
Abstract
The editing of organic molecules through single-atom modification is an enabling capability for medicinal chemistry. Although several examples of single-atom insertions into the carbon–carbon double bonds of unsaturated aromatic ring systems have been reported, heteroatom insertions into chemically inert carbon–carbon single bonds are comparatively rare. Here we report a photochemical strategy for the formal migration of oxygen atoms into carbon–carbon single bonds. This protocol is based on the ability of copper(II) salts to induce photochemical homolytic cleavage of carbon–carbon bonds adjacent to alcohols and to mediate oxidative coupling reactions of the resulting organoradical intermediates. Application of this method to cyclic alcohol substrates results in oxygen atom insertions into saturated carbocyclic rings, and its extension to linear alcohol substrates enables atomic permutation of hydroxymethyl functionalities into methyl ethers.

Similar content being viewed by others
Main
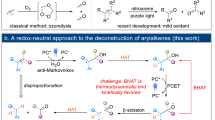
Oxidative functionalization reactions are crucial in synthetic chemistry because of their ability to introduce polar functional groups into molecules. The presence of these groups, their identity and their position profoundly alter both the physiochemical properties of bioactive compounds and their affinity towards their biological targets. Most oxidation reactions, however, introduce new polar groups only on the molecular periphery (Fig. 1a)1,2. Only a handful of robust reactions are known that insert heteroatoms into the carbon framework of an organic molecule. Classical examples include the Baeyer–Villiger oxidation3, which inserts an oxygen atom into the C–C bond of a ketone, the Beckmann rearrangement4, which is an analogous nitrogen atom insertion reaction, and the Hock process, which converts cumene to the corresponding phenol5. Such single-heteroatom insertion reactions constitute a subset of a class of transformations known as ‘skeletal editing’, which are valued in drug discovery for enabling the rapid preparation of scaffold-modified analogues without lengthy de novo synthesis6,7. Many recent reports have described new strategies to convert C(sp2)-rich arenes into heterocycles via single-atom insertion (Fig. 1b)8,9,10,11,12,13,14,15,16,17. However, in contrast, reactions that effect heteroatom insertion into fully saturated C–C bonds are quite rare18,19,20,21.

a, C–H and C–C functionalizations for peripheral and skeletal modification. b, Representative example of heteroatom insertion for sp2-(hetero)arenes. c, Design of the oxygen insertion. d, General scheme for cyclic and acyclic oxygen insertion. TBS, tert-butyldimethylsilyl; LMCT, ligand-to-metal charge transfer; Nuc, nucleophile.
One reason for the difficulty of oxidizing saturated C(sp3)–C(sp3) bonds is the relative inertness of these highly directional bonds towards exogenous chemical reactants. Common industrial alkane reformation reactions, for example, require forcing conditions, are relatively unselective for one C–C bond among many in a complex structure, and are intolerant of the high degree of functionalization common in pharmaceutical compounds22. An alternative strategy to trigger selective reactions of C–C bonds under milder conditions exploits the reactivity of functional groups already present on a molecule of interest. For example, installation of directing groups can increase the reactivity of specific C–C bonds towards transition-metal catalysts. However, the directing groups most commonly used for this strategy require the presence of a C(sp2)-hybridized functionality and have not yet been utilized in heteroatom insertion reactions, to the best of our knowledge23. Recent developments in photochemistry have provided an alternative approach to selective C–C bond cleavage that leverages the high reactivity of alkoxy radicals. Knowles has pioneered the use of photocatalytic proton-coupled electron transfer (PCET) to convert native alcohol functionalities into alkoxy radicals24, while Zuo has similarly pioneered methods utilizing ligand-to-metal charge-transfer (LMCT) photochemistry25,26 to homolyse the metal–oxygen bonds of in situ-generated metal alkoxides27. The high reactivity of the resulting oxygen-centred radical intermediates can provide a strong driving force for the β-scission of adjacent C–C bonds at ambient temperature.
