
Abstract
Three-dimensional nitrogen-rich bridged ring systems are of great interest in drug discovery owing to their distinctive physicochemical and structural properties. However, synthetic approaches towards N–N-bond-containing bridged heterocycles are often inefficient and require tedious synthetic strategies. Here we delineate an iridium-catalysed reductive approach to such architectures from C,N,N-cyclic hydrazide substrates using IrCl(CO)[P(OPh)3]2 and 1,1,3,3-tetramethyldisiloxane (TMDS), which provided efficient access to the unstabilized and highly reactive C,N,N-cyclic azomethine imine dipoles. These species were stable and isolable in their dimeric form, but, upon dissociation in solution, reacted with a broad range of dipolarophiles in [3 + 2] cycloaddition reactions with high yields and good diastereoselectivities, enabling the direct synthesis of nitrogen-rich sp3-hybridized pyrazoline polycyclic ring systems. Density functional theory calculations were performed to elucidate the origin of the diastereoselectivity of the cycloaddition reaction, and principal moment of inertia (PMI) analysis was conducted to enable visualization of the topological information of the dipolar cycloadducts.

Similar content being viewed by others
Main
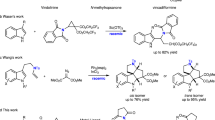
As key pharmacophores in bioactive natural products and pharmaceutical compounds, heterocycles are highly prevalent, being present in 95% of drugs on the market1. Two- (2D) and three-dimensional (3D) ring systems have been employed in drug discovery, as well as in commonly occurring scaffolds, which have often been used to improve the physicochemical profile and solubility of active pharmaceutical ingredients (Fig. 1a)2. Furthermore, structurally defined, saturated and semi-saturated heterocyclic structures are commonplace elements of drug design within medicinal chemistry programmes. One notable class of these is the nitrogen-containing sp3-rich bridged ring systems, which have been explored across the pharmaceutical industry over the past few decades. These multi-cyclic ring scaffolds have been studied and deployed as bioisosteres of commonly used functional groups such as aryls3. In particular, bridged nitrogen-containing ring systems feature in drugs approved by the Food and Drug Administration (FDA), including solifenacin (anti-muscarinic)4, varenicline (smoking cessation)5, maraviroc (anti-HIV)6 and granisetron (nausea and vomiting) (1)7. They are also commonplace in natural products (Fig. 1b) such as morphine (2)8,9,10 and dnacin A1 (3) and B1 (4)11, the latter two possessing remarkable antiproliferative activity against cancer. However, the syntheses of these bridged sp3-rich ring systems—especially those featuring N–N bonds—often require elaborate multistep routes, which are typically accompanied by low chemical efficiency, thus presenting a substantial obstacle to inclusion in drug-discovery programmes12,13. To this end, the development of enabling synthetic methodologies towards the synthesis of unexplored N,N-containing bridged ring systems from readily available starting materials with high reaction selectivity and efficiency is of importance.

a, The importance of 3D heterocycles. b, Examples of natural products possessing N-rich bridged heterocyclic ring systems. c, Classes of azomethine imines, and traditional synthetic approaches towards their preparation. d, This work, targeting the synthesis of C,N,N-cyclic azomethine imines 7 and their cycloadditions with various dipolarophiles 8. IP, intellectual property; SAR, structure–activity relationships; NPR, normalized principal moment of inertia ratios.
In parallel, the cycloaddition reactions of 1,3-dipoles (such as azomethine ylides and azomethine imines) with dipolarophiles are among the most fundamental synthetic approaches towards five-membered N-containing heterocycles with high regio- and stereoselectivity, in a single step14. Azomethine imines (Fig. 1c), in particular, have been less studied than their azomethine ylide counterparts, and are often used to access highly functionalized pyrazoline heterocycles via [3 + 2] cycloaddition with various dipolarophiles15, such as isocyanides, olefins, enones16,17 and cyclic allenes18,19,20,21. Such a class of N–N-containing ring systems has been incorporated into active pharmaceutical ingredients including antiviral, anti-inflammatory, antibacterial, antifungal, anticancer and insecticidal agents22,23,24,25. Saturated and semi-saturated pyrazoline heterocycles have also been used as proline surrogates, which play an important role in biological peptide sequences in relation to metallopeptidase activity26,27 and have been used as aza-proline derivatives to stabilize cis conformations of amide bonds in bioactive peptides28. Despite the prominent role of azomethine imines for pyrazoline synthesis, there are relatively few methods to access these important 1,3-dipoles, which include the condensation of a hydrazine and an aldehyde16,17, 1,2-prototropy of hydrazones29,30 or the opening of diaziridine rings (Fig. 1c)31. These synthetic methods, however, typically require harsh conditions, such as high temperature and/or strong acids, and are currently limited to the preparation of acyclic, C,N-cyclic and N,N-cyclic azomethine imines.
