
Abstract
Chemotherapy-induced peripheral neuropathy (CIPN) is a debilitating side effect with limited treatment options. The primary challenge in developing therapies is the lack of identified neurotoxic mechanisms. To address this, an integrated omics approach, combining transcriptomics, proteomics, and metabolomics, is essential to map the biological changes underlying the condition. Here, we show how data from these complementary approaches converge on key mechanisms involved in CIPN through a critical summary of animal and human studies. Current research highlights several key drivers of CIPN, such as inflammatory signaling and oxidative stress, mitochondrial dysfunction, and disrupted lipid metabolism. Although most data currently stem from preclinical models, the pathways identified offer promising targets for biomarker discovery and treatment. To translate these findings into clinical applications, integrated omics studies in human samples are urgently needed, focusing on a personalized approach. Future breakthroughs depend on large-scale human studies to tailor antineoplastic choices and neuroprotective treatments to individual patient needs.

Similar content being viewed by others

Introduction
Chemotherapy-induced peripheral neuropathy (CIPN) represents a particularly complex pain syndrome for which effective preventive measures and treatment options remain limited1. It significantly impacts patients’ quality of life over prolonged periods and contributes to economic burdens and increased hospitalization2. With advances in oncology leading to improved survival rates, CIPN affects several million patients worldwide annually; thus, a deeper understanding of the systemic and molecular changes underlying its development are urgently required3,4.
The heterogeneous clinical manifestations of CIPN range from sensory to motor and autonomic dysfunction. They are influenced by treatment regimens, dosage, and patient-specific factors, including age-related vulnerabilities such as reduced neuroplasticity and increased oxidative stress susceptibility5,6. Paclitaxel is among the most common chemotherapeutic drugs. Paclitaxel stabilizes microtubules and prevents their depolymerization. This stabilization prevents microtubules from disassembling, which leads to mitotic arrest, preventing normal cell division and leading to programmed cell death (apoptosis). However, this effect can affect both normal and cancer cells, resulting in side effects such as CIPN. Another class of chemotherapy agents includes proteasome inhibitors such as bortezomib and carfilzomib7,8,9. While taxane chemotherapy has been associated with dysregulation of inflammatory pathways, proteasome inhibitors primarily influence oxidative stress pathways. Bortezomib causes higher cytoskeletal damage, actin filament destabilization, and protein oxidation than carfilzomib in mouse neural stem cells7. In addition, platinum-based antineoplastics (particularly oxaliplatin and cisplatin), are also known to induce peripheral neuropathy, since they induce numerous changes either in the structure or functioning of neuronal and glial cells10.
Importantly, dose intensity and administration schedules play a crucial role, with high-dose regimens and shorter infusion times often linked to higher incidences of CIPN11.
In addition to treatment parameters, individual patient-specific factors, including comorbid conditions such as diabetes, vitamin deficiencies, and renal or thyroid dysfunction, can predispose patients to neurotoxicity12. Genetic susceptibility has also emerged as a critical determinant, with genome-wide association studies identifying single-nucleotide polymorphisms in genes related to drug metabolism, microtubule stability, and neuronal function as risk factors for CIPN13.
Recently, serum neurofilament light chain (NfL) has emerged as a promising biomarker of axonal damage in CIPN. Preclinical studies demonstrated that circulating NfL levels increase in parallel with axonal degeneration and correlate with electrophysiological and pathological evidence of peripheral nerve damage in rodent models treated with neurotoxic agents such as vincristine, paclitaxel, and cisplatin14,15. Importantly, prospective clinical studies in patients receiving paclitaxel have confirmed that serum NfL levels increase during chemotherapy and correlate with the severity of neuropathy, with early elevations predicting the development of clinically significant CIPN16. To date, NfL represents the only blood biomarker for CIPN that has undergone translational validation from preclinical models to clinical studies, supporting its potential role for early detection and monitoring of treatment-related neurotoxicity.
Despite increasing awareness of these multifactorial contributors, there is a lack of standardized biomarkers or predictive tools to guide individualized management of CIPN. This difficulty stems from the complex and multifaceted nature of CIPN, which involves diverse patient responses, varying clinical presentations, and a lack of uniform diagnostic and therapeutic guidelines17.
This gap highlights the urgent need for integrative approaches capable of decoding the molecular complexity of the syndrome. In this context, modern omics technologies, encompassing genomics, transcriptomics, proteomics, and metabolomics, offer powerful platforms to identify mechanistic drivers and patient-specific susceptibility signatures. By dissecting the molecular networks perturbed during CIPN, omics-based research may uncover novel biomarkers for early detection, prognostic assessment, and therapeutic targeting18.
In this review, we comprehensively examine the findings from various “omics” studies related to CIPN. These studies aim to identify potential biomarkers that reflect CIPN symptoms and better understand its underlying mechanisms.
Using transcriptomics to identify cellular RNA contributing to CIPN
In recent years, the rapid progress of next-generation sequencing-based technologies, such as genomics (i.e., whole genome sequencing or whole exome sequencing), epigenomics (i.e., DNA methylation analysis) and transcriptomics (i.e., microarray analysis, bulk RNA or single-cell RNA sequencing) has significantly contributed to our understanding of biological systems in both health and disease in a faster, cheaper and high-throughput manner. Unlike methods based on bulk populations, single-cell resolution technologies have become crucial for studying differentially expressed genes and biological functions at a cellular level, as well as for discovering new and potentially unique targets19,20,21,22. Applications such as single-cell RNA sequencing (scRNAseq) or single-nuclei RNA sequencing (snRNAseq), for example, have a major role in revealing rare cell features, identifying specific cell types, elucidating gene interactions and clarifying the developmental stages of cell lineages. These methodologies rely on RNA sequencing of individual whole cells or single nuclei, followed by unsupervised grouping based on shared gene expression profiles, without requiring a priori knowledge20,23.
The intricate heterogeneity of dorsal root ganglia (DRG), meticulously described using electrical properties, cell size, myelination levels, and expression of biological markers24, has been further elucidated through single-cell/nuclei transcriptomics24. scRNAseq studies on rodent sensory neurons have clarified their molecular identities, classifying them into distinct cell types25,26. Applied to various species, single-cell/nuclei transcriptomic analyses have investigated DRG heterogeneity in rodents, humans and non-human primates to identify potential similarities across species27,28,29. Remarkably, single-nuclear transcriptomics of donor-derived cells has revealed several distinct classes of human somatosensory neurons30. While human peripheral sensory neuron subtypes largely overlap with those of rodents, they exhibit unique transcriptional profiles that highlight species-specific differences30.
ScRNAseq is also used to explore how the whole DRG transcriptome can change in peripheral neuropathies31,32. However, for some neuropathies, particularly those caused by chemotherapy neurotoxicity, transcriptomic characterization of DRG is more challenging. Indeed, the plethora of chemotherapy agents capable of inducing peripheral neuropathy in animal models complicates the establishment of a unique DRG transcriptomic profile through comparative analysis across models. Using a traditional bulk RNA sequencing method, which measures the RNA expression across a large population of mixed cell types, researchers compared the transcriptome of the lumbar DRG of rodents after administration of vincristine, oxaliplatin, and cisplatin33. Vincristine treatment caused predominant upregulation of genes associated with neuroinflammatory mechanisms, neuropathic pain and nerve damage. In contrast, gene expression changes in the DRG following oxaliplatin and cisplatin treatments indicated alterations in neuronal processes33. Further studies based on bulk RNA sequencing revealed that paclitaxel induces transcriptomic changes at the sensory neuron level34, while a comparative analysis of RNA expression demonstrated that oxaliplatin primarily affects RNA levels in glial cells34,35.
However, bulk transcriptomics does not determine which cell types are involved in these changes. Although bulk RNA sequencing has become a fundamental tool in understanding cell biology36,37, it relies on total RNA expression analysis without accounting for cell heterogeneity36,37. While this approach has facilitated the identification of biomarkers for cancer diagnosis, prognosis, and prediction across various tumor types38,39,40, it misses transcriptional diversity at the single-cell scale38,39,40.
These limitations have been addressed in recent years by scRNAseq and snRNAseq, which are now considered among the most powerful genomic tools for dissecting transcriptome cell diversity, discovering rare and unique cell types and features otherwise undetectable41. For example, scRNAseq of DRG of female BALB/c mice treated with cisplatin was used to explore the effect of chemotherapy agents on the DRG transcriptome42. A cumulative dose of 42 mg/kg of cisplatin, administered over several weeks of treatment, caused upregulation of the cyclin-dependent kinase inhibitor-1a (Cdkn1a) in the DRG. scRNAseq on sorted DRG incubated with antibodies against TrkA, TrkB, and TrkC, which are markers that partially identify three major neuronal populations in the DRG, revealed seven distinct clusters, including neuronal and non-neuronal cells. Using transcriptomics, cisplatin treatment was shown to induce the upregulation of genes associated with senescence mechanisms in DRG sensory neurons.
By adopting the same methodology but employing a different model of CIPN, oxaliplatin exposure was found to induce significant modulation of inflammatory processes in both the DRG and satellite glial cells. This suggests that the complex interplay between sensory neurons and glial cells in response to oxaliplatin-induced neurotoxicity could serve as a potential target for developing therapeutic strategies to mitigate oxaliplatin-induced peripheral neuropathy43.
Differentially expressed genes across all neuronal subclusters were found using snRNAseq technology in a mouse model of paclitaxel-induced neuropathy44,45. The C-low threshold mechanoreceptors (C-LTMR) neuronal subtype exhibited the most significant transcriptomic changes after paclitaxel treatment, especially in those genes linked to nerve fiber alterations and potassium-related currents44.
An alternative approach46 investigated the effect of paclitaxel in the absence of SET-Binding Factor 2 (SBF2) in CIPN46. SBF2, previously reported to be linked to Charcot- Marie-Tooth neuropathy47,48, was found to be mutated in individuals undergoing paclitaxel treatment for breast cancer, suggesting a potential correlation with a higher risk of developing CIPN47,48,49. Paclitaxel-induced axon degeneration was exacerbated by knocking down SBF2 in pluripotent stem cell-derived sensory neurons (iPSC-dSN) generated from reprogrammed peripheral blood mononuclear cells from healthy humans. This also led to a decreased sodium current46. While identifying SBF2 as a novel potential target gene46, this also suggests that a pre-existing genetic variation in SBF2 may predispose patients to a higher risk of developing CIPN. More recently, a genome-wide association study (GWAS) identified novel susceptible genomic loci in chemotherapy-treated patients that might play a role in the development or prevention of CIPN50,51
Transcriptomic methodologies, particularly scRNA and snRNA sequencing, have revolutionized our understanding of the molecular landscape of DRG and non-neuronal cells under physiological and pathological conditions, such as CIPN. Contrary to bulk RNA sequencing, which lacks single-cell resolution, scRNA and snRNA sequencing provide a detailed understanding of cell diversity, uncovering rare subtypes and distinct molecular profiles, particularly valuable in pathological conditions. An advanced evolution of scRNAseq is represented by spatial transcriptomics, which enables the spatial mapping of cellular transcriptomes52,53.
