
Abstract
Background
BRAF status is crucial for treating pediatric low-grade gliomas (pLGG) and can be assessed non-invasively from segmented tumor regions on MRI using machine learning (ML). However, there are limitations to manual and automated tumor segmentations. This study assessed the performance of automated segmentation algorithms and a segmentation-free ML classification pipeline.
Methods
Molecularly characterized tumors and whole-brain FLAIR MR images were collected from 455 patients with pLGG treated between 1999 and 2023 at a children’s hospital. Three medical segmentation models, TransBTS, MedNeXt, and MedicalNet, were evaluated. Next, we developed a model to identify BRAF status from whole-brain FLAIR MRI, without any reliance on segmentations. We then implemented a novel pretraining regimen that embedded segmentation knowledge into the whole-brain FLAIR MRI classification model. Finally, we trained and evaluated a baseline model that used semiautomatic whole tumor volume segmentations as inputs.
Results
Here we show that the MedNeXt segmentation model (mean Dice score: 0.555) outperformed MedicalNet (0.516) and TransBTS (0.449) (p < 0.05 for all comparisons). The MedNeXt classification model achieved a one-vs-rest area under the ROC curve of 0.741 using the whole brain FLAIR sequence as an input, without any segmentation knowledge. This was improved to 0.772 through pretraining on the segmentation task, which was not significantly different from the baseline semiautomatic whole tumor volume segmentation-based model (0.756, p-value: 0.141).
Conclusions
BRAF status can be assessed non-invasively using ML models based on whole-brain FLAIR sequences. Dependence on inconsistent manual or automated segmentations can be reduced by integrating tumor region information into the model through pretraining.
Plain language summary
Pediatric low-grade gliomas are the most common brain tumors in children. Many of these tumors carry mutations in a gene called BRAF. Treatment choices depend on knowing the type of mutation, termed “BRAF status.” Up until now, BRAF status could only be confirmed through brain surgery or predicted from brain scans using artificial intelligence after first outlining the tumor on the scan. Tumors can be outlined manually by radiologists or automatically by a computer model, but both methods are prone to inconsistencies. In this study, we show that BRAF status can be predicted directly from brain scans using artificial intelligence without outlining, making the method more reliable and practical. This approach could help children avoid brain surgery and may extend to other cancers and age groups.
Similar content being viewed by others

Introduction
Pediatric low-grade gliomas (pLGG) are the most common brain tumor in children, accounting for approximately 40% of pediatric central nervous system (CNS) tumors1. As a diverse set of tumors, pLGGs can occur throughout the CNS and consist of various different histopathologies2. Despite the heterogeneity of pLGGs, it has been found that these tumors are often the result of a select few molecular alterations in the mitogen-activated protein kinase pathway, most commonly KIAA1549::BRAF (BRAF Fusion) or point mutation of BRAF p.V600E (BRAF Mutation)3. BRAF status has proven to be crucial for accurate prognostication and risk assessment of pLGG and for therapeutic decision-making4,5. The importance of genetic status is highlighted in recent editions of the WHO CNS tumor classification6. While historically CNS tumor classification has been based primarily on histological findings, the more recent approaches also rely on molecular status7.
The identification of the molecular alterations driving pLGGs has been transformative for their treatment. Therapeutics targeting specific genetic alterations that can supplement or replace classic cytotoxic treatments have been developed4,8. Usage of these targeted therapies, optimal prognostication, and risk assessment require surgically acquired tumor tissue, which is used to determine BRAF status9. Currently, in cases where tissue samples cannot be retrieved, such as with difficult-to-access tumors, the disease cannot be well characterized, and it is difficult to identify an appropriate targeted therapy.
Machine learning (ML) has shown potential as an alternative, non-invasive method of detecting BRAF status in patients with pLGG. Earlier studies used ML to successfully classify patients with pLGG by genetic status based on manually segmented tumor regions of their MRIs10,11,12,13,14,15,16. While encouraging, the pipelines described in these works depend on manual tumor segmentations that have known intra-reader17 and inter-reader18 variability issues. More recent studies have aimed to eliminate the reliance on manual segmentations by introducing more reproducible pipelines incorporating automated segmentation models19,20,21. However, such an approach is not fully reliable either, given that earlier studies focused on automating the segmentation of pLGGs either excluded difficult and outlier cases22, or failed for certain patients, missing the tumor entirely23.