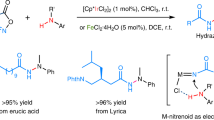
In contrast, C,N,N-cyclic azomethine imines remain inaccessible via these traditional synthetic methodologies and, accordingly, have remained unknown. Keen to access and explore the chemistry of these C,N,N-cyclic azomethine imines, and building on our expertise32,33,34,35, and that of others36,37,38,39,40,41,42,43, towards late-stage manipulation of tertiary amides and lactams, we were drawn towards the possibility of developing an iridium-catalysed reductive synthesis. We envisioned that the iridium-catalysed hydrosilylation of C,N,N-cyclic hydrazide 5 could provide the corresponding N-silylated hemiaminal 6 (Fig. 1d). Subsequently, 6 could undergo silanoate elimination and further loss of the silane on the nitrogen atom to form the elusive C,N,N-cyclic azomethine imines 7, which could then be intercepted by, for example, dipolarophiles 8, leading to the formation of diazabicyclo[3.2.1]octane 9.
Results and discussion
To test our hypothesis, we synthesized hydrazide 5a by initial alkylation of 1-(2H)-phthalazinone with MeI, followed by C=N reduction with zinc and acetic acid44. Model substrate 5a was then subjected to iridium-catalysed hydrosilylation conditions using Vaska’s complex (1 mol%) and 1,1,3,3-tetramethyldisiloxane (TMDS; 3 equiv.) at room temperature for 30 min in the hope that 7a would first be accessed via the corresponding N-silylated hemiaminal and then react in situ with the dipolarophile 8a in a [3 + 2] cycloaddition reaction. Unexpectedly, however, the homodimerization product 10a of the unstabilized azomethine imine 7a was obtained from the reaction mixture as a white, bench-stable solid in 23% isolated yield (Fig. 2a, entry 1). The solid-state dimeric structure of 10a was confirmed by single-crystal X-ray diffraction analysis (Fig. 3).

a,b, Optimization studies for dimer formation via hydrosilylation (a) and [3 + 2] cycloaddition (b) reactions. General conditions for dimer formation: 5a (0.1 mmol), [Ir]-cat (1 mol%), 1,1,3,3-tetramethyldisiloxane (3 equiv.), solvent (1.0 ml), under nitrogen atmosphere. aIsolated yield. General conditions for the [3 + 2] cycloaddition reaction: dimer 10a (0.1 mmol), vinyl sulfone 8a (y equiv.), solvent (1.0 ml), under nitrogen atmosphere. bCalculated against 1,3,5-trimethoxybenzene as an internal standard using 1H NMR spectroscopy analysis of the unpurified reaction mixture. r.t., room temperature; d.r., diastereomeric ratio.

General conditions: 5a–5h (1.0 mmol), IrCl(CO)[P(OPh3)]2 (1 mol%), 1,1,3,3-tetramethyldisiloxane (3 equiv.), Et2O (10 ml), room temperature, under nitrogen atmosphere. aDetermining the structure of the major diastereomer unambiguously using NMR spectroscopy experiments was not possible due to overlaps of key signals in the 1H NMR spectrum.
Optimization studies
With this preliminary result in hand, we turned our attention to optimizing the iridium-catalysed hydrosilylation of 5a to generate dimer 10a before exploring its downstream [3 + 2] cycloaddition reactivity. As a first approach, the catalyst loading of Vaska’s complex, the reagent stoichiometry and the reaction time were investigated. However, no significant improvement to the isolated yield of 10a when compared to the initial result (Fig. 2a, entry 1) was obtained, and substantial amounts of unreacted starting material remained in all cases. Nevertheless, this challenging hydrosilylation of hydrazide 5a was nicely overcome by employing a more active iridium complex, the phosphite derivative of Vaska’s complex (IrCl(CO)[P(OPh)3]2)45 at 1 mol% in toluene as solvent (Fig. 2a, entry 2), yielding 82% of dimer 10a. On examining different reaction solvents, diethyl ether (Et2O) was identified to be the best, affording dimer 10a in 90% yield after a 30-min reaction time (Fig. 2a, entry 3).