Application of proteomics to investigations of CIPN
Proteomics, particularly mass spectrometry (MS)-based proteomics, has emerged as a powerful technology to dissect the complex pathophysiology of CIPN and to support the development of predictive biomarkers and personalized strategies for its management.
Its main strengths are its ability to capture a dynamic and system-wide view of protein expression, modifications, interactions, and functional states, which is very valuable for understanding a multifactorial and heterogeneous condition such as CIPN. Mitochondrial dysfunction, axonal degeneration, and neuroimmune crosstalk have been shown to evolve over time during CIPN and may precede the onset of clinical symptoms54,55. Compared with transcriptomics or genomics, proteomics provides a more direct readout of the functional consequences of chemotherapeutic injury on peripheral nerves, yet its translation into clinically actionable insights remains challenging.
Modern MS-based platforms are capable of resolving wide molecular landscapes and detecting subtle biochemical perturbations, offering a translational bridge toward biomarker discovery and mechanistic insights56.
Among the various platforms, liquid chromatography tandem mass spectrometry (LC-MS/MS) has become the gold standard for proteomic profiling. Recently the introduction of ion mobility separation and high-resolution instrumentation has dramatically improved the sensitivity, accuracy, and throughput of proteomic analyses57,58,59,60. These advances enable both identification and quantification of thousands of proteins in small sample volumes within short timeframes. Importantly, this high depth of coverage increases the probability of detecting low-abundance proteins, which are often biologically and clinically relevant in early disease stages, including the prodromal phase of neuropathy61. Furthermore, the integration of machine learning and artificial intelligence into proteomic workflows enhances the ability to identify meaningful patterns, stratify patient populations, and develop clinically actionable predictive models62,63.
For these reasons, MS-based proteomics has been increasingly applied in CIPN research to elucidate drug-specific mechanisms, identify diagnostic and prognostic biomarkers, and explore therapeutic responses. Several studies, summarized in Supplementary Data 1, have adopted this approach across different chemotherapy classes and model systems.
Clinical applications of proteomics in CIPN have aimed to identify early biomarkers and explore drug-specific mechanisms. Label-free LC-MS/MS analyzed serum exosomes from 50 women with early-stage breast cancer undergoing taxane-based chemotherapy64. Neuropathy was assessed longitudinally using quantitative sensory testing, the Functional Assessment of Cancer Therapy-Neurotoxicity Subscale, and neuropathic pain scales. Over 700 proteins were identified, with significant enrichment in immune response pathways, especially the humoral response. A specific panel of 12 proteins (HBB, HP, ACTB, APOM, AGT, HRG, HPR, F9, F5, PGLYRP2, and KLKB1) was found to distinguish patients who developed neuropathy from those who did not, even at baseline, suggesting the existence of pre-existing vulnerability traits. However, the study lacked validation in independent cohorts, and the biological relevance of several identified proteins remains unclear due to their ubiquitous expression.
More recently, an untargeted plasma proteomics approach using LC-MS/MS quantified 83 proteins in a cohort of breast cancer patients receiving weekly paclitaxel (n = 36). Complement C3 levels, measured at the end of the first infusion, were significantly associated with taxane-induced peripheral neuropathy (TIPN) severity during treatment (P = 0.0002), independent of age and baseline neuropathy. Patients with higher C3 levels showed worse neuropathy trajectories and more frequent treatment discontinuation. These findings not only confirm previous suggestions of inflammatory involvement in TIPN but also propose complement activation as a potential predictive biomarker and therapeutic target in paclitaxel-treated patients65. Yet, the restricted protein panel, likely due to plasma’s wide dynamic range, highlights the persistent technical challenge of detecting low-abundance neuroimmune mediators in blood.
In vivo studies in rodent models have strengthened the evidence for an extensive proteomic dysregulation that occurs even before behavioral signs of neuropathy arise, supporting the concept of prodromal molecular events. Two independent studies investigating lumbar DRG after paclitaxel exposure in C57BL/6J mice and Sprague-Dawley rats revealed protein dysregulation linked to mitochondrial function, axonal damage/regeneration, immune signaling, and microtubule dynamics66,67.
In particular, major proteomic alterations preceded neuropathy onset (day 7 post-treatment), with 295 proteins significantly dysregulated, compared to only 15 at peak neuropathy66. Four proteins (myosin-1, AHNAK nucleoprotein, spectrin alpha chain, and integrin beta) were modulated at both timepoints, whereas spectrin beta was specifically downregulated at peak CIPN. Notably, nodal β-spectrins, which are involved in Na⁺ channel localization, may represent a convergence point between cytoskeletal remodeling and altered neuronal excitability 68. Moreover, Ingenuity Pathway Analysis identified PPARG and its regulator CTNNB1 (β-catenin) as putative upstream regulators66. Experimental validation by immunoblotting for CTNNB1 provides additional support for the involvement of these molecular pathways in the early phase of CIPN. Nevertheless, cross-study reproducibility remains limited, partly due to differences in animal strain, drug dosing, sample preparation, and data analysis pipelines.
Treatment of ND7/23 neuronal cells and RSC96 Schwann cells exposed to Paclitaxel with sonic hedgehog (Shh) protein inducers reduces senescence-related side effects69. This pattern was confirmed by in vivo studies in Paclitaxel-treated mice, which showed an improvement in nerve conduction and a decrease in axonal degeneration. Moreover, Shh activation can inhibit senescence and alleviate CIPN by stabilizing SP1 levels and blocking the TRIM25–CXCL13 axis. In particular, this latter finding involved the use of LC-MS/MS to identify proteins interacting with SP1, revealing an interaction between the E3 ubiquitin ligase TRIM25 and the SP1 protein, which was subsequently validated by a Co-IP assay.
The impact of the proteasome inhibitors bortezomib and carfilzomib on peripheral neurons has also been extensively investigated via proteomics. Both drugs altered the expression of cytoskeletal proteins, including nestin, vimentin, β-actin, ARP-2, and coronin 1C7. Notably, nestin, crucial for cell survival, was consistently downregulated by both drugs, while vimentin expression patterns diverged, possibly explaining the higher neurotoxicity of bortezomib. Lamin B1, involved in mitochondrial function and axon maintenance, was modulated only by carfilzomib.
Heat shock proteins, sensitive markers of cellular stress, were also differentially regulated. Bortezomib significantly upregulated HSP32, HSP47, HSP70, GRP78, and GRP94 in neuronal cells, while carfilzomib induced only GRP78 and GRP947,8. In iPSC-derived sensory neurons, bortezomib elicited a stronger heat shock response, with HSP70 and HSP90 being upregulated only in bortezomib-treated cells8. Additionally, mitochondrial proteins such as VDAC1 and UCP2, as well as oxidative phosphorylation components (ATP5A and UCRCQ2), were downregulated after 24 h of treatment, suggesting impaired mitochondrial function8.
Other proteins such as SQSTM1 (involved in oxidative stress and mitophagy via PINK1/Parkin) were found upregulated in both cell models and human iPSC-derived sensory neurons8,9. MAP2, a protein involved in axonogenesis and neurite morphology, was identified as a key mediator of bortezomib-induced neurotoxicity9.
Proteomics has also contributed to understanding and mitigating oxaliplatin-induced CIPN. A selective inhibitor of FABP5 (ART26.12) was tested in a rat model70. Targeted proteomic profiling using the SomaScan assay revealed systemic protein changes related to translation, protein folding, and ubiquitin-proteasome activity. Importantly, while most proteins were upregulated by both oxaliplatin and ART26.12, NSF (vesicle-fusing ATPase) was the only one reversed by the treatment, pointing to a possible protective mechanism.
An integrated platform to investigate proteome aggregation in PC12 peripheral neuropathy–related cells treated with various chemotherapeutic agents allows visualization of aggregated proteins using a fluorescent imaging method (AggStain). A more in-depth analysis was then conducted in paclitaxel-treated cells using a chemical proteomics probe (AggLink), as paclitaxel induced the most pronounced proteome aggregation. AggLink enabled the enrichment of aggregated proteins, which were subsequently characterized by LC-MS/MS. This analysis revealed an enrichment of proteins associated with the proteostasis network, particularly those involved in mitochondrial pathways (21.2%) and DNA replication/RNA transcription (23.1%). Through this platform, the authors were able to delineate the relationship between CIPN and proteome aggregation in neuronal cells, highlighting a potential role for caseinolytic mitochondrial matrix peptidase chaperone subunit B (CLPB) and heat shock protein family D member 1 (HSPD1) in disrupting cellular homeostasis in response to paclitaxel71.
Spatial omics mass spectrometry-based technologies in CIPN research
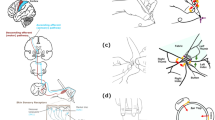
Among the previously discussed omics technologies, spatial omics mass spectrometry-based technologies merit particular attention and are emerging as pivotal tools for elucidating the intricate spatial organization of biological systems. Both mass spectrometry imaging (MSI) and imaging mass cytometry (IMC) belong to this rapidly advancing field, offering unique capabilities for studying molecular distributions within intact tissues and providing insights into cellular heterogeneity and microenvironmental interactions (Fig. 1). In fact, unlike classical proteomics, metabolomics, and transcriptomics analyses, which rely on tissue homogenization or dissociation and inevitably lose the anatomical context of the sample, spatial mass spectrometry technologies preserve the in situ localization of molecules and thereby maintain tissue architecture and the relationship between distinct cell populations. This spatial preservation is particularly relevant for the study of CIPN, where damage is known to affect specific neural compartments in a non-uniform manner. Somatosensory neurons, axons, Schwann cells, satellite glia and infiltrating immune cells each exhibit distinct susceptibilities to chemotherapeutic toxicity. Bulk omics approaches average these compartments, making it impossible to understand whether an observed molecular alteration originates from neuronal bodies in the DRG, from axonal projections in the spinal roots, or from supporting glial cells. Spatial omics directly addresses this limitation by enabling the precise localization of biochemical changes within defined microdomains of the peripheral nervous system. This ability is crucial for understanding CIPN pathogenesis, as different chemotherapeutic agents induce region-specific patterns of neurotoxicity within the peripheral nervous system, and the engagement of distinct neural circuits in nociceptive sensitization strongly influences how the condition develops and progresses72.

