
Abstract
Antiviral resistance stemming from rapid viral evolution and adaptation is a major challenge faced in treating viral infections. Here, we describe the mechanisms and factors underlying antiviral resistance and their implications to future drug development. Current improvements to conventional methods provide viable options to overcome antiviral resistance. Ongoing efforts in developing new antiviral strategies are also discussed. Examples from across virology are used to illustrate how virus evolution and antiviral therapy influence each other.
Similar content being viewed by others
Introduction
The emergence of viral pathogens with pandemic potential has been a recurring, major concern for the global community1,2. Through the relentless efforts of antiviral drug researchers and developers, numerous antivirals have been approved for use in treating infections caused by viruses such as the human immunodeficiency virus (HIV) and hepatitis C virus (HCV). However, despite these efforts, the increasing occurrence of antiviral resistance threatens to render existing antivirals ineffective3,4. The recent COVID-19 pandemic has reiterated the importance of addressing this concern, with the development of effective antiviral therapeutics being outpaced by virus evolution4.
Conventional antivirals are known as direct acting antivirals (DAAs), which are designed to disrupt virus replication cycles by binding to specific viral proteins and inhibiting their activity, while minimising cytotoxic and off-target effects. DAAs target a variety of viral proteins essential for viral replication (Fig. 1 and Table 1). The first DAA discovered was idoxuridine, a nucleoside analogue used to treat herpes simplex keratitis5. Since then, antiviral development has come a long way. Many other drugs have been developed against viruses such as oseltamivir and zanamivir against neuraminidase (NA) of influenza virus6 or enfuvirtide against gp41 of HIV7.

The various steps of the viral life cycle targeted by DAAs (purple) and HTAs (orange) are shown. A Viral binding inhibitors bind to viral glycoproteins, preventing them from binding host receptors, while host binding inhibitors act as host receptor antagonists, preventing their binding with viral glycoproteins required for viral entry. B Viral fusion inhibitors bind and disrupt the conformations of viral proteins responsible for mediating fusion between host and viral membranes. C Viral uncoating inhibitors bind to and interfere with viral proteins such as capsid or ion channels preventing capsid disassembly or endosomal acidification required for release of viral genome. D Viral nucleoside inhibitors bind to reverse transcriptases or polymerases and either cause chain termination or incorporation of a mutagen that drives a virus towards error catastrophe. E Host nucleotide depletion deprives viruses of purines and pyrimidines needed for genome replication. F Viral integrase strand transfer inhibitors bind to viral integrase active sites and displace viral DNA, preventing proviral incorporation into host genomes. G Host nuclear export disruption prevents the export of viral genomes and ribonucleoproteins out of the nucleus to their assembly site. H Host immune stimulants place host cells in an antiviral state by activating the expression of genes with antiviral activity against various steps of the viral life cycle. I Post-translational modifications are crucial for several viral proteins to function. HTAs that prevent such modifications could render these proteins non-functional, inhibiting the viral replication cycle at several different stages. J Viral assembly inhibitors interact with viral proteins, preventing their oligomerisation required for viral particle assembly. K An inhibitor that binds to and inhibits the function of a viral protein with multiple roles in the viral life cycle could simultaneously disrupt multiple stages at once. L Viral protease inhibitors bind to viral proteases and prevent the cleaving of viral polyproteins into functional proteins. M Viral egress inhibitors bind viral proteins required to separate the membranes of the host and new virions, thus preventing the budding of progeny viral particles from the host.
A common problem with DAAs is their low genetic barrier to resistance. This genetic barrier refers to the mutational threshold a viral population needs to cross, beyond which clinically meaningful resistance develops8. The first orally administrated HCV protease inhibitors to receive United States Food and Drug Administration (U.S. FDA) approval were telaprevir and boceprevir9,10. Due to their low potency, treatment had to be supplemented with pegylated interferon (IFN) and ribavirin. Both had issues such as poor safety profiles, which led to them being quickly replaced within 3 years of approval. Nevertheless, they paved the way for the development of antiviral therapeutics for HCV.
Aside from targeting viral proteins, antivirals can also be designed against host proteins, and are known as host-targeted antivirals (HTAs). HTAs target a variety of host proteins hijacked by the virus for replication (Fig. 1 and Table 2). One potential advantage of HTAs is that if they affect common pathways used by viruses of the same family, they have the potential to be broad spectrum. HTAs are theorised to have high genetic barriers to resistance because viral hosts are less prone to mutation. In some cases, becoming resistant to a HTA might require simultaneous mutations in several viral proteins, as was observed for the iminosugar UV-4B in dengue virus (DENV) and influenza viruses11,12. Of the several HTAs that have been discovered in vitro, few have generated resistance mutations. Additionally, HTAs have been found to complement the action of DAAs and mutants raised against DAAs do not show resistance to HTAs13,14.
In this review, we will discuss the development of antiviral therapeutics in light of virus adaptation and evolution. We will also review the current state of research in various alternatives to conventional antivirals.
Occurrence of resistance mutations to conventional antivirals
Treatment with an antiviral places a selection pressure on the viral population, causing a decrease in viral load. However, if complete viral suppression is not achieved, this creates a genetic bottleneck, from which drug-resistant variants are more likely to survive and replicate to replace susceptible viruses in the population15,16.
Most viruses of concern are RNA viruses, that present several traits favourable to the establishment of resistance, including: poor replication fidelity, high replication rates17, genetic diversity18, genetic reassortment19,20,21 and RNA recombination22. These factors drive high evolutionary rates and create ideal conditions for the emergence of resistant variants upon treatment with DAAs (Fig. 2A). Viruses are intracellular obligate parasites that hijack the host cell machinery to generate progeny. To disrupt viral life cycles, antivirals have to be present on the surface of or enter the cells themselves. Although DAAs are designed to target only viral proteins and not any of the host’s, early antivirals sometimes had the off-target effects of disrupting normal cellular processes whilst inhibiting viral life cycles. These undesirable side effects like cytotoxicity, restricted the dosage and frequency of treatment initially required to achieve viral suppression, and consequently provided an opportunity for the virus to develop resistance during suboptimal dosing.

A Characteristics of RNA viruses that contribute to their high evolutionary rates and adaptability. B Comparison of direct and cross-resistance. Direct resistance is when a virus exposed to a single antiviral (Antiviral A) develops resistance to it. Cross-resistance is when a virus exposed to only one antiviral (Antiviral A) acquires mutations that confer resistance to it, as well as another antiviral (Antiviral B) to which the virus was not exposed. C Resistant mutants that confer a selective advantage over the wild type viruses in the presence of antivirals quickly become prevalent in the viral population. If these mutants are fitter than wild type even in the absence of antiviral, they are transmitted more often and becomes the dominant strain. Sustained transmission of these resistant viruses can render antivirals obsolete.
The first nucleoside reverse transcriptase inhibitor (NRTI) to be developed against HIV was zidovudine (AZT) which was clinically impactful in terms of disease progression and mortality, but suffered from a variety issues, most notably, cytotoxicity in the form of mitochondrial myopathy23. AZT inhibited retroviral reverse transcriptase, but also mitochondrial DNA polymerase, causing a reduction of mitochondrial DNA over time with AZT therapy and clinically manifested as neuropathy, hepatic failure, and lactic acidosis in patients24,25. Continuous monotherapy with AZT coupled with incomplete viral suppression led to the emergence of AZT resistance.
Following this, different classes of antivirals targeting the different stages of the retroviral life cycle were discovered. However, through a combination of viral, host and antiviral factors that limited the effectiveness of the therapy, complete viral suppression was not achieved. This led to the eventual emergence of resistance mutations against all classes of antivirals. In some instances, a single substitution alone was enough to confer a high level of resistance.
A single substitution of M184V in the reverse transcriptase of HIV-1 confers 300–600 fold reduction in potency in enzymatic and cell based assays to both emtricitabine (FTC) and lamivudine (3TC), NRTIs commonly used in anti-HIV therapy26. In patients, resistance to the integrase inhibitor raltegravir can occur as three separate non-overlapping pathways with N155H, Q148R/H/K and Y143R/C substitutions in the catalytic core of HIV integrase, with each eventually leading to a high level of resistance27,28. Lenacapavir, a HIV capsid inhibitor, had the potential to treat multidrug-resistant HIV that developed from other antiretroviral regimens, and was anticipated to have no pre-existing resistance mutations, given that it was the first of its novel class of inhibitors29,30. However, several mutations discovered in vitro such as M66I, Q67H and N74D conferred over a 1000-fold reduction in susceptibility to lenacapavir29. The N74D substitution which developed in as few as 3 weeks in patients demonstrated its low genetic barrier of resistance30.
