
Abstract
The discovery of crystalline reticular materials remains largely trial-and-error despite their societal importance. We introduce our algorithmic iterative reticular synthesis (AIRES) cycle, which integrates automated synthesis, image recognition, single-crystal X-ray diffraction and, crucially, customized algorithmic decision-making, to maximize distinct crystal discoveries rather than optimizing single targets. Demonstrated on zeolitic imidazolate frameworks (ZIFs), AIRES achieves twice the discovery rate of random exploration, crystallizing 10 new linkers into diverse ZIF topologies and expanding the single-linker Zn-ZIF library by one-third. By transforming reticular synthesis from an empirical process to a systematic exploration, AIRES provides a scalable and efficient blueprint for accelerating materials discovery.

Similar content being viewed by others

Main
Crystallization of reticular materials, including metal–organic frameworks, covalent organic frameworks and zeolitic imidazolate frameworks (ZIFs), is crucial because it: (1) creates the well-ordered porous architectures that enable important societal applications, such as water harvesting1,2 and CO2 capture3,4, and (2) enables definitive atomic-precise structure determination using single-crystal X-ray diffraction (SCXRD), which provides essential understanding of the structure–property relationships that guide further material development. Yet despite decades of advances in reticular chemistry, crystallizing new frameworks remains largely empirical. Although building blocks can be rationally designed based on geometric principles of reticular synthesis5, determining which combinations will crystallize and under what specific conditions still relies heavily on trial-and-error. This inefficiency leaves vast chemical space unexplored. Beyond missing potentially transformative materials, this gap limits fundamental understanding of crystallization landscapes, hindering human intuition and the development of targeted machine-learning (ML) prediction models. Systematically exploring this space to maximize discoveries within limited experimental resources presents a fundamental challenge distinct from synthesis optimization. Instead of refining conditions for one target, discovery requires efficiently allocating experiments across many candidates, immediately pivoting from success to new possibilities. This creates a unique objective: maximizing the number of distinct crystals discovered, not success rates or material properties.
Given the scale and complexity of this challenge, computational and ML-driven methods offer great opportunity because they can process vast data and identify patterns beyond human capability. However, current implementations of these approaches in chemistry and materials science, while powerful for their intended purposes, are mismatched to this discovery objective. For instance, some approaches fit ML models to existing high-throughput or lab notebook data for classifying materials, yet make limited efforts to demonstrate how these models can extensively explore new systems or adapt to new conditions6,7,8. Bayesian optimization and genetic algorithms excel at finding optimal conditions but concentrate resources on single targets9,10,11,12,13. Experimental screening of computationally predicted or ML-generated structures treats each candidate independently, missing cross-system learning opportunities and failing to adapt when predictions prove incorrect14,15,16. The discovery challenge demands a fundamentally different approach: one that simultaneously learns crystallization patterns across diverse chemical systems and strategically allocates experimental resources based on this evolving understanding.
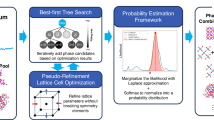
Here we present AIRES (algorithmic iterative reticular synthesis), an integrated cycle that transforms crystal discovery from empirical exploration to systematic decision-making focused on maximizing the number of distinct new structures found. AIRES combines automated synthesis, computer vision for crystal identification, and SCXRD, with the critical addition of an algorithmic framework that decides subsequent experiments (Fig. 1). AIRES uses probabilistic ML models trained on carefully designed chemical descriptors to predict crystallization outcomes across diverse systems, enabling cross-system learning where patterns from one chemical family accelerate discovery in others. These predictions feed a theoretically grounded selection strategy: we prove that for discovery objectives, greedy selection of highest-probability candidates outperforms exploration-heavy strategies optimal for property optimization. AIRES dynamically allocates resources, immediately excluding successful building blocks to focus on undiscovered ones, while continuously learning from both successes and failures to refine predictions across the chemical space. To implement this strategy in high-throughput experimentation, we developed a principled batch selection algorithm that identifies experiment sets maximizing expected discoveries while accounting for conditional dependencies. This prevents the severe redundancy that arises from simply selecting the top-k candidates, which often share similar features and provide limited new information.

a, Automated synthesis set-up showing a robotic liquid handler (left) and heating unit (right). b, Microscope imaging station (top) and optical image collected (bottom) with computer vision classification results (blue, crystal probability; green, non-crystal probability). c, SCXRD set-up. d, A two-dimensional demonstration of binary reaction outcome maps for different linker systems, with successful reactions in dark blue and failed ones in white. e, A two-dimensional demonstration of ML-predicted crystallization probability maps for different linker systems, with higher predicted probabilities in darker red. The decision-making algorithm, which aims to maximize the number of distinct discoveries, suggests candidates based on predictions for subsequent experimental validation.
We demonstrated AIRES on ZIFs, discovering ZIF crystals made of 10 out of 48 new linkers with twice the efficiency of random exploration (700 versus 1,400 experiments). This expanded the single-linker Zn-ZIF library by one-third, breaking a decade-long discovery drought. ZIFs present unique challenges: (1) although ZIFs share common tetrahedral building block geometry, linker–linker interactions control adjacent building block conformations, making it challenging to create isoreticular structures and to predict which structure will actually form because multiple arrangements can be thermodynamically stable17; (2) ZIFs’ single-point metal junctions create competing synthesis pathways between four- and six-coordination states, with solvents and counterions competing for metal sites to form unwanted side products. This combination—structural diversity emerging from subtle functionality variations coupled with complex characterization requirements—establishes ZIFs as an ideal yet rigorous system for AIRES. The platform’s success is particularly noteworthy when contrasted with carboxylate metal–organic frameworks or covalent organic frameworks, in which functionality variations typically produce isoreticular structures (new compositions with identical topologies), resulting in more predictable crystallization landscapes that are inherently easier to navigate. That AIRES could uncover new discoveries in this mature ZIF field, where over a hundred known structures had suggested exhausted possibilities18, demonstrates its remarkable ability to reveal hidden opportunities in ostensibly well-explored chemical spaces. AIRES establishes a blueprint for making reticular synthesis more systematic and efficient, with broader implications for accelerating materials discovery in general.
Results
Curation of initial ZIF synthesis dataset
Here we describe the AIRES component where the dataset was collected and annotated. Specifically, reactions were carried out in a robotic platform, and the products underwent optical imaging for crystal identification, followed by SCXRD analysis and data labelling (Fig. 1a–d).
