
Abstract
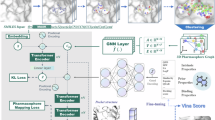
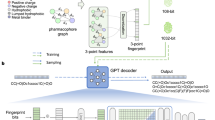
Molecular generation is a cutting-edge technology with the potential to revolutionize intelligent drug discovery. However, currently reported ligand-based or structure-based molecular generation methods remain unpractical for real-world drug discovery. Here we propose an explicit pharmacophore-oriented 3D molecular generation method, termed PhoreGen. PhoreGen employs asynchronous perturbations and updates on both atomic and bond information, coupled with a message-passing mechanism that incorporates prior knowledge of ligand–pharmacophore mapping during the diffusion–denoising process. Evaluations revealed that PhoreGen efficiently generates 3D molecules well aligned with pharmacophores, maintaining good chemical reasonability, diversity, drug-likeness and binding affinity and, importantly, produces feature-customized molecules at high frequency. By using PhoreGen, we successfully identified new bicyclic boronate inhibitors of evolved metallo-β-lactamase and serine-β-lactamases, which potentiate meropenem against clinically isolated superbugs. Moreover, we identified inhibitors of metallo-nicotinamidases, emerging targets for insecticides. This work explores an explicitly constrained mode for molecular generation and demonstrates its potential in feature-customized drug discovery.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

Data availability
The LigPhore, CpxPhore and DockPhore datasets for training and evaluation are available via Zenodo at https://zenodo.org/records/15518867 (ref. 62). The PDBBind dataset is available at http://pdbbind.org.cn. The CrossDocked2020 dataset is available at https://bits.csb.pitt.edu/files/crossdock2020/. The pharmacophore models for molecular generation can be created via our web server at https://phoregen.ddtmlab.org. The complex crystal structures (PDB codes 9KSA and 9U8M) are available via PDB at https://www.rcsb.org. Source data are provided with this paper.
Code availability
The source code of PhoreGen is available on our web server at https://phoregen.ddtmlab.org, via GitHub at https://github.com/ppjian19/PhoreGen and via Zenodo at https://zenodo.org/records/15518867 (ref. 62) under an open-source license.
References
Berdigaliyev, N. & Aljofan, M. An overview of drug discovery and development. Future Med. Chem. 12, 939–947 (2020).
Goodnow, J. R. A. Hit and lead identification: integrated technology-based approaches. Drug Discov. Today 3, 367–375 (2006).
Sadybekov, A. V. & Katritch, V. Computational approaches streamlining drug discovery. Nature 616, 673–685 (2023).
Catacutan, D. B. et al. Machine learning in preclinical drug discovery. Nat. Chem. Biol. 20, 960–973 (2024).
Du, Y. et al. Machine learning-aided generative molecular design. Nat. Mach. Intell. 6, 589–604 (2024).
Cheng, Y. et al. Molecular design in drug discovery: a comprehensive review of deep generative models. Brief. Bioinformatics. 22, bbab344 (2021).
Bian, Y. & Xie, X. Q. Generative chemistry: drug discovery with deep learning generative models. J. Mol. Model. 27, 71 (2021).
Hoogeboom, E. et al. Equivariant diffusion for molecule generation in 3D. In International Conference on Machine Learning (eds Chaudhuri, K. et al.) (PMLR, 2022).
Xu, M. et al. Geometric latent diffusion models for 3D molecule generation. In International Conference on Machine Learning (eds Chaudhuri, K. et al.) (PMLR, 2022).
Huang, L. et al. MDM: molecular diffusion model for 3D molecule generation. In Association for the Advancement of Artificial Intelligence (AAAI Press, 2022).
Luo, S. et al. A 3D generative model for structure-based drug design. In Conference on Neural Information Processing Systems (eds Ranzao, M. et al.) (OpenReview.net, 2021).
Peng, X. et al. Pocket2Mol: efficient molecular sampling based on 3D protein pockets. In International Conference on Machine Learning (eds Chaudhuri, K. et al.) (PMLR, 2022).
Guan, J. et al. 3D equivariant diffusion for target-aware molecule generation and affinity prediction. In International Conference on Learning Representations (OpenReview.net, 2023).
Huang, L. et al. A dual diffusion model enables 3D molecule generation and lead optimization based on target pockets. Nat. Commun. 15, 2657 (2024).
Zhung, W., Kim, H. & Kim, W. Y. 3D molecular generative framework for interaction-guided drug design. Nat. Commun. 15, 2688 (2024).
Wu, P. et al. Guided diffusion for molecular generation with interaction prompt. Brief. Bioinformatics 25, bbae174 (2024).