We imagined that LMCT photochemistry could provide an ideal entry into the oxidative functionalization of saturated C–C bonds, for three reasons. First, a variety of metal alkoxides have been demonstrated to undergo LMCT photoactivation and subsequent β-scission; these reactions occur under mild and operationally convenient conditions, and the selectivity of the bond cleavage is well understood and highly predictable. Second, because the LMCT generation of alkoxy radicals is a unimolecular process, this activation mode avoids the application of the deeply oxidizing potentials required for outer-sphere single-electron oxidation of alcohols and thus is readily tolerant of the oxidatively sensitive functional groups common in pharmaceutical chemistry. Finally, our laboratory, among others, has shown that the Cu(II) and Fe(III) salts used in LMCT photoactivation can subsequently promote oxidative coupling reactions with diverse nucleophilic partners to forge a range of carbon–heteroatom bonds, also with high functional-group compatibility28,29,30,31.
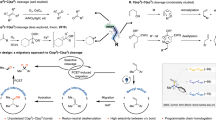
These features suggested the reaction design outlined in Fig. 1c. The in situ reaction of a saturated cycloalkanol with an appropriate redox-active first-row transition metal would result in a photoactive metal alkoxide. LMCT photoactivation would generate an alkoxy radical that is poised to undergo rapid β-scission. We imagined that metal-mediated oxidative radical coupling with the liberated carbonyl group might generate an oxocarbenium ion, which could readily be trapped with an exogenous nucleophile. Overall, this reaction would constitute a one-step protocol for the selective oxidative migration of an alcohol into a saturated carbocyclic ring. Very recently, a conceptually similar β-scission and oxidative cyclization sequence was reported using electrochemical generation of an alkoxy radical32. However, consistent with the deeply oxidizing potentials required for direct alcohol oxidation, this reaction utilized a highly positive applied potential (+2.1 V versus saturated calomel electrode), and the demonstrated functional-group compatibility of the electrochemical process was correspondingly somewhat modest.
Results
An empirical optimization campaign resulted in the identification of the reaction conditions shown in Fig. 2a. Under optimized conditions, cycloalkanol 1 is converted smoothly to ring-expanded tetrahydropyran 3 upon irradiation with 427-nm light in the presence of 2.5 equiv. of Cu(OTf)2, 3.0 equiv. of pyridine, 5.0 equiv. of isobutyronitrile (iPrCN) and 3.0 equiv. of EtOH (2) as a convenient, inexpensive nucleophilic trapping agent. The identity of the Cu(II) reagent proved to be important, as substitutions for Cu(OTf)2 afforded only trace product (Fig. 2a, entry 2). Alterations to the base and solvent led to diminished, albeit synthetically appreciable yields (entries 3 and 4). Consistent with our previous studies of oxidative LMCT photochemistry25, exclusion of the weakly coordinating iPrCN ligand results in a decrease in reactivity (entry 5). We believe the nitrile ligand improves the solubility of Cu(OTf)2 and remains bound to Cu(II) in the photoactive complex (details are provided in Supplementary section I). The reaction scaled well, with only a negligible loss of yield observed when run at a 1.00 mmol scale (entry 6). In the absence of either Cu(OTf)2 or light, no product is generated, supporting the hypothesis that this reaction proceeds through a Cu-mediated photochemical process (entries 7 and 8). Finally, we also investigated a catalytic variant of this transformation. Optimal catalytic conditions, however, required highly reactive N-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate (NFTPT) as the terminal oxidant; the lower yields and higher costs associated with this organic oxidant render the use of stoichiometric Cu(OTf)2 more attractive in this case (Supplementary Section B).

a, Reaction optimization for saturated cyclic scaffolds. b, Acetal diversification. c, Cycloalkanol scope. d, Scope of nucleophiles for direct trapping. All reactions were run at 0.20 mmol scale, prepared in a nitrogen-filled glovebox, and irradiated with two 427-nm Kessil lamps with a cooling fan. AY, NMR spectroscopy assay yield with CH2Br2 as an internal standard. IY, isolated yield. Diastereomeric ratios (d.r.) were determined by 1H NMR spectroscopy analysis using CH2Br2 as an internal standard. aCycloalkanol (1.0 equiv.), Cu(OTf)2 (2.5 equiv.), pyridine (3.0 equiv.), iPrCN (5.0 equiv.), EtOH (3.0 equiv.) and CH2Cl2 (0.1 M) for 16 h. bFollowing oxygen insertion, the acetal was reduced with BF3•Et2O (2.15 equiv.) and Et3SiH (2.0 equiv.) in CH2Cl2 (0.1 M) for 4 h. cCycloalkanol (1.0 equiv.), Cu(OTf)2 (2.5 equiv.), pyridine (3.0 equiv.), iPrCN (5.0 equiv.), nucleophile (1.0 equiv.) and CH2Cl2 (0.1 M) for 4.5 h. dSame conditions as in a, but larger alcohols were employed (3.0 equiv.) instead of EtOH. TMS, trimethylsilyl; OTf, triflate; d.r., diastereomeric ratio.