In this study, we propose a non-invasive BRAF status identification pipeline that aims to remedy the weaknesses of earlier studies that relied on manual or automated segmentations. Previous works attempting to automate the segmentation of pLGGs19,23 used deep learning (DL) models based on the U-Net24. These DL architectures, which rely on convolutional neural networks (CNN), were state-of-the-art until recently. Now, however, more advanced models, such as those relying fully or partially on transformer layers25,26, or the ConvNeXt27 architecture, have proven more effective for adult brain tumor segmentation. We aimed to evaluate the performance of each of the aforementioned DL architectures for pLGG segmentation, to develop a more reliable automated segmentation model capable of consistently identifying the tumor region. We also aimed to develop and evaluate an ML pipeline to determine BRAF status from whole-brain fluid-attenuated inversion recovery (FLAIR) MRI sequences, relying on semiautomatic whole tumor volume segmentations and features derived from them, for pretraining only, rather than using segmentations as model inputs. Once deployed, this pipeline would have no reliance on tumor segmentations whatsoever.
We find that modern DL architectures such as MedNeXt outperform earlier CNN and transformer-based models for pLGG segmentation, although automated segmentations remain unreliable as direct inputs for downstream classification. Furthermore, we show that a segmentation-free whole-brain FLAIR MRI classification model achieves performance comparable to manual segmentation-based approaches and improves through pretraining. These results demonstrate that pLGG genetic status can be assessed without reliance on tumor segmentations, providing a practical and non-invasive alternative to tissue sampling.
Methods
This study conformed to the guidelines and regulations of the research ethics board of The Hospital for Sick Children, which approved this retrospective study and waived the need for informed consent due to the retrospective nature of the study. All data were deidentified after being extracted from the electronic health record database.
Data
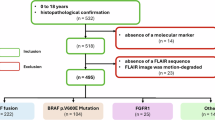
A total of 513 patients treated for pLGGs located in the brain parenchyma from 1999 to 2023 at The Hospital for Sick Children (SickKids) (Toronto, Canada) were identified using the SickKids electronic health record database. We excluded 15 patients for whom genetic information was unavailable. We excluded 43 additional patients who did not have a 2D axial or coronal FLAIR MR sequence available. We used FLAIR as our primary imaging sequence because it is widely regarded as the most sensitive method of detecting brain tumors28, and thus was the MRI sequence that was available for the most patients in our retrospective dataset. Furthermore, FLAIR is sensitive to leptomeningeal spread and non-contrast-enhancing lesions29, and depicts the tumor and surrounding area better than contrast-enhanced T1-weighted images30. The demographics of the 455 patients included in this study are summarized in Table 1.
Molecular subtyping
We followed the same stepwise process for molecular characterization as previous works, which relied on subsets of the data we used in this study11,13,16. Formalin-fixed paraffin-embedded tissue obtained during biopsy or resection was used for most patients. If not available, frozen tissue was used. BRAF mutations were detected using immunohistochemistry, sequencing, or droplet digital PCR. BRAF fusions were identified using either fluorescence in situ hybridization or an nCounter Metabolic Pathways Panel (NanoString Technologies). Patients not found to be positive for BRAF mutation or fusion were determined to be “non-BRAF altered”.
Image acquisition, segmentation, and preprocessing
Patients underwent brain MRI at field strengths of 1.5T or 3T using MRI scanners of different vendors (Signa, GE Healthcare; Achieva, Philips Healthcare; Magnetom Skyra, Siemens Healthineers). Whole tumor volumes were segmented semiautomatically using the level tracing-effect tool of 3D Slicer31,32 (Version 4.10.2) by a pediatric neuroradiology fellow (MS) and validated by a fellowship-trained pediatric neuroradiologist (MWW). To standardize images and reduce variability due to differences in acquisition methods over time, as well as other sources of heterogeneity, all scans were bias-corrected, resampled to a resolution of 240 × 240 × 155, z-score normalized, and registered to the SRI24 atlas33 using 3D Slicer.
ML pipelines for pLGG genetic status classification
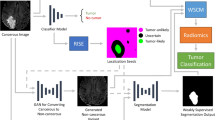
At a high level, we explored two approaches to designing a pipeline capable of classifying pLGGs into three classes: BRAF Fusion, BRAF Mutation, and non-BRAF altered. First, we assessed the potential for advanced DL models to improve the performance of automated segmentation models, making these segmentations more reliable. We then introduced a whole-brain FLAIR-based segmentation-free classification model. Figure 1 outlines the experiments that were used to assess both approaches, details of which can be found below.