ZIF single crystals are typically synthesized by solvothermal reactions in which metal salts and linkers are heated in a sealed solvent container. The reaction outcome depends in a complex manner on both the chemicals used and the reaction conditions. While literature reports typically document only successful synthesis conditions for each ZIF, the reality is that successful conditions exist as regions within a broader space of failed attempts. This limited and biased nature of published data necessitates generating a more comprehensive dataset for ML model training6,19. To address this, we first built an initial ZIF synthesis dataset by conducting high-throughput experiments using Zn(II) and linkers from previously reported ZIF structures.
In this study, we focused on Zn-ZIFs constructed from one kind of linker. Despite having 16 different linkers yielding over 30 structures, this category has seen no new additions in nearly a decade. From this limited pool, we selected eight starting linkers (Fig. 2a)—imidazole (IM), 2-methylimidazole (2mIM), 2-nitroimidazole (2nIM), 4,5-dichloroimidazole (45dcIM), benzimidazole (bIM), 9-purine (9pur), 5-chlorobenzimidazole (5cbIM) and 5-methyl-4-imidazolecarboxaldehyde (4ad5mIM)—featuring different functional groups at distinct positions on the imidazole ring. Each linker was reacted with zinc nitrate hexahydrate in N,N-dimethylformamide (DMF) under various reaction conditions grid-sampled from a predetermined condition space (Supplementary Section 2). We identified three primary reaction parameters: total concentration, log linker-to-metal ratio and reaction temperature. By sampling six, eight and seven values, respectively, from these parameters (while holding others, such as time and volume, constant), we generated 336 reactions per linker, totalling 2,688 reactions for our initial dataset.

a, Chemical structures and abbreviations of eight starting linkers: IM, 2mIM, 2nIM, 45dcIM, bIM, 9pur, 5cbIM and 4ad5mIM. Abbreviations denote neutral linkers when referring to reactants, but represent their deprotonated forms in ZIF chemical formulae unless specified otherwise. b, Representative optical images showing different reaction outcomes: clear solution, precipitate, non-ZIF crystals (red frames, failures) and ZIF single crystals (blue frame, success). Scale bars, 100 μm. Crystal structures represented by ball-and-stick models (right) show Zn5(bIM)6(HCOO)4(DMF)2 (b-axis view) and Zn(bIM)2 (c-axis view) with zinc polyhedra in pink. c, Success rate landscapes for each starting linker as a function of temperature and log linker-to-metal ratio, averaged over total concentration. Increasing shades of red indicate higher success rates.
Our high-throughput experimentation platform operates in two stages: reaction/crystallization and characterization. A robotic liquid-handling system prepares reaction mixtures in well plates, which are sealed and heated in isothermal ovens for 5 days. Initial screening uses automated optical microscopy, which reveals three outcomes: clear solutions, precipitates or single crystals. Crystal identification relies on multiple criteria: geometric shape, colour homogeneity and sufficient size (∼20 μm in the shortest dimension). The process began with expert researchers providing binary classifications (crystals versus non-crystals, where non-crystals encompassed both clear solutions and precipitates) for the optical images. We then used the EfficientNetV2-S convolutional neural network20, training it on these labelled images. The model achieved a 0.88 recall rate for crystal detection (the proportion of actual images containing crystals correctly identified by the model), demonstrating high reliability (Supplementary Fig. 26). Although the model streamlines screening by filtering out uninteresting images, human experts still validate model-identified crystal candidates for SCXRD analysis. This automated prescreening substantially reduces the time researchers spend reviewing optical images, given that most do not contain viable crystals. The importance of SCXRD analysis for finalizing the reaction outcome is illustrated by the bIM linker case, where two distinct crystal morphologies emerged: rhombic dodecahedra and non-merohedral twinned plates (Fig. 2b). SCXRD analysis revealed the former as the known ZIF-7, Zn(bIm)2, while the latter was identified as a second new crystalline product with a two-dimensional structure Zn5(bIm)6(HCOO)4(DMF)2.
To integrate SCXRD into high-throughput experimentation efficiently, we adopted two practical assumptions. First, morphologically similar crystals from the same linker are presumed to share identical crystal structures, because ZIF macroscopic symmetry probably reflects microscopic arrangement. Second, crystals matching unit cell parameters of known ZIF structures are classified as ZIFs. This approach allows rapid screening by measuring only unit cell parameters, bypassing full structure determinations. The reaction outcomes were ultimately assigned binary labels: ‘1’ for successful ZIF single-crystal formation and ‘0’ for all other outcomes.
To visualize the complex relationships between reaction conditions and outcomes, we projected the binary results onto two-dimensional parameter spaces (Fig. 2c and Supplementary Figs. 18 and 19). These visualizations reveal non-monotonic relationships interpreted from two aspects. First, classical nucleation theory dictates that optimal crystal growth requires carefully balancing external (temperature, concentration) and intrinsic (supersaturation, viscosity) parameters to control kinetics and yield large crystals for SCXRD21. Second, coordination chemistry influences ZIF formation because key intermediate species—zinc cations coordinated by two or three linkers—form extended structures via unsaturated metal centres or labile groups22. The projections show that each of the eight linkers exhibits complex, distinct crystallization regions. These observations reveal two insights. First, with a 25% ± 16% success rate, random search for crystallization is inefficient, especially for challenging ZIF linkers. Second, the distinct and complex crystallization regions defy simple patterns, highlighting the necessity for ML approaches to capture these intricate relationships.
ML-guided discovery methodology
Our discovery methodology integrates ML prediction models with automated experimentation in an iterative cycle (Fig. 1). Each iteration involves three steps: (1) model-guided selection of promising candidates, (2) automated synthesis and characterization and (3) incorporation of new results to refine future predictions. This process continues until the experimental budget (designated amount of chemicals, instrument time and researcher availability) is exhausted or all viable linker candidates have been explored.
To evaluate our ML-guided discovery methodology, we chose a space of new linkers. The selection was largely unbiased, with only two criteria applied: (1) linkers must have good solubility in DMF at ambient temperature, due to robotic liquid handler constraints; (2) linkers with coordinating functionalities (for example, hydroxyl and amino groups) were excluded to prevent competition with imidazole nitrogens for metal coordination, thus avoiding undesirable side reactions. Following these criteria, we selected 48 new linkers, split evenly between imidazole and benzimidazole cores (Extended Data Fig. 1). Among these, 2-bromoimidazole (2BrIM), 2-cyanoimidazole (2cyIM) and 2-propylimidazole (2pIM) were previously reported in Zn-ZIF synthesis using alkyl-alcohols instead of the DMF used in this study23,24. The remaining 45 linkers have not been independently synthesized into ZIF single crystals.