A Imaging Mass Cytometry (IMC) is a highly multiplexed imaging technique that uses metal-tagged antibodies (1) to detect over 40 protein markers simultaneously at subcellular resolution (~1 μm). A pulsed laser ablates the tissue at discrete locations (2), and the vaporized material is analyzed by time-of-flight mass spectrometry to identify the metal isotopes associated with each antibody. The image is constructed by raster-scanning the tissue and assigning the detected signals to spatial coordinates, generating high-dimensional maps where each pixel represents the abundance of specific protein markers (3). While IMC provides detailed spatial and phenotypic information about the tissue microenvironment, it is a destructive technique, meaning the same tissue section cannot be used for subsequent analyses, such as histological staining. B Mass Spectrometry Imaging (MSI) techniques, including Matrix-Assisted Laser Desorption/Ionization (MALDI) and Desorption Electrospray Ionization (DESI), enable label-free, untargeted imaging of a broad range of molecules, such as lipids, metabolites, and drugs, directly from tissue sections. MALDI-MSI requires matrix deposition (1) and uses a laser to desorb and ionize molecules from the coated surface (2), typically under vacuum, and can achieve high spatial resolution, down to 5–10 μm under optimized conditions. DESI-MSI operates under ambient conditions using a charged solvent spray to ionize molecules without the need for matrix application (2). This method involves minimal sample preparation and is compatible with in situ or near-real-time analysis. Depending on the specific configuration, DESI approaches can range from moderate to relatively high spatial resolution (<10 μm with nano-DESI MSI). MSI generates a three-dimensional data cube, where each x–y coordinate corresponds to a tissue location, and the third dimension represents the mass-to-charge (m/z) axis of the acquired spectrum. From this data, ion images can be reconstructed by selecting specific m/z values, producing spatial distribution maps of individual molecules. Each image reflects the localization and relative abundance of a given ion across the tissue, allowing direct correlation with morphological features (3). Importantly, MSI is generally minimally destructive, allowing subsequent histological staining (e.g., Haematoxilin and eosin staining) and enabling direct correlation between molecular and morphological features within the same tissue section.
IMC combines mass spectrometry with cytometry principles and is a label-based approach using metal-tagged antibodies to simultaneously acquire 40-plus proteins of interest within tissue sections, while having the drawback of being a destructive technology that does not allow further cell sorting analysis afterward73. MSI, by contrast, preserves tissue integrity after analysis and supports both post-acquisition histological staining and qualitative/quantitative label-free investigation. Its ability to analyze diverse molecular classes, including proteins, lipids, metabolites, N-glycans and xenobiotics, at a spatial resolution of 5–10 μm makes it an exceptionally versatile platform for peripheral nerve research. Within the domain of MSI, two ion sources stand out: MALDI (Matrix Assisted Laser Desorption/Ionization) and DESI (Desorption Electrospray Ionization). These techniques represent distinct approaches to spatially resolved molecular analysis, each offering unique advantages and applications. MALDI relies on the automatic and homogeneous deposition of an organic acidic molecule (matrix) which co-crystallizes with the analytes and a laser emitting at 355 (Nd:YAG) or 337 nm (N2 laser) for the desorption and ionization of molecules directly from a sample surface, allowing for high sensitivity and the analysis of a wide range of biomolecules, including proteins, lipids, and small molecules. In contrast, DESI is a matrix-free approach, which operates by directing a charged solvent spray onto the sample surface, causing desorption and ionization of analytes, significantly reducing sample preparation steps, but it is limited to the analysis of small molecules such as metabolites and lipids74. Recent technological advances have expanded the use of MSI toward integrated, multimodal applications74,75. MALDI-MSI enables sequential imaging of multiple molecular classes from a single tissue slice, an approach that is particularly advantageous when tissue availability is limited, as is often the case for nerve biopsies and DRG specimens, which are small structures and yield only minimal amounts of tissue76. Additionally, MALDI-MSI offers the flexibility to analyze various sample types, such as fresh-frozen, formalin-fixed paraffin-embedded tissues, as well as cytological samples. Moreover, while MSI is mostly based on an untargeted approach, the last two years have seen the development of a top-down, high-plex, multimodal, and multi-omic spatial imaging approach known as MALDI mass spectrometry-based high-plex immunohistochemistry (MALDI HiPLEX-IHC)77. This method utilizes photocleavable mass tags conjugated to antibody probes, allowing for the simultaneous imaging of intact proteins, N-glycans, and small molecules from a single tissue slice. Interestingly, the targeted MALDI-IHC analysis can be followed by label-free MALDI-MSI in a global sequential workflow, thus enabling a comprehensive molecular evaluation from a single specimen.
Despite the potential of these technologies, relatively few studies have applied spatial omics directly to CIPN models. Nonetheless, related work in neuropathic pain and peripheral nerve biology demonstrates the transformative value of spatial localization in understanding disease mechanisms. Spatial omics makes it possible not only to visualize the biochemical changes induced by chemotherapeutic agents within DRG, spinal nerve roots and the spinal cord but also to determine how these changes are distributed across neuronal bodies, axons or glial compartments. In addition, the ability to map the penetration and persistence of chemotherapeutic agents within nerve tissues provides critical information for interpreting compartment-specific toxicities.
Since 2008, there has been interest in developing MSI methods with the primary aim of investigating the native composition and distribution of proteins, neuropeptides and phospholipids in the spinal cord, facilitating the investigation of neural transmission, especially in pain-related research78,79.
In rat spinal cord tissue three groups of pain-related neuropeptides with distinguishable distribution patterns were identified using a MALDI-MSI approach: protachykinin-1-derived peptides (Neurokinin A and Substance P), prodynorphin-derived peptides (Alpha-neoendorphin, Dynorphin A (10–17) and Dynorphin B (1–13)) and proenkephalin-derived peptide (Proenkephalin (220–229), Proenkephalin (197–208), and Proenkephalin (219–229))79. In particular, substance P was exclusively localized in the superficial layer of the dorsal region of the spinal cord. In contrast, all other neuropeptides were found in both the right and left ventral and dorsal horns, with proenkephalin and neurokinin co-localizing with substance P.
These findings align with observations in CIPN models, where prodynorphin expression is strongly upregulated following treatment with chemotherapeutic agents such as paclitaxel or oxaliplatin. The prodynorphin (pDYN) gene relative intensity has been observed to be overexpressed in rodent models treated with paclitaxel or with oxaliplatin80,81. pDYN is upregulated in the cervical, thoracic and lumbar portions of the rat spinal cord of the oxaliplatin-treated group compared to the vehicle group. Moreover, when the rats in the oxaliplatin group were also treated with the β5 subunit proteasome inhibitor oprozomib, pDYN expression was reverted80. On the other hand, while substance P expression was found to remain unaltered in oxaliplatin-induced peripheral neuropathy, it was observed to be upregulated in both DRG and spinal cord tissue of rat models treated with paclitaxel alone or when in combination with other chemotherapies (vincristine and cisplatin)82. Its expression was, however, reverted after puerarin, duloxetine, or gabapentin treatments83,84,85,86. Thus, the spatial dimension could strengthen the interpretation of these results by situating molecular changes within the exact spinal microcircuits responsible for pain processing.
The relevance of MSI also extends to the investigation of peripheral nerve lipidomics. Lipids play crucial structural and signaling roles within the peripheral nervous system, and their spatial organization can be effectively explored using MSI. Although lipid dysregulation is well established in CIPN, most MSI-based lipidomic studies to date have focused on general models of nerve injury or neuropathic pain rather than CIPN itself87,88,89,90.
Using MALDI- and DESI-MSI, it has been shown that peripheral nerve injury is associated with region-specific alterations in phospholipids and lysophospholipids within the DRG, spinal cord, and nerve roots. These investigations revealed injury-dependent changes in selected phosphatidylcholines, phosphatidic acids, and lysophosphatidylcholines, highlighting the capacity of MSI to resolve lipid alterations with anatomical specificity in the peripheral nervous system87,88,89.
Beyond injury models, MSI has been applied to characterize the baseline lipidomic composition of peripheral nerves. These studies demonstrated that distinct lipid signatures can discriminate between different nerve subtypes, as well as between nerve tissue and surrounding connective or adipose tissue91,92,93. Moreover, MSI has shown that individual anatomical compartments within a single nerve, such as nerve fibers, connective tissue, and surrounding adipose tissue, exhibit markedly different lipid class distributions, particularly in their relative content of glycerolipids and sphingolipids92.
Together, these studies underscore the strength of MSI for mapping the spatial lipid organization of the peripheral nervous system and for identifying region- and tissue-specific lipid alterations following nerve injury. While these findings provide an important methodological and biological framework, their application to CIPN remains largely unexplored, highlighting a significant gap and an opportunity for future MSI-driven investigations in this field.
Indeed, despite the widespread clinical use of neurotoxic chemotherapeutic agents, MSI has so far been applied only marginally to chemotherapy-related toxicities, such as in studies investigating cisplatin-induced nephrotoxicity in animal models94. In contrast, no MSI studies are currently available that address the spatial molecular alterations at the cellular level within peripheral neuronal tissues of subjects experiencing CIPN. Addressing this gap could provide deeper mechanistic insights into CIPN and support the development of targeted therapeutic strategies.
Spatial mass spectrometry is also highly informative for the study of chemotherapeutic drug distribution. To this end, IMC has recently been employed to investigate the persistence of oxaliplatin in skin tissues of colorectal cancer patients, shedding light on chronic peripheral sensory neuropathy95. Despite the small sample cohort (only six patients), extensive deposition of platinum (195Pt) is seen in skin biopsies up to 60 months after oxaliplatin administration, with the platinum colocalizing with collagen fibers in dermis layers, thus suggesting that the persistence of 195Pt in the skin may contribute to oxaliplatin-induced neuropathy. Complementary MALDI-MSI analysis has been used to monitor the spatial localization of oxaliplatin metabolites, such as oxaliplatin-monomethionine and oxaliplatin-monocysteine complexes, in various tissues (i.e., ovary tissue, kidney tissues and multicellular tumor spheroids from colon carcinoma cell line HCT 116). These studies demonstrated that MSI can resolve isobaric platinum species with sub-ppm mass accuracy and ultra-high resolving power (>200,000)96,97,98,99.
These approaches highlight that spatial omics are capable not only of detecting chemotherapeutic agents within tissues but also of defining their chemical forms, their distribution and their potential association with long-term toxicity. The penetration, distribution, and persistence of metal-based chemotherapy drugs in non-target tissues are likely to play a key role in the onset and maintenance of CIPN. Mapping their distribution in neural tissues may help elucidate underlying mechanisms and ultimately guide the development of strategies to prevent or mitigate this debilitating side effect of antineoplastic therapy.
Taken together, these observations clearly demonstrate that spatial omics technologies provide essential insights that cannot be obtained through conventional bulk omics alone. As these technologies continue to evolve toward higher resolution, greater molecular coverage and integrated multimodal workflows, they hold exceptional promise for advancing our mechanistic understanding of CIPN and ultimately guiding the development of targeted therapeutic strategies.
