
Fish were raised similarly to the atlas cohorts with the following modifications. After collection, embryos were rinsed several times with embryo solution (Ringer’s solution with 0.01% methylene blue) instead of mild iodine, placed in fresh embryo solution, and incubated at 26–28 °C. Approximately 2 weeks after collection, embryos were placed on moist coconut fiber and incubated at 27 °C. Two weeks later, fish were hatched in 60 × 15-mm Petri dishes (VWR, 25384-168) containing 10 ml of cold 1 g l−1 humic acid solution and placed at room temperature on the benchtop. After the fish were hatched, they were put into the 26 °C circulating water systems in 0.8-l tanks at a density of 10–20 fish and fed brine shrimp twice daily. After one week, the fish were split and housed at a density of four fish per 0.8-l tank for one week, then two fish per 0.8-l tank for another week, and then one fish per 0.8-l tank for one week. At the 5th week after hatching, fish were upgraded to 2.8-l tanks, sexed and randomly assigned to their tank positions. All sexually mature fish were fed using the custom-made manual feeder as in the atlas cohorts (18 mg of dry pellets twice a day, for a total of 36 mg per day). We note that the validation cohort was run as the control for another experiment, which aimed to understand how mating affects killifish aging. The validation fish were the ‘unmated control’. Thus, the validation animals were housed with sand trays (which were used as the mating bedding for the mated group) for 4 h twice a week (8 h total per week). The male fish experienced a ‘mock cross’ twice a week, where the male fish were netted and placed back in their own tanks to mimic the ‘crossing’ of the mated group.
Lifespan analysis, including Kaplan–Meier curve plotting
First, animals with missing data (for example, sex or death date) or those harvested for RNA-seq were excluded from the analysis. The remaining animals (the animals designated for lifespan analysis and those that died of natural causes) were used to plot Kaplan–Meier survival curves. Data were entered into Prism using the defaults for survival analysis, with ‘1’ indicating a censored sample and ‘0’ indicating a sample died. Kaplan–Meier curves were plotted individually for males and females, separated by enrollment cohort.
While females were shorter-lived than males in our study (Extended Data Fig. 1a), we note that female lifespans can vary across animal facilities and different laboratories. For example, females can have a similar lifespan to males20,21,102, a longer lifespan than males34, or a shorter lifespan than males23. Environmental and dietary factors (for example, some labs feed bloodworms, whereas we fed dry pellets) likely contribute to the variability in female lifespans.
Fish length measurement
On the days of tissue collection, an image of the fish was taken using an iPhone after the fish were killed with tricaine overdose (see ‘Killifish perfusion’). Each fish was laid flat (the anterior side facing left and dorsal side facing up) along a ruler on ice, and the image was taken from above. Each image was converted from the iPhone format (HEIC) to a JPEG format (parameter: quality, 100%) using an online software (http://heic.online/). Each converted image was imported into FIJI. A straight line was drawn for the ruler to mark the length of 1 cm, and the pixel length for this line was measured by FIJI (‘the ruler length in pixels’). Next, a straight line was drawn for the fish along its anteroposterior axis, from the mouth of the fish to the end of the spine (right before the tail fin starts). The pixel length of this line was measured by FIJI (‘the fish length in pixels’). We calculated the fish length in centimeters as the ratio of ‘the fish length in pixels’ over ‘the ruler length in pixels’.
Atlas cohort tissue collection
The harvest dates were randomly assigned to each fish within each cohort. On a harvest day, each fish was fed 18 mg of Otohime fish pellets at 7:30–8:00. At ~10:30, the fish were transported from the animal facility to the lab space in their own tanks. Typically, four fish (two males and two females) were dissected on each harvest day (~30 min to dissect each fish). Dissection began with perfusion (see details below) and then tissue collections on top of ice-cold Sylgard-coated Petri dishes (filled with wet ice and covered in plastic wrap) by three operators (E.K.C., J.C. and I.H.G.). Dissected tissues were placed in 1.5-ml tubes (Fisherbrand, 02-681-320), snap frozen in liquid nitrogen and stored at −80 °C until RNA extraction. The same operator dissected the same tissues for all fish in this study (see Supplementary Table 1 for details). Muscle samples were collected from a ~1-cm region immediately anterior to the caudal fin, with skin removed and cuts made above and below the horizontal septum to remove the spinal cord and vertebrae. The spinal cord was collected by dissecting out the vertebrae and gently pulling the spinal cord from the vertebral foramen. Skin samples corresponded to the caudal fin’s most posterior ~0.5-cm portion. The retina and RPE were dissected from the eye together (by M.-R.W.). In some cases, the retina/RPE samples were dissected from individual eyes of the same animal. In other cases, samples were pooled across animals (as indicated in Supplementary Table 2 wherever relevant). Only the head kidney was collected for the kidney samples. For the liver samples, the pale-green gallbladder was removed whenever it was visible. Total visceral fat was collected (without regional distinction). All oocytes were collected for ovaries, including those that had fallen out of the organ during dissection.
Perfusion device setup
Killifish perfusion made use of a syringe pump (KD Scientific, Legato 210 Series, 788210) that permitted hands-free depression of the perfusion syringe and included the following feature: a 20-ml disposable syringe with Luer Lock tip (‘sterile syringe only with Luer Lock tip’, Amazon, B08FJCSLFC) attached to a 30-gauge, metal hub, blunt-end Luer needle (Hamilton syringe, custom needle, 7748-16; 30-gauge, metal hub needle; point style, 3; needle length, 0.375 inches). The blunt-end Luer needle was connected to ~0.25 m of BD Intramedic PE Tubing (BD, 427400), which terminated in a 30-gauge, hubless needle with a point style 4 bevel (Hamilton syringe, custom needle, 22030-01; 30-gauge, hubless needle, no hub; length, 1.5 inches; point style, 4 (12°)). The 20-ml syringe was filled with nuclease-free 0.25 M EDTA diluted in 1× PBS (Corning, 21-040-CV) and fitted into the syringe pump.
Killifish perfusion
The killifish was first deeply anesthetized in tricaine (100 mg l−1 system water, pH ~7, titrated with sodium bicarbonate) until operculum movement slowed, and the fish was unresponsive to touch. Once deeply anesthetized, the fish was placed on top of a Sylgard-coated Petri dish filled with wet ice covered in plastic wrap. The fish was secured on its side with two dissection pins—one pin piercing the muscle immediately anterior to the caudal fin and one pin piercing the gill operculum that lay in contact with the plastic wrap.
First, we exposed the gill by cutting off the operculum with scissors. Operculum removal helped visualize the gill and evaluate perfusion completion, as the gill would be flushed of blood and turn white with successful perfusion. Next, using a scalpel, a small ~1-mm incision was made through the skin immediately anterior to the urogenital opening. We then inserted a scissor at the incision site and cut along the ventral side of the fish to the gill, only cutting through the skin. Next, using the ventral incision as a starting point, a ‘window’ was created using scissors to remove the body wall covering the liver and heart. Once the heart was visible, Iris forceps were used to gently remove the transparent membrane that partially covers the heart and connects the heart to the body wall. Removal of this membrane provided complete visibility of the heart during perfusion. Next, we used spring scissors to cut the atrium, creating a blood flow outlet. Immediately after cutting the atrium, the hubless needle of the perfusion device was inserted ~1 mm into the apex of the ventricle (or as deep as the bulbus arteriosus), and the syringe pump was switched on to depress the plunger of the syringe at a rate of 3.5 ml min−1 to initiate perfusion. The needle was steadily held in place until the gill and liver were visibly devoid of blood.
RNA isolation
To reduce within-tissue batch effects, we processed all samples of the same tissue type on the same day unless otherwise noted. Due to the large number of samples, RNA extraction was performed in 2–3 batches for each tissue, with the order of samples randomized and a roughly equal assignment of age and sex combinations to each batch. The processing order of each sample within a tissue type was randomized using the ‘Randomize Range’ option in Google Sheets. After randomization, tissue samples were assigned unique numerical ‘RNA_IDs’ and split into batches of 12–24 samples for processing.
The RNA isolation protocol was based on the RNeasy Mini RNA extraction protocol from QIAGEN and was largely consistent across tissues, except as noted below. The general RNA extraction protocol is as follows. First, tissue sample tubes were removed from −80 °C storage, placed in liquid nitrogen and transferred to a 4 °C cold room to prevent tissue thawing. We put the sample tubes on a pre-chilled (−20 °C) TissueLyser 2-ml tube adaptor (QIAGEN, 69982) on dry ice in the cold room. Then, to each tube, we added ~100 µl of zirconia/silica beads (BioSpec Products, 11079105z, 0.5-mm diameter) pre-chilled at 4 °C. Next, we quickly transferred the sample tubes, which remained on the adaptor, to wet ice and added 700 µl of 4 °C QIAzol lysis reagent (QIAGEN, 79306) to each tube. The sample tubes were placed between the pre-chilled (4 °C) metal plates of the TissueLyser tube adaptor and homogenized on a TissueLyser II machine (QIAGEN, 85300) at 25 Hz and room temperature for 5 min. After the first round of breakage, we swapped the left and right adaptors before initiating the second round. Swapping the adaptors ensures that all samples receive uniform disruption and homogenization because samples closer to the TissueLyser chamber vibrate more slowly than those further away. After disruption/homogenization, the sample tubes were placed at room temperature for 3–5 min (this step helps dissolve lipids and membranes into the organic phase). Next, the lysed samples were transferred to 1.5-ml DNA LoBind tubes (Eppendorf, 0030108051) that contained 200 µl chloroform, vortexed for 15 s and incubated at room temperature for 2–3 min. Samples were centrifuged at 12,000g for 15 min at 4 °C. For each tube, a total of 350 µl aqueous phase (175 µl × 2) was transferred to another 1.5-ml DNA LoBind tube that contained 350 µl 70% ethanol. The tubes were inverted ten times to mix, followed by a brief centrifuge to collect all liquid. A total of 700 µl of each sample was transferred to an RNeasy Mini spin column (reagent from QIAGEN, 74536), centrifuged at 10,000g for 30 s at room temperature (all subsequent wash steps used this centrifugation condition). The column was washed with 350 µl of RW1 (reagent from QIAGEN, 74536) and incubated in 80 µl of DNase I solution (prepared as instructed by the manufacturer) at room temperature for 15 min. To stop the DNase I treatment, we added 350 µl of RW1 directly to the column, which was then centrifuged and washed twice with 500 µl of RPE buffer (reagent from QIAGEN, 74536) with a 2-min centrifugation step for the last RPE wash. RNA was eluted in 50 µl of nuclease-free water (Invitrogen, 10977023) in a 1.5-ml DNA LoBind tube, aliquoted and stored at −80 °C. RNA concentration was checked for all samples using a Thermo Fisher Varioskan LUX microplate reader μDrop plate (Thermo Fisher, N12391). Eight to ten RNA samples from each tissue were randomly selected to check RNA quality using an Agilent TapeStation 4200 (Agilent, G2991BA) and the TapeStation RNA ScreenTapes (Agilent, 5067-5576).
Liver
The tissues were first transferred from the collection tubes into 1.2-ml Collection Microtubes (QIAGEN, 19560) on dry ice in a 4 °C cold room. A single autoclaved and pre-chilled (on dry ice) 5-mm stainless-steel bead (QIAGEN, 69989) was added to each microtube. The microtubes were then quickly moved to wet ice, and 700 µl of QIAzol lysis reagent (QIAGEN, 79306) was added. Two rounds of homogenization were performed on a QIAGEN TissueLyser II at room temperature and 25 Hz for 5 min each. After the lysate was transferred to new 1.5-ml DNA LoBind tubes, 200 µl chloroform (Fisher Scientific, C298-500) was added, and the tubes were vortexed for 15 s and incubated at room temperature for 2–3 min. The subsequent RNA extraction protocol was performed as stated above. We note that good-quality RNA can be isolated using zirconium beads, which were used for the other tissues. This protocol was implemented due to a limited supply of reagents at the time. Lastly, the RNA from the liver samples of cohort 1 was isolated separately from the other liver samples of cohort 2.
Brain, gonads and skin
All steps involving the RNeasy Mini spin columns were performed on the QIACube HT robotic workstation (QIAGEN, 9001896) according to the manufacturer’s instructions, with the following program: (1) add 350 µl 70% ethanol to each sample aqueous phase in S-Block deep well plate, mix and transfer sample lysate into RNeasy 96 format vacuum columns (QIAGEN, 74104); (2) clear the columns using a vacuum at 25 kPa for 3 min; (3) add 400 µl RWT buffer; (4) clear the columns using a vacuum at 25 kPa for 1 min; (5) add 80 µl DNase I solution and incubate at room temperature for 15 min; (6) add 400 µl RWT; (7) clear the columns using a vacuum at 35 kPa for 1 min; (7) add 400 µl 100% ethanol and incubate at room temperature for 2 min; (8) clear the columns using a vacuum at 35 kPa for 1 min and then 25 kPa for 5 min; (9) add 45 µl nuclease-free water and incubate at room temperature for 4 min; (10) clear the columns using vacuum at 35 kPa for 1 min; (11) add 45 µl fresh nuclease-free water and 30 µl of the top elute fluid to the RNeasy 96-well plate and incubate at room temperature for 1 min; and (12) clear the columns using a vacuum at 70 kPa for 2 min. The eluted RNA samples were aliquoted and stored at −80 °C. We note that for some lipid-rich or debris-rich tissues, phase separation may be difficult (formation of the aqueous phase), making downstream processing challenging. To avoid this issue, for the ovary samples with high lipid content, the QIAzol lysate was split into 2–3 aliquots after disruption/homogenization, topped off with QIAzol to 700 µl, and then processed individually until the column steps. Before these steps, the aliquots were pooled and passed over the same column. Several ovary samples were unfortunately not recoverable with this splitting method and were lost.
Bone
To facilitate tissue lysis, we ground the bone samples before the bead-beating step of the RNA extraction protocol. Briefly, an agate mortar, pestle, metal spatula and a piece of aluminum foil were pre-chilled in liquid nitrogen. Bone samples were removed from −80 °C (tubes stored in liquid nitrogen while awaiting processing) and placed on the chilled aluminum foil, which was then folded over in half to cover the bone sample. Covering the sample prevents larger chunks of the tissue from breaking apart and ‘flying’ out of the mortar. A pestle was used to press on the foil and grind the tissues into a powder. The powder was scooped using a pre-chilled spatula and placed into a 1.5-ml tube that had been pre-chilled on dry ice. The bone ‘powder’ was stored at −80 °C until RNA extraction.
We followed the same RNA extraction protocol for the bone ‘powder’ as stated in the ‘RNA extraction’ section. Briefly, the bone powder was lysed in 700 µl of QIAzol lysis reagent and ~100 µl of pre-chilled zirconia/silica beads on the pre-chilled (4 °C) metal plates for the TissueLyser tube adaptor and homogenized on a TissueLyser II machine (QIAGEN, 85300) at 25 Hz at room temperature, for two rounds of 5-min breakage. After homogenization, we placed the sample tubes at room temperature for 3–5 min, added 200 µl chloroform, vortexed for 15 s and incubated the samples at room temperature for 2–3 min. After centrifuging at 12,000g for 15 min at 4 °C, a total of 350 µl of the aqueous phase (175 µl × 2) was transferred to a new 1.5-ml DNA LoBind tube that contained 350 µl of 70% ethanol, and the tubes were inverted ten times. A total of 700 µl of each sample was transferred to an RNeasy Mini spin column, centrifuged (10,000g, room temperature, 30 s), washed with 350 µl RW1 and then treated with DNase I as stated above. The column was washed twice with 500 µl RPE buffer. RNA was eluted in 50 µl nuclease-free water, aliquoted and stored at −80 °C.