For the ML models to effectively learn chemical patterns, we encoded the linker structures and reaction conditions into quantitative descriptors. Our linker encoding strategy encompasses four key perspectives (Fig. 3a): (1) functional group counts, (2) linker dimensions (to account for steric effects), (3) quantum mechanical properties of both neutral and deprotonated linkers and (4) quantum mechanical properties of a crucial prenucleation species—a monomer in which Zn(II) is coordinated by two neutral linkers and two nitrates. Principal component analysis of these features revealed some clustering between imidazole- and benzimidazole-based linkers (Fig. 3b). However, the relatively low explained variance (50.8% captured by the first two principal components) highlights the inherent complexity of the chemical space, which simple linear combinations of features cannot capture (Supplementary Fig. 28).

a, Linker features used for ML models, combining functional group counts (five features, blue), linker dimensions (four features, magenta), and quantum mechanical (QM) properties of neutral/deprotonated linkers (five features, yellow) and prenucleation species (seven features, green). b, Two-dimensional principal component analysis projection of the chemical space, showing the distribution of 8 starting linkers (red diamonds), 24 new IM-based (grey triangles) and 24 new bIM-based linkers (purple circles). c, Reward (number of distinct discoveries) of AIRES equipped with RFC (red) and GPC (magenta), and random search baseline (RS, grey) under the sequential setting. d, Reward of AIRES (RFC) (red) and RS (grey) under batch experimentation with 24 experiments per iteration. In c and d, solid lines represent the mean, and shaded areas represent the range of ±1 s.d. (16 repetitions for RFC/GPC; 100 repetitions for RS). The red dashed line indicates the maximum achievable reward. e, Kernel density estimation showing the distribution of the number of experiments required to discover new ZIF crystals for 10 successful linkers using AIRES (RFC) (red) versus RS (grey). The linkers analysed are: 2cyIM, 2pIM, 5-bromoimidazole (5BrIM), 4-cyanoimidazole (4cyIM), 4-methyl-5-nitroimidazole (4m5nIM), 4-bromo-5-methylimidazole (4Br5mIM), 6-fluorobenzimidazole (6fbIM), 6-methoxybenzimidazole (6mhbIM), 5-bromobenzimidazole (5BrbIM) and 6-methylbenzimidazole (6mbIM). Horizontal bars beneath each distribution indicate individual discovery events across multiple repetitions.
Our methodology treats crystallization as a binary outcome (success or failure) drawn from an underlying probability distribution reflecting inherent chemical variability and experimental precision. This approach combines two key components: a prediction model to estimate crystallization probabilities, and a greedy selection strategy that always chooses the experiment with the highest predicted probability of yielding new ZIF crystals. This strategy is model agnostic, working with any probabilistic prediction model. We explored two specific models: random forest classification (RFC)25 and Gaussian process classification (GPC)26,27. The RFC captures complex, non-linear relationships through an ensemble of decision trees. However, a key challenge arose: each initial training linkers had more than 300 data points, while new linkers were explored one experiment at a time. This data imbalance, combined with inherent differences between training and new linkers, could cause the model to overlook crucial patterns in new data. We addressed this through strategic data augmentation and dynamic hyperparameter optimization that increased the weight of new experimental results. Separately, the GPC provides naturally calibrated probability estimates and we adopt a composite product kernel that captures different patterns of variation in linker features versus reaction conditions. Although neural network-based prediction models could be incorporated into our decision-making strategy, our dataset characteristics made simpler models more appropriate: with only eight training linkers (hence eight data points for the linker representation), we had insufficient data to effectively train neural networks. We therefore selected RFC and GPC for their uncertainty quantification properties and ability to perform well with limited data.
Building on these predictive models, we developed a greedy selection strategy that prioritizes experiments with the highest predicted probability of success. Critically, our objective is explicitly defined as maximizing the number of unique successful crystallizations, not identifying optimal reaction conditions. Once a linker yields a ZIF crystal, the algorithm excludes further experiments with that linker, focusing resources on unexplored candidates. This objective fundamentally differs from traditional optimization formulations such as Bayesian optimization28 or multiarmed bandits29, which aim to maximize success probability and cumulative (non-unique) successes. Although such traditional methodologies often favour exploration over exploitation, we demonstrated through both theoretical analysis (Supplementary Section 8) and empirical validation (Supplementary Section 9) that our greedy strategy of selecting the highest-probability candidates is particularly well suited for crystal discovery, where each success represents a unique scientific advance. Furthermore, to fully utilize high-throughput capabilities, we extended this methodology to batch experimentation, developing a novel principled batch selection algorithm. In each iteration, this strategy works with the trained ML model to identify the set of experiments that collectively maximize the expected number of discoveries, accounting for potential interactions between experiments targeting the same linker through conditional probability estimation.
To rigorously compare our ML methodology with random exploration, multiple trials are necessary to account for inherent randomness. Because conducting repeated reactions is resource intensive, we adopted an equivalent validation strategy: we first experimentally executed a comprehensive grid of reaction conditions across all 48 new linkers, generating a dataset of 1,728 reactions. This allowed us to simulate both ML-guided and random exploration by iteratively suggesting experiments and retrieving the corresponding outcomes from this new dataset, rather than repeatedly conducting experiments. Different approaches revealed distinct discovery patterns (Fig. 3c). Random search (RS) required around 1,400 experiments to discover all new ZIF crystals from the 10 successful linkers, showing a steadily diminishing discovery rate characteristic of exhaustive exploration. The GPC approach showed promising early performance with rapid discoveries, but its effectiveness plateaued at levels comparable to RS, probably reflecting the challenge of extrapolating from the initial dataset to substantially different linkers. RFC emerged as the most effective approach, discovering all structures in approximately 700 experiments, a twofold acceleration compared to RS. The batch implementation of our methodology maintained the efficiency of RFC, consistently requiring approximately half the RS experiments to achieve the same number of discoveries (Fig. 3d), an advantage robust across various batch sizes (Supplementary Fig. 46). This is a non-trivial achievement because simply selecting the top-k most promising candidates would introduce severe redundancy, resulting in discovery performance substantially worse than batch RS (Supplementary Fig. 47). This demonstrates that our batch selection strategy, which accounts for dependencies between related experiments through conditional probability estimation, successfully extends the advantages of ML-guided discovery to high-throughput settings.
Analysis of distributions of experiments needed for discovery for each successful linker provides insights into both the chemical space and our algorithmic approach (Fig. 3e). For linkers such as 2cyIM, both RS and RFC achieved early discovery of the corresponding ZIFs, although the latter still provided modest acceleration. This suggests robust crystallization conditions that are relatively insensitive to precise reaction parameters. In contrast, more challenging targets such as 6mbIM showed dramatically different trajectories: while RS discovered these structures at a roughly uniform rate through blind exploration, RFC greatly accelerated their discovery through pattern recognition. Interestingly, 2pIM proved an exception where RS outperformed RFC. Despite 2pIM having moderate overall success rates (explaining RS’s earlier discovery), its unique structural features and resulting ZIF crystal topology differed substantially from other positive examples in our training data. These unique characteristics caused our greedy algorithm to prioritize other candidates with more familiar patterns, rather than selecting based on intrinsic success rates that RS relies upon. Nevertheless, overall AIRES (RFC) clearly outperformed RS, leading to a global acceleration of new ZIF discovery.