Supplementary Data 2 summarizes MSI-detectable neuropeptides, peptides, phospholipids in dorsal root ganglia and spinal cord tissues relevant to CIPN. Although it lists highly specific m/z values, this level of detail is particularly valuable given the limited number of studies applying MSI to CIPN models. By consolidating signals that have been successfully detected in these tissues, Supplementary Data 2 provides a practical reference for researchers, especially those approaching MSI in CIPN for the first time, aiding target identification, method selection, and hypothesis generation.
Role of metabolomics in biomarker assessment and mechanistic insights in CIPN
Metabolomics, a pivotal field within systems biology, involves the comprehensive analysis of small metabolites in biological systems. By examining the complete set of metabolites, or the metabolome, metabolomics provides critical insights into physiological and pathological states, reflecting the direct biochemical responses to genetic modifications, environmental changes, or disease conditions100. Since metabolites are the end products of cellular processes, their study offers a real-time snapshot of biochemical activity, enabling a functional assessment of the phenotype that complements genetic and proteomic data.
Technological advances have boosted metabolomics forward, with MS emerging as the primary tool for its sensitivity and ability to detect and quantify a vast array of metabolites. High-resolution MS, coupled with separation techniques such as gas chromatography for volatile compounds and LC-MS for a broader range of metabolites, has significantly enhanced metabolite detection across complex biological matrices. The development of ion mobility spectrometry-mass spectrometry (IMS-MS) has further refined metabolomics workflows by improving separation efficiency and structural elucidation of metabolites101. In parallel, nuclear magnetic resonance (NMR) spectroscopy remains a valuable tool, offering a non-destructive means to quantify metabolites with high reproducibility, albeit at the cost of lower sensitivity compared to MS-based approaches102.
A major challenge in metabolomics is the processing of complex datasets to extract biologically meaningful information. Raw MS and NMR data undergo extensive pre-processing, including noise reduction, peak alignment, and normalization. Bioinformatics pipelines integrate these datasets with multivariate statistical tools, such as PCA, partial least squares-discriminant analysis, and machine learning models, to identify metabolite signatures associated with specific biological conditions103. Further, pathway enrichment analyses contextualize altered metabolite levels within known metabolic networks, enabling the interpretation of dysregulated biochemical pathways in disease states104. A widely used platform for metabolomics data analysis is MetaboAnalyst, which provides a comprehensive suite of statistical and pathway analysis tools, allowing researchers to perform data processing, visualization, and functional interpretation of metabolic changes in a user-friendly and accessible format105.
Lipidomics, a specialized branch of metabolomics focusing on lipid profiling, has undergone rapid advances due to improvements in high-resolution MS techniques such as LC-MS/MS, shotgun lipidomics, and IMS106. Lipids are essential regulators of cell signaling, membrane integrity, and energy metabolism, making them critical targets for biomarker discovery. Alterations in lipid profiles have been linked to neuropathic conditions, metabolic disorders, and cancer progression, underscoring the importance of lipidomics in translational research.
In oncology, metabolomics plays a key role in biomarker discovery, treatment response monitoring, and the identification of metabolic vulnerabilities that can be targeted therapeutically107. As metabolomics technologies continue to advance, their application in clinical decision-making, drug development, and therapeutic monitoring will further enhance the ability to tailor treatments to individual metabolic profiles, leading to precision medicine107. Over the last few years, a crucial area of investigation has been the underlying mechanisms of CIPN and the evaluation of changes in metabolite homeostasis as fluid biomarkers during the onset and progression of disease. A consistent area of consensus is the prominent involvement of lipid metabolism, particularly alterations in lysophosphatidylcholines, oxidized linoleic acid derivatives, and sphingolipid pathways. Recent research has significantly advanced our understanding, particularly highlighting the crucial role of lipid mediators both in preclinical and clinical settings.
In 2013,108 using immunohistochemistry and behavioral assays in rodents, LPC administration to the median nerve was found to induce mechanical allodynia, thermal hyperalgesia, and nerve demyelination108. Mechanistically, LPC treatment increased neuronal nitric oxide synthase (nNOS) expression in both the DRGs and cuneate nucleus, leading to heightened pain responses and neuroinflammatory changes. The application of nitric oxide synthase inhibitors, such as L-NAME and 7-nitroindazole, significantly alleviated neuropathic pain behaviors, suggesting that nNOS upregulation plays a critical role in LPC-induced neuropathy108.
In another study109, the role of oxidized lipid metabolites in paclitaxel-induced peripheral neuropathic pain (PIPN)109 was investigated by applying the LC-MS/MS approach to profile oxidized lipid metabolites in the sciatic nerve, DRGs, and spinal cord of paclitaxel-treated mice. 9,10-epoxy-12Z-octadecenoic acid (9,10-EpOME), a cytochrome P450-derived linoleic acid metabolite, was significantly elevated in DRGs of paclitaxel-treated mice and contributed to TRPV1 ion channel sensitization. This led to increased excitatory synaptic activity in nociceptive spinal cord neurons and elevated calcitonin gene-related peptide release from sciatic nerves. Furthermore, a drug repurposing screen identified telmisartan, an angiotensin II receptor antagonist, as an inhibitor of CYP2J2, the human ortholog of murine CYP2J6. Telmisartan administration reduced EpOME concentrations and reversed mechanical hypersensitivity in paclitaxel-treated mice, suggesting a potential therapeutic strategy for CIPN through CYP2J2 inhibition109. These results suggested a novel and potentially effective strategy for treating paclitaxel-induced neuropathic pain and underscored the critical role of lipidomic analysis in understanding and intervening in complex biological mechanisms related to CIPN. In parallel, Wen-Luo-Tong (WLT), a traditional Chinese medicine ointment, was applied to a rat model of PIPN110. By performing a metabolomics approach with ultra-performance liquid chromatography-electrospray ionization-mass spectrometry, 19 significant metabolite variations were identified that correlated with PIPN. These findings disclosed alterations in linoleic acid metabolism and glycerophospholipid metabolism as critical in the development of neuropathy. WLT treatment could recalibrate these metabolic disturbances, unveiling a potential therapeutic effect of WLT on PIPN. Additionally, the molecular docking analysis of WLT components suggested that specific ingredients in WLT, such as hydroxysafflor yellow A, icariin, epimedin B, and 4-dihydroxybenzoic acid, have high affinity to proteins involved in the disturbed metabolic pathways, further supporting the therapeutic relevance of traditional formulations in modern medical settings.
Addressing the issue of lipid-based therapies111, the role of sphingosine 1-phosphate (S1P) and its receptor S1PR1 was investigated in paclitaxel-induced neuropathy using a rat model. S1P is a bioactive sphingolipid that plays a critical role in axonal growth and Schwann cell function112 and contributes to neuropathic pain by amplifying glial activation and cytokine release113,114,115. It is synthesized by the phosphorylation of sphingosine via sphingosine kinases (SPHK1 and SPHK2) and exerts its effects through five G-protein-coupled receptors (S1PR1–5), each with distinct functions. S1PR1 has been implicated in promoting inflammation and pain sensitization, whereas S1PR2 has been suggested to play a neuroprotective role and inflammatory modulation116,117,118. Paclitaxel administration (2 mg/kg intraperitoneally every other day for four doses) led to increased S1P levels within the spinal dorsal horn, activating S1PR1 and triggering pro-inflammatory signaling cascades, including NFκB and MAPK phosphorylation. These changes were accompanied by increased expression of TNF-α and IL-1β, key cytokines involved in neuroinflammation. Pharmacological inhibition of S1PR1 using W146 significantly reversed mechanical allodynia without affecting paclitaxel’s anticancer efficacy, highlighting S1PR1 as a potential therapeutic target.
Another emerging trend involves the expanding role of sphingolipid signaling, particularly S1P/S1PR1 pathways, across taxane- and proteasome inhibitor–induced neuropathies. Evidence that modulating S1P receptors can attenuate neuropathic pain without compromising anticancer efficacy elevates sphingolipid metabolism from a descriptive signature to a mechanistically actionable target.
A complementary study119 examined the effects of bortezomib on S1P signaling in a mouse model. Bortezomib (0.4 mg/kg subcutaneously, twice weekly for 4 weeks) caused an increase in S1P and dihydro-S1P levels in the spinal dorsal horn, particularly in astrocytes. Genetic deletion of S1PR1 in astrocytes prevented the development of neuropathic pain, while treatment with fingolimod (FTY720), an FDA-approved S1PR1 modulator, reversed bortezomib-induced hypersensitivity. This suggests that targeting S1PR1 could be beneficial across different chemotherapy-induced neuropathies.
More recently120, the modulation of S1P signaling pathways was examined as a strategic intervention to mitigate neuropathic side effects without undermining the anticancer efficacy of the drugs by clinically analyzing human patients treated with oxaliplatin, efficacy testing in rodent models treated with cisplatin, and undertaking mechanistic studies in cellular systems. The S1P2 receptor was proposed as an approach to manage CIPN, enhancing overall patient tolerance to chemotherapeutic regimens and improving quality of life by mitigating side effects. The study underlined the importance of receptor-specific interventions in the pharmacological landscape of cancer therapy.
Beyond S1P signaling, another class of sphingolipids, 1-deoxysphingolipids, has been implicated in chemotherapy-induced neurotoxicity in both in vitro and clinical studies examining their role in paclitaxel-induced neuropathy121. In vitro experiments using SH-SY5Y neuronal cells showed that paclitaxel (250 nM–1 µM) significantly increased 1-deoxysphingolipid levels by upregulating serine palmitoyltransferase activity, the key enzyme in sphingolipid biosynthesis. Unlike canonical sphingolipids, 1-deoxysphingolipids are resistant to normal degradation pathways, leading to toxic accumulation. A clinical analysis of 27 breast cancer patients receiving paclitaxel revealed that increased plasma levels of very-long-chain 1-deoxyceramides, particularly C24, were associated with greater motor neuropathy severity.
Building on these findings, it was found using a mouse model (45 mg/kg cumulative docetaxel dose over three weeks) that docetaxel significantly elevated 1-deoxysphingolipid levels in DRG neurons, leading to neuronal damage122. In primary DRG cultures, treatment with 1-deoxysphingosine (1 µM) induced neurite swellings, but co-treatment with S1P reversed these toxic effects, suggesting that modulating S1P levels could counteract taxane-induced neurotoxicity.