Lee, J., Zhung, W. & Kim, W. NCIDiff: non-covalent interaction-generative diffusion model for improving reliability of 3D molecule generation inside protein pocket. In International Conference on Machine Learning (OpenReview.net, 2024).
Zhavoronkov, A. et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 37, 1038–1040 (2019).
De Cao, N. & Kipf, T. MolGAN: an implicit generative model for small molecular graphs. In International Conference on Machine Learning (eds Dy, J. et al.) (PMLR, 2018).
Zhang, O. et al. ResGen is a pocket-aware 3D molecular generation model based on parallel multiscale modelling. Nat. Mach. Intell. 5, 1020–1030 (2023).
Liu, M. et al. Generating 3D molecules for target protein binding. In International Conference on Machine Learning (eds Chaudhuri, K. et al.) (PMLR, 2022).
Luo, S. & Hu, W. Diffusion probabilistic models for 3D point cloud generation. In IEEE Conference on Computer Vision and Pattern Recognition (IEEE, 2021).
Sun, J. et al. A critical revisit of adversarial robustness in 3D point cloud recognition with diffusion-driven purification. In International Conference on Machine Learning (eds Krause, A. et al.) (PMLR, 2023).
Lyu, Z. et al. A conditional point diffusion-refinement paradigm for 3D point cloud completion. In International Conference on Learning Representations (OpenReview.net, 2022).
Schneuing, A. et al. Structure-based drug design with equivariant diffusion models. Nat. Comput. Sci. 4, 899–909 (2024).
Guan, J. et al. DecompDiff: diffusion models with decomposed priors for structure-based drug design. In International Conference on Machine Learning (eds Krause, A. et al.) (PMLR, 2023).
Huang, Z. et al. Interaction-based retrieval-augmented diffusion models for protein-specific 3D molecule generation. In International Conference on Machine Learning (OpenReview.net, 2024).
Alakhdar, A., Poczos, B. & Washburn, N. Diffusion models in de novo drug design. J. Chem. Inf. Model. 64, 7238–7256 (2024).
Boike, L., Henning, N. J. & Nomura, D. K. Advances in covalent drug discovery. Nat. Rev. Drug Discov. 21, 881–898 (2022).
Schaller, D. et al. Next generation 3D pharmacophore modeling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 10, e1468 (2020).
Imrie, F., Hadfield, T. E., Bradley, A. R. & Deane, C. M. Deep generative design with 3D pharmacophoric constraints. Chem. Sci. 12, 14577–14589 (2021).
Zhu, H., Zhou, R., Cao, D., Tang, J. & Li, M. A pharmacophore-guided deep learning approach for bioactive molecular generation. Nat. Commun. 14, 6234 (2023).
Bush, K. & Bradford, P. A. Interplay between β-lactamases and new β-lactamase inhibitors. Nat. Rev. Microbiol. 17, 295–306 (2019).
Yang, Y. et al. Metallo-β-lactamase-mediated antimicrobial resistance and progress in inhibitor discovery. Trends Microbiol. 31, 735–748 (2023).
Brem, J. et al. Imitation of β-lactam binding enables broad-spectrum metallo-β-lactamase inhibitors. Nat. Chem. 14, 15–24 (2022).
Qiao, X. et al. An insecticide target in mechanoreceptor neurons. Sci. Adv. 8, eabq3132 (2022).
Satorras, V. G., Hoogeboom, E. & Welling, M. E(n) equivariant graph neural networks. In International Conference on Machine Learning (eds Meila, M. et al.) (PMLR, 2021).
Buttenschoen, M., Morris, G. M. & Deane, C. M. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chem. Sci. 15, 3130–3139 (2024).
Schneuing, A. et al. Multi-domain distribution learning for de novo drug design. In International Conference on Learning Representations (OpenReview.net, 2025).
Adams, K. et al. ShEPhERD: diffusing shape, electrostatics, and pharmacophores for bioisosteric drug design. In International Conference on Learning Representations (OpenReview.net, 2025).
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 61, 3891–3898 (2021).
Francoeur, P. G. et al. Three-dimensional convolutional neural networks and a cross-docked data set for structure-based drug design. J. Chem. Inf. Model. 60, 4200–4215 (2020).
Du, H. et al. CovalentInDB 2.0: an updated comprehensive database for structure-based and ligand-based covalent inhibitor design and screening. Nucleic Acids Res. 53, D1322–D1327 (2025).
Hecker, S. J. et al. Discovery of cyclic boronic acid QPX7728, an ultrabroad-spectrum inhibitor of serine and metallo-β-lactamases. J. Med. Chem. 63, 7491–7507 (2020).