Further investigation analysing the AIRES (RFC) discovery trajectory (Supplementary Fig. 35) reveals that the model effectively evaluates ZIF crystallization tendency, prioritizing promising candidates. This is evidenced by the non-uniform frequency of suggested linkers, a clear departure from RS. This prioritization is rooted in the model’s ability to learn structural features; for example, it suggests linkers with bulky substituents (at the 2 and 4/5 positions) less often due to steric hindrance around the metal centre. A deeper analysis of predicted crystallization landscapes reveals two distinct model behaviours (Supplementary Figs. 36–45): AIRES (RFC) quickly provided a reliable landscape estimate for some linkers (for example, 2cyIM, 4m5nIM), leading to early discovery. Conversely, for challenging linkers (for example, 6mhbIM, 6mbIM), the model struggled to generate a reliable landscape (persistent flatness) but still identified them as promising, allocating more resources and accelerating their discovery compared with RS.
Description of ZIFs discovered
Despite their common tetrahedral building block geometry, ZIFs display diverse network topologies, denoted by three-letter codes. Through AIRES, 10 successful linkers yielded 11 new crystals belonging to 7 distinct topologies (Fig. 4a). SOD (ZIF-A1) and ANA (ZIF-A4) topologies, obtained from 2cyIM and 2pIM respectively, match structures originally discovered in alkyl-alcohol solvents, contrasting with the DMF used here. Additional SOD structures (ZIF-A2, ZIF-A3) emerged from 4m5nIM and 6fbIM. 4cyIM generated a DFT topology structure (ZIF-A7). 4m5BrIM formed an RHO topology structure (ZIF-A8). Structures of the ykh topology (ZIF-A9, ZIF-A10, ZIF-A11) were produced by 6mhbIM, 5BrbIM and 6mbIM, previously accessible only through mixed-linker approaches30. Structural versatility was observed with 5BrIM, yielding a crb topology structure (ZIF-A5) and an unprecedented double fes topology structure (ZIF-A6). ZIF-A6 was discovered earlier than ZIF-A5 in one-sixth of attempts by AIRES (RFC), in line with their relative success rates (Supplementary Fig. 34). While a reported layered ZIF featured metal nodes in a single atomic layer31, ZIF-A6 exhibits a new double-thick-layer arrangement in which two layers of metal nodes are covalently bridged by 5BrIM, representing a new structural motif in ZIF chemistry. Constructing isoreticular structures for ZIFs is challenging because minor functional group modifications can trigger alternative frameworks by altering linker–linker interactions (for example, ZIF-7 (bIM) versus ZIF-20 (9pur)) or by introducing critical steric constraints (for example, ZIF-8 (2mIM) versus ZIF-A4 (2pIM)). Therefore, we highlight the importance of ZIF-A5 (5BrIM, crb topology) and ZIF-A7 (4cyIM, DFT topology) because these topologies were previously achieved only by unsubstituted IM linkers (ZIF-1, ZIF-2, ZIF-3). The strategic use of linkers with single functionality at the 4/5 position of the imidazole backbone creates new perspectives in ZIF structure design. Overall, AIRES’s systematic exploration expanded the known single-linker Zn-ZIF library developed over the last 20 years by one-third, breaking a decade-long discovery plateau and demonstrating untapped potential in this seemingly well-explored chemical space.

a, Crystal structures of ZIFs (ZIF-A1 to ZIF-A11) are shown and classified based on network topologies. ZIF topologies are visualized using natural tiling models and labelled with their respective three-letter topological codes, where uppercase and lowercase respectively indicate topologies known in zeolites and only in ZIFs, respectively. Disordered positions of functional groups in crystal structure are preserved in visualization. b, Crystal structures of LZIFs (LZIF-A1 to LZIF-A4) are shown and classified based on network topologies. LZIF topologies are visualized using two-dimensional tiling models.
The rich coordination chemistry of ZIF synthesis led to unexpected discoveries beyond the target structure motif. RFC also identified four new linkers (2cbIM, 2mbIM, 2m5fbIM, 2m5cbIM) that produced other types of crystals before discovering all ZIF crystals (Supplementary Section 15). Although these structures maintain tetrahedral zinc coordination, they exhibit competitive binding between linkers and auxiliary ligands at the metal centres. This competition results in layered-ZIFs (LZIFs) rather than three-dimensional frameworks (Fig. 4b). In LZIF-A1 (from 2cbIM) and LZIF-A2 (from 2mbIM), DMF and nitrate compete for coordination sites, whereas in LZIF-A3 (from 2m5fbIM) and LZIF-A4 (from 2m5cbIM), the competition comes from dimethylcarbamate, which forms through DMF oxidation. Although the metal centres deviate from ZIF-characteristic coordination, the metal–linker coordination bonds are preserved, implying that ML captured fundamental coordination principles.
Outlook
AIRES demonstrates considerable potential beyond single-linker Zn-ZIFs. The system is readily extendable to multilinker ZIFs, which require careful data labelling to capture diverse outcomes (single-linker or mixed-linker structures). Beyond ZIFs, AIRES can address crystallization challenges in other reticular or broader crystalline systems. Our core ML methodology—the greedy acquisition function—is broadly transferable, although reaction encoding requires domain-specific customization. As complexity increases, enhanced characterization is essential; supplementing SCXRD with multimodal techniques such as powder X-ray diffraction (PXRD) and Fourier transform infrared spectroscopy enables comprehensive analysis and maintains rigour. AIRES holds a unique position in the broader discovery ecosystem. First, its validated structures provide valuable data for building ML models that predict structure–property relationships. Second, AIRES outputs serve as ‘structural anchors’ for downstream, scalable materials development. For example, the initial conditions identified by AIRES, along with the simulated reference PXRD pattern from the solved structure, facilitate two critical scale-up functions: phase identification and purity assessment. Once verified, quantitative PXRD analysis (for example, peak width) yields a crystallinity index. This metric can then drive a Bayesian optimization cycle to systematically refine conditions for enhancing crystallinity and reducing impurities, thus creating a seamless connection between discovery and optimization.