We note that this protocol needs further optimization. The RNA quality of the bone samples was lower than that of the other tissues.
Fat
Fat samples are prone to RNA degradation. We used a modified RNA extraction protocol to maintain the RNA quality of the fat samples. An agate mortar, pestle and metal spatula were pre-chilled in liquid nitrogen. Fat samples were removed from −80 °C (tubes stored in liquid nitrogen during transport), transferred to the mortar and ground to a fine powder by rotating the pestle. The powder was scooped using a pre-chilled spatula, placed into a 1.5-ml tube that had been pre-chilled on dry ice and stored at −80 °C until RNA extraction.
To extract the RNA from fat samples, we placed the frozen powdered fat samples on dry ice and added ~100 µl of zirconia/silica beads to each tube. The samples were then transferred to wet ice, and 700 µl of QIAzol lysis reagent was added to each tube (the QIAzol-to-powdered tissue ratio was at least 2:1). The tissues were quickly homogenized on the TissueLyser II machine (the metal blocks from the tube holder adaptor had been pre-chilled at −20 °C) for 2.5 min at 30 Hz in a 4 °C cold room. The tissues were incubated at room temperature for 5 min, centrifuged at 12,000g for 10 min at 4 °C, and settled at room temperature for ~2–3 min. The middle pink RNA layer was transferred into new tubes containing 200 µl of chloroform, being careful not to aspirate the top lipid layer. The tubes were vortexed for 15 s and incubated at room temperature for 2–3 min. Next, we processed the fat samples in the exact method as for the other tissues, including (1) centrifuging at 12,000g for 15 min at 4 °C, (2) transferring a total of 350 µl aqueous phase to 350 µl 70% ethanol, (3) transferring 700 µl of each sample to an RNeasy Mini spin column, (3) centrifuging (10,000g, room temperature, 30 s), (4) washing with 350 µl RW1, (5) treated with DNase I and (6) washed twice with 500 µl RPE buffer. RNA was eluted in 50 µl nuclease-free water, aliquoted and stored at −80 °C.
Retina/RPE
The retina and RPE samples from each animal were dissected and processed together as one tissue. RNA was isolated from the retina/RPE samples using the RNeasy Plus Micro Kit (QIAGEN, 74034) and following the manufacturer’s instructions. Briefly, 350 µl of Buffer RLT Plus was added to each sample, and the samples were homogenized by vortexing for 30 s. The lysate was then applied to a gDNA Eliminator spin column and centrifuged at 8,000g for 30 s. The flow-through was then combined with 350 µl of 70% ethanol, mixed by pipetting and subsequently transferred to an RNeasy MinElute spin column. The column was centrifuged at 8,000g for 15 s. The column was then washed with 700 µl of Buffer RW1 and subsequently 500 µl of Buffer RPE. After applying each wash, the sample was centrifuged at the previous settings, and the flow-through was discarded. A final wash of 80% ethanol was applied to the column, and the sample tube was centrifuged for 2 min at 8,000g. Finally, the spin column membrane was dried by centrifuging the sample at full speed for 5 min. Then, the column was placed in a new 1.5-ml collection tube, and 14 µl of RNase-free water was applied to the membrane. The sample was then centrifuged for 1 min at full speed to elute the RNA.
We note that this protocol needs further optimization. The RNA quality of some of the retina/RPE samples was lower than that of the other tissues.
Tissue RNA quality and sample dropout
We note that two tissues had noticeable sample dropouts, including the retina/RPE and ovary (Extended Data Fig. 1b). This sample dropout could influence our downstream analyses (Spearman’s rank correlation and tissue aging clocks), given the lower sample size for these tissues. We note that the retina/RPE samples in the two cohorts had different animal pooling strategies and were collected at different ages due to low RNA yield. These sampling and processing differences are described in the metadata contained in Supplementary Table 2.
In the PCA plot (Fig. 1b), bone samples showed higher variability compared to other tissues, possibly due to technical difficulties in preparing high-quality RNA from the bone. This high sample variability may influence our downstream analyses, leading to a low number of age-correlated genes and poor performance of the aging clocks. We note that our normalization methods are likely inadequate in addressing the batch effects associated with RNA extraction. We recommend only using this bone dataset for identifying bone-specific genes.
cDNA library generation and sequencing
cDNA libraries were prepared using a SmartSeq-based in-house protocol. Briefly, RNA samples were thawed on ice, and the concentration was measured using the Quant-iT RNA BR kit (Thermo Fisher, Q10213) on a Varioskan LUX Multimode microplate reader (Thermo Fisher, VL0000D0). RNA sample concentrations were normalized to 25 ng µl−1, and 2 µl of each sample was used as input into the cDNA first-strand synthesis reaction. The resulting single-stranded library was amplified using nine cycles. A portion of the full cDNA library volume (6 µl) was cleaned using Agencourt AMPure XP beads (Beckman Coulter, A63881) at a 0.7× ratio following the manufacturer’s guidelines, including two washes of 10.7 µl 80% ethanol (200 Proof, Gold Shield Distributors, 412804; diluted in nuclease-free water) and elution in 4.5 µl of nuclease-free water (Invitrogen, 10977023). The concentrations of the amplified cDNA libraries were measured using a Quant-iT double-stranded DNA HS Kit (Thermo Fisher, 33120), and a subset of libraries was also measured on an Agilent TapeStation 4200 using a High Sensitivity D5000 ScreenTape (Agilent, 5067-5592).
Next, sequencing libraries were made using the Nextera XT DNA Library Preparation Kit (Illumina, FC-131-1096) and the IDT for Illumina DNA/RNA UD Index Sets A–D (Illumina, 20027213, 20027214, 20042666, 20042667), following the manufacturer’s instructions except for reducing all the reactions by half. Using half-volume reactions does not affect the performance of library preparation and conserves reagents for our large-scale experiment. The Illumina Index Sets A–D were converted into a 384-well format. Two library pools were ultimately generated, one of 322 samples and the other of 358 samples. All the samples from the same tissue type were assigned unique dual indices in the same library pool to minimize any batch effects. For tagmentation, 0.5 ng of the cDNA (2.5 µl total) was mixed with 5 µl TD buffer and then 2.5 µl ATM buffer from the Nextera kit, incubated at 55 °C for 5 min, and cooled to 10 °C. To stop the tagmentation reaction, we added 2.5 µl of NT buffer and incubated the reaction mixture at room temperature for 5 min. The cDNA library was indexed and amplified for 12 cycles in a PCR reaction containing 10 µl of tagmented DNA, 5 µl of dual indices and 7.5 µl NPM buffer. The amplified cDNA library (25 µl total) was split into two 12.5 µl aliquots, and each of which was purified using 22.5 µl of AMPure XP beads as described above. After the first set of beads was eluted in 11 µl of Buffer EB (QIAGEN, 19086), we used the eluted solution to elute the second set of beads, thereby re-pooling the samples. The final volume obtained after the second round of elution was 10 µl. We performed most pipetting steps using the Dragonfly (SPT Labtech) or Mosquito HV (SPT Labtech) robotic liquid handlers to accelerate sample processing and maintain high pipetting accuracy. We performed all steps requiring a thermocycler on a 384-well plate thermocycler (Bio-Rad). The concentration and quality of the library were measured using an Agilent TapeStation 4200 using a High Sensitivity D5000 ScreenTape (Agilent, 5067-5592). The experimental details for sequencing are provided in Supplementary Table 2.
Shallow sequencing for normalization and quality assessment
To reduce sequencing depth variability across samples, we first performed shallow sequencing to more accurately determine the amount of each sample needed in a pooled library to achieve equal representation after sequencing. First, samples were pooled (1 µl per sample) across each row of each 384-well plate, resulting in 16 pools of 18–24 µl per plate. These 32 sub-libraries were quantified using a Qubit 1× double-stranded DNA High Sensitivity Assay Kit (Thermo Fisher, Q33231) and analyzed on an Agilent 2100 Bioanalyzer (assays performed by the Stanford Protein and Nucleic Acid Facility) to determine the average library size. Then, two sequencing libraries (one per 384-well plate) were generated by pooling the 16 sub-libraries per plate in an equimolar fashion, using the Qubit concentration and average library size. All samples from the same tissue type were kept in the same pool. The two pooled libraries were sequenced separately on a NextSeq 500/550 (Illumina) machine using two 150-cycle Mid Output v2.5 kits, in 2 × 74-bp paired-end format (Illumina, 20024906). The on-instrument quality metrics, including Q30 and cluster densities, were in a suitable range for both sequencing runs.
We next ran the Bcl2fastq2 v2.20.0.422 program with 0.8 adaptor trimming stringency on the sequencing run output files to generate FASTQ files for each pooled library. Each FASTQ file was processed using Trim-galore v0.4.5 to trim adaptors and FASTQ v0.11.9 and multiqc v1.15 to assess sequencing quality. Total read counts were taken from the multiqc summary file ‘mqc_fastqc_sequence_counts_plot_1.txt’, looking only at the read 1 (R1) read counts (R1 and R2 read counts were comparable). We used the R1 read counts as input to calculate the volume of each sample required for the deep sequencing libraries (two pooled libraries, as in the shallow sequencing), using a calculation template adapted from https://github.com/kalanir/CATechopooler/blob/master/COMET384_Seq7_Echo_Calculations.xlsx, and generated the pooled libraries based on the adjusted pooling numbers. Of the 697 samples, 17 were omitted from the final deep sequencing libraries (680 samples remained) due to poor sequencing performance and library metrics.
Deep sequencing
Each pooled library was sequenced by Novogene on two (pooled library 1, which included 322 tissues) or three (pooled library 2, which included 358 tissues) lanes of an Illumina NovaSeq X 25B flow cell (2 × 150-bp paired-end) with 10% PhiX spike-in control for each lane, at a target sequencing depth of >40 million paired-end reads (20 million single-end) per sample. Novogene performed base calling, demultiplexing and FASTQ file generation.
Sequencing quality control and read mapping
Raw sequencing data (FASTQ files) were merged for each library pool (two lanes for the pooled library 1 and three lanes for the pooled library 2) and checked for quality using Trim-galore v0.5.0. The processed reads were aligned to the African turquoise killifish reference genome downloaded from NCBI (Nfu_20140520, GCF_001465895.1) using STAR v2.7.10b103 with the default parameters. Of all the sequenced samples, 14 samples had >90% of reads mapped to the genome, 252 samples had 80–90% of reads mapped, and 178 samples had 75–80% of reads mapped. Samtools (v1.16.1)104, with the parameters of MAPQ < 255 (‘samtools view --q255 --b’), was used to remove the reads mapped to multiple genomic regions. Next, we input the uniquely mapped reads into the ‘featureCounts’ program (with the default parameters) from subread (v2.0.6)105 to generate the read counts for each gene.
We identified three samples as outliers and removed them from subsequent analyses: J6 (a liver sample), L21 (a testis sample) and H19 (a skin sample). We excluded two samples (J6 and L21) because they had low total raw counts, and one sample (H19) due to low mapping performance. As a separate method, we used the gene expression connectivity analysis from the WGCNA package (v1.73)106 to detect outliers. This method computes sample-to-sample correlations and derives network connectivity for each sample, standardizes the connectivity scores and finally identifies samples with z-scores below −2 as outliers. Through this method, we verified these same three samples (J6, L21 and H19) were indeed ‘outliers’, thereby validating their removal.
PCA and quality control
All analyses of the Atlas RNA-seq data were performed in R v4.3.3 (apart from those described in ‘Calculation of transcriptomic age’, which were performed in Python), and all the scripts are publicly available via GitHub (https://github.com/emkcosta/KillifishAtlas/). First, to visualize the dataset quality, we created a DESeqDataSet object of all 677 samples using DESeq2 (v1.42.1)107. After filtering out genes for which the sum of the raw counts across all samples was 0 (15 genes), we applied the variance stabilizing transformation (‘vst’) on the raw counts stored in the whole-dataset-DESeqDataSet object and performed PCA calculation and visualization using the PCAtools package 2.14.0. The samples clustered nicely by tissue type along PC1 and PC2 (Fig. 1b).
The whole-dataset-DESeqDataSet object was then subset by tissue to generate individual tissue DESeqDataSet objects, which were stored in a list. To generate the PCA plot for a given tissue, we subset for the tissue and performed variance stabilization of the raw counts before running PCA, as described above.
Percentage variance explained
We quantified the proportion of variance that could be explained by the covariates of sex, age, cohort, RNA extraction batch, RNA extractor and the interaction of age–sex using the variancePartition package (v1.33.11)108 on a per-tissue basis. First, ages were binned into six age groups (47–52 days, 75–78 days, 102–103 days, 133–134 days, 147–155 days and 161–162 days), and age was modeled as the categorical variable ‘age_bin.’ Then, the transcripts per million (TPM) normalized count of each gene was generated for all samples. Next, the TPM count matrix was subset to include only the samples from a given tissue and prefiltered only to include genes with a TPM count greater than 0.5 in over 80% of all samples of this tissue. For most tissues, the formula ~(1 | age_bin) + (1 | sex) + (1 | cohort) + (1 | RNA_batch) + (1 | RNA_extractor) + (1 | sex:age_bin) was used to explore the respective contributions of these variables to variance. For three tissues (bone, muscle and fat), the formula ~(1 | age_bin) + (1 | sex) + (1 | cohort) + (1 | RNA_batch) + (1 | sex:age_bin) was used, as all of the RNA for these tissues had been extracted by one researcher. For retina/RPE, the formula ~(1 | age_bin) + (1 | sex) + (1 | cohort) + (1 | sex:age_bin) was used, as all the RNA for this tissue was extracted by the same researcher in one batch.
The results of the variance partition analyses for each tissue were saved in tabular format (as a CSV file) and plotted using the plotVarPart function.
Assessment of batch effect
Our dataset consisted of several technical batches, including fish cohorts (cohort 1 and 2), RNA extraction (2–3 extractions for each tissue) and sequencing (two sequencing libraries). For the fish cohorts, we harvested 2–3 animals per time point, per sex. Given the low number of samples, we did not perform batch correction, as this would reduce the power of our analysis. We assessed batch effects using PCA and found no major biases due to these technical variables. In addition, we included ‘fish cohort’ and ‘RNA batch’ as covariates in the variance partition analysis of each tissue. These technical variables accounted for the least variance (<1%), compared to age, sex and sex–age interaction. We did not perform a similar analysis for ‘sequencing batch’, as all samples of a tissue type were processed in the same sequencing batch. Based on these results, we believe that technical batches did not strongly impact data quality or our downstream analyses. All the PCA plots and variance partition analysis results can be found in Supplementary Figs. 1–3 and Extended Data Fig. 1e,f.