After identifying the optimized conditions for the formation of dimer 10a, our attention then turned to its use in the [3 + 2] cycloaddition reaction with vinyl sulfone 8a. Interestingly, by simply stirring a solution of dimer 10a and 3.0 equiv. of vinyl sulfone 8a (1.5 equiv. relative to the putative monomeric azomethine imine) in CH2Cl2 at room temperature over 20 h, the desired cycloadduct 9a was obtained in 68% yield as a single diastereoisomer (Fig. 2b, entry 1). Toluene and CH3CN were also examined, but 80 °C was required to improve the solubility of dimer 10a, resulting in slight increases in yield, to 70% and 75%, respectively (Fig. 2b, entries 2 and 3). Increasing the amount of vinyl sulfone 8a to 5 equiv. (2.5 equiv. relative to the putative monomeric azomethine imine) gave the desired cycloadduct 9a in an improved 87% yield (Fig. 2b, entry 4).
Scope development
With the optimal reaction conditions established, the scope of the reaction with respect to hydrazides 5 for accessing several azomethine imine dimers was investigated (Fig. 3). Hydrazides 5a–5h were prepared by an alkylation of 1-(2H)-phthalazinone, followed by C=N reduction with zinc and acetic acid44. The reactions proceeded in good yields when modifying the substitution on the nitrogen atom, such as linear (10a–10e) and ring-containing side chains (10f and 10g). Interestingly, similar to the optimized yield of 10a, N-protected benzyl 10b and n-butyl 10c were also amenable to this methodology. In addition, allyl- (10d) and ether-containing (10e) dimers were tolerated, as well as cyclopropane 10f and cyclopentane 10g. However, introducing a benzyl substituent at the C4 position, as shown in dimer 10h, diminished the yield slightly to 63%, and resulted in a mixture of three diastereomers.
Having successfully established the scope of the reductive formation of dimers 10a–10h from hydrazides 5a–5h, we turned our attention to the [3 + 2] cycloaddition reaction of dimer 10a and in particular the scope of it with respect to the dipolarophile (Fig. 4). A range of electron-deficient alkenes 8a–8h were explored as coupling partners. Pleasingly, the desired [3 + 2] cycloadduct 9b was furnished in 70% yield and as a single diastereoisomer when vinyl sulfone 8b was used. The structure of cycloadduct 9b was determined by single-crystal X-ray diffraction analysis. Dimethyl fumarate 8c was reactive towards dimer 10a, although a reduced yield of cycloadduct 9c was obtained. Methyl acrylate 8d and acrylonitrile 8e were compatible and afforded the respective cycloadducts 9d and 9e in good yields, albeit with imperfect regioselectivity and diastereoselectivity (1.4:1 d.r., 1:1 d.r. and 8.5:1 r.r., 2.3:1 r.r., respectively). Furthermore, the use of oxazolidinone 8f as the dipolarophile resulted in a smooth reaction, forming the desired cycloadduct 9f in excellent 95% yield and as a 2:1 mixture of endo and exo isomers. The structures of the major and minor diastereoisomers were both established by single-crystal X-ray diffraction analysis. Maleimide 8g and N-phenyl maleimide 8h provided 9g and 9h in 67% and 92% yields, respectively, with modest endo diastereoselectivity. In all examples, it should be noted that the endo and exo diastereoisomers could be readily separated by flash column chromatography. The electron-deficient alkenes 8a–8h underwent the [3 + 2] cycloaddition reaction with 10a in good to excellent yields. However, non-activated dipolarophiles, such as styrene and 2-cyclopenten-1-one, were not tolerated.

General conditions: 10a (0.1 mmol), dipolarophiles 8a–8h (0.5 mmol, 5 equiv.), CH3CN, 80 °C. a10 equiv. at 60 °C. bNeat in 8e. c2 equiv. of 8f at r.t. in CH2Cl2. The dashed box shows that all structures within the box were generated from the same reaction, that is, a mixture of two diastereoisomers.
Following exploration of the reaction scope with respect to the dipolarophiles, we then investigated the scope of the [3 + 2] cyclization with respect to dimers 10a–10h using vinyl sulfone 8a (5 equiv.) as the dipolarophile (Fig. 5). Interestingly, we were pleased to witness that all dimers 10a–10h underwent cycloaddition to give products 9i–9o in good to excellent yields and excellent diastereoselectivities (>20:1 d.r.) at 80 °C in CH3CN for 20 h. However, a slight decrease in yield was observed when dimer 10h was employed.