Direct resistance to a single antiviral drug is easily identified from screening patients with virological failure, which refers to the failure to keep viral load below a certain threshold31. Less commonly identified is the emergence of cross-resistance, which refers to a virus developing resistance to a drug it has never been exposed to, because of mutations that were selected for by a different drug (Fig. 2B). Ganciclovir-cidofovir cross-resistance during the treatment of cytomegalovirus (CMV) infection is one of the few well-described cases in the literature. Ganciclovir resistance mostly arises in the viral gene UL97, which codes for a viral protein kinase which converts ganciclovir into its active triphosphate form that stops the elongation of viral DNA by viral polymerase. However, during ganciclovir treatment, a rarer mutation, in the viral polymerase itself (UL54) confers ganciclovir–cidofovir cross-resistance32. Similarly in HIV, doravirine is a non-nucleoside reverse transcriptase inhibitor (NNRTI) that demonstrated efficacy against common NNRTI-associated mutations such as K103N, G190A and Y181C, at clinically relevant concentrations33 and was evaluated for use as treatment for NNRTI-resistant HIV. However, mutations such as V106M or Y188L which confer resistance to doravirine can often be observed in patients that underwent first-line NNRTI regimens with drugs such as efavirenz or nevirapine34, thus requiring genotypic antiviral resistance screening for patients with NNRTI resistance before administration of doravirine.
While any newly arising resistance mutation is a rare event, the speed at which it can accumulate and become fixed in a viral population varies greatly, and for some antiviral treatments can be a major concern. For instance, during the treatment of herpes simplex virus (HSV-1) with acyclovir and foscarnet, subpopulations which were resistant to both drugs became prevalent in the viral population in as little as 20 days35,36. In HCV, a sustained virologic response refers to the inability to detect virus in blood for at least 24 weeks post-treatment, although an endpoint of 12 weeks post-treatment has become prevalent in most clinical studies37. Failure to achieve this response is almost always accompanied by treatment-emergent drug resistance mutations, as demonstrated for chronic HCV patients undergoing grazoprevir therapy38.
Once a resistance mutation to DAAs is established in an individual, there is risk that this mutation spreads across the population. Transmission of antiviral resistance mutations can occur both in the absence and presence of the antiviral treatment39. Sustained transmission of such mutants, and genetic recombination with other virus strains can lead to fixing resistance mutations40 rendering the antiviral treatment obsolete (Fig. 2C). This is the case for the influenza A virus (IAV) M2 ion proton channel inhibitors amantadine and rimantadine, to which most IAV has since become resistant worldwide. The S31N mutation in M2 of IAV confers high resistance at a low fitness cost, and became present in a majority of IAV cases, resulting in both drugs no longer being clinically used41.
Worse still, antiviral therapies may also have the unintended consequence of helping to drive the evolution of viruses to the benefit of the virus. Treatment of HCV resulted in R155K mutation in the NS3/4A protease which not only conferred resistance to multiple protease inhibitors, but also allowed the virus to escape from HLA-A*68-restricted CD8+ T cells42. Similarly, acyclovir treatment of herpes resulted in the emergence of non-cytopathic HSV-1 whose mutations also better evade the immune system43.
Factors influencing the emergence of antiviral resistance
Antiviral-associated factors
There are several characteristics of an antiviral which contribute to how easily resistance develops, including genetic barriers, type of mutation, potency and duration of treatment, as well as pharmacokinetics/pharmacodynamics (Fig. 3). The genetic barrier to resistance of an antiviral refers to the number of mutations or ‘steps’ required to achieve resistance. Resistant variants requiring fewer mutations are more likely to occur. Additionally, the type of mutation also augments the genetic barrier, with transition mutations occurring more frequently than transversion mutations44. Viral polymerases have a mutational bias towards transitions, since in the process of RNA elongation, it is easier to mis-incorporate a purine with another purine rather than a pyrimidine and vice versa. The effect of this mutational preference is that resistance mutations requiring transversions are less likely to occur than those requiring transitions45.

These different factors contribute to a suboptimal dose of antiviral being used during therapy, resulting in incomplete viral suppression which eventually leads to the emergence of antiviral resistance. A Virus-associated factors include reduced susceptibility to antivirals as a result of pre-treatment baseline resistance mutations, polymorphisms and variation in viral subtypes. B Host factors include genetic variation in hosts that affect their ability to respond to treatment or tolerate an antiviral. Various factors such as side effects, high pill burdens and frequent administration of antivirals contribute to poor adherence to antiviral regimens. C Antiviral factors include poor potency or the pharmacokinetic/pharmacodynamic profile of the antiviral that limits their effectiveness to suppress viruses. A low genetic barrier to resistance enables the viruses to gain resistance quickly.
At the same time, the potency of the antiviral also determines how much time the virus has to develop resistance. Highly potent antivirals sharply reduce the viral load, therefore usually only requiring a short exposure duration, minimising the probability for resistance mutations to develop. Low or intermediate potency drugs do not effectively decrease viral load and likely require a longer duration of treatment, allowing the virus more time to generate resistant mutants. Low potency together with a low genetic barrier to resistance provides the ideal conditions for the development of antiviral resistance. The relatively low potency and genetic barrier to resistance of 3TC is why up to 70% of patients with hepatitis B virus infections develop resistant variants after 5 years of treatment. Adefovir is also of relatively low potency but has a higher genetic barrier to resistance than 3TC and therefore only has 29% of patients with resistance after 5 years. Entecavir and tenofovir are considered high potency drugs with high genetic barriers, which is why only 1% of patients develop resistance after 5 years46,47.
Highly potent antivirals might also develop resistance if they have poor pharmacokinetic and pharmacodynamic profiles. Many current antivirals such as ribavirin, adefovir, entecavir, acyclovir, AZT, 3TC and oseltamivir have hydroxyl groups in their structures which makes them polar and consequently, have poor permeability. On the other hand, drugs like chloroquine, lopinavir, arbidol, ritonavir, peramivir, baloxavir marboxil and letermovir have poor solubility due to the presence of hydrophobic groups in their structure48. These limitations reduce the bioavailability of the antiviral at their target sites, such that they are below the concentration required for effective viral suppression.
Host-associated factors
The host receiving the antiviral also has an important role in the development of antiviral resistance. Even with the most potent of antivirals, adherence of the host during an antiviral treatment regimen often affects the outcome49,50. For chronic infections such as HIV, a common problem is that the antiviral treatment has several issues such as high pill burdens, inconvenient drug administration, strict dieting, dose-limiting toxicity, and undesirable drug interactions, which result in suboptimal adherence (Fig. 3).
Interpatient variability in terms of genetics, particularly in immune-associated genes, also affects the effectiveness of treatment and the likelihood of antiviral resistance development. On human chromosome 19, single nucleotide polymorphisms (SNPs) in the IL28B gene which encodes for IFN-λ3, a type III IFN, were found to affect pegylated IFN/ribavirin treatment for HCV outcomes in patients coinfected with HCV/HIV. This information can be used to optimise treatment with IFN/ribavirin for those that respond well, while simultaneously minimising risk of exposure in patients that do not respond well51. Abacavir is a NRTI used to treat HIV, however, patients with HLA-B*5701 alleles tend to develop hypersensitivity from abacavir treatment52. Treating such patients with abacavir puts them at risk of poor adherence, and they should switch to an abacavir-free regimen to avoid development of abacavir resistance.
Environmental factors of the hosts also affect the adherence to an antiviral regimen. Surveillance studies on monkeypox virus-infected patient cohorts in Los Angeles reported up to 8.5% of patients studied were co-infected with HIV and exhibited low CD4 cell counts. The treatment of monkeypox virus infection typically involves oral tecovirimat, which requires high fat meals twice daily to ensure optimal absorption. Patient compliance to treatment regimen was affected by the poor’s accessibility to food, further highlighting the unlikelihood of meeting such dietary requirements in outbreak-prone regions, mostly in developing countries in Africa53,54,55. Strict adherence to tecovirimat treatment may also be rendered unavailing or further exacerbated by pre-existing conditions, as was observed in a subset of the HIV-infected cohort that completion of a course of oral tecovirimat did not improve clinical outcome55.In addition, recurrence of clinical symptoms is also possible and remains a concern even with good patient compliance, making extended therapy necessary56,57,58.