We started by testing 3 different architectures on the task of pLGG segmentation (1). The architecture that performed best on this task was used for all ensuing experiments. We then tested whether transfer learning from an adult brain tumor dataset could help improve segmentation performance (2). Next, the segmentation architecture was converted into a classification model by adding some layers and removing others. The classification model was trained and evaluated with semiautomatic whole tumor volume segmentations as input (3) to generate baseline results. Finally, the classification model was trained and tested with whole-brain FLAIR MRIs as input using three different initialization schemes: random (4), transfer learning (5), and pretraining (6).
Advanced DL models for more accurate tumor segmentation
We evaluated the performance of three different DL architectures for tumor segmentation on our pLGG dataset: (1) a CNN based on the MedicalNet architecture34, (2) a CNN, transformer hybrid similar to TransBTS26, and (3) a MedNeXt35 style ConvNeXt-based27 model. A review and comparison of these architectures is provided in the Supplementary Information. We relied on the GitHub repository of the referenced publications where these architectures were introduced for implementation. The number and size of layers were adjusted to obtain a similar number of trainable parameters (approximately 400k) within each model for a fair comparison between architectures. We ran all subsequent experiments with the architecture that was found to perform best on this segmentation task because it showed the highest potential to derive insights from MRIs of pLGGs (Fig. 1).
Next, we explored whether transfer learning (TL) from the Brain Tumor Segmentation (BraTS) 2020 challenge dataset, which consists of MRIs of adult patients with brain tumors, could improve the performance of our segmentation models (Fig. 2). TL aims to improve performance on one task by leveraging knowledge learned from a related task in advance36. It involves pretraining a model on a large dataset before fine-tuning it on a smaller target dataset, with the model learning to extract useful features on the larger dataset. With this knowledge transferred to the smaller target dataset, the model is less likely to overfit compared to when training from scratch. To assess the effects of TL, the model was first pretrained on the BraTS dataset, where the provided FLAIR images and segmentations were used as model inputs and outputs, respectively. Subsequently, the model was fine-tuned and evaluated on our pLGG dataset.

The input to all models was the whole-brain FLAIR MRI from the respective dataset. The model was first trained on the BraTS dataset to identify tumor regions and HGG vs LGG, for the segmentation and classification tasks, respectively. The pretrained weights were then transferred over and trained on the corresponding task using the pLGG data.
Segmentation-free whole-brain FLAIR classification
The architecture that performed best for pLGG segmentation through the experiments detailed above was reconfigured for classification. The decoder was discarded, and a fully connected layer with three outputs, one for each of the three classes (BRAF Mutation, BRAF Fusion, and non-BRAF altered), was added on top of the encoder. The resultant model was randomly initialized and then trained to identify pLGG genetic subtype from whole-brain FLAIR MRI sequences, rather than using the tumor region alone. Next, similar to the experiments detailed above for segmentation, we attempted to improve classification model performance through transfer learning using the BraTS dataset (Fig. 2). The model was first trained to differentiate between high-grade and low-grade gliomas using whole-brain FLAIR MRI sequences from the BraTS dataset, then fine-tuned on our BRAF status classification task.
In another attempt to improve classification performance, we implemented a pretraining regimen (Fig. 3) that relied on a set of tasks we hypothesized would give the model a good understanding of the tumor region. The tasks were tumor segmentation, location identification, and radiomic feature value prediction, and the model was trained on them simultaneously using the sum of the three loss functions. The tumor segmentation task was the same as described in the first set of experiments. The location identification task required the model to predict whether the tumor was located in the supratentorial or infratentorial region of the brain. The label for this binary classification task was manually identified through analysis of the FLAIR MRI sequence by a pediatric neuroradiologist (MWW). Radiomics involves extracting quantitative features from medical images37; it has been shown that these features are predictive of BRAF status11,13. We extracted radiomic features from the semiautomatic whole tumor volume segmentations and used them to train a BRAF status random forest prediction model for patients in the training and validation sets according to the open-radiomics protocol38. The top 10 radiomics features were selected according to permutation importance; the values of these features were used for pretraining. If pretraining worked as expected, the model would recognize both where the tumor was located and characteristics within the tumor that are correlated with BRAF status. As illustrated in Fig. 3, after pretraining, the model was converted into a classification model, with certain pretrained layers kept intact, before the entire model was finetuned on the pLGG classification task.