The acetal functionality embedded in the oxidized product is an attractive linchpin for further manipulation. The ethoxy substituent can be tracelessly deleted by Lewis acid-mediated silane reduction, revealing the trivial product of formal oxygen atom migration into the ring (4). Similarly, other classical Lewis or Brønsted acid-mediated diversification reactions were successful, including hydrolysis (5), allylation (6), alkylation (7) and cyanation (8), affording the corresponding products in high yields with good to excellent diastereoselectivity (Fig. 2b).
Figure 2c summarizes studies exploring the scope of this transformation with respect to the cycloalkanol. To facilitate analysis of the products, in most cases the oxidative insertion reaction was followed by hydride reduction of the acetal. Notably, the Cu salts are easily removed by aqueous work-up, and no chromatographic purification is necessary between the steps, highlighting the operational simplicity of this telescoped reaction sequence. Assay yields (AY) were reported for previously reported compounds that were potentially volatile. Cyclopentanols featuring electron-poor (9), electron-rich (10), o-substituted (11) and p-substituted (12) arenes were all tolerated in good yields. Arenes with vinyl (13) and allylic substitution (14) afforded products without evidence of the respective 5-endo-trig or 5-exo-trig radical cyclization by-products. 2-Phenylcyclobutanol was smoothly transformed to tetrahydrofuran 15, showcasing the O-insertion into a smaller ring scaffold. Both α-amino (16) and allylic (17) substrates were also effectively converted to product; however, cyclopentanols lacking a radical-stabilizing group failed to react, indicating the need for electronic stabilization in the β-scission and oxidative cyclization events. To study the regioselectivity of the β-scission, we utilized substrates that have two sites of potential radical stabilization. In each case we observed highly regioselective bond scission, favouring cleavage and functionalization at benzylic sites over oxygen- and nitrogen-stabilized positions (18,19). Finally, as skeletal editing reactions are most impactful in the context of late-stage functionalizations, we utilized our reaction conditions to synthesize oestrone derivative 20 and the oxo-analogue of a known NK1 receptor antagonist (21)33.
In our preliminary studies, ethanol was selected as an arbitrary and inexpensive trap for the electrophilic oxocarbenium intermediate. However, the facility by which diverse nucleophiles add to oxocarbenium cations suggested that variation of the trapping reagent could offer a strategy to elaborate the ring-expanded heterocycles with valuable functionalities in a single step (Fig. 2d). Using only 1.0 equiv. of exogenous nucleophile, the oxocarbenium was readily functionalized with electron-rich (22), electron-poor (23) and alkyl (24) sulfonamides in good to excellent yields and diastereoselectivities. The complex sulfonamide celecoxib was also installed in high yields (25), demonstrating the tolerance of this reaction towards the N-heterocycles commonly found in pharmaceutical compounds. Benzyl carbamate was incorporated in good yield and diastereoselectivity (26), despite its comparatively lower nucleophilicity, as was a more complex alcohol (27), showing that the oxocarbenium can be directly trapped by a range of nucleophiles. Smaller ring sizes were also readily elaborated with N-nucleophiles, furnishing amide-trapped tetrahydrofuran product 28. Substrates functionalized with substituted arenes (29), vinyl groups (30) and α-amino functionalities (31) were also readily ring-expanded in good yields.
To gain insight into the mechanism, we sought experimental and computational evidence for the key LMCT activation, β-scission and oxidative cyclization steps of the reaction cascade. To support the involvement of an LMCT event, we conducted a UV–vis study probing the formation of the proposed photoactive complex (Fig. 3a). Solutions of Cu(OTf)2 and pyridine were prepared and titrated with cyclopentanol 1. As the concentration of 1 is increased, we observe a bathochromic shift of the absorbance into the visible region, supporting the generation of a photoactive complex that involves 1 as a ligand. Notably, features assigned as LMCT transitions in the UV–vis spectra of previously reported Cu(II) alkoxide species also appear in this range34.