Viral-associated factors
Baseline viral load and resistance
Patients with a higher baseline viral load, or copy number, are statistically shown to accumulate a higher absolute number of minority variants, which may confer antiviral resistance. In an Influenza Resistance Information Study, children aged 1–5 years old had higher viral loads and longer viral shedding compared to other groups of children aged 13 years and below in the study. These children also had the highest frequency of antiviral resistance mutations detected59. A study to understand the factors contributing to failure to achieve SVR12 in HCV, found higher pretreatment viral loads increased the risk of treatment failure across different DAA regimens such as ledipasvir/sofosbuvir and sofosbuvir/velpatasvir60.
In some viruses, certain genotypes or polymorphisms in the genomes confer a reduced susceptibility to antivirals, even if the host is treatment naive. These are referred to as baseline resistance associated mutations or polymorphisms (Fig. 3). This lowered susceptibility means that treatment doses that are effective against the usually susceptible virus are now sub-optimal, which can increase the likelihood of antiviral resistance emergence, since viral suppression is not achieved. Patients infected with subtype CRF02_AG/G HIV and a lack of known resistance-associated mutations, appeared to have lower baseline susceptibility to protease inhibitors such as lopinavir/ritonavir, compared to other HIV subtypes. Consequently, this increases the risk of resistance emerging should monotherapy be employed or a lack of adherence to antiviral regimen occur61.
Prior to treatment, HCV genomes containing resistance mutations are sometimes already present, although in relatively low abundance. These mutants could prevent complete viral suppression, allowing time for the virus to develop other mutations that further increase resistance62. Several baseline resistance-associated mutations and polymorphisms have been discovered in the different genotypes and subtypes of HCV which negatively affect treatment outcomes and contribute to the emergence of antiviral resistance and virological failure63,64. The highly polymorphic residue 30 on NS5A of HCV has many substitutions across the different HCV genotypes which are associated with resistance to NS5A inhibitors. In HCV genotype 1a, glutamine is the prevalent residue, but substitutions with arginine or histidine residues occur naturally at a rate of 1%. In particular, Q30R substitutions confer a 1000-fold increase in resistance to daclatasvir65.
The presence of pre-treatment resistance, as well as breakthrough mutations that emerge during treatment, resistance screening before and after (in the event of virological failure) is necessary in order to optimise treatment regimens66.
Fitness costs
Aside from genetic barriers, viral fitness also determines whether the new genetic adaptations take hold in the population. While genetic barriers refer quantitatively to mutational steps required to achieve resistance, fitness refers qualitatively to mutants in terms of the selective advantage they bring to the virus under these growth conditions67. Mutants with low genetic barriers are readily generated but are not always fit (Fig. 4A).

A With multiple routes to develop resistance, both genetic barriers and fitness will determine which mutants will dominate. The higher the genetic barrier, the less likely resistance will emerge. Low genetic barrier and low fitness mutants (Mutant 1) are readily generated but selected against. Mutants with high fitness (Mutant 2), although unlikely to generate because of higher genetic barriers are likely to dominate the viral population if they emerge. Mutants with a very high genetic barrier (Mutant 3) are unlikely to appear because mutations incurring high fitness costs are required. B Resistance mutations confer a fitness advantage in the presence of an antiviral, but not in its absence. A double mutant with both resistance and compensatory mutations has better fitness than both wild type and resistant mutant alone in the presence of an antiviral, although not always in its absence. Compensatory mutations can occur stepwise with each compensatory mutant additively restoring the fitness lost by the resistance mutation. C Different mutations (Mutations A and B) work in synergy by conferring resistance to different antivirals (Antivirals 1 and 2). Synergistic mutations may also compensate for each other’s fitness cost (eg. low polymerase efficiency, low enzyme processivity etc.). D Compensatory mutations allow for the survival of resistance mutants which are otherwise non-viable by sufficiently restoring fitness (denoted by +) lost by the resistance mutation.
In general, once a virus is established in the human population, it reaches a local fitness maximum such that most mutations result in a fitness cost and very few would improve viral fitness any further. Thus, upon treatment with an antiviral, most resistant mutations will confer a fitness advantage in the presence of the drug; but will come with a trade-off fitness cost, in the original growth conditions without the drug. For example, the A335V mutation in non-structural protein (nsp)-13 helicase of coronaviruses confers partial resistance to remdesivir, at the cost of reduced helicase activity68. These mutants would have a disadvantage compared to wild type in the absence of the antiviral69, which explains the lack of transmission of these resistant viruses and why these mutants disappear upon cessation of treatment.
Under the selection pressure of an antiviral, if multiple routes to resistance are possible, the one with the least fitness costs, and thereby conferring the highest net fitness gain, will become dominant (Fig. 4A). As an example, both M184V and M184I substitutions in HIV reverse transcriptase confer resistance to both FTC and 3TC, with M184I usually preceding M184V. However, M184V has higher polymerase activity and will rise to become dominant over M184I over time70,71.
In HSV infection, nucleoside analogues acyclovir and famciclovir are commonly used. These drugs target the viral polymerase, but require phosphorylation by the viral thymidine kinase (TK), which is encoded by the UL23 gene to become active. Mutations in UL23 can confer resistance to both drugs. This necessitates a switch to polymerase inhibitors that do not require phosphorylation by viral (TK), such as cidofovir and foscarnet. Mutations in the viral polymerase gene (UL30), however, confer resistance to all four drugs72. Despite this apparent advantage in broad resistance, most observed resistance mutations to acyclovir occur in UL23, suggesting these mutants bear lower fitness costs, rather than better fitness advantages. Indeed, HSV requires a functional DNA polymerase to replicate but can survive without TK under the right conditions, as evidenced by the existence of TK-negative and TK-low-producer mutant viruses73. HSV also shows higher tolerance for mutations in UL23. Half of the mutants are insertions and deletions that render the entire enzyme non-functional. UL30, on the other hand, can only tolerate single nucleotide substitutions that occur in the domains of the polymerase responsible for proofreading and substrate binding, without rendering the enzyme non-functional74. Nevertheless, the existence of HSV with mutations in both TK and DNA polymerase with similar replication fitness to wild type demonstrates the ability of viruses to alleviate fitness costs36.
Compensatory mutations
Compensatory mutations are mutations that arise to alleviate or off-set the fitness costs incurred by mutations that confer antiviral resistance.
Compensation of the fitness cost of a primary mutation, by a subsequent secondary mutation, allows the double mutant to be fitter than both wild type and primary mutation viruses in the presence of the antiviral (Fig. 4B).
In influenza viruses, resistance to baloxavir, an inhibitor that targets the PA segment of the influenza replicase, is conferred by mutations I38L, I38T, or E199D for IAV and I38T for influenza B virus (IBV). These mutations result in lower polymerase activity and replication. Serial passage of these viruses revealed a D394N compensatory mutation for IAV and E329G for IBV that improved the replication of resistant viruses75. If secondary mutations sufficiently compensate for fitness costs of the primary resistant mutation, the resulting double mutant may allow for the fixing and transmission of antiviral resistance in the viral population.
Resistance to the NA inhibitor oseltamivir is caused by the H274Y mutation in the NA gene, which comes at the cost of impeding proper folding of the enzyme, resulting in initial poor viral replication. However, secondary mutations restored viral fitness, enabling the H274Y mutant to become dominant and circulate76. The L528M mutation in HBV not only restored replicative competence in drug resistant HBV, but further increased antiviral resistance77. Compensatory mutations can sometimes work in synergy (Fig. 4C). E138K and K101E mutations in HIV RT which confer resistance to rilpivirine (RPV), have been demonstrated to compensate for reduction of replicative capacity and enzyme processivity (the ability of an enzyme to remain attached to the template when copying it) as a result of the M184I/V mutation, which confers FTC resistance. M184I in turn, compensates for the lower DNA polymerisation efficiency (the ability of the polymerase to generate products) caused by K101E78,79.
Compensatory mutations appear after the antiviral resistance mutation, often in a stepwise manner on the way to their optimum (Fig. 4B). Mutations in the HBV virus reverse transcriptase M204V/I confer resistance to 3TC at the cost of lower in vitro replication efficiency. A L180M mutation emerges after M204V/I, partially restoring replication fitness back to around ~40% that of wild type, while also contributing to antiviral resistance. A third mutation V173L occurs following the first two, further increasing replication efficiency back to ~90% wild type levels80.