The top of the image describes the pretraining phase, where the whole MRI is used as the input. In red is the architecture that was found to be best for tumor segmentation, which processes the image and outputs the predicted tumor region (top right, in green). During pretraining, a fully connected layer was added to the segmentation architecture. The output of the bottleneck flowed into the fully connected layer, which output radiomic feature values and tumor location (middle right, in green). The fully connected layer consisted of 11 neurons (10 radiomics features, one location feature). The bottom of the figure describes the fine-tuning phase, which relied on the pretrained weights in the blue box (encoder and bottleneck layers). These layers were transferred over directly from the pretrained model and used as the starting point for training the final BRAF status classification model. A new fully connected layer consisting of just three neurons (one for each class) was added, to take in the bottleneck output, and generate a BRAF status prediction (bottom right, in green). The same pipeline described at the bottom of the figure for fine-tuning was used for the experiment where we aimed to identify BRAF status from whole MRIs without pretraining, except the model parameters were randomly initialized.
Classification based on tumor region alone
Most previous studies aiming to identify pLGG genetic status used manually segmented tumor regions as inputs to their ML models. Thus, a final set of experiments, which used semiautomatic whole tumor volume segmentations alone as input to the same classification architecture described above, was run and served as a baseline.
Statistics and reproducibility
To train and evaluate all models, patients were divided into train (80%), validation (10%), and test (10%) sets 25 times using a stratified split to ensure representative proportions of each class within each split. In each of these 25 trials, three separate models were trained on the training set using three different learning rates (0.00001, 0.0001, 0.001). Validation loss was calculated on the validation set at the end of each epoch. The optimal model selected for final evaluation on each trial was chosen according to the learning rate and epoch that resulted in the lowest validation loss. Reported performance metrics were based on the unseen test set, which was only used for model evaluation. All experiments for all models used identical data splits, allowing for direct comparisons of performance across architectures and initialization strategies. The “corrected resampled t-test”39,40 was used to test for significant differences in model performance. The use of nested-cross validation violates the independence assumption of the traditional t-test41, which could not be used here.
Experiment configuration
Kaiming Normal weight initialization was used to initialize all model parameters, where pretraining was not employed. Dropout was used to train all models (0.75 for fully connected layers, 0.25 for convolutional layers where entire channels were zeroed out) and turned off for interference. Cosine annealing was used to decay the learning rate over a maximum of 200 epochs for classification, and 50 for segmentation, which was slower. Training was stopped early to conserve resources if validation loss did not fall for 10 epochs in a row. The batch size was 8 images for classification experiments, but only 2 for segmentation experiments, which required more memory. Gradient accumulation was used for the segmentation experiments to effectively increase the batch size by only updating model parameters after every other batch.
Cross-entropy loss, weighted by the proportion of patients in each class, was used to train classification models. Classification models were primarily evaluated according to the One-Vs-Rest area under the receiver operating characteristic curve (AUC). Segmentation models were trained using the sum of soft dice loss and binary cross-entropy loss and evaluated by the Dice score. During pretraining, the radiomic feature value prediction and location identification tasks were trained using a smooth L1 loss and binary cross-entropy loss, respectively. Python 3.11.0 was used to run all experiments, and the PyTorch 1.13.042 library was used for DL.
Results
Segmentation
The segmentation results across 25 trials for all three DL architectures are shown in Fig. 4. The MedNeXt segmentation model (mean Dice score: 0.555, 95% confidence interval (CI) for the mean Dice score: [0.529, 0.574]) outperformed both the CNN-based MedicalNet (0.516, [0.492, 0.537]) and the CNN-transformer hybrid TransBTS (0.449, [0.420, 0.482]). The differences between the performance of each of the models were statistically significant. The p-values of the corrected resampled t-tests when comparing the performance of the MedNeXt model to that of MedicalNet and TransBTS were 0.045 and 0.00014, respectively, while comparing the latter two models resulted in a p-value of 0.017.

The solid orange line is the median of each distribution, while the dashed red line is the mean.
Classification
The mean three-class AUCs for all classification models are listed in Table 2. With weights initialized from scratch, the MedNeXt style classification model achieved a mean AUC of 0.747 (95% CI for the mean: [0.719, 0.774]) on the whole brain FLAIR MRI BRAF status classification task, where no segmentation was used. Performance improved (p-value: 0.0466) through pretraining on the segmentation, location, and radiomic feature value prediction tasks, resulting in a mean AUC of 0.779 (95% CI for the mean: [0.759, 0.799]), which was found to be the best classifier according to AUC. Additional performance metrics for this best model are listed in Table 3. Notably, despite a relatively high AUC, accuracy was determined to be relatively low overall (57%). The confusion matrix and per-class metrics in Table 3 suggest that this is because the model lacks the ability to differentiate between BRAF Mutation and non-BRAF altered tumors. Overall, the model is best at identifying BRAF Fusion: the AUC (0.867), precision (0.746), and recall (0.747) for this class were the highest.