a, UV–vis titration study with Cu(OTf)2 (25 mM), pyridine (30 mM) and increasing amounts of 1. The UV–vis study was conducted in MeCN for improved solubility. b, Investigating the reversibility of bond scission starting from 1 and cis-1. RSM, returned starting material determined by 1H NMR spectroscopy analysis using CH2Br2 as an internal standard. c, Left: hypothesized reaction pathways for oxidative cyclization. Right: computed potential energy surfaces (PESs) for radical cyclization and polar cyclization pathways. Calculated Gibbs free energies (ΔG) ((PCM(CH2Cl2) using B3LYP/6-31G(d)) are reported in units of kcal mol−1. INT, intermediate; TS, transition state; DFT, density functional theory.
To better understand the β-scission event, we questioned whether ring-opening of the alkoxy radical might be reversible, in analogy to recent observations on the LMCT-promoted stereochemical isomerization of similar cycloalkanols35. Stereochemically pure cis and trans isomers of 2-phenylcyclopentanol (1 and cis-1) were prepared and subjected to the conditions for oxidative migration (Fig. 3b). In this experiment, both substrates converged to the same diastereomer with similar yields and product ratios, supporting the formation of an achiral intermediate in which the stereochemical information of the initial state is lost. More importantly, the unreacted starting materials retain their original configuration without loss of fidelity. We infer from these results that the ring-opening process is irreversible under these conditions.
Finally, we imagined two mechanistic possibilities for formation of the oxocarbenium intermediate: radical cyclization to generate an α-oxy radical that can be oxidized by Cu(II) to afford the oxocarbenium (Fig. 3c, pathway A) or direct Cu(II)-mediated oxidation of the benzylic radical to its corresponding carbocation before cyclization of the aldehyde (Fig. 3c, pathway B). To probe this question, we performed a computational analysis comparing the relative energies of the two reaction pathways. All structures were optimized and minima confirmed on the potential energy surface using B3LYP with the 6-31G(d) basis set, and a polarizable continuum model for dichloromethane was used. All calculations were performed with Gaussian 1636. DFT calculations concluded that the radical cyclization onto the aldehyde would have a transition state (TS1) of 22.6 kcal mol−1 and be endergonic by 7.8 kcal mol−1. A high activation energy would result in substantially longer reaction times than those observed in our experiments, and the lack of evidence for intramolecular reactions with appropriately positioned alkenes (Fig. 2c, 13 and 14) requires that the lifetime of the radical must be short. In contrast, the cationic cyclization was calculated to be exergonic by 10.4 kcal mol−1, with a minimal barrier for the cyclization step (3.0 kcal mol−1, TS2), consistent with rapid cyclization upon oxidation of the radical. Because Cu(II) salts are known to oxidize organoradicals at diffusion-controlled rates37, we propose that pathway B is probably the dominant mechanism for product formation.
Together, the spectroscopic, experimental and computational evidence supports the mechanistic proposal outlined in Fig. 1c: in situ self-assembly of a photoactive Cu(II) alkoxide species enables a visible-light-promoted LMCT transition. This results in dissociative homolysis to generate a high-energy alkoxy radical, which induces an irreversible β-scission of the adjacent C(sp3)–C(sp3) bond. The ring-opened organoradical species is rapidly oxidized by Cu(II), and ensuing oxidative cyclization of the aldehyde followed by trapping of the oxocarbenium intermediate affords the oxygen migration product.
Much of the recent interest in heteroatom insertion reactions has focused on transformations of ring systems. Novel methods for molecular editing of acyclic substructures could be equally enabling in medicinal chemistry, but of the few examples reported so far38,39,40, there has been only one isolated example of a heteroatom insertion into a C(sp3)–C(sp3) bond41. We imagined that the oxidation of C–C bonds in acyclic alcohols could be accomplished using a mechanistic logic similar to that described above to provide a complementary synthetic outcome (Fig. 4a). In a linear alcohol, LMCT-promoted β-scission results in the liberation of formaldehyde as a small-molecule by-product. Although the Cu(II)-mediated oxidative cyclization of the tethered aldehydes was facile, trapping of this liberated aldehyde presents a more formidable challenge. In the presence of an exogenous nucleophilic reaction partner, however, metal-mediated oxidative coupling of the resulting organoradical species would lead to formation of a new carbon–heteroatom bond.