DESeq2 differential expression analysis
To explore the age–sex interactions in our dataset, we performed differential expression analysis using DESeq2 on the tissue-specific DESeqDataSet objects (see ‘PCA and quality control’). We first performed differential expression analysis using the design ‘~sex + age_bin + sex:age_bin,’ with ‘female’ and ‘age_bin1’ being the reference levels for sex and age_bin, respectively. The age_bin variable was modeled as a categorical variable (so as not to assume linearity), and we limited age_bin to bins 1–5 to focus on age bins for which we had sufficient sex balance (no female samples were collected in the 6th age_bin).
We next performed differential expression analysis between males and females in age_bin (1–5) using the design ‘~sex’ (with ‘female’ as the reference sex). For the sex-differentially expressed genes (sex-DEGs) identified from this analysis, a positive log2-fold change indicates that the expression level for a gene is higher in males than in females. A negative log2-fold change indicates that the expression level for a gene is higher in females than in males. We plotted the prevalence of sex-DEGs (including both positive and negative DEGs) as a percentage of the total genes expressed in each tissue and each age_bin (Fig. 1e).
The analysis in Fig. 1e revealed that in tissues where variance partition analysis identified a contribution of sex to gene expression variance (sex term: gonad, skin, fat, kidney; sex–age interaction term: liver, bone, skin, gonad), the proportion of genes differentially expressed between males and females varied across age bins and followed distinct temporal trajectories in each tissue.
Identification of age-correlated genes
Age-correlated genes were identified on a per-tissue basis. First, a DESeq2 dds object was generated using the raw count matrix, and the sample metadata table was subset for a given tissue and sex. Then, the raw count matrix was normalized using the ‘median of ratios’ method of DESeq2. To accelerate the identification of genes most correlated with age, we prefiltered this count matrix to include only genes with a TPM count greater than 0.5 in over 80% of all samples in each tissue. These criteria exclude the genes with low counts, which are sensitive to noise in detection. After prefiltering, we used the processed normalized count matrix as input to calculate Spearman’s rank correlation between gene expression (normalized counts) and age, where age is the independent variable. A gene with an absolute value of Spearman’s rank correlation |ρ| > 0.5 was considered an ‘age-correlated’ gene. While Spearman’s rank correlation captures monotonic behaviors, we used other methods (see ‘Gene expression trajectory analysis’ below) to study the genes with other dynamics during aging (for example, expressed in only one age, cyclic expression).
To identify age-correlated genes for both sexes combined, we used both male and female samples as one input for a given tissue before performing the same DEseq2 normalization, prefiltering (TPM count greater than 0.5 in over 80% of all samples for the given tissue) and Spearman’s rank correlation calculation.
To identify the age-correlated genes shared across tissues and both sexes, we first subset the atlas data by tissue, but we analyzed both male and female samples together when calculating Spearman’s rank correlation for each gene. Next, we found the intersection of the age-correlated genes (an absolute Spearman’s rank correlation of at least 0.5) in at least six tissues and plotted the Spearman’s rank correlation of each tissue as a heat map (Fig. 2a). The Spearman’s rank correlations for each tissue are listed in Supplementary Tables 4 (sex-split) and 5 (sex-combined).
GSEA
To perform GSEA109 on the age-correlated genes for each sex and tissue, we first calculated a ranked score for each gene by multiplying the Spearman’s rank correlation with the ‘−log10(P value)’ and sorted all transcripts in descending order based on this score. Next, we used Protein Blast (best-hit protein with BLASTp E-value < 1 × 10−3) to identify the human ortholog for each killifish gene. The average of the ranked scores was calculated if multiple killifish paralogs were blasted to the same human gene. A killifish gene was removed if no human ortholog was found. Lastly, we ran the enrichment analysis via clusterProfiler (v4.2.2)110,111 and the Bioconductor annotation data package (org.Hs.eg.db v3.13.0). The P values of the enriched pathways were corrected for multiple hypothesis testing using the Benjamini–Hochberg method. A GO term (all three categories—biological process, cellular component and molecular function—were tested) was considered significantly enriched if it had an FDR < 0.05. The top GO terms significantly altered by age in both males and females were graphed as a dot plot in Fig. 1f and Extended Data Fig. 2.
In Extended Data Figs. 2–4 and Supplementary Figs. 4 and 5, the GO terms are grouped by the similar functions of the GO terms, such as ‘telomere maintenance’ and ‘telomere organization’. For Extended Data Fig. 2, the tissues were clustered hierarchically by ‘NES × −log(FDR)’, separately for males and females. Extended Data Fig. 4 and Supplementary Figs. 4 and 5 plot the GO terms significantly altered with age in only one sex and that differed in the direction of change between the two sexes. In Extended Data Fig. 4, the tissues were alphabetically ordered, and the male and female results for each tissue were juxtaposed. The complete GSEA data are listed in Supplementary Table 6.
For selected GO terms (Fig. 1g), heat maps were generated using Spearman’s rank correlations from males and females when the two sexes were analyzed separately (‘sex-split’). The above GSEA analysis outputs the human ortholog genes that drive each GO term. The gene lists of the same GO terms from males and females were merged, and the killifish genes corresponding to these human ortholog genes were identified (one human gene name could correspond to multiple killifish genes, and all the killifish genes were plotted). The heat maps were generated using pheatmap v.1.0.12, with a defined scale from −1 to 1 (because Spearman’s rank correlations do not exceed this boundary) and with the genes clustered.
Hypergeometric GO enrichment
We used the GOstats v2.68.0 package for this analysis.
We first performed this analysis focusing on the pathways shared across tissues (Extended Data Fig. 3a,b). To do so, we separated the age-correlated genes into the upregulated gene set (Spearman’s rank correlation ρ > 0.5) and downregulated gene set (Spearman’s rank correlation ρ < −0.5) for each tissue and sex. Next, we ran the enrichment analysis for each tissue using the ‘universe’ gene set, which included all the genes except those with ‘NA’ FDR-adjusted P values. ‘Biological processes’ were used as the ontology, and at least five genes were used to define a GO term. We selected the same GO terms found in GSEA to plot the hypergeometric GO enrichment results. The enrichment scores (Extended Data Fig. 3a,b) were capped at 50 (that is, scores higher than 50 were set to 50) to enhance visualization of all the GO terms. The full results were included in Supplementary Table 7.
Overall, similar GO terms were found in both GSEA and hypergeometric GO analysis, although the results were weaker for hypergeometric GO analysis. For example, we plotted the hypergeometric GO enrichment of the main GO terms identified by GSEA that were shared across tissues (see Extended Data Fig. 2 for these terms). The best example where GSEA and hypergeometric GO analysis matched well was inflammation-related terms (for example, adaptive immune response, cytokine production, innate immune response), which were significantly enriched in age-upregulated genes in females across multiple tissues (bone, brain, fat, gonad, kidney, heart, liver, spinal cord and spleen). A few other examples showed stronger results by GSEA than hypergeometric GO analysis. One example was the inflammation-related terms for males, which were significantly upregulated with age in retina/RPE, skin and spinal cord by both analyses; however, only GSEA identified enrichment in bone. Another example was that the GO terms related to ribosome biogenesis and translation were significantly upregulated with age in female skin and heart by GSEA, but not by hypergeometric GO analysis. The stronger patterns in GSEA may reflect the presence of additional genes within a specific biological pathway that were altered with age in the same direction—although more subtly affected by age—as those with a Spearman’s rank correlation greater than 0.5.
Next, we performed hypergeometric GO enrichment analysis on genes that showed different directions of change with age between the two sexes (‘sex-divergent’; Supplementary Figs. 4 and 5). Twelve of the 13 tissues had fewer than 10 age-correlated genes (that is, the absolute value of Spearman’s rank correlation greater than 0.5) that differed in the direction of change between the two sexes. Only the gonads had more than 10 of such genes, but still fewer than 30. These gene sets were too low for performing hypergeometric GO enrichment analysis. Therefore, we expanded the criteria to include genes that were significantly changed with age in at least one sex but showed the opposite sign of change in the other sex. Specifically, for each tissue, we identified the genes belonged to each of the following four categories: (1) Spearman’s rank correlation ρ < −0.5 for males and ρ > 0 for females; (2) ρ < −0.5 for females and ρ > 0 for males; (3) ρ > 0.5 for males and ρ < 0 for females; and (4) ρ > 0.5 for females and ρ < 0 for males. Next, we performed the enrichment analysis as described above (ontology denotes biological processes; universe indicates all genes with non-NA FDR-adjusted P values; at least five genes for a GO term). The results were plotted for each of the four categories separately. Supplementary Fig. 4 plotted the results for categories 1 and 4 together (that is, upregulated with age in females but downregulated in males), and Supplementary Fig. 5 plotted the results for categories 2 and 3 together (that is, downregulated with age in females but upregulated in males). The full results are included in Supplementary Table 8.
For the sex-divergent genes, three GO terms were notably consistent between GSEA and hypergeometric GO analysis. All these terms were upregulated in females but downregulated in males for specific tissues. They were related to (1) extracellular matrix (for the liver), (2) lysosome and autophagy (for the gonads, fat and kidney) and (3) neutrophil activation (for the kidney, fat and spleen). Because these pathways were robust enough to be identified by both GSEA and hypergeometric GO analysis, it would be interesting to confirm the expression of the genes underlying the enriched GO terms and perform functional validation in follow-up studies.
For Fig. 2b, we used genes shared across five or more tissues for this analysis, as the gene set shared by six or more tissues (Fig. 2a) was too small a set for this analysis. The upregulated and downregulated genes shared across five tissues (derived from ‘sex-combined’ analysis and listed in Supplementary Table 9) were separately used for the hypergeometric test implemented in GOstats (v2.68.0). The background genes (‘universe’) were defined as all the genes with a non-NA value for the FDR for a given comparison. The full GO analysis results are given in Supplementary Table 10.
Gene expression trajectory analysis
Hierarchical clustering was performed on gene expression trajectories for genes expressed in all tissues. A gene was considered expressed if greater than 80% of the samples for a tissue type had a TPM of greater than 0.5. The intersection of expressed genes in each tissue resulted in 10,847 genes expressed in all tissues. For each tissue analyzed, the third age bin (102–103 days) was removed to avoid the lower sample number at this time point from driving the gene expression trajectory trend.
For each gene in each tissue, LOESS regression was performed to find a ‘trajectory’ for the z-scaled normalized gene counts over age. These gene expression trajectories were then grouped into clusters using hierarchical clustering (see the gene list for each expression cluster in Supplementary Table 11).
Optimal cluster number, k, was identified based on a balance of two criteria: (1) cluster robustness (as measured by silhouette score) and (2) gene-set size appropriateness for enrichment tests. Average silhouette scores were computed for k = 2–20 using the ‘silhouette’ function in the R package cluster v2.1.8. For the brain (Supplementary Fig. 6), for example, silhouette scores declined from ~0.31 (k = 2) to ~0.16 (k = 5) and then remained nearly constant (0.12–0.11) through k = 10 before falling steeply to <0.10 at k = 11 and beyond. The inflection at k = 10 marked the largest k that did not compromise cluster cohesion and separation. For pathway enrichment, we typically require a gene set of ~100 to 1,000 genes (~10% of the expressed genes) as input, given the annotation coverage of the killifish genome version we used. At k = 5, for three of the five clusters, each cluster’s gene-set size constituted 23–31% of the expressed genes. By contrast, at k = 10, for eight of the ten clusters, each cluster’s gene-set size constituted <11% of the expressed gene (one cluster’s gene-set size constituted 24%). Thus, at k = 10, most of the gene-set sizes fell more within the typical bounds for enrichment analysis, providing adequate statistical power while preserving biological granularity. We used k = 10 for our analysis, as it represented the optimal trade-off between cluster robustness and functional interpretability.
Genes that made up each cluster were then analyzed using hypergeometric GO enrichment to identify enriched biological pathways. The complete GO analysis results are provided in Supplementary Table 12.
Conservation analysis for ncRNA-3777
The cDNA sequence of ncRNA-3777 was input into the ‘sequence search’ in the RNACentral database (https://rnacentral.org/), which performed local alignment using nhmmer112 with default parameters. All the human, mouse and zebrafish sequences returned by the search function were downloaded and examined for their genomic locations and known functions. The input sequence and the search results were summarized in Supplementary Table 13.
Protein sequence alignment and AlphaFold analysis
We measured the protein sequence similarity between killifish and human IGF2BP3 proteins by aligning the two sequences using Clustal Omega (https://www.ebi.ac.uk/jdispatcher/msa/clustalo/)113. The percentage identity matrix and the sequence alignment file were output files from this alignment analysis (Supplementary Table 14).
The protein sequence encoded by the killifish IGF2BP3 gene (LOC107383282) was placed in the ‘Search’ function of the AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/). This search returned the prediction data for the sequence named ‘AF-A0A1A8U593-F1-v4’ in the AlphaFold database. Using the MMseqs2 clustering algorithm in the AlphaFold database, the orthologous human sequence was identified (AF-Q9NZI8-F1). We downloaded the Protein Data Bank files for both the killifish and human proteins and imported these files into the ChimeraX software (v1.9rc202412100036)114,115,116 to adjust the display color of the protein sequences and orientation and to perform superimposition between the killifish and human sequences using the ‘Matchmaker’ function (parameter: ‘best pairing of chains’, reference sequence is the human sequence). The protein sequence images were exported and saved as ‘png’ files (resolution, 2,000 × 1,920; supersample, none).
Conservation analysis for DEGs
We selected one comprehensive human RNA-seq dataset and one mouse RNA-seq dataset to assess gene-level and pathway-level aging signatures. For the human dataset, we used the GTEx v10 dataset. Raw .gct files and accompanying sample metadata were obtained from the GTEx portal. Differential expression analysis was performed on each tissue-specific DESeq2 object (v1.42.1)107, using age and sex as covariates (design = ~age + sex). For the mouse dataset, we used the Tabula Muris Senis RNA-seq dataset9 and obtained tissue-level DESeq2 results (age and sex as covariates) from P.M.-L. Killifish differential expression analyses were performed as described in ‘DESeq2 differential expression analysis’.
For each species, we identified DEGs between young and old individuals. In humans, this comparison included 20–29 years (~100% survival) versus 70–79 years (~60–80% survival, the oldest age group)117. For mice, we used 3 months (100% colony survival) and 24 months (50–75% colony survival; https://www.nia.nih.gov/research/dab/aged-rodent-colonies/). For killifish, we compared 47–52 days (~100% colony survival) to 133–134 days (~30% colony survival), due to under-sampling at the ~50% survival time point.
To assess cross-species conservation of aging-associated transcriptional changes, we compared DEGs from each killifish tissue to DEGs from anatomically analogous tissues in humans and mice. In cases where multiple anatomical matches were possible (for example, human gut regions), comparisons were performed separately (Supplementary Table 15). Only genes that were significantly differentially expressed (FDR-adjusted P value < 0.05) and changed in the same direction with age in both species were included in the overlap analysis. Killifish gene names were mapped to mouse and human orthologs as appropriate for each comparison. Overlapping DEGs were visualized using log2-fold change distributions and box plots in ggplot2. Species-specific and shared DEG lists are provided in Supplementary Table 15.