General conditions: 10a–10h (0.1 mmol), dipolarophile 8a (0.5 mmol, 5 equiv.), CH3CN, 80 °C.
Mechanistic investigation
Control experiments
To probe the mechanistic origin and solution behaviour of dimers 10a–10h, control experiments were conducted. Based on our working hypothesis, following rapid N–H silylation, the carbonyl of the hydrazide is readily hydrosilylated under iridium-catalysed reductive conditions to the corresponding N-silylated hemiaminal, which subsequently forms the key C,N,N-cyclic azomethine imine via silanoate elimination. However, due to the high reactivity of these cyclic azomethine imines, homodimerization led to the formation of bench-stable dimers 10a–10h. Although no direct spectroscopic evidence of the azomethine imine could be found, these dimers are presumably in dynamic equilibrium with their monomers in solution, as supported circumstantially by their behaviour in undergoing [3 + 2] cycloaddition reactions with dipolarophiles. To investigate this proposed hypothesis, we studied the solution behaviour of the methyl (Me) dimer 10a (2.0 equiv.) with the benzyl (Bn) dimer 10b (1.0 equiv.) in CD2Cl2 at 40 °C for 16 h. Pleasingly, the crossover dimer 11 was indeed formed in a ratio of 1.8:1.6:1 (10a:11:10b) (Fig. 6). This ratio changed to 1.8:1.0:1.4 (10a:11:10b) when the ratio of dimers 10a and 10b was adjusted to 1:1. Thus, these data indicate that the dimers are indeed in equilibrium with their monomers in solution and then undergo [3 + 2] cycloaddition in the presence of dipolarophiles. To further confirm this result, hydrazides 5a and 5b in a ratio of 2:1 were treated with the optimal reductive hydrosilylation reaction conditions, revealing the formation of mixed dimer 11, along with homodimers 10a and 10b. This supports that 5a and 5b were indeed reduced under the reaction conditions and formed the corresponding C,N,N-cyclic azomethine imines 7a and 7b, which then dimerized to form dimer 11, along with their homodimers 10a and 10b.

Dimers 10a (2 equiv.) and 10b (1 equiv.), CD2Cl2, 40 °C. The structures are grayed out in order to place more emphasis on structure 11.
Computational analysis
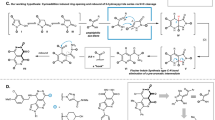
To investigate the origin of the diastereoselectivity in the [3 + 2] cycloaddition of a C,N,N-cyclic azomethine imine and a dipolarophile, density functional theory analysis was performed (Fig. 7a)16,46,47,48. As mentioned above, a remarkable diastereoselectivity was observed when vinyl sulfone 8a was used as a dipolarophile (d.r. > 20:1), while almost equal amounts of diastereomers were obtained with methyl acrylate 8d (d.r. = 1.4:1). These differences were studied by calculating the [3 + 2] cycloaddition transition-state (TS) structures between the azomethine imine monomer 7a and the corresponding dipolarophiles. The key TSs with vinyl sulfone 8b indicated that TS1-2, which leads to the experimentally obtained cyclized product 9b, is kinetically more feasible (∆∆G‡ = 1.8 kcal mol−1). On the other hand, the energy difference between the two cycloaddition TSs with methyl acrylate 8d is minimal as expected (∆∆G‡ = 0.3 kcal mol−1). An activation strain analysis that decomposes an electronic activation barrier into the strain and interaction energies revealed that TS1-1 is more destabilized than TS1-2 due to the increased strain energy (∆∆E‡strain = 6.0 kcal mol−1)49,50,51,52,53,54,55. Despite the fact that TS1-1 is more asynchronous, which should relieve the strain of a TS56, the increased strain energy originates from the steric repulsion between the ethyl group of the dipolarophile and the aromatic ring of the dipole. This is evidenced by the higher degree of pyramidalization (sum of angles, SoA) at the bond-forming α-carbon of vinyl sulfone34. This atom creates a new C−C bond at a later stage of the cyclization process, and a much smaller SoA of 353.7° for TS1-1 compared to 358.8° for TS2-1 implies that the strain localization around this atom dominates the trend in the activation energy barrier.

a, Transition-state structures for the 1,3-dipolar cycloaddition between the azomethine imine 8a and dipolarophiles computed at SMD(MeCN)/M06-2X/6-311+G(d,p)//M06-2X/6-31G(d) level of theory. b, PMI analysis of various accessible dimers and bicyclic pyrazoline compounds (P1–P14). Geometry optimizations of all compounds were performed at the M06-2X/6-31G(d) level of theory in the gas phase. Energies (kcal mol−1) and forming bond lengths (Å) of TS geometries are provided. ΔG, Gibbs free energy.