Expanding on the role of lipid alterations in CIPN, a recent study on a PIPN model sheds new light on how age influences the susceptibility to and severity of paclitaxel-induced neurotoxicity, through detailed metabolomic profiling123. The impact of age on the metabolic disruptions caused by paclitaxel was systematically examined in Wistar rats. Older rats experienced more significant weight loss, greater nerve damage, and severe metabolic disruptions compared to their younger counterparts. The importance of energy utilization and potential mitochondrial dysfunction in CIPN animals was demonstrated, with significant changes in triglycerides, diglycerides, acylcarnitines, carnosine, long-chain ceramides, sphingolipids, and bile acids. These findings suggest that age plays a significant role in PIPN development and metabolic adaptation to paclitaxel neurotoxicity and offer insights into why older patients might experience more severe side effects. This understanding suggests a shift towards more targeted and personalized treatment modalities in older cancer patients.
In the clinical setting, multiple studies have identified different metabolic biomarkers that may predict the risk of CIPN. A comprehensive serum lipidomics study was undertaken in multiple myeloma patients receiving bortezomib at 1.3 mg/m² subcutaneously or intravenously on days 1, 4, 8, and 11 of each 21-day cycle, in combination with low-dose dexamethasone (20 mg/day on the same days)124. LC-MS/MS analyzed 385 lipid species from serum samples collected pre-treatment, during treatment, and post-treatment. Statistical analyses revealed that lower levels of glycerophospholipids, sphingolipids, and cholesteryl esters were significantly correlated with poor treatment response, whereas increased lysophosphatidylcholines, phosphatidylcholines, ceramides, and oxidative fatty acids were associated with severe bortezomib-induced neuropathy, suggesting a link between lipid metabolism dysregulation and neurotoxicity.
A pharmacometabolomics approach assessed metabolic predictors of paclitaxel-induced peripheral neuropathy in whole blood samples collected from 60 female breast cancer patients at three different time points: pre-treatment (baseline), immediately before the end of the first paclitaxel infusion, and 24 h post-infusion125. Patients received paclitaxel at either 80 mg/m² weekly for 12 cycles or 175 mg/m² every three weeks for 4 cycles. One-dimensional 1D-¹H NMR spectroscopy was used to quantify plasma metabolite concentrations, which were then correlated with neuropathy severity assessed using the EORTC CIPN20 questionnaire. Lower pre-treatment concentrations of histidine, phenylalanine, and threonine were associated with increased neuropathy severity, indicating a potential role for amino acid metabolism in CIPN susceptibility.
These lipidomic signatures, demonstrated in rodent models and corroborated clinically, converge on mechanisms of membrane destabilization, mitochondrial stress, and neuroinflammatory signaling, suggesting that lipid remodeling represents a shared metabolic axis driving peripheral nerve vulnerability. Likewise, studies reporting linoleic acid and glycerophospholipid pathway disruption reinforce this recurring lipid-based phenotype.
Beyond the recognized patterns, metabolomics has revealed emerging trends that broaden the conceptual framework of CIPN pathophysiology. Most notably, several recent studies implicate bile acid dysregulation, particularly increases in secondary bile acids such as deoxycholic and taurocholic acid, in driving or predicting CIPN, mediated through gut microbiota alterations and activation of pro-inflammatory signaling pathways. This gut-liver-nerve axis represents a systemic mechanism that extends beyond neuron-intrinsic injury and highlights how host-microbiome interactions may modulate chemotherapy toxicity.
A second area of convergence involves amino acid homeostasis, where shifts in histidine, phenylalanine, threonine, and other amino acids correlate with CIPN severity or susceptibility; although findings vary by cohort and drug class, the overall trend indicates that altered amino acid metabolism may reflect impaired redox balance or reduced metabolic resilience prior to neuropathy onset.
Amino acid metabolism was further investigated126 in a large cohort study within the SWOG S0221 clinical trial, which enrolled 1,191 breast cancer patients receiving paclitaxel at either 80 mg/m² weekly for 12 weeks or 175 mg/m² every three weeks for 4 cycles. Targeted quantitative LC-MS/MS was performed to measure pre-treatment levels of 20 amino acids. Logistic regression models were used to examine associations between amino acid concentrations and CIPN occurrence, adjusting for age, race, body mass index, and paclitaxel dosing schedule. Contrary to prior findings, histidine was not confirmed as a predictive biomarker. However, higher pre-treatment levels of glutamate, phenylalanine, tyrosine, and valine were associated with increased neuropathy severity, though these findings lost statistical significance after multiple comparison adjustments.
A metabolomics approach to identify early predictors of vincristine-induced peripheral neuropathy (VIPN) in pediatric leukemia patients analyzed plasma samples from 36 children with acute lymphoblastic leukemia undergoing treatment with vincristine at 1.5 mg/m² intravenously (maximum dose: 2 mg) weekly for 4 weeks during induction, followed by biweekly doses during consolidation and maintenance, according to the AALL0932 protocol127. Using LC-MS/MS, metabolite profiling was conducted at three treatment time points: day 8, day 29 (end of induction), and 6 months into therapy (consolidation phase). Patients were classified into high neuropathy and low neuropathy groups based on Total Neuropathy Score–Pediatric Vincristine assessments. Machine learning algorithms identified a small set of metabolites capable of accurately distinguishing high-risk patients. These findings led to the development of VIPN, a prediction tool for early identification of patients at high risk for VIPN, allowing for potential dose modifications to minimize neurotoxicity.
A microbiome and metabolome analysis on fecal and serum samples from 170 breast cancer patients undergoing paclitaxel treatment at cumulative doses exceeding 800 mg/m examined the role of gut microbiota-derived bile acids PIPN128. Secondary bile acids, particularly deoxycholic acid (DCA), were significantly elevated in patients with severe PIPN due to gut microbiota alterations favoring Clostridium species overgrowth. In a rodent PIPN model, rats received paclitaxel at 2 mg/kg intraperitoneally every other day for four doses and exhibited increased DCA accumulation in the DRGs, elevated levels of C-C motif ligand 5 (CCL5), and overexpression of CCR5, a chemokine receptor involved in neuroinflammation. CCR5 blockade with maraviroc (2 mg/kg intravenous) significantly reduced neuropathic pain behaviors and neuronal hyperexcitability, suggesting that DCA-CCR5 signaling is a key driver of PIPN. These findings identify gut microbiota-derived bile acid dysregulation as a novel mechanism underlying chemotherapy-induced neuropathy and propose CCR5 as a potential therapeutic target.
Complementing these findings, in a prospective, multicenter pharmacometabolomics study in 142 colorectal cancer patients treated with oxaliplatin-based chemotherapy129, plasma samples were collected at baseline, post-cycle one, and at symptom onset. Using UPLC-quadrupole time of flight-MS, untargeted metabolomic profiling followed by multivariate statistical analysis identified that decreased plasma LPC (18:2) and increased taurocholic acid levels were associated with the development of CIPN. Pathway enrichment analysis further implicated disruptions in bile acid biosynthesis, glycerophospholipid metabolism, and fatty acid oxidation, reinforcing the clinical value of pharmacometabolomics in tailoring individualized therapeutic approaches.
Together, metabolomics studies move beyond isolated metabolite lists to reveal CIPN as a multifaceted metabolic disorder involving intersecting disturbances in lipid remodeling, bioenergetic stress, amino acid imbalance, and bile acid signaling. The convergence of lipid-centered pathways across models provides a strong foundation for biomarker development, whereas the emergence of gut-derived bile acids and receptor-specific sphingolipid signaling highlights new mechanistic directions and potential therapeutic opportunities. At the same time, drug-specific metabolic fingerprints, age-related susceptibility, and heterogeneous amino acid signatures emphasize the need for larger, harmonized, and mechanistically integrated metabolomics studies to translate these findings into personalized strategies.
Application of multiple omics techniques in CIPN
Since single-omics-based technology is limited in capturing the complex biochemical and molecular changes underlying chemotherapy-induced toxicity, the integration of multi-omics provides a more comprehensive understanding of the mechanisms involved in CIPN. A recent study proposed a multimodal biomarker discovery approach combining genomics, transcriptomics, proteomics, and metabolomics with functional neurophysiological assessments and patient-reported outcomes to identify predictors of TIPN130.
By integrating multiple omics layers, this study aimed to identify genetic variations, altered gene expression profiles, dysregulated protein pathways, and metabolic disruptions associated with TIPN onset and severity. In particular, genomic and transcriptomic analyses focused on detecting risk-associated variants and differential gene expression patterns, while proteomics and metabolomics targeted circulating biomolecules linked to nerve damage. Additionally, nerve conduction studies and small fiber function tests provided physiological validation, ensuring that molecular findings correlated with functional deficits in peripheral nerves.
The systems biology approach employed in this study offers several advantages. Indeed, the inclusion of multiple molecular layers increases the likelihood of identifying clinically relevant biomarkers that could eventually translate into diagnostic or predictive tools.
Further supporting the role of multi-omics in elucidating chemotherapy-induced neurotoxicity, another recent study investigated the antinociceptive effects of a Cannabis sativa L. extract on PIPN in rats131. By combining transcriptomic, metabolomic, and gut microbiota analyses, this study identified neuroactive ligand-receptor interactions, sphingolipid metabolism alterations, and gut microbiome dysregulation as key contributors to neuropathy pathophysiology. The administration of a cannabidiol-rich hemp extract significantly reversed paclitaxel-induced mechanical allodynia and thermal hyperalgesia, while also restoring metabolic imbalances and gut microbiota composition. These findings underscore the importance of multi-omics profiling in both mechanistic exploration and the identification of potential therapeutic strategies for CIPN.
However, several challenges remain. CIPN is influenced by multiple host-related factors, including age, genetic susceptibility, metabolic status, and co-existing neuropathic conditions, which may complicate biomarker generalizability. Moreover, the wide variety of symptoms in CIPN, including neuropathic pain, sensory loss and itch, further complicates the mechanistic understanding of this condition and limits the development of effective treatments1,132,133. Although well-established animal models that mimic CIPN features have contributed to discoveries on drug-induced neurotoxicity, they often fail to represent the clinical situation due to differences in species physiology, drug dosing and administration routes134,135,136. Preclinical studies of CIPN overlook factors such as sexual dimorphisms and the biological complexity of human tissues, which are missing in cell culture or nerve explant models137,138. Despite decades of research and extensive preclinical models, no effective treatments have been translated into the clinic, suggesting the need for more sophisticated and integrated approaches that overcome these limitations. Access to peripheral biological samples from patients with CIPN, such as blood or skin used to generate iPSCs, provides a valuable platform for identifying predictive biomarkers and temporal molecular changes. However, human samples also present challenges, as they must usually be limited to noninvasive areas and are subjected to patients’ heterogeneity influenced by drug type, dosage and possible comorbidities.