For the brain, we further compared the set of significant killifish DEGs (FDR < 0.05) to a previously defined set of 82 genes associated with aging across multiple mouse brain regions—termed the ‘common aging score’57. The log2-fold changes of the intersecting genes were plotted for both species as a paired bar plot in Fig. 2j.
We next assessed conservation of age-associated expression changes at the pathway level. GSEA was independently performed on each tissue-specific DEG set. For human and mouse, species-specific GO terms were used. For killifish, gene names were converted to mouse or human orthologs and analyzed accordingly. Significant GO terms (FDR < 0.10) were identified in each species and overlapped across analogous tissues to detect conserved aging-associated pathways. To summarize results, significant GO terms were reduced into parent categories using the rrvgo package v1.14.2, and representative terms were visualized in bubble plots. Complete lists of species-specific and overlapping GO terms are available in Supplementary Table 15.
Identification of cell-type-specific immune cell genes
The data exploration application (https://alanxu-usc.shinyapps.io/nothobranchius_furzeri_atlas/) associated with the publication59 was used to identify cell-type-specific expression of immune genes. The ‘Bubbleplot/Heatmap’ tab was used to generate the gene expression dot plot for cell types (Extended Data Fig. 9a). The ‘CellInfo versus GeneExpr’ tab was used to generate uniform manifold approximation and projection plots with single gene expression overlaid (Extended Data Fig. 9g). The ‘Gene coexpression’ tab was used to generate gene coexpression uniform manifold approximation and projection plots (Extended Data Fig. 9h). For all plots generated using this dataset for this publication, plots were downloaded as PNGs and edited slightly for figure clarity in Adobe Illustrator.
Single-cell deconvolution of head kidney RNA-seq
We used the R package ‘granulator’ (v1.10.0)118 and a published single-cell dataset59 as the single-cell reference to perform single-cell deconvolution of our bulk kidney dataset. Granulator is a unified testing interface that enables rapid execution and benchmarking of seven single-cell deconvolution methods.
Granulator runs all deconvolution algorithms sequentially using the same input data: (1) bulk expression sample profiles represented as TPM and (2) a single-cell expression reference profile represented as TPM. To generate TPM-normalized bulk sample profiles, the raw RNA-seq counts for kidney samples were normalized by gene length and scaled by sample to per million. To generate the TPM-normalized single-cell reference, we first split the raw counts for male-derived and female-derived single cells. According to Teefy et al.59, some cell types in the kidney exhibit sex dimorphism in both their transcriptional profile and cell-type proportion (Supplementary Fig. 10). These sex-specific gene matrices were then normalized to get TPM values. Because the R package granulator requires a single expression profile per cell type, TPM values were averaged across individual cells of a cell type to get a unified cell-type expression profile, which served as input into granulator.
For some cell types, granulator outputted negative estimates of their cell-type proportions. According to the package vignette, a negative estimate can result from a mismatch between the single-cell reference with the bulk data, due to technical factors (for example, noise, tight expression collinearity of two cell types and undetected cell types by the single-cell dataset). The package vignette recommends refining the single-cell reference by benchmarking across several single-cell datasets, which are currently unavailable for the killifish kidney. Therefore, we set any negative value to zero, so that these cell types did not contribute to the cell-type proportion calculation. For the remaining cell types with nonzero estimates, the estimates were summed, and the sum was scaled to 100%. Additionally, we observed that the estimated cell-type proportions generated by granulator varied across the seven deconvolution algorithms (Supplementary Fig. 11). Thus, we presented the predicted proportions from three main models. The resulting proportions were then plotted by cell type across age using ggplot2. In Extended Data Fig. 8b, we defined the estimated proportion of lymphoid cells as the summed proportions of natural killer/T cells and B cells, and that of myeloid cells as the summed proportions of thrombocytes, neutrophils, mast cells and macrophages. The ‘young’ group was from the age group of 47–52 days, and the ‘old’ group was from the age group of 133–134 days for females and 161–162 days for males. The cell proportion data are listed in Supplementary Table 24.
One limitation of our analysis was that some differences between our dataset and the published reference dataset may have contributed to reduced model performance. The published single-cell dataset59 was generated under somewhat different conditions: both head and tail kidney were used, and samples were pooled from multiple young animals. With these caveats in mind, our deconvolution results nevertheless supported an age-associated alteration in kidney immune cell composition in female killifish.
In situ validation of the age-related gene expression changes
Tissues were collected from validation cohort animals (see ‘African turquoise killifish husbandry’) and placed directly into ~6 ml 4% paraformaldehyde (Santa Cruz Biotechnology, CAS 30525-89-4). Metadata for the animals used in each experiment are listed in Supplementary Table 16. Samples were fixed for 16–24 h, washed with cold ~12 ml nuclease-free PBS (Corning, 21-040-CM) for four 1-h washes, and then incubated in a nuclease-free methanol/PBS buffer series with each wash on ice for at least 10 min: 66% methanol/33% PBS, 100% methanol and 100% methanol (Sigma-Aldrich, 34860-1L-R). Samples were then stored in fresh 100% methanol at −20 °C until cryo-sectioning.
To prepare samples for cryo-sectioning, the samples were removed from −20 °C storage and put through a reverse nuclease-free methanol/PBS buffer series to rehydrate the samples: 75% methanol/25% PBS, 50% methanol/50% PBS, 25% methanol/75% PBS and 100% PBS. Samples were incubated in 1 ml of each buffer for 15–30 min on ice. After the methanol/PBS series, an additional wash in 1× PBS was performed for 15 min, and then samples were placed in 1 ml 30% nuclease-free sucrose solution (sucrose dissolved in nuclease-free 1× PBS, then filter-sterilized) and stored at 4 °C overnight.
Tissue-specific embedding and sectioning strategies
The day after, samples were removed from the sucrose solution and dissected to prepare them for embedding. For each tissue, the dissection strategy was unique: kidney marrow ‘lobes’ were dissected away from the muscle wall, and gut samples were cut lengthwise from the posterior to the anterior end to create a flat sheet. After dissection, samples were preincubated in the Neg-50 Frozen Section Medium (Fisher Scientific, 22-110-617) in individual wells of a 24-well plate (Corning, 353046) at room temperature for 10–15 min. Before placing gut samples in Neg-50, they were gently rinsed in sucrose solution using a Pasteur pipette to wash away residual food debris from the lumen.
Following preincubation, each tissue type required unique embedding strategies in Neg-50. Kidney marrow lobes were embedded side-by-side, maintaining left–right and anteroposterior orientation. Gut samples were rolled using the ‘Swiss roll’ technique, with the anterior intestinal bulb’s luminal surface toward the center of the spiral and the posterior intestine toward the outside119. Samples were placed into a cryomold containing a thin (~1–2 mm) sheet of frozen Neg-50 Medium on dry ice. Additional Neg-50 Medium was added, and the sample was left on dry ice to freeze fully. Frozen blocks were placed at −20 °C until sectioning.
Samples were sectioned in batches by sex and age group so that the same section plane for each animal in the group was mounted on the same slide. All animals were given unique blinding IDs and deconvolved after quantification of mRNA spot count data. Samples were sectioned (30 µm) on a cryostat (Leica, CM3050 S), mounted on charged glass slides (Fisher Scientific, 22-037-246) and stored at −20 °C until staining.
HCR
To validate mRNA expression from the atlas, a fluorescence in situ hybridization technique named HCR was used120. The probes for each mRNA were designed using a custom-made Python script120, purchased from IDT as ‘oPools’ and listed in Supplementary Table 17. The following HCR amplifiers were purchased as solutions from Molecular Instruments and are listed in the format of ‘Amplifier-fluorophore’: B1-647, B3-546 and B5-488.
HCR was performed according to a protocol from Molecular Instruments (‘HCR RNA-FISH, fresh/fixed frozen tissue sections’). Briefly, tissue sections were equilibrated to room temperature from −20 °C, rehydrated in 0.5–1 ml PBS for 5–10 min, and residual Neg-50 was gently washed off using PBS (‘Neg-50-free’). To reduce autofluorescence in the kidney samples, we photobleached the slides by incubating the Neg-50-free kidney slides in 1 ml 1× PBS under an intense LED light (‘RAYHOO 18 W LED’, Amazon, B0CR1CHP7X) in an opaque chamber (cardboard box) at 4 °C for at least 45 min. For the gut (and kidney, optionally), the Neg-50-free sections were first baked at 60 °C for 1 h in an in situ hybridization oven to increase adhesion between the tissue samples and the glass slides. After baking, the samples were rehydrated in 500 µl of 100% ethanol, 500 µl of 70% ethanol and 500 µl of 50% ethanol for 5-min incubations each. Next, the sections were post-fixed using 4% paraformaldehyde (diluted from 32% paraformaldehyde (Electron Microscopy Sciences, 15714-S) in PBS) at room temperature for 15 min, followed by four 500 µl PBST washes with a 5-min incubation between each wash, and then prehybridized.
After prehybridization, the buffer was removed, and 100 µl hybridization buffer (for each HCR probe, use 1 µl of the 0.5 pmol µl−1 stock per 100 µl hybridization buffer) was added to each slide, followed by a 37 °C incubation for 16–20 h. After hybridization, each slide was washed with 500 µl HCR probe wash buffer (Molecular Instruments, buffer type: tissue section), 500 µl 75% wash buffer (75% HCR probe wash buffer, 25% 5× SSCT), 500 µl 50% wash buffer (50% HCR probe wash buffer, 50% 5× SSCT), 500 µl 25% wash buffer (25% HCR probe wash buffer, 75% 5× SSCT) and 500 µl 5× SSCT (diluted from 20× SSCT (Ambion, AM9770) with nuclease-free water) at 37 °C with a 15-min incubation for each wash. Next, each slide was incubated in 200 µl HCR amplification buffer (Molecular Instruments, buffer type: tissue section) for 30 min to 4 h before switching to 100 µl amplification buffer supplemented with the fluorescent hairpin pairs (prepared according to the manufacturer’s instructions) for 20–24-h incubation at room temperature in the dark. Lastly, each sample was washed twice in 500 µl of 5× SSCT/DAPI (10 µg ml−1 DAPI), with a 30-min incubation for each wash, followed by an optional 5-min 500 µl 5xSSCT wash. The slides were mounted with ProLong Gold Antifade reagent (Thermo Fisher, P36934) and sealed with nail polish.
The slides were imaged using a Zeiss LSM 900 confocal laser scanning microscope (Zeiss) equipped with Zen 3.0 (blue edition) software, a Zeiss Plan-Apochromat ×40/1.4 oil objective and Zeiss Immersol oil 518 F (Zeiss, 4Y00-R0DY-1007-3VF3) as the immersion medium. The imaging conditions were the following: 9-slice z-stacks with a step size of 0.75 µm; the Alexa Fluor 546 channel (laser at 1%; detector gain, 775 V; detector offset, 256; detector digital gain, 1.0); the Alexa Fluor 488 channel (laser at 2.5%; detector gain, 650 V; detector offset, 256; detector digital gain, 1.0); the Alexa Fluor 647 channel (laser at 8.0%; detector gain, 650 V; detector offset, 512; detector digital gain, 1.0); and DAPI (laser at 0.5%; detector gain, 650 V; detector offset, 256; detector digital gain, 1.0). Four fields of view per tissue section and four animals per condition were imaged. All images were taken in comparable regions across biological replicates, specifically along the caudal–rostral axis of the ‘Swiss roll’ for the gut (using individual villi as landmarks) and along the caudal–rostral axis for the kidney (in interstitial and kidney tubule epithelial regions).
Quantification of HCR images
All samples were blinded and randomized after tissue harvest. Each sample was assigned a unique sample ID, which was used for sample processing and imaging. The sample information was not revealed until after image quantification. To quantify IGF2BP3 (LOC107383282), LOC107373777 (ncRNA-3777) and irf4a (LOC107383908) mRNA levels, we first performed maximum-intensity projection in the z-direction for all images using a FIJI121 macro script (z-planes 4–6 were used for IGF2BP3 and ncRNA-3777, and all nine z-planes were used for irf4a). Maximum projection images were then loaded into QuPath software (v.0.5.1, https://qupath.github.io/) to quantify mRNA spots. First, the cells were segmented using a nuclear mask created based on the DAPI signal (DAPI threshold, 3000; sigmaMicrons, 1.5; minAreaMicrons, 10.0; maxAreaMicrons, 400.0). Then an expansion of 10 µm from the DAPI mask was used as the cell boundary. Detections of red blood cells (which exhibited strong autofluorescence in all channels) and cells located in the kidney tubule regions were manually removed to avoid false-positive subcellular spot detections. All raw images can be accessed on FigShare+122,123.
Next, we used the QuPath subcellular detection function to detect each type of mRNA using specific parameters. Because the gut images had highly variable background signals, each image required a separate threshold to detect the signal from noise for counting the IGF2BP3 and ncRNA-3777 mRNA spots. To consistently distinguish signal from background, for each fluorescent channel, we plotted the distribution of the maximum signal of each cell (k), found the mean value of k and defined ‘signal’ to be at least 0.5 standard deviations above the mean of k in the image. This method matched well with manual counting. The kidney images had mostly consistent backgrounds, so the same QuPath detection parameters were applied to most of the kidney images. However, a subset of the kidney images required different parameters to accommodate a high background (see Supplementary Table 17 for the complete parameter record and results). For irf4a quantification, only cells located in the interstitial region were counted. After QuPath detection, every cell was visually inspected to confirm that the detected spots matched the results of manual counting. A small number of false-positive spots were manually removed (these spots usually occurred in regions overlapping with red blood cells). The number of a specific type of mRNA per cell and the cell number of each image were recorded (Supplementary Table 18).
We reported the average mRNA counts per cell for each animal. For this calculation, we first determined the total number of mRNA spots by summing the counts across all cells in four fields of view imaged for an animal. We calculated the average number of mRNA counts per cell by dividing the total number of mRNA counts by the total number of cells summed across the four fields of view. When comparing the differences in average mRNA counts between young and old samples within one sex, we calculated the statistical significance using the Mann–Whitney U-test. We used two-way ANOVA (sex, age and sex–age interaction) to analyze both sexes together. We noted that all conditions except one (old female for IGF2BP3) were confirmed to have normal distributions by the Shapiro–Wilk test.
Cell dissociation and flow cytometry of killifish head kidney
Animals were randomly selected from the validation cohorts to use for flow cytometry experiments. For males, three batches of young and old animals were processed for head kidney flow cytometry (three batches per experiment, with the old males aged from 136 to 143 days old or from 151 to 179 days old). Two batches were used for females. Experimental metadata are documented in Supplementary Table 19.
Fish were anesthetized for 1.5 min in an ice slurry made using system water. Once operculum movement slowed, and the fish was no longer responsive to touch, the animal was dissected and transcardially perfused with 10 ml of ice-cold 0.25 M EDTA solution (Fisher Scientific, AAJ15694AP) in 1× PBS (Thermo Fisher, 10010049) as described above. Following perfusion, head kidney tissue was carefully dissected from the body wall and placed in 5 ml of ice-cold fetal bovine serum (FBS; Fisher, 50-152-7067) in a well of a 12-well culture plate (Celltreat, 229111). This process was repeated until all animals in the batch were perfused and dissected.