Topological analysis
To analyse the accessible dimensionality and topological features of the 3D N-rich bridged compounds, principal moment of inertia (PMI) analysis was performed (Fig. 7b)57,58. The 3D character can be obtained by calculating the PMI of a molecule along three orthogonal axes (I1, I2 and I3). Plotting the normalized values (NPR1 = I1/I3, NPR2 = I2/I3) for individual compounds onto a 2D graph within a triangular array allows the visualization of topological information of molecules. The vertices of the triangle correspond to idealized 1D, 2D and 3D molecular structures with rod-, disc- and sphere-like symmetries, respectively. As expected, the [3.2.1]-bicyclodiazaoctane core possesses a high 3D character, and the plot is located near the vertex with a sphere-like symmetry. Within the scope of computed bicyclic pyrazoline compounds using the combination of synthesized C,N,N-cyclic azomethine imines and experimentally utilized dipolarophiles, a wide range of chemical space can be accessible. For example, pyrazoline products from acrylonitrile have the rod-like symmetry (P6, P7), whereas pyrazoline products from maleimide are located between the sphere and rod symmetries (P10, P11). Interestingly, the C,N,N-cyclic azomethine imine dimers have a variety of topological features depending on the substituent on the nitrogen atom. These analyses support that the bridged heterocyclic compounds accessible using the current methodology expand the known molecular complexity into new 3D nitrogen-rich sp3 chemical space.
Conclusions
In summary, a synthetic strategy enabling access to 3D nitrogen-rich bridged ring systems from readily available starting materials has been successfully developed. Relying on the selective iridium-catalysed hydrosilylation of the carbonyl group of C,N,N-cyclic hydrazides using 1 mol% of IrCl(CO)[P(OPh)3]2 and superstoichiometric TMDS, this approach provides convenient access to unstabilized C,N,N-cyclic azomethine imines, which were obtained as bench-stable dimers. Mechanistic investigations revealed that through a dynamic equilibrium with their azomethine imine dipoles, these dimers were found to efficiently undergo [3 + 2] cycloaddition reactions with various dipolarophiles, leading to the formation of structurally complex 3D nitrogen-rich bridged ring systems with good to excellent diastereo- and regioselectivities. The diastereoselectivity of the cycloaddition reaction was elucidated by density functional theory calculations, and PMI analysis was investigated to visualize the topological information of the homodimers and the cycloaddition products.
Methods
General procedure for the preparation of dimers 10a–10h
To a stirred solution of the relevant hydrazide 5a–5h (1.0 mmol) and IrCl(CO)[P(OPh)3]2 (7.8 mg, 1 mol%) in diethyl ether (0.1 M) at room temperature was added TMDS (0.53 ml, 3.0 mmol, 3 equiv.). After 30 min, the reaction mixture was concentrated, and the resulting solid was washed with pentane once, filtered, and concentrated under reduced pressure, yielding dimers 10a–10h.
General procedure for the [3 + 2] cycloadditions of dimer 10a with dipolarophiles 8a–8h
To a stirred solution of dimer 10a (29.2 mg, 0.1 mmol) in CH3CN was added the relevant dipolarophile 8a–8h (0.5 mmol, 5.0 equiv., unless otherwise stated). The reaction was stirred at 80 °C for 20 h and then concentrated. The crude material was purified by column chromatography to afford the desired cycloadduct 9a–9h.
Data availability
The data that support the findings of this study are available within the article and its Supplementary Information. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2284573 (9b), 2284574 (9f-endo), 2284575 (9f-exo) and 2284576 (10a). These data can be obtained free of charge from the CCDC via www.ccdc.cam.ac.uk/data_request/cif. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript (AAM) version arising from this submission.
References
Taylor, R. D., MacCoss, M. & Lawson, A. D. Rings in drugs: miniperspective. J. Med. Chem. 57, 5845–5859 (2014).
Aldeghi, M., Malhotra, S., Selwood, D. L. & Chan, A. W. E. Two- and three-dimensional rings in drugs. Chem. Biol. Drug Des. 83, 450–461 (2014).
Rice, S., Cox, D. J., Marsden, S. P. & Nelson, A. Efficient unified synthesis of diverse bridged polycyclic scaffolds using a complexity-generating ‘stitching’ annulation approach. Chem. Commun. 57, 599–602 (2021).