Fyfe, P. K., Rao, V. A., Zemla, A., Cameron, S. & Hunter, W. N. Specificity and mechanism of Acinetobacter baumanii nicotinamidase: implications for activation of the front-line tuberculosis drug pyrazinamide. Angew. Chem. Int. Ed. 48, 9176–9179 (2009).
Wang, C. & Rajapakse, J. C. Pharmacophore-guided de novo drug design with diffusion bridge. Preprint at https://doi.org/10.48550/arXiv.2412.19812 (2025).
Heider, J. et al. Apo2ph4: a versatile workflow for the generation of receptor-based pharmacophore models for virtual screening. J. Chem. Inf. Model. 63, 101–110 (2023).
Seo, S. & Kim, W. Y. PharmacoNet: deep learning-guided pharmacophore modeling for ultra-large-scale virtual screening. Chem. Sci. 15, 19473–19487 (2024).
Ho, J., Jain, A. & Abbeel, P. J. A. Denoising diffusion probabilistic models. In Conference on Neural Information Processing Systems (eds Larochelle, H. et al.) (Curran Associates Inc, 2020).
Peng, X., Guan, J., Liu, Q. & Ma, J. MolDiff: addressing the atom-bond inconsistency problem in 3d molecule diffusion generation. In International Conference on Machine Learning (eds Krause, A. et al.) (PMLR, 2023).
Pearce, T., Brintrup, A., Zaki, M. & Neely, A. High-quality prediction intervals for deep learning: a distribution-free, ensembled approach. In International Conference on Machine Learning (eds Dy, J. et al.) (PMLR, 2018).
Dhariwal, P. & Nichol, A. Diffusion models beat GANs on image synthesis. In Conference on Neural Information Processing Systems (eds Ranzao, M. et al.) (OpenReview.net, 2021).
Yu, J. et al. Knowledge-guided diffusion model for 3D ligand–pharmacophore mapping. Nat. Commun. 16, 2269 (2025).
Dai, Q. et al. AncPhore: a versatile tool for anchor pharmacophore steered drug discovery with applications in discovery of new inhibitors targeting metallo-β-lactamases and indoleamine/tryptophan 2,3-dioxygenases. Acta Pharm. Sin. B 11, 1931–1946 (2021).
Liu, Z. et al. Forging the basis for developing protein–ligand interaction scoring functions. Acc. Chem. Res. 50, 302–309 (2017).
Su, M. et al. Comparative assessment of scoring functions: the CASF-2016 update. J. Chem. Inf. Model. 59, 895–913 (2019).
Yan, Y.-H. et al. Discovery of 2-aminothiazole-4-carboxylic acids as broad-spectrum metallo-β-lactamase inhibitors by mimicking carbapenem hydrolysate binding. J. Med. Chem. 66, 13746–13767 (2023).
Xiao, Y.-C. et al. Design and enantioselective synthesis of 3-(α-acrylic acid) benzoxaboroles to combat carbapenemase resistance. Chem. Commun. 57, 7709–7712 (2021).
Wang, Y.-L. et al. Structure-based development of (1-(3′-mercaptopropanamido) methyl)boronic acid derived broad-spectrum, dual-action inhibitors of metallo- and serine-β-lactamases. J. Med. Chem. 62, 7160–7184 (2019).
Van Berkel, S. S. et al. Assay platform for clinically relevant metallo-β-lactamases. J. Med. Chem. 56, 6945–6953 (2013).
Xiao, Q. et al. Upgrade of crystallography beamline BL19U1 at the Shanghai Synchrotron Radiation Facility. J. Appl. Crystallogr. 57, 630–637 (2024).
Peng, J. PhoreGen source code and data (LigPhore, CpxPhore, and DockPhore dataset and trained weights). Zenodo https://doi.org/10.5281/zenodo.15518867 (2025).
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (grant nos. 82122065, 82473845 and 82073698), the National Key R&D Program of China (grant no. 2023YFF1204901), the Sichuan Science and Technology Program (grant no. 2025YFHZ0085), the Foundation for Innovative Research Groups of the Natural Science Foundation of Sichuan Province (grant no. 2024NSFTD0026) and the Basic Research Foundation of Sichuan University (grant no. 2023SCUH0073). We thank the staff at beamline BL19U1 of the Shanghai Synchrotron Radiation Facility, National Facility for Protein Science (Shanghai, China), for their great support. We also thank the members of the Mass Spectrometry Platform (R. Wang and X. Wu) for proteomic techniques and data interpretation.