Single-cell suspensions from head kidney tissue were prepared for flow cytometry using a non-enzymatic dissociation protocol adapted from zebrafish124. Kidney marrow in FBS was pipetted 50 times with a 5-ml serological pipette to dissociate the tissue mechanically. The digestion mixture was then applied to a 100-µm cell strainer (Fisher Scientific, 07-201-432) sitting atop a 50-ml conical tube (Fisher Scientific, 1443222). Tissue clumps remaining on the mesh were gently triturated using the plunger of a 1-ml syringe (Fisher Scientific, 14-826-88), and then 5 ml of SM buffer (5% FBS in 1× PBS) was used to wash the well of the 12-well plate and the 100-µm strainer mesh. Filtered cells were then pelleted (400g, 4 min, 4 °C), and the supernatant was removed using a 10-ml serological pipette until about 200 µl remained. The pellet was then resuspended in 5 ml of SM buffer by pipette five times and then was applied to a 40-µm cell strainer (Sigma-Aldrich, CLS431750-50EA) on top of a 50-ml tube. The strainer was then washed with 2 ml of SM buffer, and cells were pelleted once more under the same conditions. The supernatant was again removed (leaving about 100 µl of SM buffer), and pellets were resuspended using 500 µl of additional SM buffer. The cell suspension was moved to a 1.5-ml Low-Adhesion Tube (USA Scientific, 1415-2600) and centrifuged at 400g for 2 min at 4 °C. Finally, the supernatant was removed until 200 µl remained. Cells were resuspended, and about 5–10 min before the sample loading onto the cytometer, 15 µl of the live/dead stain 7-AAD (BD Biosciences, 559925) was added. Right before loading onto the cytometer, the cell suspension was passed through the 35-µm strainer mesh cap of a 5-ml round-bottom FACS tube (Corning, 352235) followed by gentle vortex. Then, the sample was loaded for analysis and/or sorting on a Sony MA900 Cell Sorter (nozzle size, 100 µm; flow rate, 4).
Gates were drawn to exclude debris and to capture live, single cells. Then, gross populations of immune cells (erythroid, myeloid, lymphoid, progenitor, all leukocytes) were identified by side scatter and forward scatter based on a protocol developed for zebrafish64. The myeloid:lymphoid ratio was calculated by dividing the total number of myeloid cells by the total number of lymphoid cells for each sample. For all cytometry plots used for quantification, a total of 50,000 events were recorded. Cytometric analysis was performed using FlowJo version 10.10.0.
Bulk RNA-seq of FACS-sorted cells
Five males of various ages (67, 88 and 201 days) were used to test the gating strategies for sorting different populations of kidney-dissociated cells by FACS. The number of cells collected per sample is listed in Supplementary Table 20. Cells were sorted into 350 µl Buffer RLT Plus (reagent from the QIAGEN RNeasy Plus Micro Kit, QIAGEN, 74034) containing β-mercaptoethanol (10 µl β-ME per 1 ml Buffer RLT Plus), briefly vortexed for 30 s, and then frozen immediately on dry ice. For sorted volumes exceeding 500 µl, additional Buffer RLT Plus was added to the sorted cells before vortexing and freezing at a ratio of 350 µl Buffer RLT Plus for each additional 100 µl of cytometer sheath fluid. Frozen homogenates were stored at −80 °C until RNA extraction.
Bulk total RNA extraction of the sorted cell populations from flow cytometry was performed using the QIAGEN RNeasy Plus Micro Kit (QIAGEN, 74034) according to the manufacturer’s instructions. The frozen homogenates of sorted cells were thawed on ice for 30 min. Once completely thawed, homogenates were briefly centrifuged, then applied to a gDNA Eliminator spin column and centrifuged at 10,000g for 30 s. The flow-through was then added to a DNA LoBind tube (Sigma-Aldrich, 022431021) containing an equal volume of 70% ethanol. This process was repeated until all the remaining volume of homogenate was passed through a gDNA Eliminator spin column and mixed with an equal volume of 70% ethanol. Then, samples were pipette mixed, transferred to a RNeasy MinElute Spin Column and centrifuged at 10,000g for 15 s, discarding the flow-through. This step was repeated until the entire ethanol–homogenate mixture was applied to the RNeasy MinElute Spin Column. The column was then washed twice, first with 700 µl of Buffer RW1 and then 500 µl of Buffer RPE, centrifuging at 10,000g for 15 s and discarding the flow-through each time. A final, longer wash was performed with 80% ethanol, after which the spin column was centrifuged for 2 min at 10,000g. Then, the spin column was transferred to a new collection tube and dried by centrifuging at 12,000g for 5 min. Finally, the column was transferred to a new 1.5-ml collection tube, and 14 µl of RNase-free water was applied to the membrane. The column was then centrifuged for 1 min at 12,000g to elute the RNA. RNA was quantified using the Quant-iT RNA BR kit (Thermo Fisher, Q10213) on a Varioskan LUX multimode microplate reader, aliquoted and stored at −80 °C.
cDNA and library synthesis were performed using a modified in-house Smart-seq2 pipeline similar to that described above for whole tissues (see ‘cDNA library generation and sequencing’), with a few modifications to accommodate lower input concentrations of RNA. First, the single-stranded library was amplified using 16 cycles. Next, tagmentation was performed using 0.1 µl of the Illumina Tn5 enzyme (Illumina, 20034198), 0.26 µl of nuclease-free water and 0.64 µl of 2.5× TAPS-PEG crowding agent per sample. The crowding agent was prepared by combining filtered 40% wt/wt PEG 8000 (Promega, V3011) 1:1 vol/vol with 5× TAPS-MgCl2 (3 ml of 0.5 M TAPS-NaOH pH 8.5 (Boston Bioproducts, BB2375) combined with 750 µl of 1 M MgCl2 (Sigma-Aldrich, M1028) and 26.25 ml of nuclease-free water and adjusted to pH 8.4). The resulting tagmented library was amplified for 12 cycles using the Kapa enzyme (KAPA HiFi PCR kit, Kapa Biosystems, KK2102).
RNA-seq analysis (quality control, mapping, count generation, DESeq2 analysis) and plotting were also performed as in the atlas dataset, except using a different expression cutoff from the atlas: in this case, genes with at least one count in at least one sample were retained (rather than requiring at least 80% of samples having at least one count). The PCA plot was generated using the ‘plotPCA’ function in DESeq2 (v.1.34.0), and the heat map was generated by ‘pheatmap’ (v.1.0.12) with the parameters: rows and columns clustered, z-scale for each gene, and capping z-scale at −2 and 2 (any value below −2 or above 2 was assigned as the lowest color or the highest color, respectively).
Calculation of transcriptomic age
Training tissue clock models using BayesAge 2.0
All computations relating to the tissue clocks were performed using Python (v3.11.11) in a series of Jupyter notebooks. Transcriptomic age was calculated using the published method BayesAge 2.0 (ref. 85). This method utilizes a Bayesian framework to estimate the most likely transcriptomic age of a sample (‘tAge’) and uses locally weighted scatterplot smoothing (LOWESS) regression to model the nonlinear dynamics of gene expression, enabling age prediction between 47 and 163 days of age at day-level resolution.
Before training tissue-specific models, we first preprocessed the raw gene expression matrix. Raw gene expression counts were normalized using frequency count normalization, in which raw counts were transformed into relative frequencies by dividing the raw count for each gene by the total read count for the sample. Next, LOWESS regression was used to fit a trend for each gene across age.
After preprocessing, we performed model training. We used leave-one-sample-out cross-validation (LOSO-CV) to separate our dataset into training and test sets: For each tissue clock, we split the tissue dataset of size N into a training set of N − 1 samples and a test set of one sample (‘left out’). For each training–testing group, we first trained a reference matrix by taking the gene frequency counts for each gene for the N − 1 samples in the training set, computed a LOWESS regression fit and performed feature selection for enhanced biological interpretation. To select features of interest, we calculated the Spearman’s rank correlation between gene frequency and age for each gene. A set of genes (M genes) with the highest absolute Spearman’s rank correlation was used for age prediction. Notably, each time LOSO-CV was performed, the identities of the top Spearman’s rank-correlated genes might differ, as leaving a different sample out might slightly alter the relationship between age and gene expression. The resulting trained matrix stored the predicted gene frequency levels using a LOWESS fit across age and the Spearman’s rank correlation values for each gene in the dataset for these N − 1 samples.
Next, we performed age prediction for the ‘left out’ test sample. We selected a given number (M) of top Spearman’s rank-correlated genes (different M values were included during testing, 5, 10, 15 and so on, up to 50 genes were iteratively tested), and for each gene, we computed the probability of observing the gene expression for that gene for a particular age, assuming a Poisson distribution. The probability for the age-related gene state is given by equation (1):
where x is the specific age, \({\lambda }_{x}\) is the expected gene expression count at age x and \({k}_{g}\) is the observed gene expression count for the test sample, ϕ.
The expected gene expression count was derived from the frequency-normalized trained reference matrix, and the observed gene expression count came from the observed raw counts of a particular gene from the M genes for the test sample.
Then, for each age x, the probability of the test sample being a given age was the product of the individual probabilities for each gene as given by equation (2):
The age prediction (tAge) for the test sample was then found by computing an age-likelihood distribution and finding the maximum likelihood age as given by equations (3) and (4):
To avoid numerical underflow errors during computation, we replaced the product of individual gene probabilities at a given age with the sum of logarithms of these individual gene probabilities. We determined the maximum likelihood age from this distribution. This operation preserves the numerical relationship and avoids Python rounding errors as shown in equations (5) and (6):
We repeated this process, leaving out a different sample from the tissue dataset until each sample had been tested. After this process, we obtained age predictions for each sample in our tissue dataset. Performing LOSO-CV with different gene-set sizes (M) informed us of the optimal M that corresponded to the most concordance between chronological and predicted age. We referred to this optimal condition for a tissue clock as the ‘optimal clock’ using the BayesAge model. We calculated the Pearson correlation (r), coefficient of determination (R2) and mean absolute error (MAE) using the Python SciPy (v1.13.1) package to evaluate model performance. The results for LOSO-CV for BayesAge are summarized in Supplementary Table 21.
Comparison of BayesAge 2.0 to other models
To perform age prediction using Elastic Net, we used DESeq2-normalized counts. We z-scaled the gene expression data using the StandardScaler function in the scikit-learn Python module (v1.5.2). Elastic Net is a linear regression model that combines Lasso (L1) and Ridge (L2) regularization. To optimize model performance, Elastic Net requires tuning of two hyperparameters, α and λ, which control the trade-off between L1 and L2 regularization and the strength of the regularization, respectively. To implement hyperparameter tuning, we performed a parameter grid search using the GridSearchCV function from scikit-learn for the parameter λ (called ‘alpha’ in scikit-learn’s implementation of Elastic Net) and α (called ‘l1 ratio’ in scikit-learn). This search was performed in two steps: first, we tested alpha values from 1 × 10−5, 1 × 10−4 and 1 × 10−3, continuing up to 100, and ‘l1 ratio’ values from 0 to 1 in increments of 0.1. The maximum number of iterations was set to 10,000 for most tissue clocks (except for the brain, for which it was set to 30,000). We implemented LOSO-CV for each combination of alpha and l1 ratio parameters using the LeaveOneOut function in scikit-learn. To evaluate model performance, we used MAE. Once the optimal parameters were identified, the second step involved increasing the maximum iteration number to 100,000 for all tissues to ensure convergence of the objective function and to finalize age predictions. The ‘optimal’ tissue clock for Elastic Net used the optimal parameters for α and λ derived from hyperparameter tuning. For these analyses, random seeding was set to 42 to ensure reproducibility. The results of hyperparameter tuning and LOSO-CV are summarized in Supplementary Table 21.
PC-R is a regression technique that combines principal component analysis and linear regression, fitting a linear regression model using a subset of the principal components as predictors. As in our implementation of Elastic Net, we used the DESeq2-normalized counts to implement PC-R and then scaled the data using the ‘StandardScaler’ function. To perform PCA analysis, we used the PCA function in scikit-learn. We implemented LOSO-CV for each principal component number from 5 to 20 in increments of five using the ‘cross_val_predict’ function in scikit-learn, and we evaluated model performance using MAE. The ‘optimal’ tissue clock for PC-R occurred when the identified principal component number maximized the coefficient of determination and minimized MAE. For these analyses, random seeding was set to 1 to ensure reproducibility. Results of LOSO-CV are summarized in Supplementary Table 21.
We note that the differing normalization methods used by BayesAge 2.0 and Elastic Net/PC-R reflect the distinct modeling strategies and goals of each aging clock. BayesAge 2.0 performs normalization by dividing each gene’s raw count by the total read count of the sample, producing relative expression values. This choice supports two key objectives of the model: (1) Probabilistic modeling: BayesAge 2.0 uses a Poisson distribution to model gene counts, where the expected value is tied to age-dependent parameters. Normalizing by total counts enables comparability across samples while ensuring the count structure remains interpretable within the Poisson framework. (2) Biological signal prioritization: This normalization also helps emphasize genes that correlate most strongly with age across the dataset, which improves the model’s interpretability and focus on age-relevant biological signals. In contrast, Elastic Net and PC-R use DESeq2 normalization, which adjusts for sequencing depth and composition bias through estimated size factors. This method is more suitable for regression-based models that rely on stabilized variance across genes and assume approximately homoscedastic input. DESeq2’s approach better supports the assumptions of linear models and ensures that high-count genes do not dominate the regression simply due to scale.
To evaluate the performance of BayesAge 2.0 in comparison to other models, we compared the residuals from BayesAge 2.0, to Elastic Net and PC-R by computing the residuals in two ways: (1) as the difference between the predicted age and the line of best fit, and (2) as the difference between predicted age and true chronological age for each sample.
Sex-combined versus sex-specific clocks
Regarding the performance difference between sex-combined and sex-specific clocks, the difference might occur due to how features were selected. Since BayesAge 2.0 used correlation-based gene preselection, it was more directly tuned to identify genes that tracked with age within each sex. Thus, when the model was trained separately by sex, it was likely to capture sex-specific aging signals more effectively—genes correlated with age in males, but not females (or vice versa), would be included in one model but excluded from the other. Elastic Net and PC-R relied on either regularization or dimensionality reduction applied across all features, often after normalization. These methods did not necessarily prioritize features with high correlation to age per sex. Thus, they might be less sensitive to subtle sex-specific patterns unless those patterns had strong global predictive power and did not account for potential nonlinear changes. Therefore, BayesAge 2.0 might benefit more from sex splitting because its feature selection was already correlation aware, making it more responsive to the biological differences introduced by sex-specific aging trajectories.