Methods
Automated synthesis
The synthesis was automated using an Opentrons OT-2 robot equipped with dual pipettes: a P20 (1–20 μl) and a P300 (20–300 μl). The robot’s 11-slot platform was configured with two tip racks, a 96-well plate, a waste collection tube rack and three custom 3D-printed holders, each accommodating six 20-ml vials containing stock solutions. Stock solutions of metal salts and linkers were prepared at two concentrations (0.15 and 0.50 mol l−1), with pure DMF available for dilution. The system was controlled through the Opentrons app by Jupyter notebook, using custom Python scripts and JSON files defining labware specifications.
For the eight starting linkers, we sampled a reaction condition space comprehensively using six values of total concentration (0.05, 0.10, 0.15, 0.25, 0.35 and 0.50 mol l−1), eight values of log linker-to-metal ratio (−0.30, 0.00, 0.30, 0.48, 0.70, 0.90, 1.00 and 1.18), and seven values of temperature (65, 85, 100, 110, 120, 130 and 140 °C). The boundary of this reaction condition space was determined as described in Supplementary Section 2. For the 48 new linkers, considering their higher cost and our understanding of parameter sensitivity gained from initial dataset, we sampled fewer conditions in the same reaction condition space: two values of total concentration (0.15 and 0.40 mol l−1), six values of log linker-to-metal ratio (0.00, 0.25, 0.50, 0.75, 1.00 and 1.25), and three values of temperature (70, 100 and 130 °C). This sampling strategy resulted in 336 reactions per starting linker and 36 candidate reactions per new linker.
To execute these reactions using the robotic system, the sampled reaction parameters need to be converted into specific volumes of solutions. As mentioned, the robotic system used three components: a stock solution of metal salt (concentration cmetal_stock), a stock solution of linker (concentration clinker_stock) and pure solvent. Given target parameters of total concentration (ctotal) and log linker-to-metal ratio (log LMR), and a fixed total volume (Vtotal), the required volumes of each component can be calculated using equations (1)–(3).
The reactions were conducted in a TOPAS polymer 96-well plate with 0.5-ml borosilicate glass inserts purchased from Chemglass Life Sciences. A CSV file read by the Python script guided dispensing volumes for each well, combining metal salt solution, linker solution and DMF as needed. While maintaining a constant 300-μl total volume, we varied total concentrations and linker-to-metal ratios across the reactions on a plate. To prevent cross-contamination, pipettes discarded and reloaded fresh tips between different stock solutions. The complete plate preparation took approximately 30 min. Following automated dispensing, the plate was sealed with PTFE/silicone stoppers and secured between steel bars using aluminium clamps before heating in isothermal ovens at the sampled temperature for that plate.
Post-reaction, the cooled plate was unsealed and returned to the robot for supernatant removal. A separate Python script controlled the pipette to aspirate solution 3 mm above the bottom of each well, leaving only a thin liquid layer and solid products for imaging.
Optical imaging and crystal recognition model
Optical image acquisition was performed using a HRX-01 digital microscope (Hirox-USA), featuring a multi-illumination high-resolution auto turret zoom lens and a motorized 100 mm × 100 mm automated xy stage. Images were captured in transmission mode, with light passing through the well plate and samples from below the xy stage. For each well, we captured five images: one at 50× magnification centred on the well, and four at 400× magnification focusing on each quadrant (top left, top right, bottom left, bottom right). The microscope automatically determined optimal focus by scanning a predefined z-axis range to maximize contrast-based profiles. Well coordinates were manually measured and stored in a CSV file for consistent microscope positioning across imaging sessions.
A crystal recognition model to identify suitable single crystals for SCXRD analysis was developed using 20,365 images collected from reactions of eight starting linkers. The image dataset, comprising 7,184 crystal images and 13,181 non-crystal images, was manually labelled by one crystallographer and verified by another. Images were processed from their original resolution of 2,040 × 1,530 × 3 to 512 × 512 × 3 for model input. The image dataset was randomly split into 18,327 training images and 2,038 test images, with representative examples shown in Supplementary Figs. 23 and 24.
The model architecture utilized EfficientNetV2-S implemented in PyTorch32. We used transfer learning with ImageNet-1K pretrained weights to leverage established feature extraction capabilities. The model’s output layer was configured for binary classification to distinguish between crystal and non-crystal images. The network comprises 20.18M trainable parameters with a computational cost of 14.87G multiply–add operations. We fine-tuned the model using the ADAM optimizer33 with a batch size of 64 under the cross-entropy loss. All layers remained trainable during fine-tuning to allow the network to adapt to the specific visual features of single crystals. To balance the dataset, we applied oversampling to crystal images only through data augmentation techniques including flipping images, applying CLAHE contrast enhancement, introducing defocus and motion blur, adjusting brightness and contrast randomly, and shifting RGB values. Training was conducted for 300 epochs with a learning rate of 0.001 and a weight decay of 0.0001. The best model that maximized the validation accuracy was evaluated on the test set. Training and validation loss curves are presented in Supplementary Fig. 25, and the confusion matrix on the test set is shown in Supplementary Fig. 26.
In the stage of exploring new linker systems, where the trained recognition model is used to predict outcomes from optical images, candidates with higher predicted probabilities were prioritized for human examination and subsequent SCXRD characterization. This approach enabled the earlier verification of new discoveries by focusing human effort on the most promising candidates.
Reaction encoding
Reaction data were encoded by concatenating linker features with experimental features into comprehensive feature vectors. The experimental features included three primary reaction parameters plus metal and linker concentrations, with the latter two derived from total concentration and log linker-to-metal ratio. The linker features were computed through two sets of DFT calculations in Gaussian 16 (ref. 34). For linkers, geometry optimization was performed for both neutral and deprotonated forms using the B3LYP exchange-correlation functional with the 6-31g* basis set, accounting for DMF solvent effects through the polarizable continuum model35. From these calculations, we extracted linker dimensions (using imidazolate nitrogen coordinates as reference points) and electronic properties, including the energy difference between neutral and deprotonated forms and atomic electrostatic potentials. To represent the prenucleation species, additional DFT calculations were performed using the M06-L functional with the def2-SVP basis set. These calculations examined clusters containing one zinc centre coordinated by two neutral linkers and two nitrates. For each linker type, up to 16 different clusters with varying linker orientations were optimized. Single-point energy calculations using the SMD implicit continuum solvent model36 identified the most thermodynamically stable configurations, from which we extracted formation energies, structural parameters, and atomic polar tensor charges on zinc and nitrogen atoms. A complete list of features and their meanings is provided in Supplementary Section 7.
Decision-making methodology
Our discovery methodology uses the greedy principle: always choose the candidate reaction most likely to yield a new crystal structure. We developed greedy methods for both individual reaction and parallel batches, supported by ML models that predict crystallization success.