Kobayashi, S., Ikeda, K., Suzuki, M., Yamada, T. & Miyata, K. Effects of YM905, a novel muscarinic M3-receptor antagonist, on experimental models of bowel dysfunction in vivo. Jpn. J. Pharmacol. 86, 281–288 (2001).
Coe, J. W. et al. Varenicline: an α4β2 nicotinic receptor partial agonist for smoking cessation. J. Med. Chem. 48, 3474–3477 (2005).
Woollard, S. M. & Kanmogne, G. D. Maraviroc: a review of its use in HIV infection and beyond. Drug Des. Dev. Ther. 9, 5447–5468 (2015).
Hsu, E. S. A review of granisetron, 5-hydroxytryptamine3 receptor antagonists, and other antiemetics. Am. J. Therap. 17, 476–486 (2010).
Swerdlow, M. General analgesics used in pain relief: pharmacology. Br. J. Anaesth. 39, 699–712 (1967).
Bilfinger, T. V. & Kushnerik, V. The use of morphine in surgery: an overview. Adv. Neuroimmunol. 4, 133–144 (1994).
Przewłocki, R. & Przewłocka, B. Opioids in chronic pain. Eur. J. Pharmacol. 429, 79–91 (2001).
Scott, J. D. & Williams, R. M. Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics. Chem. Rev. 102, 1669–1730 (2002).
Blakemore, D. C. et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394 (2018).
Campos, K. R. et al. The importance of synthetic chemistry in the pharmaceutical industry. Science 363, eaat0805 (2019).
Nair, V. & Suja, T. Intramolecular 1,3-dipolar cycloaddition reactions in targeted syntheses. Tetrahedron 63, 12247–12275 (2007).
Mish, M. R., Guerra, F. M. & Carreira, E. M. Asymmetric dipolar cycloadditions of Me3SiCHN2. Synthesis of a novel class of amino acids: azaprolines. J. Am. Chem. Soc. 119, 8379–8380 (1997).
Najera, C., Sansano, J. M. & Yus, M. 1,3-Dipolar cycloadditions of azomethine imines. Org. Biomol. Chem. 13, 8596–8636 (2015).
Oppolzer, W. Intramolekulare cycloadditionen von azomethiniminen, teil I: reaktion von ungesaettigten aldehyden mit N-acyl-N′-alkylhydraziden. Tetrahedron Lett. 11, 3091–3094 (1970).
Lofstrand, V. A. & West, F. G. Efficient trapping of 1,2‐cyclohexadienes with 1,3‐dipoles. Chem. Eur. J. 22, 10763–10767 (2016).
Barber, J. S. et al. Diels–Alder cycloadditions of strained azacyclic allenes. Nat. Chem. 10, 953–960 (2018).
Yamano, M. M. et al. Cycloadditions of oxacyclic allenes and a catalytic asymmetric entryway to enantioenriched cyclic allenes. Angew. Chem. 131, 5709–5713 (2019).
Almehmadi, Y. A. & West, F. A mild method for the generation and interception of 1,2-cycloheptadienes with 1,3-dipoles. Org. Lett. 22, 6091–6095 (2020).
Mishra, V. K., Mishra, M., Kashaw, V. & Kashaw, S. K. Synthesis of 1,3,5-trisubstituted pyrazolines as potential antimalarial and antimicrobial agents. Biorg. Med. Chem. 25, 1949–1962 (2017).
Rana, D. N., Chhabria, M. T., Shah, N. K. & Brahmkshatriya, P. S. Discovery of new antitubercular agents by combining pyrazoline and benzoxazole pharmacophores: design, synthesis and insights into the binding interactions. Med. Chem. Res. 23, 2218–2228 (2014).
Shin, S. Y. et al. Colorectal anticancer activities of polymethoxylated 3-naphthyl-5-phenylpyrazoline-carbothioamides. Bioorg. Med. Chem. Lett. 26, 4301–4309 (2016).
Joshi, R. S., Mandhane, P. G., Diwakar, S. D., Dabhade, S. K. & Gill, C. H. Synthesis, analgesic and anti-inflammatory activities of some novel pyrazolines derivatives. Bioorg. Med. Chem. Lett. 20, 3721–3725 (2010).
Simon, J. A. & Schreiber, S. L. Grb2 SH3 binding to peptides from Sos: evaluation of a general model for SH3–ligand interactions. Chem. Biol. 2, 53–60 (1995).