a, Left: reaction design for oxygen insertion into acyclic scaffolds. Right: reaction optimization for oxygen insertion into saturated acyclic scaffolds. b, Select scope examples for methanol trapping. c, Scope examples for trapping with larger alkyl alcohols. All reactions were run at the 0.30 mmol scale, prepared in a nitrogen-filled glovebox, and irradiated with two 427-nm Kessil lamps with a cooling fan. Diastereomeric ratios were determined by 1H NMR spectroscopy analysis of the crude mixture using CH2Br2 as an internal standard. aAlcohol substrate (1.0 equiv.), MeOH (10.0 equiv.), Cu(OTf)2 (2.5 equiv.), pyridine (3.0 equiv.), MeCN (15.0 equiv.) and CH2Cl2 (0.1 M) for 48 h bAlcohol substrate (1.0 equiv.), MeOH (20.0 equiv.), FeCl3 (5.0 equiv.), DMAP (6.0 equiv.) and MeCN (0.05 M) for 48 h cAlcohol substrate (1.0 equiv.), corresponding alcohol coupling partner (5.0 equiv.), Cu(OTf)2 (2.5 equiv.), pyridine (3.0 equiv.), MeCN (15.0 equiv.) and CH2Cl2 (0.1 M) for 48 h. dAlcohol substrate (1.0 equiv.), corresponding alcohol coupling partner (8.0 equiv.), Cu(OTf)2 (4.0 equiv.), pyridine (4.8 equiv.), MeCN (24.0 equiv.) and CH2Cl2 (0.1 M) for 48 h. DMAP, 4-dimethylaminopyridine.
We became particularly interested in the case where the trapping nucleophile would be methanol. The product of this reaction would be a positional isomer of the starting alcohol. Overall, one might consider this transformation a novel type of molecular edit in which the order of the atoms in a functional group is exchanged, but the molecular constitution is preserved. Such a transformation would be a functional-group permutation42 that would enable the polarity and hydrogen-bonding ability of a bioactive molecule to be altered with only a minimal perturbation of its shape and steric profile.
Reaction optimization showed that conditions akin to those of the cyclic skeletal editing reaction could be utilized to convert 2-phenylethanol (32) to the isomeric methyl ether (34) in good yield (Fig. 4a, entry 1). The reaction was similarly sensitive to Cu(II) and base identity (entries 2 and 3), and a change in the nitrile additive and its stoichiometry proved to have a beneficial effect on reactivity (entry 4). Control experiments again showed that the reaction does not proceed in the absence of light or Cu(II), which suggested that a similar LMCT mechanism initiates the reaction (entries 5 and 6). Once optimized conditions were developed, we turned our attention to the scope of the reaction. The electronics of the arene radical-stabilizing group had a modest influence on the reaction outcome, with electron-poor (35) and electron-rich (36) arenes providing comparable yields. Aryl bromides (37) and aryl chlorides (38) were tolerated with different substitution patterns on the arene. Secondary (39) and tertiary (40) substitution of the benzylic position was tolerated, as was a thiophene stabilizing group (41). A tetrahydroisoquinoline-derived substrate (42) reacted smoothly in this reaction, as did more complex alcohols derived from flurbiprofen (43) and fenoprofen (44). A switch in metal oxidant from Cu(II) to Fe(III) enabled the high-yielding coupling of amine-stabilized substrates including oxazolidine- (45), pyrrolidine- (46) and morpholine- (47) containing substrates. Finally, benzimidazole product 48 was obtained in moderate yield, showcasing the method’s ability to tolerate N-heterocycles. We imagined that this novel transformation might be most obvious when applied to hydroxymethyl substituents, but the formal positional isomerization of more complex acyclic alcohols is also feasible. For example, secondary alcohol substrates could be trapped with EtOH (49) and n-pentanol (50). Ring-containing benzyl alcohol (51) and cyclohexylmethanol (52) gave similar yields. Cyclic and acyclic tertiary alcohols could also be employed, furnishing oxygen-inserted products (53–56) in good yields.