The baseline model, which used the tumor region alone as an input, performed similarly to the whole-brain FLAIR MRI models. The mean baseline AUC of 0.756 (95% CI for the mean: [0.735, 0.778]) was only slightly higher than the randomly initialized whole FLAIR MRI model, and the difference between the performance of the two models was not statistically significant (p-value: 0.342). The best-performing whole-brain FLAIR MRI model, pretrained on pLGG data, produced a higher AUC than the baseline, though again the difference was found to not be statistically significant (p-value: 0.141).
Transfer learning
TL from BraTS did not have a statistically significant impact on segmentation or classification performance. For the segmentation task, pretraining the MedNeXt style model on BraTS before fine-tuning it on our pLGG dataset resulted in a mean Dice score of 0.539 (95% CI for the mean: [0.525, 0.553]). This was lower than when training from scratch (0.555), and not significantly different (p-value: 0.180). Similarly, pretraining the MedNeXt classification model on BraTS prior to fine-tuning resulted in lower AUC (mean: 0.725, 95% CI for the mean: [0.704, 0.746]) than when training from scratch (0.747). Statistical testing showed that the difference was not significant (p-value: 0.215).
Discussion
In this study, we introduced a segmentation-free MRI-based pipeline to assess molecular status in patients with pLGG. Our whole-brain FLAIR MRI classification approach reduces the reliance of ML models used for pLGG genetic status classification on tumor segmentations. Our results illustrate that even without pretraining, and thus without any knowledge about tumor location whatsoever, the whole-brain FLAIR MRI classification model performs similarly to the baseline model that takes the tumor region as an input. Pretraining on pLGG tumor segmentations and features derived from them before fine-tuning on the classification task was found to improve model performance. This pretraining approach introduces a novel way of incorporating tumor information into a classification pipeline that does not rely on segmentations after training. The pretrained whole-brain FLAIR MRI classification model was able to accurately identify tumors with a BRAF Fusion but had difficulties differentiating between BRAF Mutations and non-BRAF altered tumors. These results suggest identification of BRAF Fusion, the most common genetic alteration in pLGGs, is the most promising use case for the whole-brain FLAIR MRI classification pipeline. We emphasize that once deployed, the pretrained model does not have any need for tumor segmentations.
We also explored the potential for advanced DL architectures to accurately and reliably segment MRIs of patients with pLGGs. Previous PLGG segmentation studies have relied on CNN-based models19,23; we found that a modern architecture, based on the MedNext architecture, performed better. The MedNeXt model was designed using ConvNeXt27 layers, which combine the inherent inductive biases of CNNs with the ability of transformers to scale efficiently and capture long-range dependencies35. It appears that MedNeXt combines the best of CNNs and transformers; it was previously shown to outperform both CNNs and transformers for adult brain tumor segmentation35. Our results suggest MedNeXt is better at pediatric brain tumor segmentation, too. However, for some patients, segmentations were poor (Fig. 5), suggesting that automated segmentations are too unreliable for use in downstream tasks although they were sufficiently accurate to be helpful in pretraining. Although expert manual segmentations are currently considered the gold standard, they pose practical challenges as classification model inputs due to known inter- and intra-rater variability, as well as scalability concerns. Variability in model inputs may reduce model reliability and harm clinician confidence when predictions are sensitive to subtle differences in input delineations.

Notably, for the two topmost cases in the right column, the segmentation is completely off, with no tumor identified at all in the slice depicted. A downstream classification model will struggle to identify the genetic status of a tumor accurately if the automated segmentation model it depends on does not correctly identify the tumor.
Notably, TL from BraTS did not improve classification or segmentation model performance. TL has successfully boosted model performance for numerous medical tasks36. However, it has not always proven useful, likely due to domain shift, which is when there are differences between the dataset involved in pretraining and the dataset the model is later fine-tuned on43,44. For example, as described in ref. 45, previous studies focused on TL from BraTS for pLGG automatic tumor segmentation23,46 did not find a consistent, substantial improvement in model performance using TL. Considering the significant differences between adult and pediatric brain tumors47,48,49, it is perhaps unsurprising that knowledge does not transfer between the two patient groups. We hypothesize that our custom pretraining scheme involving semiautomatic whole tumor volume segmentation, tumor location, and radiomic features was successful because it did not rely on any additional data and thus was not susceptible to domain shift.