Compensatory mutations augment the fitness of resistant viral mutants, allowing for pathways of escape which are normally selected against because of fitness costs. When designing broad spectrum antivirals, highly conserved regions of the viral genome are popular targets because they are present across several viral strains, genus and even families. These regions are conserved because they play essential roles in viral life cycles and any change to them comes at high fitness costs. One such example is the lysine residue in the F1 motif of polymerases in many single-stranded RNA viruses. This includes K229 in the PB1 subunit of influenza, K291 in nsp4 of chikungunya virus (CHIKV) and K159 in the RNA-dependent RNA polymerase (RdRp) of Coxsackie virus B3 (CVB3). Mutation of this in response to the antiviral favipiravir confers resistance with fitness costs of varying degrees (low in CHIKV, moderate in IAV and complete loss of viability in CVB3. As a result, respective compensatory mutations have emerged to offset these fitness costs (P653L mutation in the PA subunit of influenza polymerase, A239G in the RdRp of CVB3)81,82,83 (Fig. 4D). Targeting of conserved sites in the viral genome places the virus in a difficult position of selecting between resistance to antiviral activity and impairment of function. However, the emergence of compensatory mutations in less conserved regions, offsets some of the fitness costs. This provides an escape for viruses in which they can keep their costly resistance mutations but with an overall smaller loss of fitness.
Understanding which mutations emerge over others, the genetic barriers to their development, the severity of their fitness costs and what compensatory mutations are possible could better inform on which compounds are worth pursuing. Along this thread, treatment with virus inhibitory peptide-based HIV entry inhibitors have been found to have a high genetic barrier to resistance and crossing that barrier required several mutations that came at high fitness costs in infectivity, fusogenicity, and replication, thereby supporting the development of this class of antivirals84.
Existing strategies to overcome resistance to conventional antivirals
Combination therapeutics
Antivirals are often used in combination to prevent emergence of resistant variants of the virus, by increasing the genetic barrier to resistance or eliminating the infection before resistance can develop.
The long history of antiretroviral therapy provides many examples of this. As mentioned above, the first anti-HIV drug zidovudine was a NRTI and it was quickly followed by several other NRTIs: zalcitabine, didanosine, stavudine which all suffered from the same limitations because they shared a similar mechanism of action.
The subsequent discovery of two new classes of anti-HIV medications: protease inhibitors (PIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs), allowed for combination antiretroviral therapy (cART) with drugs of different classes, also known as highly active antiretroviral therapy (HAART). HAART significantly improved treatment outcomes in terms of morbidity and mortality. Other classes of HIV drugs were later discovered such as entry and integrase inhibitors, providing more options for HAART.
Combination therapy has also been applied to other viruses. Combination of daclatasvir with sofosbuvir and/or ribavirin have achieved high rates of sustained virologic response as the NS5A mutations do not affect sofosbuvir and ribavirin85,86. A combination of tenofovir, sofosbuvir, daclatasvir, and ribavirin successfully treated HBV, HCV and hepatitis E virus87.
Antiviral HTA and DAA combinations can display synergy particularly when they target different stages of the virus life cycle (Fig. 1). A combination of the mutagenic DAA molnupiravir (targeting viral replication), with TMPRSS2-inhibiting HTAs such as camostat, avoralstat, or nafamostat (targeting virus entry) synergistically suppressed SARS-CoV-2. Addition of brequinar, a HTA pyrimidine biosynthesis inhibitor, resulted in the three drugs further suppressing the virus88. Combinations of a HTA inhibitor of the Raf/MEK/ERK cellular signalling pathway, ATR-002, used by many viruses during replication with molnupiravir, remdesivir or Paxlovid demonstrated synergy against SARS-CoV-2 in vitro89. A combination of UV-4B and IFN against DENV were found to work in synergy. IFN stimulates the suppression of DENV in SK-N-MC (neuronal), HUH-7(liver) and HFF-1 (skin) cells, at therapeutically feasible concentrations; however, SK-N-MC cells are less responsive to IFN, possibly because they express fewer IFN receptors. On the other hand, UV-4B was cytotoxic in HUH-7 cells and cytostatic in HFF-1 cells at concentrations ≥100 µM. Combination of the two antivirals enabled complementary viral suppression across different tissue types at lower concentrations (Fig. 5A), which is particularly useful when treating pantropic viruses like DENV90.

A The benefits of using antiviral combinations. Antiviral combinations can be used to exploit antagonistic resistance mutations which increase susceptibility to each other when they occur. Additionally, combinations lower doses of each constituent antiviral required for therapeutic effectiveness. Different antivirals (Antivirals A and B) also penetrate tissues to different degrees, which is especially important in pantropic viral infections. B Pharmaco-enhancements refer to modification of a drug to improve traits such as bioavailability and half-life. Boosting with a secondary antiviral (Antiviral B) slows the metabolism of the first antiviral (Antiviral A), prolonging exposure to the primary antiviral at the target site. C Mechanism of nucleotide analogue lethal mutagenesis. The analogue is incorporated into viral genomes during replication and increases the error rate of viral RNA-dependent RNA polymerase (RdRp), by allowing ambiguity in base pairing in subsequent rounds of replication, leading to an accumulation of erroneous mutations. Once the error rate exceeds the error threshold, fit viral genomes are no longer being replicated, resulting in viral extinction. D Resistance to mutagens can occur by several methods, such as mutations in the polymerase that prevent mutagen incorporation, mutations that increase fidelity to counter the increase in error rate, and the excision of mutagens by proofreaders which are present in some viruses. Mutagen efficacy is different for different viruses because of variation in mutational robustness which is the ability to express a phenotype despite deleterious mutations. Mutagens must be used at an optimal concentration, excessively high concentrations are genotoxic and toxic to the host, while sub-optimal concentrations may accelerate viral evolution.
Antiviral resistance mutations are sometimes antagonistic to each other, a feature which may be exploited in combination therapy. HCV A92K and P32del mutants demonstrated high resistance to ledipasvir, velpatasvir and elbasvir, but were susceptible to grazoprevir, ribavirin and sofosbuvir91. The K65R mutation in HIV is a common mutation that emerges during treatment with tenofovir, causing a clinically significant 2-fold reduction in susceptibility. However, when a K65R mutant gains an additional M184V substitution, which confers resistance to 3TC/FTC, it only had a 1.5-fold reduction in tenofovir susceptibility92. This further encourages the use of antiviral combinations, particularly with pairings that have resistance mutations that are antagonistic to one another (Fig. 5A). Drugs which were ineffective because of resistant mutations can be effective in combination93. It should be noted that HTAs do not always prevent the emergence of resistance to DAAs when used in combination, as demonstrated by the emergence of oseltamivir-resistant influenza during treatment involving combinations of the HTA nitazoxanite with oseltamivir in vitro and in vivo13.
Pharmaco-enhancements
Some antivirals face the issue of limited bioavailability which refers to the extent of which the drug is in circulation inside the body and thereby accessible at the target site. This could be due to a number of reasons like poor solubility, quick metabolism or undesired drug interactions. One way to get around this problem is to modify the drug itself to have more favourable characteristics (Fig. 5B). The antiviral acyclovir is a viral DNA polymerase chain terminator and is the first line treatment for HSV encephalitis and has limited effectiveness against other DNA viruses like cytomegalovirus, Epstein Barr virus and varicella zoster virus. Acyclovir is limited by its poor bioavailability and short half-life, requiring frequent dosing to achieve therapeutic concentrations. The modification of acyclovir to make valacyclovir, a L-valyl ester from acyclovir improved bioavailability at least three times94, allowing for a less frequent regimen which would improve adherence95. Subsequent modifications to improve the half-life and spectrum of antiviral activity against other DNA viruses, resulted in the synthesis of ganciclovir, penciclovir and famciclovir96,97,98,99.
If modification of a drug is not possible, or comes at the cost of reduced antiviral activity, another option is the addition of a pharmacokinetic enhancer. Pharmacokinetic enhancement, sometimes referred to as “boosting”, refers to the use of one drug to increase the effectiveness of another, the latter having antiviral activity (Fig. 5B). Early protease inhibitors against HIV such as saquinavir, indinavir, nelfinavir, and ritonavir were limited by their poor bioavailability, requirement of frequent dosing, high pill burdens and accompanying toxicity at therapeutic concentration. Ritonavir, although a protease inhibitor itself, was poorly tolerated in patients, but had the additional effect of being a potent inhibitor of cytochrome P450 CYP3A4 isoenzyme which is responsible for the metabolism of some other HIV protease inhibitors100. This led to the development of ritonavir boosted protease inhibitors as a treatment, allowing for smaller doses (compared to monotherapeutic concentrations) of both ritonavir and the primary protease inhibitor administered, reducing toxicity and improving pill burden and adherence to the regimen101. Boosted protease inhibitors also achieved better viral suppression, reducing the chance of resistance emerging102. Which is why all protease inhibitors recommended for treating HIV are also recommended to be boosted with ritonavir, with lopinavir even being co-formulated with ritonavir as a pill (Meltrex)100. In HCV ritonavir is part of the Viekira Pak regimen, a combination of ombitasvir a HCV NS5A inhibitor; paritaprevir, a HCV NS3/4A protease inhibitor; dasabuvir, a HCV non-nucleoside NS5B palm polymerase inhibitor and ritonavir co-packaged together. Ritonavir prolonging paritaprevir exposure in blood plasma103.