Sequential algorithm
In the sequential discovery process, candidates were selected individually, with each subsequent choice informed by previous outcomes. Our algorithm used a greedy strategy compatible with any probabilistic prediction model. At each iteration, the algorithm selected the candidate with the highest predicted probability of success. Upon successful crystallization, all remaining candidates for that linker were removed from consideration to focus resources on unexplored candidates. The model was retrained after each iteration to incorporate new reaction data.
The sequential greedy algorithm operates as follows:
-
1.
Initialization phase
-
Construct initial model \({{\mathcal{M}}}\) using training data \({{{\mathcal{D}}}}_{{{\rm{train}}}}\)
-
Define experiment space \({{\mathcal{X}}}\) containing all possible reaction candidates
-
-
2.
Discovery cycle (repeated until budget exhaustion)
-
Selection: choose experiment xt maximizing predicted success probability from \({{\mathcal{M}}}\)
-
Execution: conduct experiment and record outcome yt
-
Update:
-
Incorporate new datapoint (xt, yt) into \({{{\mathcal{D}}}}_{{{\rm{train}}}}\)
-
For successful outcomes (yt = 1), remove all remaining experiments with the same linker from \({{\mathcal{X}}}\)
-
Retrain model \({{\mathcal{M}}}\) using updated dataset
-
-
The greedy approach emphasizes exploitation (selecting promising experiments) over exploration (selecting uncertain experiments), departing from conventional strategies in Bayesian optimization and multiarmed bandits that typically balance both aspects. While greedy methods can potentially become fixated on initially promising candidates in traditional optimization settings, our distinct objective—maximizing the number of unique discoveries—naturally avoids this limitation. Upon discovering a successful crystallization for a linker, the algorithm automatically moves on to explore other candidates. Conversely, each negative result decreases the predicted success probability for the corresponding linker, naturally shifting focus to more promising alternatives. Indeed, theoretical analysis demonstrated the optimality of the greedy method under conditions where the posterior knowledge of each linker is independent (Supplementary Section 8).
Although the sequential greedy algorithm required model retraining at each iteration, the computational costs remained manageable in our implementation. While online variants of the prediction models could offer a trade-off between computational efficiency and statistical performance, we maintained the full retraining approach.
Batch algorithm
The batch algorithm extended our greedy methodology to select groups of size NB that collectively maximize the expected number of new discoveries. Although finding the globally optimal batch would be computationally intractable due to combinatorial complexity, we developed an efficient sequential batch construction method that preserved the greedy principle.
For each batch, candidates were selected iteratively by computing their marginal contribution to the batch’s expected reward. This approach accounted for dependencies between candidates within the same batch, particularly when multiple candidates targeted the same linker. The algorithm is the following.
-
1.
Initialization phase
-
Input: training data \({{{\mathcal{D}}}}_{{{\rm{train}}}}\) and batch size NB
-
Train initial model \({{\mathcal{M}}}\) on \({{{\mathcal{D}}}}_{{{\rm{train}}}}\)
-
Define experiment space \({{\mathcal{X}}}\)
-
-
2.
Batch construction cycle (repeated until budget exhaustion)
-
Initialize: create empty batch \({{{\mathcal{B}}}}_{t}=\{\}\)
-
Batch assembly: for b = 1 to NB:
-
Calculate expected reward R(x) for all remaining candidates \(x\in {{\mathcal{X}}}\)
-
Select candidate \({x}_{t}^{b}\) maximizing R(x)
-
Add selected candidate to batch: \({{{\mathcal{B}}}}_{t}\leftarrow {{{\mathcal{B}}}}_{t}\cup \{{x}_{t}^{b}\}\)
-
Update reward function R using equation (4)
-
Remove selected candidate from consideration: \({{\mathcal{X}}}\leftarrow {{\mathcal{X}}}\setminus \{{x}_{t}^{b}\}\)
-
-
Execution and update:
-
Run all experiments in batch \({{{\mathcal{B}}}}_{t}\) and observe outcomes \(\{{y}_{t}^{b}\}\)
-
Add new data to training set: \({{{\mathcal{D}}}}_{{{\rm{train}}}}\leftarrow {{{\mathcal{D}}}}_{{{\rm{train}}}}\cup {\{({x}_{t}^{b},{y}_{t}^{b})\}}_{b = 1}^{{N}_{B}}\)
-
Retrain model \({{\mathcal{M}}}\) on updated dataset
-
-
The key challenge in batch selection is accurately estimating the marginal reward of adding a new reaction outcome to the current batch. For any candidate reaction xnew = (xl, xe), where xl denotes the linker feature and xe the experimental feature, we compute the marginal reward R(xnew) as:
where \({{{\mathcal{B}}}}_{t}^{l}\) represents the set of already-selected candidates in the current batch using the same linker l, and Y(x) denotes the random outcome of candidate x. Hence, estimating the expected reward reduces to estimating two probabilities: a conditional probability and a joint probability. The joint probability can be computed using the chain rule, decomposing it into a product of conditional probabilities that can be estimated individually:
To estimate the conditional probabilities, we can temporarily augment the training set with virtual observations of failures for previously selected candidates \({{{\mathscr{B}}}}_{t}^{l}\) and then retrain the model. While such estimation is exact for pure probabilistic models such as the GPC, it serves as a practical heuristic for other prediction models such as the RFC.
The computational cost remained similar to the sequential approach. Within each batch, selecting a candidate required updating the expected reward contributions for other candidates under the same linker. This update involved refitting the model with the training set augmented by virtual labels to estimate conditional probabilities. By storing these conditional probabilities, we efficiently computed the joint probabilities. The total number of model fits equalled the total suggested experiments, matching the sequential case.
Our batch algorithm naturally accommodated practical experimental constraints, such as the requirement for all reactions in a batch to share the same temperature due to equipment limitations. While the underlying greedy principle remained unchanged, we adapted the algorithm by restricting the search space during batch construction. For example, to handle temperature constraints imposed by our experimental set-up (in which multiple reactions in a well plate shared an oven temperature), we evaluated the algorithm’s selected batch across a discrete set of temperatures and selected the configuration with the largest expected reward.
Prediction models
While our methodology required probabilistic estimates of crystallization success, it remained model agnostic. We implemented and compared two distinct approaches: RFC and GPC.
RFC
RFC combines predictions from multiple decision trees, each trained on bootstrap samples of the training data. We used the proportion of trees predicting success as our probability estimate. The RFC implementation in the scikit-learn library37 was used in this study. Two key modifications were necessary to adapt RFC for our discovery process.