Currently, the genetic status of pLGG is ascertained through the analysis of tumor tissue acquired through either resection or biopsy9. However, in more than one-third of cases, neither option is possible or clinically advisable20. This is particularly true for tumors in anatomically challenging locations, such as midline pLGG45. As a result, the genetic status of these difficult-to-access tumors often remains unknown. Our model offers a potential non-invasive solution for these patients. Even in cases where biopsy is feasible, it presents limitations: samples can be inadequate for molecular analysis, and the procedure itself is expensive and carries inherent risks. Reported rates of permanent and transient complications are 0.7% and 5.8%, respectively50. Therefore, even when biopsy is an option, our non-invasive approach provides a potentially safer, more accessible alternative for determining the genetic status of pLGG.
Our work is not without limitations. Our observations about the relative performance of different DL models and training schemes are specific to the situations in which we evaluated them, namely, on our hospital’s pLGG dataset using a finite set of hyperparameters. Further tests on different tasks, datasets, and model configurations need to be run to determine the generalizability of these claims. However, the fact that similar results have been found for related tasks35 supports our claims. Additionally, our model was best suited for identifying BRAF Fusion and had difficulties differentiating BRAF Mutations from non-BRAF altered tumors. This could potentially be addressed by data augmentation techniques such as synthetic image generation, which has proven successful in improving pLGG genetic status classification51, but was not applied here. Furthermore, uncertainty quantification techniques such as evidential DL52, or dropout as Bayesian approximation53, could be applied to identify uncertain predictions, potentially facilitating the use of our model to distinguish between all 3 classes more accurately, or even to identify uncertain segmentations, enabling the use of automated segmentations for certain cases. Notably, despite these limitations, our findings suggest that the model in its current form already holds clinical utility. Specifically, it could be deployed as a binary classifier, distinguishing BRAF Fusion, the most common genetic alteration in pLGG, from all other genetic subtypes, with a high degree of accuracy.
Conclusion
We introduced the first model for pLGG genetic status assessment that does not require tumor segmentations as inputs. Our training pipeline uses segmentations and features derived from them for pretraining, but once deployed, the final model does not require any segmentations to determine the genetic status of pLGG.
Data availability
Due to its sensitive nature, the medical data used in this study has not been made publicly available. Data will be made available upon reasonable request to the corresponding author, pending approval of the institution and trial/study investigators who contributed to the dataset. Source data for figures can be found in the supplementary material.
Code availability
All code used to design ML models, run experiments, and analyze results is available on GitHub54.
References
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013-2017. Neuro. Oncol. 22, iv1–iv96 (2020).
Sievert, A. J. & Fisher, M. J. Pediatric low-grade gliomas. J. Child Neurol. 24, 1397–1408 (2009).
Ryall, S. et al. Integrated molecular and clinical analysis of 1000 pediatric low-grade gliomas. Cancer Cell 37, 569–583.e5 (2020).
Ryall, S., Tabori, U. & Hawkins, C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol. Commun. 8, 30 (2020).
AlRayahi, J., Alwalid, O., Mubarak, W., Maaz, A. U. R. & Mifsud, W. Pediatric brain tumors in the molecular era: updates for the radiologist. Semin. Roentgenol. 58, 47–66 (2023).
Louis, D. N. et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 23, 1231–1251 (2021).
Fangusaro, J. et al. Pediatric low-grade glioma: state-of-the-art and ongoing challenges. Neuro Oncol. 26, 25–37 (2024).
Manoharan, N., Liu, K. X., Mueller, S., Haas-Kogan, D. A. & Bandopadhayay, P. Pediatric low-grade glioma: targeted therapeutics and clinical trials in the molecular era. Neoplasia 36, 100857 (2023).
Bennett, J. et al. Canadian pediatric neuro-oncology standards of practice. Front. Oncol. 10, 593192 (2020).
Wagner, M. W. et al. Dataset size sensitivity analysis of machine learning classifiers to differentiate molecular markers of paediatric low-grade gliomas based on MRI. Oncol. Radiother. 16, 01–06 (2022).
Wagner, M. W. et al. Radiomics of pediatric low-grade gliomas: toward a pretherapeutic differentiation of BRAF-mutated and BRAF-fused tumors. AJNR Am. J. Neuroradiol. https://doi.org/10.3174/ajnr.A6998 (2021).
Liu, Z. et al. Radiomic features from multiparametric magnetic resonance imaging predict molecular subgroups of pediatric low-grade gliomas. BMC Cancer 23, 848 (2023).