Identification of the top genes underlying each tissue clock
We identified the key genes contributing to each aging clock model and quantified their feature importance (FI) as follows: (1) BayesAge 2.0: Genes were selected for model inclusion based on their absolute Spearman rank correlation with age. Once selected, each gene contributed equally to the probabilistic inference of biological age. We reported the set of genes and their corresponding feature importance scores for each optimal sex-combined tissue clock. (2) Elastic Net: Gene contributions were defined by the learned regression coefficients. We reported the nonzero coefficients for genes expressed in each tissue, as these directly indicated both the magnitude and direction of each gene’s effect on predicted age. (3) Principal component regression: Because this approach relied on multiple principal components to predict age, we derived a composite feature importance score for each gene. Specifically, we multiplied each gene’s principal component loading by the regression coefficient of the corresponding principal component, summed across the top N principal components included in the model. Then the feature importance score for gene g, denoted the feature importance \({{FI}}_{g}\), is given by equation (7):
where g is a given gene, j is a principal component index, N is the number of top principal components used in the regression model, \({L}_{g,j}\) denotes the loading of gene g on principal component j and \({\beta }_{j}\) denotes the regression coefficient for principal component j.
After calculating the feature importance scores for each gene in each clock model, we examined the overlap of genes across approaches. We began with the genes comprising the BayesAge 2.0 optimal sex-combined tissue clocks and compared these to those contributing to the Elastic Net and principal component regression clocks. To highlight potential shared drivers of age prediction, we focused on the subset of BayesAge 2.0 genes that also ranked within the top decile of absolute feature importance scores in both the Elastic Net and principal component regression models.
The top genes underlying each tissue aging clock for the three machine-learning models are listed in Supplementary Table 22.
Age prediction in other datasets
To demonstrate the generalizability of our tissue-specific clocks to other datasets, we performed age prediction in five additional published RNA-seq datasets. These datasets include: (1) Xu et al.36: wild-type aging in brain, heart, muscle and spleen; (2) McKay et al.23: a liver transcriptomic dataset from male and female killifish fed on ad libitum and dietary-restriction diets from sexual maturity (4 weeks) to 9 weeks of age; (3) Astre et al.20: a liver dataset from a heterozygous AMP biosynthesis mutant APRT+/− and wild-type fish on ‘fed’ and ‘fasted’ diets; (4) Ripa et al.21: a fat (visceral adipose tissue) transcriptomic dataset from a constitutive AMPKγ1 mutant (UBI:γ1(R70Q)) and wild-type fish on ‘fed’ and ‘fasted’ diets; and (5) Smith et al.25: a gut dataset involving microbiota ‘transfers’ from 6-week-old male donors to 9.5-week-old male recipients (‘Ymt’) or from 9.5-week-old donors to 9.5-week-old male recipients (‘Omt’).
We applied three distinct modeling approaches for age prediction: BayesAge 2.0, Elastic Net and principal component regression (PC-R). For each dataset, both sex-combined and sex-split models were evaluated. FASTQ files and associated metadata were downloaded via the SRA Run Selector and processed using the same alignment and quantification pipeline as the Atlas dataset, accounting for whether reads were paired-end or single-end. Frequency-normalized raw feature counts were used as input for BayesAge 2.0, while DESeq2-normalized counts were used for Elastic Net and principal component regression.
Transfer calibration strategy and hold-out policy
After preprocessing, for BayesAge 2.0 and principal component regression, we performed a transfer calibration strategy due to the strong batch effects between the published datasets and our atlas data. We defined calibration samples as untreated control animals (for example, fed wild-type young and old fish) and selected two young and two old animals for each permutation of calibration. We used these calibration samples along with the atlas data for training. Importantly, we performed the following hold-out policy: (i) no test sample was used to fit the model predicting itself; (ii) all calibrations were repeated across all permutations (we rotated the selection of calibration samples to ensure that all control samples were used); (iii) uncalibrated performance was shown (Extended Data Fig. 10b) to demonstrate the need for transfer. We provided representative results from one rotation shown in Fig. 6.
For BayesAge 2.0, after selecting calibration samples, we constructed a reference matrix specific to the tissue in the query dataset. This reference matrix included all samples of the relevant tissue in the Atlas dataset and the calibration samples from the query dataset. For the remaining test (non-calibration) samples from the query dataset, the model predicted tAge of each sample by computing the age-likelihood distributions using the expected gene frequencies from the reference matrix and the observed expression from the query dataset. Gene-set sizes (M) from 5 to 200 (in increments of 5) were tested, and predictions for each gene number were reported. For the optimal clock, we selected the gene number at which predictions became stable for the first time (see Supplementary Fig. 9 for an example of gene number selection using the McKay et al. dataset23). We did not perform calibration for the McKay et al. dataset due to low batch effect (likely because this dataset was generated in our lab), and the BayesAge 2.0 age prediction for all the gene numbers tested for this dataset were included in Supplementary Fig. 9.
For PC-R, we applied PCA to the training data (Atlas + calibration samples) and performed linear regression to predict age on the remaining test samples from the query dataset (that is, all query samples except those used for calibration). We tested principal component dimensions from 5 to 20 (in increments of five) and evaluated performance to determine optimal dimensionality. We defined the optimal number of principal components as the point where the Mann–Whitney U-test P values stabilized across prediction outputs. As with BayesAge 2.0, calibration samples were rotated, and representative predictions are shown in Fig. 6.
For Elastic Net, we used a different calibration strategy based on the LOSO-CV approach using a combined dataset of atlas and query samples. Optimal parameters for λ and α were first determined through hyperparameter tuning, as described in ‘Comparison of BayesAge 2.0 to other models’, separately for the sex-combined, male-specific and female-specific clocks. Using these optimized parameters, we performed LOSO-CV by iteratively leaving out one sample (from either the atlas or query dataset), training the model on all remaining samples and predicting the age of the held-out sample. The nonzero coefficients, which represent the genes used by the model for age prediction, were reported. We noted that the current models used non-nested cross-validation for hyperparameter tuning, which might lead to a small level of ‘leakage’ from training to prediction. Future use of nested cross-validation will be valuable in overcoming this limitation.
Our modeling approaches produced generally robust age predictions, except in the case of three samples from Xu et al.36. These samples included two spleen samples (spleen 2, 6-week-old male; spleen 6, 6-week-old female) and one brain sample (brain 6, 6-week-old female). We performed a principal component-based outlier analysis on all spleen and brain samples in the dataset to identify samples whose values of PC1 were greater than two standard deviations away from the mean. Spleen 6 was detected to be an outlier along PC1 (26.4% of the variation), as was brain 6 along PC1 (21.83% of the variation in the brain dataset). Thus, we removed these samples from the dataset before BayesAge 2.0 and PC-R modeling. Spleen 2 was not identified as an outlier by this method but was removed from the plot due to poor model performance, as indicated in the figure legend. Outliers were not excluded for the Elastic Net model, as it was less sensitive to batch effects. It will be valuable to test whether performing batch correction methods before applying the aging clocks could improve model performance in future studies.
Either sex-combined or sex-specific predictions were plotted in Fig. 6. We selected the models to plot generally based on their better model performance in the original atlas model (Fig. 5b). The predicted age data for all datasets (both sex-combined and sex-specific models for BayesAge 2.0, Elastic Net and PC-R) are listed in Supplementary Table 23.
Statistics and reproducibility
All statistical tests were two sided. All experiments were randomized, and the investigators were blinded to allocation during experiments and outcome assessment. No statistical method was used to predetermine sample size, but our sample sizes were close to those reported in previous publications20,21,22,23,28,30,31,32,33,34,35,36,37,38,39. Data exclusion for lifespan and RNA-seq analyses was indicated in the respective method subsections. For differential expression analysis by DESeq2, we used a two-sided Wald test followed by the Benjamini–Hochberg method to correct for multiple hypothesis testing, generating an adjusted P value (FDR). DESeq2-normalized count distributions were assumed to be normal, despite not being formally tested. Unless otherwise noted, we defined statistical significance at FDR < 0.05. For identifying conservation of the age-associated expression changes at the pathway level, we reported the results at FDR < 0.1, as indicated in the text. The exact test names and number of animals per group are provided in the figure legends. We defined a biological replicate to be an individual fish. The nonparametric Mann–Whitney U-test was used for most analyses unless specified. Two-way ANOVA was performed if data and residual normality were confirmed using the Shapiro–Wilk test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw FASTQ files can be accessed in the Sequence Read Archive (SRA) under BioProject ID PRJNA1274512. The normalized expression data matrix is available on SRA and for exploration through an R-based Shiny application (https://twc-stanford.shinyapps.io/atlas/). Raw images can be accessed in FigShare+ (https://doi.org/10.25452/figshare.plus.29983765.v1 and https://doi.org/10.25452/figshare.plus.29983678.v1)122,123.
Code availability
All code has been shared in the public GitHub repository (https://github.com/emkcosta/KillifishAtlas/).
References
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell 186, 243–278 (2023).
Yousefzadeh, M. J. et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 19, e13094 (2020).
Jin, J., Yang, X., Gong, H. & Li, X. Time- and gender-dependent alterations in mice during the aging process. Int. J. Mol. Sci. 24, 12790 (2023).
Hagg, S. & Jylhava, J. Sex differences in biological aging with a focus on human studies. Elife 10, e63425 (2021).
Serre-Miranda, C. et al. Age-related sexual dimorphism on the longitudinal progression of blood immune cells in BALB/cByJ mice. J. Gerontol. A Biol. Sci. Med. Sci. 77, 883–891 (2022).
Zhu, D. et al. Sex dimorphism and tissue specificity of gene expression changes in aging mice. Biol. Sex Differ. 15, 89 (2024).
Menees, K. B. et al. Sex- and age-dependent alterations of splenic immune cell profile and NK cell phenotypes and function in C57BL/6J mice. Immun. Ageing 18, 3 (2021).
Oh, H. S. et al. Plasma proteomics links brain and immune system aging with healthspan and longevity. Nat. Med. 31, 2703–2711 (2025).
Schaum, N. et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583, 596–602 (2020).
Valdesalici, S. & Cellerino, A. Extremely short lifespan in the annual fish Nothobranchius furzeri. Proc. Biol. Sci. 270, S189–S191 (2003).
Kim, Y., Nam, H. G. & Valenzano, D. R. The short-lived African turquoise killifish: an emerging experimental model for ageing. Dis. Model Mech. 9, 115–129 (2016).
Cellerino, A., Valenzano, D. R. & Reichard, M. From the bush to the bench: the annual Nothobranchius fishes as a new model system in biology. Biol. Rev. Camb. Philos. Soc. 91, 511–533 (2016).
Harel, I. & Brunet, A. The African turquoise killifish: a model for exploring vertebrate aging and diseases in the fast lane. Cold Spring Harb. Symp. Quant. Biol. 80, 275–279 (2015).
Hu, C. K. & Brunet, A. The African turquoise killifish: a research organism to study vertebrate aging and diapause. Aging Cell 17, e12757 (2018).
Poeschla, M. & Valenzano, D. R. The turquoise killifish: a genetically tractable model for the study of aging. J. Exp. Biol. 223 (Suppl. 1), jeb209296 (2020).
Platzer, M. & Englert, C. Nothobranchius furzeri: a model for aging research and more. Trends Genet. 32, 543–552 (2016).
Kirschner, J. et al. Mapping of quantitative trait loci controlling lifespan in the short-lived fish Nothobranchius furzeri–a new vertebrate model for age research. Aging Cell 11, 252–261 (2012).
Gerhard, G. S. et al. Life spans and senescent phenotypes in two strains of Zebrafish (Danio rerio). Exp. Gerontol. 37, 1055–1068 (2002).
Boos, F., Chen, J. & Brunet, A. The African turquoise killifish: a scalable vertebrate model for aging and other complex phenotypes. Cold Spring Harb. Protoc. 2024, 107737 (2024).
Astre, G. et al. Genetic perturbation of AMP biosynthesis extends lifespan and restores metabolic health in a naturally short-lived vertebrate. Dev. Cell 58, 1350–1364 (2023).
Ripa, R. et al. Refeeding-associated AMPKγ1 complex activity is a hallmark of health and longevity. Nat. Aging 3, 1544–1560 (2023).
Moses, E. et al. The killifish germline regulates longevity and somatic repair in a sex-specific manner. Nat. Aging 4, 791–813 (2024).
McKay, A. et al. An automated feeding system for the African killifish reveals the impact of diet on lifespan and allows scalable assessment of associative learning. Elife 11, e69008 (2022).
Terzibasi, E. et al. Effects of dietary restriction on mortality and age-related phenotypes in the short-lived fish Nothobranchius furzeri. Aging Cell 8, 88–99 (2009).
Smith, P. et al. Regulation of life span by the gut microbiota in the short-lived African turquoise killifish. Elife 6, e27014 (2017).
Valenzano, D. R. et al. Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate. Curr. Biol. 16, 296–300 (2006).
Yu, X. & Li, G. Effects of resveratrol on longevity, cognitive ability and aging-related histological markers in the annual fish Nothobranchius guentheri. Exp. Gerontol. 47, 940–949 (2012).
Baumgart, M. et al. Longitudinal RNA-seq analysis of vertebrate aging identifies mitochondrial complex I as a small-molecule-sensitive modifier of lifespan. Cell Syst. 2, 122–132 (2016).
Wei, J. et al. Effects of metformin on life span, cognitive ability, and inflammatory response in a short-lived fish. J. Gerontol. A Biol. Sci. Med. Sci. 75, 2042–2050 (2020).
Baumgart, M. et al. RNA-seq of the aging brain in the short-lived fishN. furzeri- conserved pathways and novel genes associated with neurogenesis. Aging Cell 13, 965–974 (2014).
Reichwald, K. et al. Insights into sex chromosome evolution and aging from the genome of a short-lived fish. Cell 163, 1527–1538 (2015).
Cencioni, C. et al. Aging triggers H3K27 trimethylation hoarding in the chromatin of Nothobranchius furzeri skeletal muscle. Cells 8, 1169 (2019).
Kelmer Sacramento, E. et al. Reduced proteasome activity in the aging brain results in ribosome stoichiometry loss and aggregation. Mol. Syst. Biol. 16, e9596 (2020).
Teefy, B. B. et al. Dynamic regulation of gonadal transposon control across the lifespan of the naturally short-lived African turquoise killifish. Genome Res. 33, 141–153 (2023).
Mazzetto, M. et al. RNA-seq analysis of brain aging in wild specimens of short-lived turquoise killifish: commonalities and differences with aging under laboratory conditions. Mol. Biol. Evol. 39, msac219 (2022).
Xu, A. et al. Transcriptomes of aging brain, heart, muscle, and spleen from female and male African turquoise killifish. Sci. Data 10, 695 (2023).
Van Houcke, J. et al. Aging impairs the essential contributions of non-glial progenitors to neurorepair in the dorsal telencephalon of the killifish Nothobranchius furzeri. Aging Cell 20, e13464 (2021).
Vanhunsel, S. et al. The killifish visual system as an in vivo model to study brain aging and rejuvenation. NPJ Aging Mech. Dis. 7, 22 (2021).
Bergmans, S. et al. Age-related dysregulation of the retinal transcriptome in African turquoise killifish. Aging Cell 23, e14192 (2024).
Benayoun, B. A. et al. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res. 29, 697–709 (2019).
Di Fraia, D. et al. Altered translation elongation contributes to key hallmarks of aging in the killifish brain. Science 389, eadk3079 (2025).
Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014).
Oliva, M. et al. The impact of sex on gene expression across human tissues. Science 369, eaba3066 (2020).
Consortium, R. N. et al. RNAcentral: an international database of ncRNA sequences. Nucleic Acids Res. 43, D123–D129 (2015).