First, we addressed the substantial data imbalance between initial and newly collected data. While each starting linker had 336 samples, new linker data accumulated incrementally. We implemented a data augmentation procedure that injected multiple copies of newly collected data during iterative experimentation. The optimal degree of data augmentation depends on the chemical similarity between new and training linkers. When a new linker closely resembles a training linker, each new experimental result should carry similar weight to the training data, requiring minimal augmentation. Conversely, if a new linker’s synthesis mechanism differs substantially from all training linkers, the initial dataset is merely a distraction, necessitating stronger augmentation to prioritize learning from new experimental outcomes. We selected an augmentation number of 10, accounting for the ratio between the number of experiments for each initial (336) and new linker (36). This choice provided a balance between leveraging previous knowledge and adapting to novel chemical space. Additional results for other augmentation numbers can be found in Supplementary Fig. 32.
Second, we implemented dynamic hyperparameter optimization to accommodate the evolving training set. Every 100 iterations we performed five-fold cross-validation on newly collected data: for each fold, we trained on all data (including the initial training set) except the validation fold, selecting the maximum allowed tree depth that minimized classification cross-entropy loss. We kept other hyperparameters as default in the scikit-learn library.
GPC
GPC models the probability of success through a latent Gaussian process, providing naturally calibrated probability estimates with uncertainty quantification. Our initial dataset revealed distinct patterns: outcomes varied smoothly across reaction conditions for fixed linkers, but showed less continuity across linker features at fixed conditions, reflecting the underlying chemical complexity. To capture these characteristics, we designed a composite kernel function:
where kRBF represents a radial basis function kernel operating on experimental features xe, capturing smooth variations across experimental features, and kMat52 denotes a Matérn-5/2 kernel operating on linker features xl, allowing less smooth transitions across molecular structures. We computed posterior probabilities and determined the hyperparameters using the variational inference approximation implemented in the GPyTorch library38.
Data availability
The crystal structures are available from the Cambridge Crystallographic Data Centre (CCDC) under reference numbers: 2411199 (ZIF-A1), 2411197 (ZIF-A2), 2411200 (ZIF-A3), 2411198 (ZIF-A4), 2411067 (ZIF-A5), 2411068 (ZIF-A6), 2411070 (ZIF-A7), 2411069 (ZIF-A8), 2411079 (ZIF-A9), 2411078 (ZIF-A10), 2411080 (ZIF-A11), 2411239 (LZIF-A1), 2411238 (LZIF-A2), 2411236 (LZIF-A3), 2411237 (LZIF-A4), 2411077 (Zn(2nIM)(HCOO)), 2411076 (Zn5(bIM)6(HCOO)4(DMF)2), and 2411075 (Zn2(9pur)2(HCOO)2(DMF)). These data can be obtained free of charge from the CCDC via www.ccdc.cam.ac.uk/data_request/cif. Reaction data, labelled optical images, computational data and ML results are available via Zenodo at https://doi.org/10.5281/zenodo.14511016 (ref. 39).
Code availability
Codes for training the crystal recognition model and the sequential and batch algorithms are available at https://doi.org/10.5281/zenodo.14511016 (ref. 39).
References
Furukawa, H. et al. Water adsorption in porous metal–organic frameworks and related materials. J. Am. Chem. Soc. 136, 4369–4381 (2014).
Fathieh, F. et al. Practical water production from desert air. Sci. Adv. 4, eaat3198 (2018).
Lin, J.-B. et al. A scalable metal–organic framework as a durable physisorbent for carbon dioxide capture. Science 374, 1464–1469 (2021).
Rohde, R. C. et al. High-temperature carbon dioxide capture in a porous material with terminal zinc hydride sites. Science 386, 814–819 (2024).
Yaghi, O. M. et al. Reticular synthesis and the design of new materials. Nature 423, 705–714 (2003).
Raccuglia, P. et al. Machine-learning-assisted materials discovery using failed experiments. Nature 533, 73–76 (2016).
Xie, Y. et al. Machine learning assisted synthesis of metal–organic nanocapsules. J. Am. Chem. Soc. 142, 1475–1481 (2019).
Li, Z. et al. Robot-accelerated perovskite investigation and discovery. Chem. Mater. 32, 5650–5663 (2020).
Mekki-Berrada, F. et al. Two-step machine learning enables optimized nanoparticle synthesis. NPJ Comput. Mater. 7, 55 (2021).
Voznyy, O. et al. Machine learning accelerates discovery of optimal colloidal quantum dot synthesis. ACS Nano 13, 11122–11128 (2019).
Xie, Y. et al. Accelerate synthesis of metal–organic frameworks by a robotic platform and Bayesian optimization. ACS Appl. Mater. Interfaces 13, 53485–53491 (2021).
Moosavi, S. M. et al. Capturing chemical intuition in synthesis of metal–organic frameworks. Nat. Commun. 10, 539 (2019).
Domingues, N. P. et al. Using genetic algorithms to systematically improve the synthesis conditions of Al-PMOF. Commun. Chem. 5, 170 (2022).
Szymanski, N. J. et al. An autonomous laboratory for the accelerated synthesis of novel materials. Nature 624, 86–91 (2023).
Wu, Y. et al. Universal machine learning aided synthesis approach of two-dimensional perovskites in a typical laboratory. Nat. Commun. 15, 138 (2024).
Zheng, Z. et al. Shaping the water-harvesting behavior of metal–organic frameworks aided by fine-tuned GPT models. J. Am. Chem. Soc. 145, 28284–28295 (2023).
Baburin, I., Leoni, S. & Seifert, G. Enumeration of not-yet-synthesized zeolitic zinc imidazolate MOF networks: a topological and DFT approach. J. Phys. Chem. B 112, 9437–9443 (2008).
Zheng, Z., Rong, Z., Nguyen, H. L. & Yaghi, O. M. Structural chemistry of zeolitic imidazolate frameworks. Inorg. Chem. 62, 20861–20873 (2023).
Huo, H. et al. Machine-learning rationalization and prediction of solid-state synthesis conditions. Chem. Mater. 34, 7323–7336 (2022).
Tan, M. & Le, Q. Efficientnetv2: smaller models and faster training. Proc. Mach. Learn. Res. 139, 10096–10106 (2021).
Kashchiev, D. Nucleation (Elsevier, 2000).
Lim, I. H., Schrader, W. & Schüth, F. Insights into the molecular assembly of zeolitic imidazolate frameworks by ESI-MS. Chem. Mater. 27, 3088–3095 (2015).
Li, K. et al. Zeolitic imidazolate frameworks for kinetic separation of propane and propene. J. Am. Chem. Soc. 131, 10368–10369 (2009).
Tian, Y.-Q. et al. Cadmium imidazolate frameworks with polymorphism, high thermal stability, and a large surface area. Chem. Eur. J 16, 1137–1141 (2010).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Williams, C. K. & Barber, D. Bayesian classification with Gaussian processes. IEEE Trans. Pattern Anal. Mach. Intell. 20, 1342–1351 (1998).