Conclusions
In summary, we have developed an approach towards skeletal editing through the selective functionalization of saturated carbon–carbon bonds. The LMCT strategy for alcohol activation enables native functionalities commonly found in pharmaceutical lead compounds to be exploited as triggers for formal oxygen atom migration. These reactions occur without the use of strongly oxidizing photoredox catalysts or the application of deeply oxidizing redox potentials, and the tolerance for oxidatively sensitive functional groups is high. This new method thus enables the rapid preparation of cyclic and acyclic ethers from elaborated alcohol substrates and adds important new capability to the growing library of skeletal editing methods. Moreover, this study demonstrates that LMCT activation is ideally poised to address selectivity and reactivity challenges associated with the functionalization of C(sp3)–C(sp3) bonds and could thus provide an alternative entry into the design of new atom-insertion reactions.
Methods
General procedure for the oxygen insertion of cycloalkanols
An oven-dried 8-ml vial equipped with a stir bar was placed in a nitrogen-filled glovebox. The vial was charged with Cu(OTf)2 (181 mg, 0.50 mmol, 2.5 equiv.), iPrCN (89.8 μl, 1.00 mmol, 5.0 equiv.), pyridine (48.3 μl, 0.60 mmol, 3.0 equiv.) and cycloalkanol (0.20 mmol, 1.0 equiv.). The reaction components were diluted with anhydrous CH2Cl2 (2 ml, 0.1 M), and EtOH (35.0 μl, 0.60 mmol, 3.0 equiv.) was added. The vial was capped and taken out of the glovebox, then placed in front of two 427-nm Kessil lamps and a fan for cooling. The reaction was irradiated for 16 h. On completion, the reaction was diluted with EtOAc, and the organic layer was washed with NH4Cl and NaHCO3 before being dried by Na2SO4 and concentrated under reduced pressure. The crude mixture was purified by flash chromatography on silica gel to afford the pure product.
General procedure for the functional-group permutation of acyclic alcohols
An oven-dried 8-ml vial equipped with a stir bar was placed in a nitrogen-filled glovebox and charged with Cu(OTf)2 (271 mg, 2.5 equiv., 0.75 mmol). A solution of the starting material alcohol (1.0 equiv., 0.30 mmol) in anhydrous CH2Cl2 (3.0 ml, 0.1 M) was added, followed by methanol (121 µl, 10 equiv., 3.0 mmol), pyridine (72.8 µl, 3.0 equiv., 0.90 mmol) and acetonitrile (235 µl, 15 equiv., 4.50 mmol). The vial was sealed with a septa cap and removed from the glovebox. The vial was then irradiated on a stir plate between two 427-nm Kessil lamps and a fan for cooling. After 48 h, the crude reaction was diluted with 5 ml Et2O and washed with 4 ml of 1.0 M HCl. The organic layer was filtered through a Na2SO4 and silica plug. The crude mixture was purified by flash chromatography on silica gel to afford the pure product.
Data availability
All data are available in the main text or the Supplementary Information.
References
Davies, H. M. L. & Morton, D. Recent advances in C–H functionalization. J. Org. Chem. 81, 343–350 (2016).
Liang, Y. et al. Carbon–carbon bond cleavage for late-stage functionalization. Chem. Rev. 123, 12313–12370 (2023).
Baeyer, A. & Villiger, V. Einwirkung des Caro’schen Reagens auf Ketone. Ber. Dtsch. Chem. Ges. 32, 3625–3633 (1899).
Beckmann, E. Zur Kenntniss der Isonitrosoverbindungen. Ber. Dtsch. Chem. Ges. 19, 988–993 (1886).
Hock, H. & Lang, S. Autoxydation von Kohlenwasserstoffen, IX. Mitteil.: Über Peroxyde von Benzol-Derivaten. Ber. Dtsch. Chem. Ges. 77, 257–264 (1944).
Joynson, B. W. & Ball, L. T. Skeletal editing: interconversion of arenes and heteroarenes. Helv. Chim. Acta. 106, e202200182 (2023).
Jurczyk, J. et al. Single-atom logic for heterocycle editing. Nat. Synth. 1, 352–364 (2022).
Siddiqi, Z., Wertjes, W. C. & Sarlah, D. Chemical equivalent of arene monooxygenases: dearomative synthesis of arene oxides and oxepines. J. Am. Chem. Soc. 142, 10125–10131 (2020).