Baljon, K. J. et al. LncRNA PVT1: as a therapeutic target for breast cancer. Pathol. Res. Pract. 248, 154675 (2023).
Liu, S. et al. Long noncoding RNA PVT1 promotes breast cancer proliferation and metastasis by binding miR-128-3p and UPF1. Breast Cancer Res. 23, 115 (2021).
Tang, J. et al. LncRNA PVT1 regulates triple-negative breast cancer through KLF5/beta-catenin signaling. Oncogene 37, 4723–4734 (2018).
Dong, L., Wang, H., Gao, Y., Wang, S. & Wang, W. Long non-coding RNA PVT1 promotes the proliferation, migration and EMT process of ovarian cancer cells by regulating CTGF. Oncol. Lett. 25, 71 (2023).
Wu, Y., Gu, W., Han, X. & Jin, Z. LncRNA PVT1 promotes the progression of ovarian cancer by activating TGF-beta pathway via miR-148a-3p/AGO1 axis. J. Cell. Mol. Med. 25, 8229–8243 (2021).
Li, L., Chen, J., Wang, A. & Yi, K. ALKBH5 regulates ovarian cancer growth via demethylating long noncoding RNA PVT1 in ovarian cancer. J. Cell. Mol. Med. 28, e18066 (2024).
Chen, Y., Li, F., Li, D., Liu, W. & Zhang, L. Atezolizumab and blockade of lncRNA PVT1 attenuate cisplatin resistant ovarian cancer cells progression synergistically via JAK2/STAT3/PD-L1 pathway. Clin. Immunol. 227, 108728 (2021).
Mao, Z., Xu, B., He, L. & Zhang, G. PVT1 promotes angiogenesis by regulating miR-29c/vascular endothelial growth factor (VEGF) signaling pathway in non-small-cell lung cancer (NSCLC). Med. Sci. Monit. 25, 5418–5425 (2019).
Lederer, M., Bley, N., Schleifer, C. & Huttelmaier, S. The role of the oncofetal IGF2 mRNA-binding protein 3 (IGF2BP3) in cancer. Semin Cancer Biol. 29, 3–12 (2014).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 52, D368–D375 (2024).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Hahn, O. et al. Atlas of the aging mouse brain reveals white matter as vulnerable foci. Cell 186, 4117–4133 e4122 (2023).
Bjorgen, H. & Koppang, E. O. Anatomy of teleost fish immune structures and organs. Immunogenetics 73, 53–63 (2021).
Teefy, B. B. et al. Widespread sex dimorphism across single-cell transcriptomes of adult African turquoise killifish tissues. Cell Rep. 42, 113237 (2023).
Liang, Y., Van Zant, G. & Szilvassy, S. J. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood 106, 1479–1487 (2005).
Norddahl, G. L. et al. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell 8, 499–510 (2011).
Dykstra, B., Olthof, S., Schreuder, J., Ritsema, M. & de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 208, 2691–2703 (2011).
Carneiro, M. C. et al. Short telomeres in key tissues initiate local and systemic aging in zebrafish. PLoS Genet. 12, e1005798 (2016).
Mahony, C. B. & Monteiro, R. Protocol for the analysis of hematopoietic lineages in the whole kidney marrow of adult zebrafish. STAR Protoc 5, 102810 (2024).
Mittrucker, H. W. et al. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science 275, 540–543 (1997).
Hagman, J. Critical functions of IRF4 in B and T lymphocytes. J Immunol 199, 3715–3716 (2017).
Consortium, I. R. F. I. et al. A multimorphic mutation in IRF4 causes human autosomal dominant combined immunodeficiency. Sci. Immunol. 8, eade7953 (2023).
Wang, S., He, Q., Ma, D., Xue, Y. & Liu, F. Irf4 regulates the choice between T lymphoid-primed progenitor and myeloid lineage fates during embryogenesis. Dev. Cell 34, 621–631 (2015).
Rutledge, J., Oh, H. & Wyss-Coray, T. Measuring biological age using omics data. Nat. Rev. Genet. 23, 715–727 (2022).
Palmer, R. D. Aging clocks & mortality timers, methylation, glycomic, telomeric and more. A window to measuring biological age. Aging Med. 5, 120–125 (2022).
Horvath, S. & Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 19, 371–384 (2018).
Field, A. E. et al. DNA methylation clocks in aging: categories, causes, and consequences. Mol. Cell 71, 882–895 (2018).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Levine, M. E. et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 10, 573–591 (2018).
Yang, Y. et al. Metformin decelerates aging clock in male monkeys. Cell 187, 6358–6378 (2024).
Petkovich, D. A. et al. Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab. 25, 954–960 (2017).
Meyer, D. H. & Schumacher, B. Aging clocks based on accumulating stochastic variation. Nat. Aging 4, 871–885 (2024).
Meyer, D. H. & Schumacher, B. BiT age: a transcriptome-based aging clock near the theoretical limit of accuracy. Aging Cell 20, e13320 (2021).
Oh, H. S. et al. Organ aging signatures in the plasma proteome track health and disease. Nature 624, 164–172 (2023).
Fitzgerald, K. N. et al. Potential reversal of epigenetic age using a diet and lifestyle intervention: a pilot randomized clinical trial. Aging 13, 9419–9432 (2021).
Quach, A. et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 9, 419–446 (2017).
Thompson, M. J. et al. A multi-tissue full lifespan epigenetic clock for mice. Aging 10, 2832–2854 (2018).
Lu, Y. et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 588, 124–129 (2020).
Martin-Herranz, D. E. et al. Screening for genes that accelerate the epigenetic aging clock in humans reveals a role for the H3K36 methyltransferase NSD1. Genome Biol. 20, 146 (2019).
Mboning, L., Costa, E. K., Chen, J., Bouchard, L. S. & Pellegrini, M. BayesAge 2.0: a maximum likelihood algorithm to predict transcriptomic age. Geroscience 47, 5389–5399 (2025).
Fong, S. et al. Principal component-based clinical aging clocks identify signatures of healthy aging and targets for clinical intervention. Nat. Aging 4, 1137–1152 (2024).
Kawaguchi, D., Furutachi, S., Kawai, H., Hozumi, K. & Gotoh, Y. Dll1 maintains quiescence of adult neural stem cells and segregates asymmetrically during mitosis. Nat. Commun. 4, 1880 (2013).
Ayana, R. et al. Single-cell sequencing unveils the impact of aging on the progenitor cell diversity in the telencephalon of the female killifish N. furzeri. Aging Cell 23, e14251 (2024).
Nieuwenhuis, T. O., Rosenberg, A. Z., McCall, M. N. & Halushka, M. K. Tissue, age, sex, and disease patterns of matrisome expression in GTEx transcriptome data. Sci. Rep. 11, 21549 (2021).
Leote, A. C., Lopes, F. & Beyer, A. Loss of coordination between basic cellular processes in human aging. Nat. Aging 4, 1432–1445 (2024).
Vafaie, F. et al. Collagenase-resistant collagen promotes mouse aging and vascular cell senescence. Aging Cell 13, 121–130 (2014).
Gutierrez-Fernandez, A. et al. Loss of MT1-MMP causes cell senescence and nuclear defects which can be reversed by retinoic acid. EMBO J. 34, 1875–1888 (2015).
Ewald, C. Y., Landis, J. N., Porter Abate, J., Murphy, C. T. & Blackwell, T. K. Dauer-independent insulin/IGF-1-signalling implicates collagen remodelling in longevity. Nature 519, 97–101 (2015).
Selenou, C., Brioude, F., Giabicani, E., Sobrier, M. L. & Netchine, I. IGF2: development, genetic and epigenetic abnormalities. Cells 11, 1886 (2022).
Constancia, M. et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 417, 945–948 (2002).
DeChiara, T. M., Efstratiadis, A. & Robertson, E. J. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345, 78–80 (1990).
Abi Habib, W. et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet. Med. 20, 250–258 (2018).
Livingstone, C. IGF2 and cancer. Endocr. Relat. Cancer 20, R321–R339 (2013).
Sciacca, L. et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene 18, 2471–2479 (1999).
Ziegler, A. N. et al. IGF-II promotes stemness of neural restricted precursors. Stem Cells 30, 1265–1276 (2012).
Chen, J., Khondker, R. C. & Brunet, A. Breeding and reproduction of the African turquoise killifish Nothobranchius furzeri. Cold Spring Harb. Protoc. 2023, pdb prot107816 (2023).
Valenzano, D. R. et al. The African turquoise killifish genome provides insights into evolution and genetic architecture of lifespan. Cell 163, 1539–1554 (2015).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Hoffman, G. E. & Schadt, E. E. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinfs17, 483 (2016).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Wheeler, T. J. & Eddy, S. R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489 (2013).
Madeira, F. et al. The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 52, W521–W525 (2024).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Arias, E., Minino, A., Curtin, S. & Tejada-Vera, B. U.S. Decennial Life Tables for 2009–2011, United States Life Tables. Natl Vital Stat. Rep. 69, 1–73 (2020).
Pfister, S., Kuettel, V. & Ferrero, E. granulator: rapid benchmarking of methods for in silico deconvolution of bulk RNA-seq data. R package version 1.19.0 https://doi.org/10.18129/B9.bioc.granulator (2025).
Moolenbeek, C. & Ruitenberg, E. J. The “Swiss roll”: a simple technique for histological studies of the rodent intestine. Lab Anim. 15, 57–59 (1981).
Choi, H. M. T. et al. Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, dev165753 (2018).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Chen, J., Costa, E. K., Schmahl, N. & Tsenter, A. HCR-IF of the head kidney of young and old killifish males and females. Figshare+ https://doi.org/10.25452/figshare.plus.29983765.v1 (2025).
Chen, J., Costa, E. K., Schmahl, N. & Tsenter, A. HCR of the gut of young and old killifish males and females. Figshare+ https://doi.org/10.25452/figshare.plus.29983678.v1 (2025).
Stachura, D. L. & Traver, D. Cellular dissection of zebrafish hematopoiesis. Methods Cell. Biol. 133, 11–53 (2016).
Acknowledgements
We thank all members of the labs of T.W.-C. and A.B., particularly F. Boos, N. Rappoport, E. Sun and A. Tsai for scientific discussion and feedback on the paper. We thank F. Boos, N. Rappoport and E. Sun for help with independent code validation. We thank J. You for discussion on image quantification parameters. We also thank M. Housh, S. V. Perry, S. Boyle, J. Chung, R. Khondker and R. Barajas, for their assistance with killifish husbandry over the years. We thank D. Channappa, K. Dickey and H. Zhang for administrative support. This work was supported by The Phil and Penny Knight Initiative for Brain Resilience (to T.W.-C.), the Simons Foundation (to A.B. and T.W.-C.), the Michael J. Fox Foundation (to T.W.-C), the NOMIS Foundation (to T.W.-C. and A.B.), the Milky Way Research Foundation (to A.B.), the Glenn Foundation for Medical Research (to A.B.) and the CZ Biohub Investigator Program (to A.B.), a National Institutes of Health (NIH) New Innovator Award (DP2AG086979; to P.P.S.), the Esther A. & Joseph Klingenstein Fund (to P.P.S.) and Chan Zuckerberg Initiative Collaborative Pairs Awards (to P.P.S.). E.K.C. was supported by the NIH Training Grant (T32MH020016) awarded to the Leland Stanford Junior University and the Fondation Bertarelli Graduate Fellowship Fund. J.C. is a Jane Coffin Childs fellow, Stanford Katharine McCormick fellow and a Stanford Jump Start Award recipient. I.H.G. is funded by an NIH Pathway to Independence Award (K99AG088304) and an MAC3 Dementia and Ageing Fellowship. L.M. is a Eugene V. Cota-Robles fellow, Warren Alpert Computational Biology and Artificial Intelligence Network scholar and National Science Foundation Research Traineeship (NRT) fellow. This work was additionally supported by the NIH Training Grant in Genomic Analysis and Interpretation (T32HG002536) (to L.M.) and the National Science Foundation UCLA Quantum Science and Engineering PhD Fellowship (to L.M.).
Author information
Authors and Affiliations
Contributions
E.K.C., J.C., I.H.G., A.B. and T.W.-C. conceptualized the study. E.K.C., J.C. and I.H.G. raised animals for cohorts, designed the collection strategy and harvested all atlas tissues. I.H.G. optimized the transcardial perfusion protocol and perfused each animal in this study. E.K.C., J.C. and I.H.G. performed RNA extractions. E.K.C. and J.C. performed library preparations and all the computational analysis except the tissue clocks. L.M. designed the computational method BayesAge 2.0 under the supervision of M.P. and L.S.B. and worked with E.K.C. to refine the clocks. E.K.C. curated independent query datasets for implementation of age prediction using tissue clocks. J.C. performed validation experiments and collected tissue samples. E.K.C. and N.S. performed histological sectioning. E.K.C., N.S., A.T. and J.C. performed HCR staining, imaging and analysis. E.K.C., R.N. and J.C. performed the head kidney FACS experiment. M.-R.W. performed retina/RPE dissections and RNA extractions under the supervision of S.W. P.M.S. made the Shiny App for data exploration and advised on data preprocessing. P.P.S. provided the general RNA-seq analysis pipeline (quality control, mapping, DESeq2 and GSEA analysis) and provided computational analysis advice. E.K.C., J.C., A.B. and T.W.-C. wrote the original manuscript draft. A.B. and T.W.-C. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Aging thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Metadata for the multi-tissue killifish transcriptomic aging atlas.
(a) Kaplan-Meier survival curves for the two cohorts (left, middle) from which samples for RNA-sequencing were derived (left, 16 females, 22 males; middle, 25 females, 31 males). On the right is the survival curve for both cohorts combined (41 females, 53 males). Blue, male survival curve; pink, female survival curve. Yellow highlights and orange ticks on the x-axis denote sample collection windows. F, female; M, male. (b) Number of samples analyzed for each tissue, sex, and age group in this study. The red numbers denote incidences of sample dropout. ‘—’ indicates ‘not applicable.’ (c) Fish length (cm) across age (days post-hatching). Each dot represents the caudal-rostral length of a single individual animal. Symbol color, biological sex (F, female; M, male). Sample sizes were: n = 23 for female (n = 6 for 47-52 days, n = 6 for 75-78 days, n = 3 for 102-103 days, n = 4 for 133-134 days, and n = 4 for 147-152 days) and n = 32 for male (n = 6 for 47-52 days, n = 6 for 75-78 days, n = 4 for 102-103 days, n = 6 for 133-134 days, n = 4 for 147-152 days, and n = 6 for 161-162 days). Box plots show the median, 25th (Q1) and 75th (Q3) percentiles; whiskers extend to Q1 − 1.5×(Q3 − Q1) and Q3 + 1.5×(Q3 − Q1). (d, e) Bar plots of the median percent variance explained across all genes expressed in a tissue for the covariates of age, sex, and the interaction term sex:age (panel d) and for the technical covariates of cohort batch and RNA batches (panel e). (f) Example per-tissue PCA (brain) colored by RNA batches. See Supplemental File 3 for all other tissues.
Extended Data Fig. 2 Cross-tissue pathways enriched for the genes correlated with age.