Pharmacokinetic enhancement was also applied for COVID-19, such is the case of Paxlovid, which is the combination of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) 3C-like protease inhibitor nirmatrelvir and ritonavir, which also reduced the metabolism of nirmatrelvir in the liver, allowing for prolonged nirmatrelvir antiviral effect104 (Fig. 5B).
Another way of improving the pharmacokinetic profile of a drug is by changing the delivery system such as by delivery to the target site. Antivirals against respiratory infections such influenza, respiratory syncytial virus (RSV) and coronaviruses had better bioavailability at the target site, when delivered by inhalation through devices like nebulisers and aerosol inhalers, and also lower systematic exposure and toxicity105. Also being explored are delivery methods like biocompatible targeted nanoparticles carrying drugs which have improved antiviral effect when compared to the same drug administered in vitro without nanoparticle delivery. Such delivery systems offer advantages of site specificity, controlled and sustained release, and compound stability. Additionally, hydrophobicity composition, size and surface to volume ratio of the particle can be modified to suit targeting needs, reducing off-target and undesirable side-effects, while improving bioavailability and lowering frequency of administration106,107.
Lethal mutagenesis
Many rapidly replicating RNA viruses operate near an error threshold, which is a limit on the mutational rate that, if exceeded, results in the accumulation of deleterious mutations such that fit viral genomes cannot replicate in the viral population, therefore driving the virus to extinction108. The strength of their high mutation rates in enabling rapid evolution and escape109,110,111 could be exploited as a weakness using base analogues to cause lethal mutagenesis. When incorporated into viral RNA, antiviral nucleoside analogues, such as ribavirin and favipiravir, and more recently molnupiravir, allow ambiguous base pairing in subsequent rounds of replication, resulting in an accumulation of mutations in the viral population driving it towards non-viability (Fig. 5C).
Some viruses with high error rates have evolved a proof-reader which corrects replication errors. The RdRp of coronaviruses has the characteristically low fidelity of other RNA viruses, and despite their large genome size (~30 kb), replicates its genome with relatively high fidelity. This is due to the presence of nsp14, an enzyme that has the dual function of viral RNA cap methyltransferase and 3′ to 5′ exoribonuclease (ExoN) ability to excise exogenous nucleotides112. This exonuclease activity, originally evolved to ensure sufficient fidelity for replication and mediate RNA recombination113, was even demonstrated to excise ribavirin from the viral genome (Fig. 5D), and is thought to explain the poor efficacy of ribavirin against coronaviruses in vivo114. Not all mutagens are excised. Favipiravir115,116 and molnupiravir117 have been demonstrated to evade coronavirus ExoN proofreading function.
For viruses that do not have proofreading activity, mutagenic nucleotide analogues can be countered with mutations. A single G64S substitution in the polymerase of poliovirus conferred resistance to ribavirin by increasing the fidelity of the viral polymerase118. Although high fidelity polymerase poliovirus can replicate similarly to wild type viruses, the ability of the virus to adapt to new antiviral compounds or environmental changes is reduced. Prolonged ribavirin treatment of HCV in patients revealed several mutations in the NS5B polymerase that conferred resistance, possibly through improved replicative fidelity119. Another method of mutagen resistance is through specific discrimination against them by viral polymerases. FMDV polymerases containing the M296I mutation had a lowered capacity to incorporate ribavirin itself120. In influenza, resistance to favipiravir is conferred by a K229R mutation in the PB1 segment of the polymerase. K229R does not increase fidelity, but reduces the incorporation of favipiravir (but not ribavirin) during RNA replication81 (Fig. 5D).
Despite these benefits, lethal mutagenesis has several limitations. Efficacy of the mutagen can depend on the mutational robustness of a virus which refers to the ability of the virus to maintain a phenotype while tolerating deleterious mutations121. For instance, CVB3 is less mutationally robust than poliovirus, resulting in the former being more susceptible to mutagens122. Treatment with mutagens requires high concentrations of drug, and intermediates of these drugs, may be utilised by host cellular processes, making them cytotoxic and or genotoxic to the host123,124,125. On the flipside, sublethal doses of nucleoside analogues are thought to provide a narrow window for viral persistence and the eventual emergence of resistance81,126. Worse still, these resistant viruses, coupled with their respective compensatory mutants, have the potential for transmission127. Aside from resistance, if doses of the drug are not sufficient to clear the viral load, the drug could have the opposing action of accelerating viral evolution rather than viral extinction14,128 (Fig. 5D).
Potential alternatives to conventional antivirals
Monoclonal antibodies
Monoclonal antibodies (mAbs) are mostly synthetic antibodies characterised by their monovalent affinity for a single epitope, rendering them an attractive tool as therapeutics129. Since the advent of hybridoma technology, mAbs have hitherto been utilised as therapeutics for several diseases including cancer, neurodegenerative disorders and infectious diseases130,131,132,133,134. Viral infections are no exception to this, with mAbs targeting viral glycoproteins (Fig. 6). Examples include nirsevimab, which targets respiratory syncytial virus (RSV) fusion protein and inmazeb (a mAb cocktail comprising atoltivimab, odesivimab and maftivimab), which targets the Zaire ebolavirus (EBOV) transmembrane glycoprotein. Both have already been approved by the U.S. FDA and are in active use135.

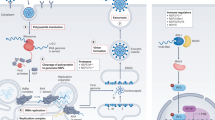
Schematic depicting the general life cycle of a RNA or DNA virus, starting from viral attachment to release of new viruses (Steps 1 to 7). In RNA viruses, replication of their RNA genome occurs directly in the cytoplasm (Step 3a) whereas in DNA viruses, the DNA genome is first transported into the nucleus before being transcribed into mRNA (Step 3b). Figure shows the targets inhibited by each antiviral alternative, denoted by the red lines. A Monoclonal antibodies target the surface glycoproteins of viruses, preventing its binding to cell surface receptors and attachment of virus to the cell. B RNA interference is comprised mainly of siRNA and miRNA, each of which can be assembled with the RNA-induced silencing complex (RISC). siRNA associated with RISC induce gene silencing by targeting viral RNA from RNA virus genomes or the transcribed mRNA of DNA virus genomes. In contrast, miRNA associated with RISC repress the translation of RNA, thereby preventing the synthesis of any structural proteins required for the assembly of new viruses. C Defective viral genomes (DVGs) are typically present in RNA virus populations and bear mutations which can occur at any part of the genome. DVGs compete with wild type genomes for replication resources such as RNA-dependent RNA polymerase, or for newly synthesised structural proteins essential for virus assembly. D Locked-nucleic acids (LNAs) anti-sense oligonucleotides are comprised of nucleotide subunits modified by a methylene bridge within their ribose sugar LNAs can bind to specific, functional RNA motifs critical for viral replication.
Unfortunately, viruses with rapid evolution rates counter efforts in utilising mAbs as antivirals. This was evident during the recent COVID-19 pandemic, when casirivimab-imdevimab, previously effective against the SARS-CoV-2 Delta variant, had poor neutralising activity against the later Omicron and XBB variants136,137,138,139. While casirivimab-imdevimab was initially effective against the Omicron variant during its inception, its efficacy waned against the later Omicron sub-variants, such as BA.2.75.2, BA.4.6 and BQ.1.1. This observation was consistent with other mAbs, such as sotrovimab and amubarvimab–romlusevimab140. Similarly, It was reported that a substitution of glutamic acid on residue 484 of SARS-CoV-2 spike protein arose after treatment with bamlanivimab and casirivimab-imdevimab, reducing mAb efficacy141,142,143,144. These prominent examples strongly suggest that the rise of immune escape mutants can be induced by mAbs.
How can we improve the efficacy of mAb therapy? Similar to DAAs, combinatory treatment with mAbs may present as an effective therapy option by widening the scope of epitopes that mAbs may target145,146. mAb cocktails confer broad-spectrum antiviral effects either against closely related variants of the same virus or to viruses within the same family. For instance, a cocktail of two mAbs conferred protection against different variants of SARS-CoV-2145, although further optimisation in vivo was suggested. In another example, two different EBOV-targeting mAbs derived from mice immunised with filovirus-based vaccines conferred protection to EBOV in mice and Sudan virus in guinea pigs. On the contrary, protection against EBOV in guinea pigs was not observed, suggesting that mAb cocktail efficacy may be dependent on the host species and adaptation of the virus147.