Kini, R. M. & Evans, H. J. A hypothetical structural role for proline residues in the flanking segments of protein–protein interaction sites. Biochem. Biophys. Res. Commun. 212, 1115–1124 (1995).
Zhang, Y., Malamakal, R. M. & Chenoweth, D. M. A single stereodynamic center modulates the rate of self‐assembly in a biomolecular system. Angew. Chem. Int. Ed. 54, 10826–10832 (2015).
Grigg, R., Kemp, J. & Thompson, N. X=Y–ZH systems as potential 1,3-dipoles. Tetrahedron Lett. 19, 2827–2830 (1978).
Le Fevre, G., Sinbandhit, S. & Hamelin, J. Addition d’hydrazone aux oléfines en milieu acide: cycloaddition polaire cationique [3+ + 2]. Tetrahedron 35, 1821–1824 (1979).
Molchanov, A., Sipkin, D., Koptelov, Y. B. & Kostikov, R. Thermolysis of 6-aryl-1,5-diazabicyclo [3.1.0] hexanes in the presence of N-arylmaleimides. Russ. J. Org. Chem. 37, 841–851 (2001).
Matheau-Raven, D. et al. Catalytic reductive functionalization of tertiary amides using Vaska’s complex: synthesis of complex tertiary amine building blocks and natural products. ACS Catal. 10, 8880–8897 (2020).
Xie, L.-G. & Dixon, D. J. Iridium-catalyzed reductive Ugi-type reactions of tertiary amides. Nat. Commun. 9, 2841 (2018).
Yamazaki, K. et al. General pyrrolidine synthesis via iridium-catalyzed reductive azomethine ylide generation from tertiary amides and lactams. ACS Catal. 11, 7489–7497 (2021).
Gabriel, P., Almehmadi, Y. A., Wong, Z. R. & Dixon, D. J. A general iridium-catalyzed reductive dienamine synthesis allows a five-step synthesis of catharanthine via the elusive dehydrosecodine. J. Am. Chem. Soc. 143, 10828–10835 (2021).
Katahara, S. et al. An iridium-catalyzed reductive approach to nitrones from N-hydroxyamides. J. Am. Chem. Soc. 138, 5246–5249 (2016).
Yoritate, M. et al. Unified total synthesis of stemoamide-type alkaloids by chemoselective assembly of five-membered building blocks. J. Am. Chem. Soc. 139, 18386–18391 (2017).
Yang, Z.-P., He, Q., Ye, J.-L. & Huang, P.-Q. Asymmetric total synthesis and absolute configuration determination of (−)-verrupyrroloindoline. Org. Lett. 20, 4200–4203 (2018).
Katahara, S. et al. Five-step total synthesis of (±)-aspidospermidine by a lactam strategy via an azomethine ylide. Org. Lett. 23, 3058–3063 (2021).
Takahashi, Y., Sato, T. & Chida, N. Iridium-catalyzed reductive nucleophilic addition to tertiary amides. Chem. Lett. 48, 1138–1141 (2019).
Yang, Z.-P., Lu, G.-S., Ye, J.-L. & Huang, P.-Q. Ir-catalyzed chemoselective reduction of β-amido esters: a versatile approach to β-enamino esters. Tetrahedron 75, 1624–1631 (2019).
Hugelshofer, C. L., Palani, V. & Sarpong, R. Calyciphylline B-type alkaloids: total syntheses of (−)-daphlongamine H and (−)-isodaphlongamine H. J. Am. Chem. Soc. 141, 8431–8435 (2019).
Hugelshofer, C. L., Palani, V. & Sarpong, R. Calyciphylline B-type alkaloids: evolution of a synthetic strategy to (−)-daphlongamine H. J. Org. Chem. 84, 14069–14091 (2019).
Sugimoto, A. et al. 7-(Ethoxycarbonyl)-6,8-dimethyl-2-phenyl-1(2H)-phthalazinone derivatives: synthesis and inhibitory effects on platelet aggregation. J. Med. Chem. 27, 1300–1305 (1984).
Rogova, T. et al. Reverse polarity reductive functionalization of tertiary amides via a dual iridium-catalyzed hydrosilylation and single electron transfer strategy. ACS Catal. 10, 11438–11447 (2020).
Hamlin, T. A. et al. Elucidating the trends in reactivity of aza‐1,3‐dipolar cycloadditions. Eur. J. Org. Chem. 2019, 378–386 (2019).