Author information
Authors and Affiliations
Contributions
J.P., J.-L.Y., Z.-B.Y. and Y.-T.C. contributed equally to this work. G.-B.L. conceived, planned and supervised this study. J.P. and J.-L.Y. designed and trained the model supervised by G.-B.L.; J.P., J.-L.Y. and Y.-G.W. performed model validations. Z.-B.Y. and Y.-T.C. performed chemical synthesis. S.-Q.W., F.-B.M. and Y.-T.C. conducted protein production and bioactivity testing. J.P., J.-L.Y., Z.-B.Y., Y.-T.C. and G.-B.L. wrote the manuscript. G.-B.L. revised and polished the manuscript. All authors contributed to the final draft and approved the final version for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Computational Science thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Kaitlin McCardle, in collaboration with the Nature Computational Science team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 View of the top-10 ranked generated molecules with their corresponding pharmacophore models.
It reveals that for pharmacophore models with common, balanced feature compositions, PhoreGen can generate well-mapped, high-quality 3D molecules. Boron atoms, nitrogen atoms, oxygen atoms, phosphorus atoms, sulfur atoms, and chlorine atoms are pink, blue, red, darkorange, goldenrod, and limegreen respectively, while the other colors are carbon atoms.
Extended Data Fig. 2 Distributions of the pharmacophore feature and exclusion sphere counts versus the atom counts of generated molecules.
a,b For the complex-derived pharmacophore models (that is, the CpxPhore test set), the atom counts of the generated molecules show correlation with the pharmacophore feature and exclusion sphere counts. c,d For the ligand-derived pharmacophore models (that is, the LigPhore test set), the generated molecule’s atom counts also correlate with the feature and exclusion sphere counts. The sample size can be found in the Source Data file. Shadings indicate the feature or exclusion sphere counts, with darker shades representing higher counts. The box is the interquartile range (IQR), the line inside the box is the median, the whiskers represent data within ±1.5 × IQR, and the dots outside the whiskers are outliers. The r value refers to the Pearson correlation coefficient.
Extended Data Fig. 3 Correlation between the cavity sizes of pharmacophore models and the atom counts of generated molecules.
a,b For both the complex-derived (a) and ligand-derived (b) pharmacophore models, the atom counts of the generated molecules show a strong correlation with the corresponding cavity sizes, as indicated by the average distance between the pharmacophore features and exclusion spheres. The r value refers to the Pearson correlation coefficient.
Extended Data Fig. 4 CN1 and CN2 form a covalent bond with the catalytic cysteine of Naam.
a, The LC–MS/MS spectrum of CN1-modified peptide Y266DVC269VGATAVDALSAGYR283 from Drosophila melanogaster Naam. b, The LC–MS/MS spectrum of CN2-modified peptide M253KGATDIYVC262(Carbamidomethyl)GLAYDVC269 from Drosophila melanogaster Naam. The results demonstrated that CN1 and CN2 form a covalent bond with the catalytic residue Cys269 of Drosophila melanogaster Naam. CN1/CN2 (120 μM) was incubated with Drosophila melanogaster Naam (25 μM) at 4 °C for 2 h and the mixture was digested by trypsin.
Extended Data Fig. 5 CN1 and CN2 show inhibitory activity against the pests’ Naam enzymes.
a, b IC50 curves are obtained by incubating compounds CN1 and CN2 with the pests’ Myzus persicae Naam (15 nM) and Bemisia tabaci Naam (30 nM) enzymes, respectively, for durations ranging from 0 to 120 minutes, revealing that both compounds are time-dependent inhibitors of these Naam enzymes; data are presented as mean values ±SEM from n = 3 biological replicates, with error bars representing the SEM. c, d The melting curves (first-derivative of dissociation) of Myzus persicae Naam (10 μM) and Bemisia tabaci Naam (10 μM) in presence or absence of CN1 (50 μM) or CN2 (50 μM), reveal that these two compounds bind to and stabilize these enzymes; data are presented as mean values ±SEM from n = 3, 2, 3, 3 for c and n = 3, 2, 3, 1 for d biological replicates, with error bars representing the SEM.
Supplementary information
Supplementary Information (download PDF )
Supplementary Notes, Tables 1–29, and Figs. 1–29.
Supplementary Data 1 (download ZIP )
Source data for Supplementary Figs. 1–5 and 7–10.
Supplementary Data 2 (download ZIP )
Source data for Supplementary Tables 5–21.
Source data
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Peng, J., Yu, JL., Yang, ZB. et al. Pharmacophore-oriented 3D molecular generation toward efficient feature-customized drug discovery. Nat Comput Sci 5, 898–914 (2025). https://doi.org/10.1038/s43588-025-00850-5
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s43588-025-00850-5