Male (M) vs. female (F) gene set enrichment analysis (GSEA) results, identifying the shared or unique pathways enriched for the genes upregulated or downregulated with age in the 13 tissues. GSEA was performed using a permutation-based enrichment test implemented in clusterProfiler with Gene Ontology annotations. For females (F) and males (M) separately, tissues are clustered by similarity of enrichment as calculated by the product of the NES and –log(FDR). NES, normalized enrichment score. Dot size, –log10 of the FDR-adjusted P value (that is, false discovery rate [FDR] after multiple hypothesis testing).
Extended Data Fig. 3 Cross-tissue hypergeometric GO enrichment analysis for genes correlated with age.
For males (M) and females (F) separately, age-upregulated genes (Spearman’s rank correlation ρ > 0.5; panel a) or age-downregulated genes (ρ < −0.5; panel b) from each tissue were used as input for Gene Ontology (GO) enrichment analysis. Enrichment significance was assessed using a hypergeometric test implemented in GOstats, with the background (‘universe’) defined as all genes with non-NA FDR-adjusted P values for the corresponding tissue and sex. GO terms consistent with the GSEA analysis were plotted. Enrichment scores were capped at 50 for visualization. Dot size represents −log10 of the FDR-adjusted P value (that is, false discovery rate [FDR] after multiple hypothesis testing). Empty slots indicate GO terms that were not detected.
Extended Data Fig. 4 Sex-specific pathways enriched for the genes correlated with age.
Male (M) vs. female (F) gene set enrichment analysis (GSEA) results in the 13 tissues, identifying the GO terms showing opposite signs of upregulation or downregulation with age in the two sexes, as well as those for which the change with age was significant in only one sex (‘sex-divergent’). GSEA was performed using a permutation-based enrichment test implemented in clusterProfiler with Gene Ontology annotations. Statistical significance was assessed using permutation-derived P values and adjusted for multiple hypothesis testing using the Benjamini–Hochberg method; GO terms with FDR < 0.05 were considered significant. NES, normalized enrichment score. Dot size, –log10 of the FDR-adjusted P value (that is, false discovery rate [FDR] after multiple hypothesis testing). Boxes indicate the main sex-divergent GO terms in each tissue.
Extended Data Fig. 5 Conservation analysis between the killifish and other species.
(a) Summary table of RNAcentral search results for sequence conservation of ncRNA-3777 in human, mouse, and zebrafish ncRNA catalogs44. The number and the type of ncRNA that were high-confidence hits are shown. (b) IGF2BP3 protein structure prediction using AlphaFold for the killifish (left) and human (middle)54,55,56. The killifish IGF2BP3 protein was 75.91% identical to the human protein by alignment and largely overlapped structurally with the human protein (right). (c) Distribution of log2 fold change values for the genes that were differentially expressed during aging by DESeq2 and showed a conserved direction of change between analogous tissues in 1) killifish (pink) and human (blue), or in 2) killifish (pink) and mouse (green). DESeq2 results were from comparing between 133-134 days and 47-52 days in killifish (corresponding to ~30% and ~100% colony survival, respectively), between 27 months and 3 months in mouse (corresponding to 50-75% and 100% colony survival, respectively), or between 70-79 years and 20-29 years in human (corresponding to ~60-80% and ~100% survival, respectively). Statistical significance was assessed using the two-sided Wald test implemented in DESeq2, and P values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method. Differential expression analyses were performed using RNA-seq samples; sample sizes for each tissue, species, and age group are reported in Supplementary File 15. Human or mouse tissue names are shown; see Supplementary File 15 for the corresponding killifish tissues used in each comparison. Each point represents a gene that was significantly differentially expressed in both species (FDR-adjusted P value < 0.05) and showed a consistent change in the same direction (either up or down-regulated with age). Colored points indicate the genes with a Spearman’s rank correlation coefficient ρ > 0.5 or < –0.5 in killifish, while gray points represent genes that did not meet this correlation threshold. Box plots show the median, 25th (Q1) and 75th (Q3) percentiles; whiskers extend to Q1 − 1.5×(Q3 − Q1) and Q3 + 1.5×(Q3 − Q1).
Extended Data Fig. 6 Conservation analysis between killifish and other species.
(a) Bar plot showing the number of overlapping Gene Ontology (GO) biological process terms between killifish and mouse tissues. GO terms were derived independently for killifish and mouse tissues using gene set enrichment analysis (GSEA) with a permutation-based enrichment test, and statistical significance was assessed using permutation-derived P values adjusted for multiple hypothesis testing with the Benjamini–Hochberg method. GO terms meeting a significance threshold of FDR-adjusted P value < 0.1 in both species were included in the overlap analysis. No statistical hypothesis testing was performed for the overlap counts shown; the bar plot represents a descriptive summary of shared enriched GO terms. Bar color indicates killifish tissue. (b) Bar plot of the number of overlapping GO terms between killifish and human tissues, with analysis and data presented as described for panel a. (c) Killifish vs. human and killifish vs. mouse GSEA results, identifying pathways shared between species that are enriched among genes upregulated or downregulated with age in each tissue. GSEA was performed using a permutation-based enrichment test implemented in clusterProfiler with Gene Ontology annotations. Statistical significance was assessed using permutation-derived P values and adjusted for multiple hypothesis testing using the Benjamini–Hochberg method. Pathways with an FDR-adjusted P value < 0.1 were considered significant. NES, normalized enrichment score. Dot size represents −log10 of the FDR-adjusted P value (that is, false discovery rate [FDR] after multiple hypothesis testing).
Extended Data Fig. 7 Tissue-specific gene expression dynamics for the gut and muscle.
(a) Hierarchical clustering of the gene expression trajectories for the gut (sex-combined), highlighting cluster 8. Hierarchical clustering was performed on locally estimated scatterplot smoothing (LOESS) regression trajectories of gene expression across age. Clustering and trajectory fitting were descriptive and did not involve statistical hypothesis testing. Each line is the trajectory of an individual gene, and the average trajectory for the cluster is depicted by the black line. The number of genes in each cluster is indicated by ‘n.’ The most highly significant GO term from Hypergeometric GO enrichment (terms related to Biological Processes) is listed, along with the number of genes that comprise the cluster. (b) Hypergeometric GO enrichment (terms related to Biological Processes) for the genes in gut cluster 8. Enrichment significance was assessed using a hypergeometric test implemented in GOstats, with the background (‘universe’) defined as all genes with non-NA FDR-adjusted P values used in the clustering analysis. P values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method. Select GO terms significantly enriched (FDR-adjusted P value < 0.05) for each cluster are plotted. The dot color represents the enrichment score of each GO term, with the maximum value of the scale adjusted to 15 to improve the color resolution of GO terms with lower enrichment. Dot size, –log10 of the FDR-adjusted P value (that is, false discovery rate [FDR] after multiple hypothesis testing). (c) Hierarchical clustering of the gene expression trajectories for the muscle (sex-combined), highlighting cluster 7. Analysis and graphics, as in panel a. (d) Hypergeometric GO enrichment for muscle cluster 7, analysis conducted as in panel b.
Extended Data Fig. 8 Estimated cell-type proportions over age.
(a) Cell-type proportions by sample in Teefy et al., 202359 kidney single cell dataset, separated by sex. Symbol color, biological sex (F, female; M, male). Bar length, mean cell-type proportion across samples. Each dot corresponds to a sample in the single cell reference dataset, plotting the percent composition of a cell-type in that given sample. (b, c) Estimated cell-group proportions (panel b) and cell-type proportions (panel c) across age for lymphoid cells and myeloid cells, split by sex. Data derived from single-cell reference matrices that represented the averaged expression across all cells of a specific cell type (for example, B cells, neutrophils). Statistical comparisons were performed at the sample level using a two-sided Mann–Whitney U test between the indicated groups. Each data point represents one fish. Sample sizes were n = 6 (female young), n = 4 (female old), n = 6 (male young), and n = 6 (male old). Symbol color, biological sex (F, female; M, male). N.a., not available.
Extended Data Fig. 9 The aging killifish kidney marrow changes in gene expression and cell-type composition.
(a) Dot plot of gene expression for genes in Fig. 4b, showing cell-type-specific enrichment. The human ortholog (uppercase) or the zebrafish ortholog (lowercase) is written before the ‘/’ symbol, and the killifish gene name is written after the ‘/’ symbol. Dot color indicates the level of expression, and dot size indicates the percentage of cells expressing the gene. (b) Flow cytometry gating scheme, showing representative gating workflow from raw event data to live cells. (c) Principal Component (PC) Analysis of the dissociated male head kidney cell populations that were FACS-sorted based on the gating strategy as in panel b. Each dot is an individual animal (myeloid population: 3 fish; lymphoid population: 2 fish). These males were harvested at different ages (67, 88, and 201 days) to test whether the gating strategy could be applied to different age groups. (d) Heatmap showing the expression of myeloid- and lymphoid-specific markers (see panel a), clustered by samples. The expression of each gene is plotted as Z-scaled, DESeq2-normalized counts. The human ortholog (uppercase) or the zebrafish ortholog (lowercase) is written before the ‘/’ symbol, and the killifish gene name is written after the ‘/’ symbol. (e) Quantification of myeloid: lymphoid ratio (total myeloid events: total lymphoid events) from flow cytometry data. Each dot is an animal. The young males were 55-62 days old (n = 6), and the old males were 136-143 days old (n = 6) (approximately the same chronological age as the females in Fig. 4f). Significance was assessed using a two-sided Mann–Whitney U test. (f) Scatterplot of the counts normalized by DESeq2 for irf4b (killifish gene name: irf4) in the head kidney transcriptome of the atlas dataset. Each dot is the expression of irf4b in an individual sample, with n as reported in Extended Data Fig. 1b. Red, female (F). Blue, male (M). (g) UMAP (uniform manifold approximation and projection) plots of data from a killifish single-cell RNA-sequencing tissue atlas59, with overlayed expression levels for irf4a (left) and irf4b (right). The number of cells expressed irf4a or irf4b is indicated by ‘n.’ (h) Co-expression UMAP showing the expression level of irf4a and ptprc. Data were derived from the single-cell tissue atlas59. The irf4ahigh ptprclow cells are red (1368 cells in the source dataset), irf4alow ptprchigh cells are blue (11,635 cells), and irf4ahigh ptprchigh cells are purple (904 cells). (i) Example single-z-plane HCR image of young male head kidney tissue, illustrating the cytoarchitecture of renal tubules (outlined by white dashed lines) and surrounding interstitial space containing hematopoietic tissue. Quantification of the irf4a transcripts was restricted to the interstitial space. This tissue organization was consistently observed across head kidney samples from n = 16 animals, and a representative image is shown. Scale bar, 10 µm.
Extended Data Fig. 10 Tissue-specific transcriptomic aging clocks can predict age in a published aging dataset.
(a) A published aging transcriptomic dataset (Xu dataset36) of four tissues (brain, heart, muscle, and spleen) obtained from male and female killifish at young and old ages. (b) Age prediction for hearts using sex-combined models when the atlas clocks were directly applied to the query samples, without calibration. Each dot in the box plots represents the predicted tAge for the heart transcriptome of an individual fish. Male, green; female, purple. Sample sizes are reported in Supplementary File 23. Statistical significance between young and old samples for each model was assessed using a two-sided Mann–Whitney U test. (c) Calibration strategies for each machine-learning model. For BayesAge 2.0 and Principal Component (PC) regression, two young and two old samples from the Xu dataset were selected to be part of the ‘training’ set, which also included the atlas data. The remaining samples from the Xu dataset were tested using the calibrated clocks. We rotated the query samples used for calibration and performed all permutations of calibration. Here, one example permutation was plotted in panels d-g. For Elastic net, the ‘Leave One Sample Out Cross Validation’ (LOSO-CV) pipeline was applied, where the calibration training set included all the atlas and query samples except the test sample (N-1), and age was predicted for the test sample after calibration. (d-g) Predicted age (after calibration) for each sample from the Xu dataset. Each dot represents one individual fish; sample sizes for each tissue, age group, and sex are reported in Supplementary File 23. Statistical testing was performed as in panel (b) using a two-sided Mann–Whitney U test when two or more samples were available per group. The graphics and the statistics were generated as in panel b. (#), one spleen sample (spleen_2) was excluded from the plot due to poor model performance. P value was not calculated when there was only one young sample in the testing data. N.a., not available.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–11
Supplementary Table 1 (download XLSX )
Metadata file for the atlas experiment.
Supplementary Table 2 (download XLSX )
Sequencing-related metadata file for the atlas dataset.
Supplementary Table 3 (download XLSX )
Tissue sex-split correlation analysis using Spearman’s rank correlation.
Supplementary Table 4 (download XLSX )
Tissue sex-combined correlation analysis using Spearman’s rank correlation.
Supplementary Table 5 (download XLSX )
Tissue GSEA GO enrichment results for the sex-split correlation results.
Supplementary Table 6 (download XLSX )
Tissue hypergeometric GO enrichment analysis results for the genes upregulated or downregulated with age for each tissue and each sex.
Supplementary Table 7 (download XLSX )
Tissue hypergeometric GO enrichment analysis results for sex-divergent genes.
Supplementary Table 8 (download XLSX )
Age-associated genes that are shared amongs five or more and six or more tissues.
Supplementary Table 9 (download XLSX )
Hypergeometric GO enrichment (biological processes) for the shared age-correlated genes across five or more tissues.
Supplementary Table 10 (download XLSX )
Gene list of each expression cluster in the hierarchical clustering trajectory analysis, by tissue.
Supplementary Table 11 (download XLSX )
Hypergeometric GO results for the gene list of each expression cluster in the hierarchical clustering trajectory analysis, by tissue.
Supplementary Table 12 (download XLSX )
ncRNA-3777 sequence search output files from RNAcentral.
Supplementary Data 13 (download TXT )
Clustal Omega protein sequence alignment results comparing killifish and human IGF2BP3.
Supplementary Table 14 (download XLSX )
Conservation analysis using DEGs, GO terms and GSEA results, by comparing analogous tissues in killifish, mouse and humans.
Supplementary Table 15 (download XLSX )
Animal metadata for the atlas validation cohort used for HCR.
Supplementary Table 16 (download XLSX )
HCR probe sequences for each of the probes used in this study.
Supplementary Table 17 (download XLSX )
HCR mRNA spot counts and cell number exported from QuPath quantification.
Supplementary Table 18 (download XLSX )
Experimental metadata for the flow cytometry experiment presented in Fig. 4.
Supplementary Table 19 (download XLSX )
Experimental metadata for the sorted cell RNA-seq experiment to validate gating.
Supplementary Data 20 (download PDF )
Model performance for all sex-combined and sex-specific clocks.
Supplementary Table 21 (download XLSX )
The top genes underlying each tissue aging clock for the three machine-learning models.
Supplementary Table 22 (download XLSX )
Age prediction results of the published datasets.
Supplementary Table 23 (download XLSX )
Single-cell deconvolution cell proportion data for head kidney.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Costa, E.K., Chen, J., Guldner, I.H. et al. Multi-tissue transcriptomic aging atlas reveals predictive aging biomarkers in the killifish. Nat Aging 6, 636–664 (2026). https://doi.org/10.1038/s43587-026-01074-6
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s43587-026-01074-6