Indeed, single-omic approaches provide valuable, yet partial views of the molecular setting underlying CIPN, as each captures only a specific layer of biological regulation and is limited by methodological constraints, incomplete tissue accessibility, and tissue-specific heterogeneity. Transcriptomics maps gene expression programs with high granularity but cannot account for post-transcriptional regulation, protein turnover, or functional protein states. Proteomics reveals dynamic cellular processes such as inflammation, cytoskeletal remodeling, and mitochondrial dysfunction, nonetheless it is restricted by low sensitivity to early or low-represented drivers, challenges in detecting post-translational modifications and protein to protein interactions, and the limited availability of disease-relevant neural tissues. Metabolomics highlights downstream biochemical perturbations but is highly susceptible to interindividual variability, treatment-related differences, species effects, and matrix-dependent inconsistencies that often produce discordant biomarker signals across studies. Spatial approaches add anatomical context but require specialized high-cost instrumentation, optimized sample handling, and substantial computational resources, and they still depend on complementary methods for confident molecular identification. Because each omics discipline operates within its own technical and biological boundaries, single-layer analyses frequently generate fragmented, non-overlapping, or difficult-to-interpret results.
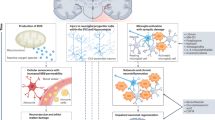
A multi-omic strategy is therefore essential to overcome these limitations by integrating molecular information across transcriptional, translational, post-translational, metabolic, and spatial dimensions. Such integrative frameworks enable the reconstruction of causal pathways rather than isolated associations, link upstream gene expression changes to downstream protein activity and metabolic consequences, and contextualize these processes within specific cellular and spatial microenvironments. This systems-level perspective enhances mechanistic inference, increases biomarker specificity and reproducibility and supports the identification of convergent pathophysiological signatures (Fig. 2). A clear example of multi-omic integration comes from a recent study in which blood-derived mRNA, miRNA and metabolites were collected from CIPN patients. These complementary approaches revealed biological changes associated with neuropathy and pathway analysis identified key signaling mechanisms involved in CIPN pathophysiology, including dysregulation of CREB and opioid signaling, as well as the neuronal endocannabinoid system139. These findings highlight the value of integrative multi-omics approaches for uncovering interconnected processes underlying pathological conditions. The successful application of multi-omics integration is also evident in other neurological disorders. In this context, the MINDSETS study in Alzheimer’s disease showed that integrating radiomics, genetic data, and clinical–cognitive assessments into a unified analytical framework markedly enhances the classification of Alzheimer’s disease, vascular dementia, and related conditions131. Importantly, multi-omics integration increased diagnostic accuracy by an average of 6.5% over neuroimaging alone, consistently outperforming single-modality approaches. Through feature-importance analyses and longitudinal evaluation, the study further demonstrated that multi-omic datasets capture subtle, temporally evolving pathological signals that remain undetectable when each modality is examined in isolation, thereby enabling earlier diagnosis and more accurate monitoring of treatment response. These findings emphasize the ability of multi-omic strategies to untie complex and overlapping phenotypes, improve mechanistic insight, and support personalized therapeutic decision-making. Such evidence strongly suggests that multifactorial and heterogeneous conditions could benefit from similar multi-omic architectures. In this regard, CIPN represents an ideal candidate for a multi-omic approach, integrating neurophysiological measures, metabolomics, proteomics, peripheral neuroimaging, and detailed clinical phenotyping to define mechanistically coherent patient subgroups, reduce inter-individual variability, and detect early molecular alterations preceding clinically evident neuropathy.

At the center, CIPN is depicted as a hub surrounded by interconnected puzzle segments representing four core omics technologies: Transcriptomics, Proteomics, Metabolomics, and Spatial Omics. These domains highlight the layered molecular complexity of CIPN, from gene expression to spatially resolved biochemical alterations. In addition, three model systems (Human, Animal, and Cellular) are shown as foundational tools for translational research. Together, this integrated approach emphasizes the value of omics-driven discovery in identifying biomarkers, understanding pathomechanisms, and developing personalized therapeutic strategies for CIPN.
Conclusions
CIPN is a complex and heterogeneous pathology, and transcriptomic methodologies, particularly scRNA and snRNA sequencing, have revolutionized our understanding of the molecular landscape of DRG and non-neuronal cells under physiological and pathological conditions140. Contrary to bulk RNA sequencing, which lacks single-cell resolution, scRNA and snRNA sequencing provide a detailed understanding of cell diversity, uncovering rare subtypes and distinct molecular profiles, particularly valuable in pathological conditions. These technologies have been fundamental in identifying transcriptomic changes in CIPN and potential differences across multiple chemotherapy agents. For instance, snRNAseq has revealed the novel role of C-LTMR fibers in paclitaxel-induced neuropathy, while the innovative use of iPSC-dSN has implicated genes such as SBF2 as possible contributors to CIPN. These findings highlight the powerful impact of these high-resolution transcriptomics techniques in advancing our knowledge of peripheral neuropathies and establishing a tool for developing new therapeutic strategies to treat CIPN.
In addition, metabolomic analysis highlights that paclitaxel treatment alters lipid metabolism significantly, suggesting a shift in energy utilization and potential mitochondrial dysfunction.
Moreover, an omics-based approach with temporal and spatial resolution of biological description could be an essential tool for understanding its underlying molecular mechanism. Challenges in analyzing huge amounts of data remain significant obstacles in omic sciences, which require a rapid and accurate integration of the results by bioinformatic analysis that is sometimes difficult to interpret. Moreover, since traditional bulk sequencing provides global information about targeted tissue, single-cell sequencing profiles the heterogeneity of each type of cell. Therefore, it will be of great importance to improve advanced individual omics in the spatial context, such as through spatial transcriptomics or untargeted MSI approaches, where these spatial approaches are overlaid with tissue images, providing insight into cell-to-cell communication and interactions. Implementation of omics would most likely offer a breakthrough in peripheral nervous system pathology, identifying susceptible patients early in their development of CIPN. Until now, omics investigations have been used to compare preclinical models with patient samples to translate drug candidates into clinical drugs, enabling researchers to define comprehensive descriptions from the origin cause to the functional consequences of the disease. Although much still needs to be discovered, the preliminary encouraging data obtained from omics sciences show that changes in lipid signaling pathways, overproduction of mitochondrial reactive oxygen species and several candidates involved in dysregulation of inflammatory pathways could be possible therapeutic targets to reduce the development of CIPN.
References
Cavaletti, G. & Marmiroli, P. Chemotherapy-induced peripheral neurotoxicity. Curr. Opin. Neurol. 28, 500–507 (2015).
Pike, C. T., Birnbaum, H. G., Muehlenbein, C. E., Pohl, G. M. & Natale, R. B. Healthcare costs and workloss burden of patients with chemotherapy-associated peripheral neuropathy in breast, ovarian, head and neck, and nonsmall cell lung cancer. Chemother. Res. Pr. 2012, 913848 (2012).
Seretny, M. et al. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Pain 155, 2461–2470 (2014).
Karschnia, P., Nelson, T. A. & Dietrich, J. Mechanisms and treatment of cancer therapy-induced peripheral and central neurotoxicity. Nat. Rev. Cancer 25, 887–909 (2025).
Areti, A., Yerra, V. G., Naidu, V. & Kumar, A. Oxidative stress and nerve damage: role in chemotherapy induced peripheral neuropathy. Redox Biol. 2, 289–295 (2014).
Tao, Z., Chen, Z., Zeng, X., Cui, J. & Quan, M. An emerging aspect of cancer neuroscience: a literature review on chemotherapy-induced peripheral neuropathy. Cancer Lett. 611, 217433 (2024).
Karademir, B. et al. Proteomic approach for understanding milder neurotoxicity of Carfilzomib against Bortezomib. Sci. Rep. 8, 16318 (2018).
Jannuzzi, A. T. et al. Higher proteotoxic stress rather than mitochondrial damage is involved in higher neurotoxicity of bortezomib compared to carfilzomib. Redox Biol. 32, 101502 (2020).
Hrstka, S. C. L. et al. Proteomic analysis of human iPSC-derived sensory neurons implicates cell stress and microtubule dynamics dysfunction in bortezomib-induced peripheral neurotoxicity. Exp. Neurol. 335, 113520 (2021).
McKeage, M. J., Hsu, T., Screnci, D., Haddad, G. & Baguley, B. C. Nucleolar damage correlates with neurotoxicity induced by different platinum drugs. Br. J. Cancer 85, 1219–1225 (2001).
Moulder, S. L. et al. A randomized phase 2 trial comparing 3-hour versus 96-hour infusion schedules of paclitaxel for the treatment of metastatic breast cancer. Cancer 116, 814–821 (2010).
Cavaletti, G. et al. Early predictors of peripheral neurotoxicity in cisplatin and paclitaxel combination chemotherapy. Ann. Oncol. 15, 1439–1442 (2004).
Baldwin, R. M. et al. A genome-wide association study identifies novel loci for paclitaxel-induced sensory peripheral neuropathy in CALGB 40101. Clin. Cancer Res. 18, 5099–5109 (2012).
Meregalli, C. et al. Neurofilament light chain as disease biomarker in a rodent model of chemotherapy induced peripheral neuropathy. Exp. Neurol. 307, 129–132 (2018).
Meregalli, C. et al. Neurofilament light chain: a specific serum biomarker of axonal damage severity in rat models of chemotherapy-induced peripheral neurotoxicity. Arch. Toxicol. 94, 2517–2522 (2020).
Karteri, S. et al. Prospectively assessing serum neurofilament light chain levels as a biomarker of paclitaxel-induced peripheral neurotoxicity in breast cancer patients. J. Peripher Nerv. Syst. 27, 166–174 (2022).
Molassiotis, A. et al. Are we mis-estimating chemotherapy-induced peripheral neuropathy? Analysis of assessment methodologies from a prospective, multinational, longitudinal cohort study of patients receiving neurotoxic chemotherapy. BMC Cancer 19, 132 (2019).
Bonomo, R. & Cavaletti, G. Clinical and biochemical markers in CIPN: a reappraisal. Rev. Neurol. 177, 890–907 (2021).
Jung, M. et al. Cross-species transcriptomic atlas of dorsal root ganglia reveals species-specific programs for sensory function. Nat. Commun. 14, 366 (2023).
Tang, F. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
Bhuiyan, S. A. et al. Harmonized cross-species cell atlases of trigeminal and dorsal root ganglia. Sci. Adv. 10, eadj9173 (2024).
Lu, T. et al. Decoding transcriptional identity in developing human sensory neurons and organoid modeling. Cell 187, 7374–7393.e28 (2024).
Treutlein, B. et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375 (2014).
Basbaum, A. I., Bautista, D. M., Scherrer, G. & Julius, D. Cellular and molecular mechanisms of pain. Cell 139, 267–284 (2009).
Usoskin, D. et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 18, 145–153 (2015).
Sharma, N. et al. The emergence of transcriptional identity in somatosensory neurons. Nature 577, 392–398 (2020).
Kupari, J. et al. Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain. Nat. Commun. 12, 1510 (2021).
Tavares-Ferreira, D. et al. Spatial transcriptomics of dorsal root ganglia identifies molecular signatures of human nociceptors. Sci. Transl. Med. 14, eabj8186 (2022).