In the past decade, advances in protein engineering allowed the use of bispecific monoclonal antibodies (BsAbs) in treating various diseases148,149. BsAbs are specialised mAbs which target more than one epitope across different proteins, and thus might be a better alternative than conventional or combinatorial mAb therapy for viral infections150,151,152. BsAbs may be designed to target two different viral structural proteins, as demonstrated by the work of Chang et al. and Moirangthem et al. in thrombocytopenia syndrome virus and IAV, respectively153,154. In recent work on coronaviruses, BsAbs with other epitope-targeting configurations were employed as well. Lee et al. described the enhanced antiviral efficacy against Middle East respiratory syndrome coronavirus using a BsAb targeting two different epitopes on the spike protein surpassing that of parental mAb155. In contrast, Li et al. reported a broad-spectrum antiviral approach against various beta-coronaviruses using BsAbs targeting beta-coronavirus spike protein and its respective receptor, angiotensin-converting enzyme 2 which is found in humans156.
The abundance of epitopes that mAbs can target, coupled with the diversity of mAb-specificities which may be used in combination, positions mAbs as a strong class of antivirals. Despite the promising rudimentary studies, it should be noted that the generation of immune escape mutants may be closely associated with the use of mAb therapy. Resistance mutations have been reported to emerge in patients treated with mAb therapy157. and continue to complicate its efficacy. It has been postulated that new variants emerged in patients treated with mAb therapy. Taking SARS-CoV-2 as an example, spike protein mutations at residues 455 and 456 reportedly arising from mAb-induced selective pressure were shown to have contributed to the total immune escape of the XBB.1.5.70 mutant from a panel of antibodies isolated from patients post-infection with the BA.4 or BA.5 strain158. It remains crucial to be prudent in the use of mAb therapy to avoid generating novel immune escape mutants which may thwart the efficacies of existing antivirals.
RNA interference
RNA interference (RNAi) is a biological process present in mammalian and other invertebrate species, which induces gene silencing at the transcriptional or translational level159. The two main arms of the RNAi process consist of small molecules known as small-interfering RNA (siRNA) and microRNA (miRNA). Their synthesis is mediated by the RNase III Dicer enzyme and the products associated with the Argonaute (AGO) family of proteins to initiate the assembly of the RNA-induced silencing complex (RISC) which gets involved in gene silencing160,161 (Fig. 6). A third class of gene silencing RNA molecules known as piwi-interacting RNAs (piRNA) interact with the PIWI sub-group of the AGO family and are derived from Dicer-independent synthesis pathway162,163. These different types of RNA molecules drive different pathways that achieve RNAi with slightly different mechanisms.
In the siRNA pathway, siRNAs target RNA molecules by binding to complementary bases. The RISC then cleaves the target RNA, causing degradation. In the context of viral infections, double-stranded RNA (dsRNA) molecules derived from the viral genome are recognised as pathogen-associated molecular patterns and processed by the Dicer enzyme into siRNA molecules which facilitate repression of viral replication163. Upon successful loading into AGO, siRNA can bind to complementary virus-derived dsRNA strands which either exist as an inherent part of the viral genome or as intermediate by-products of viral replication. The binding mechanism driven by base-complementarity allows siRNA high target specificities to be designed as antivirals164. This enables the development of siRNAs to target both DNA and RNA viruses via their transcribed mRNA and genomic RNA respectively. Theoretically, viruses may also be targeted at different steps of their life cycle, although more studies might be required to prove this speculation165,166.
Several studies involving synthetic siRNA appear to point towards viral replication as the prime target for viral suppression. In herpes simplex virus type 1 (HSV-1), both Kalke et al. and Lasanen et al. repressed HSV-1 viral load and replication in vitro with a siRNA designed to target the HSV-1 UL29 gene, which encodes for a DNA-binding protein integral to the replication of HSV-1167,168. The interference of HSV-1 replication was successful even in acyclovir-resistant strains. Similarly, Idris et al. reported significant antiviral activity, as well as prophylactic potential, when administering various siRNA targeting different SARS-Cov-2 helicase and RNA-dependent RNA polymerase, which are involved in viral replication169. Likewise, Pei et al. have demonstrated the inhibitory effects of a recombinant siRNA targeting open-reading frame 7 (ORF7) of the varicella zoster virus (VZV) genome on virus replication170. Interestingly, ORF7 has been suggested to be crucial for proper cytoplasmic envelopment in VZV171. This might open a window of opportunity to expand the repertoire of antiviral siRNAs, although more studies are necessary to establish whether it is feasible to target any other steps of the virus life cycle besides viral replication.
On the other hand, miRNAs are mostly host-derived, although they may also virus-derived. Most of the known virus-derived miRNAs are encoded by DNA viruses such as members of the Herpesviridae family, and to a lesser extent RNA viruses172. Unlike their RNA-cleaving counterparts, miRNAs repress RNAi by regulating the expression and translation of target genes. One of the most widely studied miRNAs is miR-155, which is expressed in response to infection by a diversity of viral pathogens including influenza, SARS-CoV-2 and enterovirus A71. Following its induction, miR-155 modulates both innate and adaptive immune responses173,174.
Viruses have evolved to combat the antiviral effects of siRNA using viral suppressors of RNAi (VSR). VSRs were first described in plant viruses although in recent years, they have also been reported in several mammalian virus families175,176,177. It is interesting to note that VSRs across genetically different virus families obstruct RNAi through the same mechanism, by competing with host Dicer enzyme for dsRNA as template, thereby preventing any downstream synthesis of antiviral siRNA178. VSRs may also compete with existing siRNAs for cellular resources necessary for the assembly of RISC, as shown in the case of ZIKV where sub-genomic viral RNA can bind to the host proteins RHA and PACT and counteract RNAi179. In influenza A virus, non-structural protein 1 (NS1) functions as a VSR and represses RNAi. Moreover, NS1 is highly mutable and this has been shown to give rise to phylogenetically different variants of IAV. It is unknown whether these different phenotypes might further diversify and complicate the current pool of RNAi suppression mechanisms180,181.
In contrast, Lezcano et al. demonstrated that the 1A gene in drosophila C virus (DCV), which is typically involved in RNAi suppression, accumulated more polymorphisms in a RNAi-deficient infection model182. The team also reported increased viral fitness in DCV propagated in this model. Similarly, Samuel et al. reported increased virulence of Sindbis virus in Aedes aegypti bearing a defective Dicer-2 gene as compared to their wild type counterparts183. Overall, these studies indicate how VSR evolution may occur constantly even in the absence of any selective pressure from the host RNAi response, against the backdrop of existing rapid viral evolution. The dynamics between viral evolution and host response remain complex and this warrants more studies on the modulatory potential of various RNAi pathways before exploring its potential for antiviral therapeutic development.
Defective viral genomes
Many RNA viruses are known to generate defective interfering particles (DIPs), which are typically characterised by their truncated genomes known as defective viral genomes (DVGs), due to their high mutation rates during error-prone viral replication184. These truncated genomes may either work synergistically with wild type virus to facilitate the population’s adaptation, or antagonistically to hinder the life cycle of wild type virus when challenged with different environments109,185,186. As the diversity of DVGs and mechanisms of generation have been reviewed in detail, this review will focus on the role of DIPs in the context of antiviral therapy187.
Ever since their initial discovery in influenza viruses by Von Magnus, DIPs have been heavily described in experimental studies on influenza188,189,190. DIPs have also been described in other major virus families as naturally-occurring particles during infection, such as in flaviviruses, paramyxoviruses and coronaviruses191,192,193. Due to their smaller genomes, DIPs typically possess a competitive edge over wild type viruses within the same viral population during replication. Proposed mechanisms which confer fitness advantages to DIPs include competition with wild type virus for resources crucial for replication and assembly (Fig. 6), as well as induction of the host innate immune response194,195.
DVGs with immunostimulatory potentials were previously described by Sun et al., who showed that DVGs generated during natural infection with RSV could inhibit viral replication in vivo196. In contrast, the work conducted by Pelz et al. utilised cell lines lacking IFN activity against IAV replication. Instead, their results showed that DIPs which replicate faster exhibited stronger suppression of IAV replication in vitro, likely attributed to increased competition with wild type IAV due to its persistence197.