Kudus, K. et al. Increased confidence of radiomics facilitating pretherapeutic differentiation of BRAF-altered pediatric low-grade glioma. Eur. Radiol. https://doi.org/10.1007/s00330-023-10267-1 (2023).
Xu, J., Lai, M., Li, S., Cai, L. & Shi, C. Noninvasive prediction of histological grading in pediatric low-grade gliomas using preoperative T2-FLAIR radiomics features. World Neurosurg. https://doi.org/10.1016/j.wneu.2023.04.096 (2023).
Soldatelli, M. D. et al. Identification of multiclass pediatric low-grade neuroepithelial tumor molecular subtype with ADC MR imaging and machine learning. AJNR Am. J. Neuroradiol. 45, 753–760 (2024).
Kudus, K. et al. Beyond hand-crafted features for pretherapeutic molecular status identification of pediatric low-grade gliomas. Sci. Rep. 14, 19102 (2024).
Bø, H. K. et al. Intra-rater variability in low-grade glioma segmentation. J. Neurooncol. 131, 393–402 (2017).
Zou, K. H. et al. Statistical validation of image segmentation quality based on a spatial overlap index. Acad. Radiol. 11, 178–189 (2004).
Vafaeikia, P. et al. MRI-based end-to-end pediatric low-grade glioma segmentation and classification. Can. Assoc. Radiol. J. 75, 153–160 (2024).
Tak, D. et al. Noninvasive molecular subtyping of pediatric low-grade glioma with self-supervised transfer learning. Radiol. Artif. Intell 6, e230333 (2024).
Vafaeikia, P. et al. Improving the segmentation of pediatric low-grade gliomas through Multitask Learning. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2022, 2119–2122 (2022).
Vafaeikia, P. Deep learning methods for pediatric brain tumour diagnosis (University of Toronto, Toronto, 2022).
Boyd, A. et al. Expert-level pediatric brain tumor segmentation in a limited data scenario with stepwise transfer learning. medRxiv https://doi.org/10.1101/2023.06.29.23292048 (2023).
Ronneberger, O., Fischer, P. & Brox, T. U-Net: Convolutional Networks for Biomedical Image Segmentation. In Medical Image Computing and Computer-Assisted Intervention – MICCAI 2015 234–241 (Springer International Publishing, 2015).
Karimi, D., Vasylechko, S. D. & Gholipour, A. Convolution-free medical image segmentation using transformers. in Medical Image Computing and Computer Assisted Intervention – MICCAI 2021 78–88 (Springer International Publishing, 2021).
Wang, W. et al. TransBTS: multimodal brain tumor segmentation using transformer. in Medical Image Computing and Computer Assisted Intervention – MICCAI 2021 109–119 (Springer International Publishing, 2021).
Liu, Z. et al. A ConvNet for the 2020s. In Proc. of the IEEE/CVF Conference on Computer Vision and Pattern Recognition 11976–11986 (2022).
Gupta, T., Gandhi, T. K., Gupta, R. K. & Panigrahi, B. K. Classification of patients with tumor using MR FLAIR images. Pattern Recognit. Lett. 139, 112–117 (2020).
Haddad, A. F., Young, J. S., Morshed, R. A. & Berger, M. S. FLAIRectomy: resecting beyond the contrast margin for glioblastoma. Brain Sci. 12, 544 (2022).
Barajas, R. F. Jr. & Cha, S. Metastasis in adult brain tumors. Neuroimaging Clin. N. Am 26, 601–620 (2016).
Kikinis, R., Pieper, S. D. & Vosburgh, K. G. 3D Slicer: a platform for subject-specific image analysis, visualization, and clinical support. in Intraoperative Imaging and Image-Guided Therapy (ed. Jolesz, F. A.) 277–289 (Springer New York, New York, NY, 2014).
Fedorov, A. et al. 3D Slicer as an image computing platform for the quantitative imaging network. Magn. Reson. Imaging 30, 1323–1341 (2012).
Rohlfing, T., Zahr, N. M., Sullivan, E. V. & Pfefferbaum, A. The SRI24 multichannel atlas of normal adult human brain structure. Hum. Brain Mapp. 31, 798–819 (2010).
Chen, S., Ma, K. & Zheng, Y. Med3D: transfer learning for 3D medical image analysis. Preprint at https://doi.org/10.48550/arXiv.1904.00625 (2019).
Roy, S. et al. MedNeXt: Transformer-driven scaling of ConvNets for medical image segmentation. In Lecture Notes in Computer Science 405–415 (Springer Nature Switzerland, Cham, 2023).