Beutick, S. E., Vermeeren, P. & Hamlin, T. A. The 1,3‐dipolar cycloaddition: from conception to quantum chemical design. Chem. Asian J. 17, e202200553 (2022).
Domingo, L. R. & Ríos-Gutiérrez, M. A molecular electron density theory study of the reactivity of azomethine imine in [3 + 2] cycloaddition reactions. Molecules 22, 750 (2017).
van Zeist, W.-J. & Bickelhaupt, F. M. The activation strain model of chemical reactivity. Org. Biomol. Chem. 8, 3118–3127 (2010).
Fernández, I. & Bickelhaupt, F. M. The activation strain model and molecular orbital theory: understanding and designing chemical reactions. Chem. Soc. Rev. 43, 4953–4967 (2014).
Wolters, L. P. & Bickelhaupt, F. M. The activation strain model and molecular orbital theory. WIREs Comput. Mol. Sci. 5, 324–343 (2015).
Bickelhaupt, F. M. & Houk, K. N. Analyzing reaction rates with the distortion/interaction‐activation strain model. Angew. Chem. Int. Ed. 56, 10070–10086 (2017).
Vermeeren, P., van der Lubbe, S. C., Fonseca Guerra, C., Bickelhaupt, F. M. & Hamlin, T. A. Understanding chemical reactivity using the activation strain model. Nat. Protoc. 15, 649–667 (2020).
Ess, D. H. & Houk, K. Distortion/interaction energy control of 1,3-dipolar cycloaddition reactivity. J. Am. Chem. Soc. 129, 10646–10647 (2007).
Ess, D. H. & Houk, K. Theory of 1,3-dipolar cycloadditions: distortion/interaction and frontier molecular orbital models. J. Am. Chem. Soc. 130, 10187–10198 (2008).
Vermeeren, P., Hamlin, T. A., Fernández, I. & Bickelhaupt, F. M. Origin of rate enhancement and asynchronicity in iminium catalyzed Diels–Alder reactions. Chem. Sci. 11, 8105–8112 (2020).
Chen, S.-J. et al. Accessing three-dimensional molecular diversity through benzylic C–H cross-coupling. Nat. Synth. 2, 998–1008 (2023).
Sanchez, A. et al. A shapeshifting roadmap for polycyclic skeletal evolution. J. Am. Chem. Soc. 145, 13452–13461 (2023).
Acknowledgements
Y.A.A. thanks King Abdulaziz University (KAU) for a postgraduate scholarship. N.J.G. thanks the EPSRC for a Vacation Internship Award (grant no. EP/W524311/1). The computation was performed using the Research Center for Computational Science, Okazaki, Japan (project 23-IMS-C12). We thank B. D. A. Shennan (University of Oxford) and M. Formica (University of Oxford) for fruitful discussions.
Author information
Authors and Affiliations
Contributions
Y.A.A. and D.J.D. conceived the project. Y.A.A., J.M. and N.J.G. conducted all the experimental work and analysed the data. K.Y. conducted the computational work. K.E.C. conducted single-crystal X-ray diffraction experiments. The paper was written by Y.A.A., K.Y. and D.J.D., with proofreading from all authors. D.J.D. directed the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Peter Seavill, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Experimental details, Supplementary Figs. 1–9 and Tables 1–7.
Supplementary Data 1
Crystallographic data for compound 9b; CCDC reference 2284573.
Supplementary Data 2
Structure factors for compound 9b; CCDC 2284573.
Supplementary Data 3
Crystallographic data for compound 9f-endo; CCDC reference 2284574.
Supplementary Data 4
Structure factors for compound 9f-endo; CCDC 2284574.
Supplementary Data 5
Crystallographic data for compound 9f-exo; CCDC reference 2284575.
Supplementary Data 6
Structure factors for compound 9f-exo; CCDC 2284575.
Supplementary Data 7
Crystallographic data for compound 10a; CCDC reference 2284576.
Supplementary Data 8
Structure factors for compound 10a; CCDC 2284576.
Supplementary Data 9
XYZ coordinates for the substrate and [3 + 2] cycloaddition transition state (TS) structures and principal moment of inertia (PMI) analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Almehmadi, Y.A., McGeehan, J., Guzman, N.J. et al. Iridium-catalysed synthesis of C,N,N-cyclic azomethine imines enables entry to unexplored nitrogen-rich 3D chemical space. Nat. Synth 3, 1168–1175 (2024). https://doi.org/10.1038/s44160-024-00574-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44160-024-00574-w