In another recent study, authors demonstrated that HIV-derived therapeutic interfering particles (TIPs) engineered to replicate with a basic reproductive ratio (R0) > 1, suppressed viral load in humanised mice 5 days post-challenge with HIV-1. An extended experiment involving infant rhesus macaque models challenged with chimeric simian/human immunodeficiency virus (SHIV) was also performed. Here, the authors reported that pre-treatment with a TIP adapted to Simian immunodeficiency virus reduced SHIV RNA and proviral DNA in lymphoid tissues, accompanied with enhanced survival rates. Reduction of viral loads in TIP-treated models was not attributed to immune responses due to the absence of elevated inflammatory response198.
Currently, studies on the precise antiviral mechanism of DIPs are limited. Future work could aim to deepen the differential profiling of DVG sub-populations and characterise their phenotypes to elucidate the complex interactions between DVGs/DIPs, standard virus and host factors.
Targeting viral RNA secondary structures with locked nucleic acids
The genomes of RNA viruses are known to fold on themselves, forming what is known as secondary and tertiary RNA structures199,200. These structures play important regulatory roles in the life cycle of many RNA viruses, for instance, the frame shifting element of coronaviruses that controls the essential programmed −1 ribosomal frameshift crucial for viral replication201,202; the conserved cloverleaf-like RNA structure in enteroviruses that recruits proteins for genome replication203; some conserved RNA pseudoknot structures resistance to host Xrn1 enzyme degradation, responsible for the production of subgenomic flaviviral RNA that influences flaviviral pathogenicity204,205,206; heterogeneous RNA conformations that control splicing and gene expression in HIV207 and influencing host immune suppression in influenza208. Conservation of these structures across viral genera and families203,205,206,209,210,211 and the important roles they play make these higher order RNA structures attractive targets when designing broad spectrum antivirals.
Locked nucleic acid (LNA) anti-sense oligonucleotides refer to a type of oligonucleotides that contain nucleotides modified by a methylene bridge within their ribose sugar that confers greater stability212. This stability enables LNAs to function as very potent compounds that bind with high affinity, favourable traits for therapeutic use213. LNAs can be used to target functional RNA motifs critical for viral replication (Fig. 6), by binding to them and forming RNA duplexes that are degraded by RNase H, as demonstrated in Japanese encephalitis virus in vitro214. LNA binding can also prevent the proper folding of RNA required for viral replication, as demonstrated in HCV where LNAs blocked conformational changes in the structure of NS5B CRE SL9266 pseudoknot, leading to impairment of viral translation215.
In IAV, a RNA structure known as a packaging stem–loop 2 plays a role in packaging of virions in vitro and pathogenesis in vivo. Targeting this structure with a LNA ensured a 100% survival rate in mice challenged with a lethal inoculum of virus 2 weeks pre- and 3 days post-infection. These mice went on to develop an immune response robust enough to withstand 10-fold lethal inoculum re-challenge. Importantly, attempts at selection of resistance did not yield any mutants, indicating high genetic barrier to resistance, likely because the structure is critical for viability. This LNA sustained its potency even against IAV resistant to NA inhibitors, providing an option to treat IAV antiviral resistance. Applying the same approach to the conserved RNA structures of SARS-CoV-2 prophylactically, greatly reduced viral transmission in hamsters210.
The binding of an LNA to the 5′ leader sequence of SARS-CoV-2, caused disruption of a stem-loop structure which was essential for replication in human cells in vitro and an in vivo mouse model. The LNA was effective against several SARS-CoV-2 variants of concern in vitro and when administered daily to mice intranasally216. LNAs are flexible in the sense that they can be modified to target conserved RNA structures of other viruses which have similar essential roles in viral replication. Therefore, there is a need to comprehensively identify regions of viral RNA genome that are conserved and critical for replication beyond the proteins that they code for217,218 and design LNAs that bind to and impair their function, as demonstrated for CHIKV where LNAs designed in against individual replicative elements in viral RNA were able to interfere with replication complex and prevent production of infectious progeny219.
Current strategies to discover new antivirals
Deep mutational scanning
Deep mutational scanning (DMS) is a technique that entails the use of deep sequencing technology in conjunction with a mutant library of genes or genomes generated from reverse genetics to study the phenotypic effects of any mutation of interest. The output that can be derived from DMS has been used extensively in several applications across virology, including virus surveillance, virus evolution and prediction of immune escape mutants220,221,222,223.
One of the major areas that is worth exploring with DMS is epistasis, which is characterised by the mutual influence of two or more mutations, which typically leads to an observable phenotypic change224. These fitness-associated interactions at the genetic level provide a wealth of information on the relationships between different viral genes or proteins and their impact on viral fitness, which may be exploited for the development of antiviral strategies225.
The effects of epistasis are commonly observed in highly mutable RNA viruses such as influenza viruses of the H3N2 subtype, which gain egg-adaptive mutations during the vaccine production process226. It was established that L194P, a leucine to proline substitution on residue 194 of hemagglutinin, exacerbates antigenicity and reduces vaccine efficacy. Interestingly, L194P bears opposing effects to several other amino acid substitutions such as G186V and this incompatibility yields detrimental effects to H3N2 influenza viability in vitro227,228. Conversely, Lei et al. reported that double mutants bearing G186D and D190N substitutions were more prevalent in circulating H3N2 influenza strains and exhibited enhanced receptor-binding capabilities than their single mutant counterparts229. Such synergistic epistatic interactions have also been observed in NA substitutions, whereby circulating H1N1 strains bearing both the V241I and N369K mutations have increased prevalence and fitness230.
The consistent emergence of epistatic interactions are well-represented in SARS-CoV-2. One of the earliest mutations in the spike protein, D614G, was associated with increased infectivity and increased viral load. Virulence was further intensified with the emergence of the H655Y mutation in the gamma strain which also persisted in the later omicron strain. SARS-CoV-2 evolution remains an ongoing process, with constant emergence of new mutations further diversifying the combinatorial effects from epistatic interactions231,232,233. Similarly, in HCV, epistatic interactions in the non-structural 3 (NS3) protein stabilised its conformation and promoted drug resistance even though most of the mutations alone were non-beneficial234,235. These findings seem to suggest the favourable selection of viruses with strong epistatic interactions in nature.
The strong interplay between epistasis and virulence makes it an important factor when considering the design of antivirals. In recent years, studies utilised deep mutational scanning (DMS) to study resistance mutations in other pathogens such as bacteria and fungi in a bid to better understand their impact on antimicrobial development236,237,238. Unsurprisingly, the use of DMS for such purposes has also gained traction in antiviral research in SARS-CoV-2 and IAV for example233,239,240. Continued monitoring of resistance mutations is paramount for the exploitation of epistatic interactions in our favour.
Proteomics-based approaches
Protein-protein interactions (PPI) are ubiquitous and constitute an important part of cellular processes. In viral infections, virus proteins react with host proteins to carry out a variety of functions throughout the virus life cycle241. These virus-host interactions form the basis for the design of HTAs, such as the HIV-1 antiviral maraviroc which prevents entry of HIV-1242. Understanding these interactions can drive the discovery of new druggable targets.
Proteomics-based approaches such as mass spectrometry, coupled with advances in bioinformatics tools allowed us to identify targets within the host proteome243,244. Kinases, which catalyse phosphorylation reactions across different biomolecules, is an example of a class of potentially druggable targets that has been studied extensively with proteomics-based approaches. These enzymes play a role in regulating viral replication245,246. Haas et al. identified 13 kinases modulated by IAV from a pool of 332 IAV-host PPI and described two kinase inhibitors with pan-antiviral activity against both IAV and SARS-CoV-2247. Similarly, other studies utilising comparative proteomics to study phosphorylation profiles successfully identified kinase inhibitors with antiviral activity against enterovirus A71 and SARS-CoV-2 respectively248,249,250.
The discovery of new PPI may also uncover the mechanisms of existing drugs and their potential to be repurposed as antivirals. This was evident during the COVID-19 pandemic, where Gordon et al. discovered 69 compounds that targeted a subset of the 332 high confidence PPI identified between SARS-CoV-2 and host proteins251.
Utilising artificial intelligence
Artificial intelligence (AI) and its multitude of machine learning algorithms available, can be harnessed to reduce the costs of and accelerate the discovery of new antivirals252. Machine learning approaches such as reinforcement learning utilise artificial neural networks trained with the appropriate data to generate novel compounds with the desired properties253. Deep reinforcement learning has been demonstrated to be able to design novel chemical entities against wild type and oseltamivir resistant influenza254 which showed antiviral activity in vitro and in vivo255.