Ray, P. R. et al. RNA profiling of human dorsal root ganglia reveals sex differences in mechanisms promoting neuropathic pain. Brain 146, 749–766 (2023).
Nguyen, M. Q., von Buchholtz, L. J., Reker, A. N., Ryba, N. J. & Davidson, S. Single-nucleus transcriptomic analysis of human dorsal root ganglion neurons. Elife 10, e71752 (2021).
Hu, G. et al. Single-cell RNA-seq reveals distinct injury responses in different types of DRG sensory neurons. Sci. Rep. 6, 31851 (2016).
George, D. S. et al. The Mas-related G protein-coupled receptor d (Mrgprd) mediates pain hypersensitivity in painful diabetic neuropathy. Pain 165, 1154–1168 (2024).
Starobova, H., Mueller, A., Deuis, J. R., Carter, D. A. & Vetter, I. Inflammatory and neuropathic gene expression signatures of chemotherapy-induced neuropathy induced by vincristine, cisplatin, and oxaliplatin in C57BL/6J mice. J. Pain. 21, 182–194 (2020).
Sun, W. et al. Transcriptome analysis reveals dysregulation of inflammatory and neuronal function in dorsal root ganglion of paclitaxel-induced peripheral neuropathy rats. Mol. Pain. 19, 17448069221106167 (2023).
Sun, W. et al. Comparative transcriptome of dorsal root ganglia reveals distinct etiologies of paclitaxel- and oxaliplatin-induced peripheral neuropathy in rats. Neuroscience 516, 1–14 (2023).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Ozsolak, F. & Milos, P. M. RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 12, 87–98 (2011).
Shukla, S. et al. Development of a RNA-seq based prognostic signature in lung adenocarcinoma. J. Natl. Cancer Inst. 109, djw200 (2017).
Han, L.-O., Li, X.-Y., Cao, M.-M., Cao, Y. & Zhou, L.-H. Development and validation of an individualized diagnostic signature in thyroid cancer. Cancer Med. 7, 1135–1140 (2018).
Zhou, J.-G. et al. Development and validation of an RNA-seq-based prognostic signature in neuroblastoma. Front. Oncol. 9, 1361 (2019).
Kulkarni, A., Anderson, A. G., Merullo, D. P. & Konopka, G. Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol. 58, 129–136 (2019).
Calls, A. et al. Cisplatin-induced peripheral neuropathy is associated with neuronal senescence-like response. Neuro Oncol. 23, 88–99 (2021).
Calls, A. et al. A transient inflammatory response contributes to oxaliplatin neurotoxicity in mice. Ann. Clin. Transl. Neurol. 9, 1985–1998 (2022).
Sun, W. et al. Single-nucleus rna sequencing identifies universal camk1d upregulation and dysregulated c-ltmr subtypes as key drivers of paclitaxel-induced neuropathy. Cell Biol Toxicol. 41, 114 (2025).
Li, Y. et al. Contribution of S1pr1 -featured astrocyte subpopulation to cisplatin-induced neuropathic pain in male mice. Pain. 167, 204–217 (2026).
Cunningham, G. M. et al. The impact of SBF2 on taxane-induced peripheral neuropathy. PLoS Genet. 18, e1009968 (2022).
Senderek, J. et al. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum. Mol. Genet. 12, 349–356 (2003).
Conforti, F. L. et al. A new SBF2 mutation in a family with recessive demyelinating Charcot-Marie-Tooth (CMT4B2). Neurology 63, 1327–1328 (2004).
Schneider, B. P. et al. Charcot-Marie-Tooth gene, SBF2, associated with taxane-induced peripheral neuropathy in African Americans. Oncotarget 7, 82244–82253 (2016).
Hooshmand, K. et al. Polygenic risk of paclitaxel-induced peripheral neuropathy: a genome-wide association study. J. Transl. Med. 20, 564 (2022).
Jones, M. K. et al. Pharmacogenomics of chemotherapy induced peripheral neuropathy using an electronic health record-derived definition: a genome-wide association study. Support Care Cancer 33, 362 (2025).
Burgess, D. J. Spatial transcriptomics coming of age. Nat. Rev. Genet. 20, 317 (2019).
Moses, L. & Pachter, L. Museum of spatial transcriptomics. Nat. Methods 19, 534–546 (2022).
Doyle, T. M. & Salvemini, D. Mini-review: mitochondrial dysfunction and chemotherapy-induced neuropathic pain. Neurosci. Lett. 760, 136087 (2021).
Ollodart, J., Steele, L. R., Romero-Sandoval, E. A., Strowd, R. E. & Shiozawa, Y. Contributions of neuroimmune interactions to chemotherapy-induced peripheral neuropathy development and its prevention/therapy. Biochem. Pharm. 222, 116070 (2024).
Fingleton, E., Li, Y. & Roche, K. W. Advances in proteomics allow insights into neuronal proteomes. Front. Mol. Neurosci. 14, 647451 (2021).
Aslam, B., Basit, M., Nisar, M. A., Khurshid, M. & Rasool, M. H. Proteomics: technologies and their applications. J. Chromatogr. Sci. 55, 182–196 (2017).
Perchepied, S., Zhou, Z., Mitulović, G. & Eeltink, S. Exploiting ion-mobility mass spectrometry for unraveling proteome complexity. J. Sep Sci. 46, e2300512 (2023).
Jiang, Y. et al. The future of proteomics is up in the air: can ion mobility replace liquid chromatography for high throughput proteomics? J. Proteome Res. 23, 1871–1882 (2024).
Rozanova, S. et al. Quantitative mass spectrometry-based proteomics: an overview. Methods Mol. Biol. 2228, 85–116 (2021).
Krasny, L. & Huang, P. H. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol. Omics 17, 29–42 (2021).
Mehta, S. et al. A galaxy of informatics resources for MS-based proteomics. Expert Rev. Proteom. 20, 251–266 (2023).
Vlahou, A. Implementation of clinical proteomics: a step closer to personalized medicine? Proteom. Clin. Appl. 13, e1800088 (2019).
Chen, E. I. et al. Identifying predictors of taxane-induced peripheral neuropathy using mass spectrometry-based proteomics technology. PLoS ONE 10, e0145816 (2015).
Nguyen-Hoang, N. et al. Quantitation of plasma proteins to predict taxane-induced peripheral neuropathy. JCO Precis Oncol. 9, e2400380 (2025).
de Clauser, L. et al. Proteome and network analysis provides novel insights into developing and established chemotherapy-induced peripheral neuropathy. Front. Pharm. 13, 818690 (2022).
Hanna, R. et al. Proteomic analysis of dorsal root ganglia in a mouse model of paclitaxel-induced neuropathic pain. PLoS ONE 19, e0306498 (2024).
Liu, C. -H. et al. Nodal β spectrins are required to maintain Na channel clustering and axon integrity. Elife 9, e52378 (2020).
Zou, Y. et al. Sonic hedgehog restrains the ubiquitin-dependent degradation of SP1 to inhibit neuronal/glial senescence associated phenotypes in chemotherapy-induced peripheral neuropathy via the TRIM25-CXCL13 axis. J. Adv. Res. 68, 387–402 (2025).
Warren, G. et al. Discovery and preclinical evaluation of a novel inhibitor of FABP5, ART26.12, effective in oxaliplatin-induced peripheral neuropathy. J. Pain. 25, 104470 (2024).
Wang, Z. et al. Chemical sensors detect and resolve proteome aggregation in peripheral neuropathy cell model induced by chemotherapeutic agents. Bioorg. Chem. 148, 107491 (2024).
Burgess, J. et al. Chemotherapy-induced peripheral neuropathy: epidemiology, pathomechanisms and treatment. Oncol. Ther. 9, 385–450 (2021).
Ijsselsteijn, M. E., van der Breggen, R., Farina Sarasqueta, A., Koning, F. & de Miranda, N. F. C. C. A 40-marker panel for high dimensional characterization of cancer immune microenvironments by imaging mass cytometry. Front. Immunol. 10, 2534 (2019).
Djambazova, K. V., van Ardenne, J. M. & Spraggins, J. M. Advances in imaging mass spectrometry for biomedical and clinical research. Trends Analyt. Chem. 169, 117344 (2023).
Piga, I., Magni, F. & Smith, A. The journey towards clinical adoption of MALDI-MS-based imaging proteomics: from current challenges to future expectations. FEBS Lett. 598, 621–634 (2024).
Denti, V. et al. Spatial multiomics of lipids, N-glycans, and tryptic peptides on a single FFPE tissue section. J. Proteome Res. 21, 2798–2809 (2022).
Lim, M. J. et al. MALDI HiPLEX-IHC: multiomic and multimodal imaging of targeted intact proteins in tissues. Front. Chem. 11, 1182404 (2023).
Monroe, E. B. et al. SIMS and MALDI MS imaging of the spinal cord. Proteomics 8, 3746–3754 (2008).
Sui, P. et al. Neuropeptide imaging in rat spinal cord with MALDI-TOF MS: Method development for the application in pain-related disease studies. Eur. J. Mass Spectrom. 23, 105–115 (2017).
Caputi, F. F. et al. The active second-generation proteasome inhibitor oprozomib reverts the oxaliplatin-induced neuropathy symptoms. Biochem. Pharmacol. 182, 114255 (2020).
Meade, J. A. et al. Kappa opioid receptors mediate an initial aversive component of paclitaxel-induced neuropathy. Psychopharmacology 237, 2777–2793 (2020).
Tatsushima, Y. et al. Involvement of substance P in peripheral neuropathy induced by paclitaxel but not oxaliplatin. J. Pharmacol. Exp. Ther. 337, 226–235 (2011).
Chiba, T. et al. Paclitaxel-induced peripheral neuropathy increases substance P release in rat spinal cord. Eur. J. Pharmacol. 770, 46–51 (2016).
Wu, Y., Chen, J. & Wang, R. Puerarin suppresses TRPV1, calcitonin gene-related peptide and substance P to prevent paclitaxel-induced peripheral neuropathic pain in rats. Neuroreport 30, 288–294 (2019).
Wang, J. et al. Participation of transient receptor potential vanilloid 1 in the analgesic effect of duloxetine for paclitaxel induced peripheral neuropathic pain. Neurosci. Lett. 773, 136512 (2022).
Akhilesh, Uniyal, A., Mehta, A. & Tiwari, V. Combination chemotherapy in rodents: a model for chemotherapy-induced neuropathic pain and pharmacological screening. Metab. Brain Dis. 39, 43–65 (2024).
Mihara, Y. et al. Lysophosphatidic acid precursor levels decrease and an arachidonic acid-containing phosphatidylcholine level increases in the dorsal root ganglion of mice after peripheral nerve injury. Neurosci. Lett. 698, 69–75 (2019).