Rasmussen, C. & Williams, C. Gaussian Processes for Machine Learning (MIT Press, 2006).
Shahriari, B., Swersky, K., Wang, Z., Adams, R. P. & De Freitas, N. Taking the human out of the loop: a review of Bayesian optimization. Proc. IEEE 104, 148–175 (2015).
Lattimore, T. & Szepesvári, C. Bandit Algorithms (Cambridge Univ. Press, 2020).
Yang, J. et al. Principles of designing extra-large pore openings and cages in zeolitic imidazolate frameworks. J. Am. Chem. Soc. 139, 6448–6455 (2017).
Yang, Q.-F. et al. A series of metal–organic complexes constructed from in situ generated organic amines. CrystEngComm 10, 1534–1541 (2008).
Paszke, A. et al. Pytorch: an imperative style, high-performance deep learning library. Adv. Neural Inform. Process. Syst. 32, 8026–8037 (2019).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. In Proc. 3rd International Conference for Learning Representations (ICLR, 2015).
Frisch, M. J. et al. Gaussian 16 Revision A.03 (Gaussian, 2016).
Tomasi, J., Mennucci, B. & Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3094 (2005).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Gardner, J., Pleiss, G., Weinberger, K. Q., Bindel, D. & Wilson, A. G. Gpytorch: blackbox matrix–matrix Gaussian process inference with GPU acceleration. Adv. Neural Inform. Process. Syst. 31, 7576–7586 (2018).
Rong, Z. & Chen, Z. Algorithmic iterative reticular synthesis. Zenodo https://doi.org/10.5281/zenodo.14511016 (2025).
Acknowledgements
This research was supported by the Bakar Institute of Digital Materials for the Planet (BIDMaP) and the King Abdulaziz City for Science and Technology (Center of Excellence for Nanomaterials and Clean Energy Applications, KACST). The work of F.L. is funded by the Australian Research Council (ARC) under grant DP210100045 held by M.B. L.E.G. acknowledges financial support from the VinUniversity Innovation Fund (project number 5). Z.R. thanks M. Gao for discussion on SCXRD refinement. Z.R. thanks D. M. Proserpio for discussion on the topological classification of new ZIF structures. Z.R. thanks A. Yao and Y. Monno for assisting with the use of the automated optical microscope. Z.Z. and Z.R. thank D. Small for helping set up computational jobs on clusters. The computation of linker features used the high-performance computing resource of the Molecular Graphics and Computation Facility at the University of California Berkeley with funding number NIH S10OD034382. This research used the Savio computational cluster resource provided by the Berkeley Research Computing programme at the University of California, Berkeley (supported by the UC Berkeley Chancellor, Vice Chancellor for Research, and Chief Information Officer). This research used resources of the Advanced Light Source at Lawrence Berkeley National Laboratory, a DOE Office of Science User Facility under contract number DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
Z.R., O.M.Y. and L.E.G. conceptualized the project. Z.R. and Z.Z. determined the scope of linkers for experimental studies. Z.R., K.G., A.A.A. and Z.Z. conducted automated synthesis and collected optical images. Z.R. and K.G. labelled the optical images. H.T.N.L., K.H.B., H.H.P., T.N.-D. and D.D.L. developed the crystal recognition model, with V.B.T.P. providing guidance on data preparation. Z.R. performed SCXRD experiments and solved the crystal structures. Z.R., S.C., Z.Z. and T.J.I. designed and computed linker feature vectors, with J.S. providing guidance on quantum mechanical aspects. Z.C., M.B. and F.L. developed the ML-guided discovery methodology. Z.C., F.L. and Z.R. conducted ML experiments on ZIF reaction data. J.T.C. and C.B. provided valuable suggestions on ML data presentation. Z.R., O.M.Y. and Z.C. prepared the initial draft, and all authors contributed to revising the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks Wonyoung Choe, Jian Lin and Ben Slater for their contribution to the peer review of this work. Primary Handling Editor: Joel Cejas-Sánchez, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Chemical structures and abbreviations of 48 new linkers.
The linkers are categorized by core scaffold: Imidazole-based (left) and Benzimidazole-based (right). Full chemical names corresponding to the abbreviations are listed in Supplementary Section 1.
Supplementary information
Supplementary Information (download PDF )
Supplementary Sections 1–16, Tables 1–22 and Figs. 1–81.
Supplementary Data 1
Crystallographic data for compound LZIF-A1, CCDC 2411239.
Supplementary Data 2
Crystallographic data for compound LZIF-A2, CCDC 2411238.
Supplementary Data 3
Crystallographic data for compound LZIF-A3, CCDC 2411236.
Supplementary Data 4
Crystallographic data for compound LZIF-A4, CCDC 2411237.
Supplementary Data 5
Crystallographic data for compound ZIF-A1, CCDC 2411199.
Supplementary Data 6
Crystallographic data for compound ZIF-A2, CCDC 2411197.
Supplementary Data 7
Crystallographic data for compound ZIF-A3, CCDC 2411200.
Supplementary Data 8
Crystallographic data for compound ZIF-A4, CCDC 2411198.
Supplementary Data 9
Crystallographic data for compound ZIF-A5, CCDC 2411067.
Supplementary Data 10
Crystallographic data for compound ZIF-A6, CCDC 2411068.
Supplementary Data 11
Crystallographic data for compound ZIF-A7, CCDC 2411070.
Supplementary Data 12
Crystallographic data for compound ZIF-A8, CCDC 2411069.
Supplementary Data 13
Crystallographic data for compound ZIF-A9, CCDC 2411079.
Supplementary Data 14
Crystallographic data for compound ZIF-A10, CCDC 2411078.
Supplementary Data 15
Crystallographic data for compound ZIF-A11, CCDC 2411080.
Supplementary Data 16
Crystallographic data for compound Zn(2nIM)(HCOO), CCDC 2411077.
Supplementary Data 17
Crystallographic data for compound Zn5(bIM)6(HCOO)4(DMF)2, CCDC 2411076.
Supplementary Data 18
Crystallographic data for compound Zn2(9pur)2(HCOO)2(DMF), CCDC 2411075.
Source data
Source Data Fig. 2 (download XLSX )
Source data for reaction outcome projection.
Source Data Fig. 3 (download XLSX )
Source data for linker features and algorithm output.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rong, Z., Chen, Z., Luong, F. et al. Algorithmic iterative reticular synthesis of zeolitic imidazolate framework crystals. Nat. Synth 5, 409–419 (2026). https://doi.org/10.1038/s44160-025-00939-9
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44160-025-00939-9