Kumar, P. R. Addition of phthalimidonitrene to substituted indoles. Heterocycles 26, 1257–1262 (1987).
Reisenbauer, J. C., Green, O., Franchino, A., Finkelstein, P. & Morandi, B. Late-stage diversification of indole skeletons through nitrogen atom insertion. Science 377, 1104–1109 (2022).
Maeda, K. & Hayashi, T. The formation of triaryl-s-triazine in the chemiluminescence reaction of triarylimidazole and in the photo-oxygenation of triarylimidazole in the presence of ammonia. Bull. Chem. Soc. Jpn 44, 533–536 (1971).
Maeda, K., Mishima, T. & Hayashi, T. The formation of substituted quinazolines from substituted indoles by the sensitized photo-oxygenation in the presence of ammonium acetate. Bull. Chem. Soc. Jpn 47, 334–338 (1974).
Liu, S. & Cheng, X. Insertion of ammonia into alkenes to build aromatic N-heterocycles. Nat. Commun. 13, 425 (2022).
Finkelstein, P. et al. Nitrogen atom insertion into indenes to access isoquinolines. Chem. Sci. 14, 2954–2959 (2023).
Gupta, A., Bhatti, P., Laha, J. K. & Manna, S. Skeletal editing by hypervalent iodine mediated nitrogen insertion. Chem. Eur. J. 30, e202401993 (2024).
Wang, J., Lu, H., He, Y., Jing, C. & Wei, H. Cobalt-catalyzed nitrogen atom insertion in arylcycloalkenes. J. Am. Chem. Soc. 144, 22433–22439 (2022).
Ghosh, B. et al. Sulfenylnitrene-mediated nitrogen-atom insertion for late-stage skeletal editing of N-heterocycles. Science 387, 102–107 (2025).
Sandvoß, A. & Wahl, J. M. From cycloalkanols to heterocycles via nitrogen insertion. Org. Lett. 25, 5795–5799 (2023).
Murai, K., Kobayashi, T., Miyoshi, M. & Fujioka, H. Oxidative rearrangement of secondary amines using hypervalent iodine(III) reagent. Org. Lett. 20, 2333–2337 (2018).
Kelley, B. T., Walters, J. C. & Wengryniuk, S. E. Access to diverse oxygen heterocycles via oxidative rearrangement of benzylic tertiary alcohols. Org. Lett. 18, 1896–1899 (2016).
López, M. M., Jamey, N., Pinet, A., Figadère, B. & Ferrié, L. Oxidative ring expansion of cyclobutanols: access to functionalized 1,2-dioxanes. Org. Lett. 23, 1626–1631 (2021).
Choudhary, N. & Saraf, D. N. Hydrocracking: a review. Product RD 14, 74–83 (1975).
Souillart, L. & Cramer, N. Catalytic C–C bond activations via oxidative addition to transition metals. Chem. Rev. 115, 9410–9464 (2015).
Yayla, H. G., Wang, H., Tarantino, K. T., Orbe, H. S. & Knowles, R. R. Catalytic ring-opening of cyclic alcohols enabled by PCET activation of strong O–H bonds. J. Am. Chem. Soc. 138, 10794–10797 (2016).
Abderrazak, Y., Bhattacharyya, A. & Reiser, O. Visible-light-induced homolysis of Earth-abundant metal–substrate complexes: a complementary activation strategy in photoredox catalysis. Angew. Chem. Int. Ed. 60, 21100–21115 (2021).
Juliá, F. Ligand-to-metal charge transfer (LMCT) photochemistry at 3d-metal complexes: an emerging tool for sustainable organic synthesis. ChemCatChem 14, e202200916 (2022).
Guo, J. et al. Photocatalytic C−C bond cleavage and amination of cycloalkanols by cerium(III) chloride complex. Angew. Chem. Int. Ed. 55, 15319–15322 (2016).
Li, Q. Y. et al. Decarboxylative cross-nucleophile coupling via ligand-to-metal charge transfer photoexcitation of Cu(II) carboxylates. Nat. Chem. 14, 94–99 (2022).
Lutovsky, G. A., Gockel, S. N., Bundesmann, M. W., Bagley, S. W. & Yoon, T. P. Iron-mediated modular decarboxylative cross-nucleophile coupling. Chem 9, 1610–1261 (2023).