Kim, H. E. et al. Transfer learning for medical image classification: a literature review. BMC Med. Imaging 22, 69 (2022).
Khalvati, F., Zhang, Y., Wong, A. & Haider, M. A. Radiomics. Encycl. Biomed. Eng. 2, 597–603 (2019).
Namdar, K., Wagner, M. W., Ertl-Wagner, B. B. & Khalvati, F. Open-radiomics: a collection of standardized datasets and a technical protocol for reproducible radiomics machine learning pipelines. BMC Med. Imaging 25, 312 (2025).
Bouckaert, R. R. & Frank, E. Evaluating the replicability of significance tests for comparing learning algorithms. in Advances in Knowledge Discovery and Data Mining 3–12 (Springer Berlin Heidelberg, 2004).
Nadeau, C. & Bengio, Y. Inference for the generalization error. Mach. Learn. 52, 239–281 (2003).
Dietterich, T. G. Approximate statistical tests for comparing supervised classification learning algorithms. Neural Comput. 10, 1895–1923 (1998).
Paszke, A. et al. PyTorch: an imperative style, high-performance deep learning library. in Proc. 33rd International Conference on Neural Information Processing Systems 8026–8037 (Curran Associates Inc., Red Hook, NY, USA, 2019).
Kwasigroch, A., Grochowski, M. & Mikołajczyk, A. Self-supervised learning to increase the performance of skin lesion classification. Electronics 9, 1930 (2020).
Matsoukas, C., Haslum, J. F., Sorkhei, M., Soderberg, M. & Smith, K. What makes transfer learning work for medical images: Feature reuse & other factors. In 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR) 9215–9224 (IEEE, 2022).
Kudus, K., Wagner, M., Ertl-Wagner, B. B. & Khalvati, F. Applications of machine learning to MR imaging of pediatric low-grade gliomas. Childs. Nerv. Syst. https://doi.org/10.1007/s00381-024-06522-5 (2024).
Nalepa, J. et al. Segmenting pediatric optic pathway gliomas from MRI using deep learning. Comput. Biol. Med. 142, 105237 (2022).
Merchant, T. E., Pollack, I. F. & Loeffler, J. S. Brain tumors across the age spectrum: biology, therapy, and late effects. Semin. Radiat. Oncol. 20, 58–66 (2010).
Dias, S. F., Richards, O., Elliot, M. & Chumas, P. Pediatric-like brain tumors in adults. Adv. Tech. Stand. Neurosurg. 50, 147–183 (2024).
Pollack, I. F. Pediatric brain tumors. Semin. Surg. Oncol. 16, 73–90 (1999).
Hamisch, C. et al. Feasibility, risk profile and diagnostic yield of stereotactic biopsy in children and young adults with brain lesions. Klin. Padiatr. 229, 133–141 (2017).
Zhou, M. et al. Generating 3D brain tumor regions in MRI using vector-quantization Generative Adversarial Networks. Comput. Biol. Med. 185, 109502 (2025).
Sensoy, M., Kandemir, M. & Kaplan, L. M. Evidential deep learning to quantify classification uncertainty. In Proc. of the 32nd International Conference on Neural Information Processing Systems 3183–3193 (Curran Associates Inc., 2018).
Gal, Y. & Ghahramani, Z. Dropout as a Bayesian approximation: Representing model uncertainty in deep learning. ICML 48, 1050–1059 (2015).
Kudus, K. Code for ‘Segmentation-Free Pretherapeutic BRAF-Status Identification Of Pediatric Low-Grade Gliomas’. https://github.com/IMICSLab/pLGG_whole_image (2025).
Acknowledgements
This study was made possible by the financial support of the Canadian Institutes of Health Research (CIHR) (Funding Reference Number: 184015).
Author information
Authors and Affiliations
Contributions
K.K., B.E.W., and F.K. conceptualized and designed the study. K.K. developed the machine learning models, executed all experiments, and designed the pretraining and classification workflows. J.B., A.L., P.D., U.T., and C.H. were responsible for gathering, curating, and verifying clinical and genetic data. M.W., M.S., and B.E.W. performed imaging review and tumor segmentation validation. K.K. drafted the manuscript, and all authors contributed to its critical revision and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks Ioan Paul Voicu and Margot Lazow for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source Data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kudus, K., Wagner, M., Sheng, M. et al. Segmentation-free pretherapeutic assessment of BRAF-status in pediatric low-grade gliomas. Commun Med 5, 507 (2025). https://doi.org/10.1038/s43856-025-01204-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43856-025-01204-y