Banno, T. et al. Arachidonic acid containing phosphatidylcholine increases due to microglial activation in ipsilateral spinal dorsal horn following spared sciatic nerve injury. PLoS ONE 12, e0177595 (2017).
Xu, D. et al. Increased arachidonic acid-containing phosphatidylcholine is associated with reactive microglia and astrocytes in the spinal cord after peripheral nerve injury. Sci. Rep. 6, 26427 (2016).
Uranbileg, B. et al. Alteration of the lysophosphatidic acid and its precursor lysophosphatidylcholine levels in spinal cord stenosis: a study using a rat cauda equina compression model. Sci. Rep. 9, 16578 (2019).
Tomalty, D. et al. Molecular characterization of human peripheral nerves using desorption electrospray ionization mass spectrometry imaging. J. Anat. 243, 758–769 (2023).
Fernández, R. et al. Deciphering the lipid architecture of the rat sciatic nerve using imaging mass spectrometry. ACS Chem. Neurosci. 7, 624–632 (2016).
Rubakhin, S. S., Ulanov, A. & Sweedler, J. V. Mass spectrometry imaging and GC-MS profiling of the mammalian peripheral sensory-motor circuit. J. Am. Soc. Mass Spectrom. 26, 958–966 (2015).
Moreno-Gordaliza, E. et al. MALDI-LTQ-Orbitrap mass spectrometry imaging for lipidomic analysis in kidney under cisplatin chemotherapy. Talanta 164, 16–26 (2017).
Cao, Y. et al. Skin platinum deposition in colorectal cancer patients following oxaliplatin-based therapy. Cancer Chemother. Pharmacol. 84, 1195–1200 (2019).
Ferey, J. et al. Imaging matrix-assisted laser desorption/ionization fourier transform ion cyclotron resonance mass spectrometry of oxaliplatin derivatives in human tissue sections. Talanta 237, 122915 (2022).
Larroque, M. et al. Study of oxaliplatin penetration into ovaries of patients treated with hyperthermic intraperitoneal chemotherapy (HIPEC) for peritoneal metastases of colorectal and appendiceal origin using mass spectrometry imaging. Pleura Peritoneum 6, 67–74 (2021).
Bouslimani, A., Bec, N., Glueckmann, M., Hirtz, C. & Larroque, C. Matrix-assisted laser desorption/ionization imaging mass spectrometry of oxaliplatin derivatives in heated intraoperative chemotherapy (HIPEC)-like treated rat kidney. Rapid Commun. Mass Spectrom. 24, 415–421 (2010).
Liu, X. & Hummon, A. B. Chemical imaging of platinum-based drugs and their metabolites. Sci. Rep. 6, 38507 (2016).
Nicholson, J. K. & Lindon, J. C. Systems biology: metabonomics. Nature 455, 1054–1056 (2008).
Schrimpe-Rutledge, A. C., Codreanu, S. G., Sherrod, S. D. & McLean, J. A. Untargeted metabolomics strategies-challenges and emerging directions. J. Am. Soc. Mass Spectrom. 27, 1897–1905 (2016).
Emwas, A.-H. et al. NMR spectroscopy for metabolomics research. Metabolites 9, 123 (2019).
Dunn, W. B. et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 6, 1060–1083 (2011).
Kell, D. B. & Oliver, S. G. The metabolome 18 years on: a concept comes of age. Metabolomics 12, 148 (2016).
Pang, Z. et al. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388–W396 (2021).
Shevchenko, A. & Simons, K. Lipidomics: coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 11, 593–598 (2010).
Wishart, D. S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 15, 473–484 (2016).
Wang, H.-Y., Tsai, Y.-J., Chen, S.-H., Lin, C.-T. & Lue, J.-H. Lysophosphatidylcholine causes neuropathic pain via the increase of neuronal nitric oxide synthase in the dorsal root ganglion and cuneate nucleus. Pharm. Biochem. Behav. 106, 47–56 (2013).
Sisignano, M. et al. Targeting CYP2J to reduce paclitaxel-induced peripheral neuropathic pain. Proc. Natl. Acad. Sci. USA 113, 12544–12549 (2016).
Wu, F.-Z. et al. Wen-Luo-Tong decoction attenuates paclitaxel-induced peripheral neuropathy by regulating linoleic acid and glycerophospholipid metabolism pathways. Front. Pharm. 9, 956 (2018).
Janes, K. et al. The development and maintenance of paclitaxel-induced neuropathic pain require activation of the sphingosine 1-phosphate receptor subtype 1. J. Biol. Chem. 289, 21082–21097 (2014).
Li, C., Yamamoto, T., Kanemaru, H., Kishimoto, N. & Seo, K. Effects of sphingosine-1-phosphate on the facilitation of peripheral nerve regeneration. Cureus 16, e73784 (2024).
Sanna, M. G. et al. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 279, 13839–13848 (2004).
Choi, J. W. & Chun, J. Lysophospholipids and their receptors in the central nervous system. Biochim Biophys. Acta 1831, 20–32 (2013).
Shota Yamamoto and Nobuaki Egashira Lipid signaling in chemotherapy-induced peripheral neuropathy. Curr. Opin. Toxicol. 28, 1–6 (2021).
Maceyka, M., Harikumar, K. B., Milstien, S. & Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 22, 50–60 (2012).
Blaho, V. A. & Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 55, 1596–1608 (2014).
Dong, D., Yu, X., Tao, X., Wang, Q. & Zhao, L. S1P/S1PR1 signaling is involved in the development of nociceptive pain. Front. Pharm. 15, 1407347 (2024).
Stockstill, K. et al. Dysregulation of sphingolipid metabolism contributes to bortezomib-induced neuropathic pain. J. Exp. Med. 215, 1301–1313 (2018).
Wang, W. et al. Activation of sphingosine 1-phosphate receptor 2 attenuates chemotherapy-induced neuropathy. J. Biol. Chem. 295, 1143–1152 (2020).
Kramer, R. et al. Neurotoxic 1-deoxysphingolipids and paclitaxel-induced peripheral neuropathy. FASEB J. 29, 4461–4472 (2015).
Becker, K. A. et al. Role of 1-Deoxysphingolipids in docetaxel neurotoxicity. J. Neurochem. 154, 662–672 (2020).
Bonomo, R. et al. Effect of age on metabolomic changes in a model of paclitaxel-induced peripheral neurotoxicity. J. Peripher. Nerv. Syst. 29, 58–71 (2024).
Maekawa, K. et al. Serum lipidomics for exploring biomarkers of bortezomib therapy in patients with multiple myeloma. Cancer Sci. 110, 3267–3274 (2019).
Sun, Y. et al. Pharmacometabolomics reveals a role for histidine, phenylalanine, and threonine in the development of paclitaxel-induced peripheral neuropathy. Breast Cancer Res. Treat. 171, 657–666 (2018).
Chen, C. -S. et al. Pre-treatment amino acids and risk of paclitaxel-induced peripheral neuropathy in SWOG S0221. Res. Sq. https://doi.org/10.21203/rs.3.rs-3242513/v1 (2023).
Verma, P. et al. A metabolomics approach for early prediction of vincristine-induced peripheral neuropathy. Sci. Rep. 10, 9659 (2020).
Zhong, S. et al. Blockade of CCR5 suppresses paclitaxel-induced peripheral neuropathic pain caused by increased deoxycholic acid. Cell Rep. 42, 113386 (2023).
Hua, Y., Lv, J., Zhang, Y., Ding, Y. & Chen, J. LC-MS-based serum metabolomics analysis and potential biomarkers for oxaliplatin induced neurotoxicity in colorectal cancer. J. Pharm. Biomed. Anal. 252, 116492 (2025).
Sharma, A. et al. A multimodal approach to discover biomarkers for taxane-induced peripheral neuropathy (TIPN): a study protocol. Technol. Cancer Res. Treat. 21, 15330338221127169 (2022).
Xu, Y. et al. The potential antinociceptive effect and mechanism of Cannabis sativa L. extract on paclitaxel-induced neuropathic pain in rats uncovered by multi-omics analysis. Molecules 29, 1958 (2024).
Colvin, L. A. Chemotherapy-induced peripheral neuropathy: where are we now? Pain 160, S1–S10 (2019).
Kober, K. M. et al. Phenotypic characterization of paclitaxel-induced peripheral neuropathy in cancer survivors. J. Pain. Symptom Manag. 56, 908–919.e3 (2018).
Gadgil, S. et al. A systematic summary and comparison of animal models for chemotherapy induced (peripheral) neuropathy (CIPN). PLoS ONE 14, e0221787 (2019).
Cascella, M. Chemotherapy-induced peripheral neuropathy: limitations in current prophylactic strategies and directions for future research. Curr. Med. Res. Opin. 33, 981–984 (2017).
Currie, G. L. et al. Animal models of chemotherapy-induced peripheral neuropathy: A machine-assisted systematic review and meta-analysis. PLoS Biol. 17, e3000243 (2019).
Lehmann, H. C., Staff, N. P. & Hoke, A. Modeling chemotherapy induced peripheral neuropathy (CIPN) in vitro: prospects and limitations. Exp. Neurol. 326, 113140 (2020).
Smulders, P. S. H. et al. Chemotherapy-induced peripheral neuropathy models constructed from human induced pluripotent stem cells and directly converted cells: a systematic review. Pain 165, 1914–1925 (2024).
Sharma, A. et al. Longitudinal multi-omics analyses of chemotherapy-induced peripheral neuropathy in response to taxanes. BMC Cancer 25, 1591 (2025).
Chen, X., Gan, Y., Au, N. P. B. & Ma, C. H. E. Current understanding of the molecular mechanisms of chemotherapy-induced peripheral neuropathy. Front. Mol. Neurosci. 17, 1345811 (2024).
Acknowledgements
We acknowledge the contribution of P.P. for the conceptual model of the multi-omics framework, obtained via SciDraw.io. and Freepik graphical resources. This work was supported by Cariplo Foundation under Grant to C.M. No. 2019-1482. C.M. is also a recipient of the project PRIN 2022 PNRR with code P2022R43RA, which has received funding from NextGeneration EU—MUR—M4C2 1.1, CUP C53D23008500001.
Author information
Authors and Affiliations
Contributions
Conceptualization, C.M., I.P., and R.B.; writing—original draft preparation, I.P., R.B., C.C., L.P., and P.P.; writing—review and editing, I.P. and R.B.; visualization, I.P. and P.P.; supervision, C.M.; funding acquisition, C.M. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks Jian Hu, Mingzhu Zhai and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Piga, I., Bonomo, R., Chinello, C. et al. Using an integrated omics approach to uncover the mechanisms underlying chemotherapy-induced peripheral neuropathy (CIPN). Commun Med 6, 269 (2026). https://doi.org/10.1038/s43856-026-01622-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43856-026-01622-6