Xu, P., López-Rojas, P. & Ritter, T. Radical decarboxylative carbometalation of benzoic acids: a solution to aromatic decarboxylative fluorination. J. Am. Chem. Soc. 143, 5349–5354 (2021).
Liu, W. et al. Single-bond cleavage of alcohols. Org. Lett. 23, 8413–8418 (2021).
Zhao, L. et al. Electrochemical deconstructive and ring-expansion functionalization of unstrained cycloalkanols. Org. Lett. 26, 4882–4886 (2024).
Seabrook, G. R. et al. L-733,060, a novel tachykinin NK1 receptor antagonist; effects in [Ca2+]i mobilisation, cardiovascular and dural extravasation assays. Eur. J. Pharmacol. 317, 129–135 (1996).
Hayes, E. C. et al. Electronic structure of a CuII–alkoxide complex modeling intermediates in copper-catalyzed alcohol oxidations. J. Am. Chem. Soc. 138, 4132–4145 (2016).
Wen, L. et al. Multiplicative enhancement of stereoenrichment by a single catalyst for deracemization of alcohols. Science 382, 458–464 (2023).
Frisch, M. J. et al. Gaussian 16 revision a.03.2016 (Gaussian, 2016).
Kochi, J. K. & Subramanian, R. V. Kinetics of electron-transfer oxidation of alkyl radicals by copper(II) complexes. J. Am. Chem. Soc. 87, 4855–4866 (1965).
Kennedy, S. H., Dherange, B. D., Berger, K. J. & Levin, M. D. Skeletal editing through direct nitrogen deletion of secondary amines. Nature 593, 223–227 (2021).
Yamakoshi, W., Arisawa, M. & Murai, K. Oxidative rearrangement of primary amines using PhI(OAc)2 and Cs2CO3. Org. Lett. 21, 3023–3027 (2019).
Brägger, Y. et al. Oxidative amination by nitrogen atom insertion into carbon–carbon double bonds. Science 387, 1108–1114 (2025).
Harnedy, J., Maashi, H. A., El Gehani, A. A. M. A., Burns, M. & Morrill, L. C. Deconstructive functionalization of unstrained cycloalkanols via electrochemically generated aromatic radical cations. Org. Lett. 25, 1486–1490 (2023).
Roure, B. et al. Photochemical permutation of thiazoles, isothiazoles and other azoles. Nature 637, 860–867 (2025).
Acknowledgements
Funding for this research was provided by the NIH (GM144129) and Pfizer. C.R.L. is the recipient of a postdoctoral fellowship from the NIH (F32 GM145089). F.W. was funded by Cusanuswerk. NMR spectroscopy and MS facilities at UW–Madison are funded by the NIH (S10 OD012245), NSF (CHE-930456) and a generous gift from the Paul J. and Margaret M. Bender Fund. We additionally thank the SynCat Center at UW–Madison for providing access to a parallel photochemical reactor.
Author information
Authors and Affiliations
Contributions
T.P.Y. conceived the concept. C.A.M. discovered and developed the reaction with the cyclic substrates. C.A.M. and F.W. developed the scope of the cyclic substrates. C.R.L. and M.F.M. discovered and developed the reaction with the acyclic substrates. C.R.L. and M.F.M. developed the scope of the acyclic substrates. C.A.M., M.F.M. and D.M.B. conducted the mechanistic experiments and computations. C.A.M. and T.P.Y. wrote the manuscript. S.W.B. contributed substantially to development of the project scope. All authors reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
S.W.B. is an employee and shareholder of Pfizer, Inc. The other authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Joel Cejas-Sánchez, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Experimental details, Sections A–P, Tables 1–11 and Figs 1–15.
Supplementary Data 1 (download XLSX )
Source data for UV–vis studies.
Source data
Source Data Fig. 3 (download XLSX )
Source data for UV–vis titration.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
MacAllister, C.A., Lacker, C.R., Maciejewski, M.F. et al. Oxygen migration into carbon–carbon single bonds by photochemical oxidation. Nat. Synth 5, 349–356 (2026). https://doi.org/10.1038/s44160-025-00937-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44160-025-00937-x
This article is cited by
-
Skeletal editing of C(sp3)–C(sp3) bonds via photoinduced oxidative oxygen migration
Nature Synthesis (2025)