AI can also assist in repurposing existing antivirals. Conventional drug-target prediction involves using available information in a “one-sided view”, but an approach called matrix completion with multi‐view side information (MVMC), utilises multiple available structural and chemical information about a given drug, as well as sequential or structural information of a target to complete an interaction matrix. MVMC demonstrated not only greater accuracy to previous methods of predictions, but also unveiled novel drug-target interactions for previously approved drugs256. Such approaches to drug repositioning during events like the COVID-19 pandemic, could assist clinicians in selecting prospective drug candidates to be further validated, streamlining the drug repurposing process in order to respond quickly to rapidly mutating viruses like SARS-CoV-2257. Versatility of such drug prediction tools allows application for many other human pathogenic viruses.
Conventionally, antiviral resistance is discovered through phenotypic and genotypic techniques, which are time consuming and costly. AI can be employed to study viral evolution258 and predict the emergence of antiviral resistance before they occur. The massive amount of sequencing performed on the genome of SARS-CoV-2 can be leveraged to study viral evolution by extensively mapping the effects of viral mutations on their fitness259. Mapping mutations in the viral genome regarding their benefits, fitness costs, and compensatory mutations, could reveal pathways of viral evolution and patterns of constraint which can guide antiviral drug design260. Although not as extensively done for SARS-CoV-2, mapping of fitness landscapes based on collective sequence data has been attempted (on the genome, but more often on specific genes) for many other pathogenic human viruses such as polio261,262, HIV263, DENV264, influenza265 and HCV266.
Viruses are constantly evolving and as their genomes change, so do their fitness landscapes and even evolutionary constraints267. As such, there is a need to constantly build on and update previously generated maps, so that they provide updated information268. Recently, a new AI tool called EVEscape has been developed to study viral evolution, using a deep-learning approach and trained with available historical sequences and structural data, is shown to predict the potential of escape mutations to emerge269.
Discussion and concluding remarks
In this review, we outlined the current challenges that are imposed on conventional antivirals by virus evolution and the emergence of antiviral resistance. We also discussed the current alternatives which are being explored to DAAs. Amongst these alternative approaches, monoclonal antibodies and siRNAs are the only ones which have been implemented as therapeutics. Currently, there is a lack of information on proposed alternatives such as harnessing epistatic interactions, DIPs or LNAs. More studies are warranted to elucidate specific mechanisms and assess the safety of utilising these alternatives. Until then, we remain reliant mostly on existing DAAs as the main class of antivirals.
Despite the limitations of conventional antivirals and the challenges posed by viral evolution, new potential DAAs are continuously being discovered and developed. These drugs have greater potency, safety profile and specificity towards target tissues than their predecessors. Existing helicase-primase inhibitors like Pritelivir and Amenamevir are used to treat nucleoside analogue resistant HSV270, but lack sufficient penetration into neuronal tissue to affect viral latency271. Newer helicase-primase inhibitors such as IM-250 have improved tissue exposure that allow for treatment of latent HSV and with low frequency of resistance272. Nucleoside analogues and helicase inhibitors have been demonstrated to work synergistically against HSV273 providing more options for combination therapy. Nucleoside analogues against CMV often suffer from dose-limiting cytotoxicity. Filociclovir, a new nucleoside analogue has been demonstrated to not only have a better safety profile, but also be effective against some resistance mutants of previous nucleoside analogues274. A potential new combination treatment regimen against HIV, islatravir and lenacapavir with multiple mechanisms of action and target multiple stages of the life cycle, respectively, have high genetic barriers to resistance and demonstrated no antagonism nor cross resistance in vitro275.
We also discussed the potential of proteomics-based approaches and AI to study viral-host interactomes251,276,277,278. Even though these approaches are new with rudimentary progress, they present opportunities to fill the current knowledge gaps in virus-host interactions for the discovery of new antivirals.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- AGO:
-
Argonaute
- AI:
-
Artificial intelligence
- BsAb:
-
Bispecific monoclonal antibody
- CHIKV:
-
Chikungunya virus
- CMV:
-
Cytomegalovirus
- COVID-19:
-
Coronavirus disease 2019
- CVB3:
-
Coxsackie virus B3
- DAA:
-
Direct acting antiviral
- DCV:
-
Drosophila C virus
- DENV:
-
Dengue virus
- DIP:
-
Defective interfering particle
- DMS:
-
Deep mutational scanning
- DVG:
-
Defective viral genome
- EBOV:
-
Ebolavirus
- ExoN:
-
Exoribonuclease
- FTC:
-
Emtricitabine
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- HIV:
-
Human immunodeficiency virus
- HSV:
-
Herpes simplex virus
- HTA:
-
Host-targeted antiviral
- IAV:
-
Influenza A virus
- IBV:
-
Influenza B virus
- IFN:
-
Interferon
- LNA:
-
Locked nucleic acid
- mAb:
-
Monoclonal antibody
- miRNA:
-
MicroRNA
- MVMC:
-
Multi‐View Matrix Completion
- NA:
-
Neuraminidase
- Nsp:
-
Non-structural protein
- NRTI:
-
Nucleoside reverse transcriptase inhibitor
- NNRTI:
-
Non-nucleoside reverse transcriptase inhibitor
- PPI:
-
Protein-protein interaction
- RdRp:
-
RNA-dependent RNA polymerase
- RNAi:
-
RNA interference
- RSV:
-
Respiratory syncytial virus
- SARS-CoV-2:
-
Severe acute respiratory syndrome virus 2
- siRNA:
-
Small interfering RNA
- TIP:
-
Therapeutic interfering particle
- TK:
-
Thymidine kinase
- TMPRSS2:
-
Transmembrane serine protease 2
- U.S. FDA:
-
United States Food and Drug Administration
- VSR:
-
Viral suppressors of RNAi
References
Li, G., Hilgenfeld, R., Whitley, R. & De Clercq, E. Therapeutic strategies for COVID-19: progress and lessons learned. Nat. Rev. Drug Discov. 22, 449–475 (2023).
Torreele, E. et al. From private incentives to public health need: rethinking research and development for pandemic preparedness. Lancet Glob. Health 11, e1658–e1666 (2023).
Karim, M., Lo, C.-W. & Einav, S. Preparing for the next viral threat with broad-spectrum antivirals. J. Clin. Invest. 133, e170236 (2023).
Giovanetti, M. et al. Epidemic history and evolution of an emerging threat of international concern, the severe acute respiratory syndrome coronavirus 2. J. Med. Virol. 95, e29012 (2023).
Prusoff, W. H. Synthesis and biological activities of iododeoxyuridine, an analogue of thymidine. Biochim. Biophys. Acta 32, 295–296 (1959).
Parra-Rojas, C., Nguyen, V. K., Hernandez-Mejia, G. & Hernandez-Vargas, E. A. Neuraminidase inhibitors in influenza treatment and prevention—is it time to call it a day? Viruses 10, 454 (2018).
Alaofi, A. L. The Glu143 residue might play a significant role in T20 peptide binding to HIV-1 receptor gp41: an in silico study. Molecules 27, 3936 (2022).
Luber, A. D. Genetic barriers to resistance and impact on clinical response. MedGenMed Medscape Gen. Med. 7, 69 (2005).
Scheel, T. K. H. & Rice, C. M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 19, 837–849 (2013).
Holmberg, S. D., Spradling, P. R., Moorman, A. C. & Denniston, M. M. Hepatitis C in the United States. N. Engl. J. Med. 368, 1859–1861 (2013).
Plummer, E. et al. Dengue virus evolution under a host-targeted antiviral. J. Virol. 89, 5592–5601 (2015).
Warfield, K. L. et al. Lack of selective resistance of influenza A virus in presence of host-targeted antiviral, UV-4B. Sci. Rep. 9, 7484 (2019).
Tilmanis, D. et al. Host-targeted nitazoxanide has a high barrier to resistance but does not reduce the emergence or proliferation of oseltamivir-resistant influenza viruses in vitro or in vivo when used in combination with oseltamivir. Antiviral Res. 180, 104851 (2020).
Schreiber, A. et al. The host-targeted antiviral drug Zapnometinib exhibits a high barrier to the development of SARS-CoV-2 resistance. Antiviral Res. 225, 105840 (2024).
Bull, R. A. et al. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog 7, e1002243 (2011).
Wang, D. et al. Evolution of drug-resistant viral populations during interruption of antiretroviral therapy. J. Virol. 85, 6403–6415 (2011).
Mason, S., Devincenzo, J. P., Toovey, S., Wu, J. Z. & Whitley, R. J. Comparison of antiviral resistance across acute and chronic viral infections. Antiviral Res. 158, 103–112 (2018).
Bagaglio, S., Uberti-Foppa, C. & Morsica, G. Resistance mechanisms in hepatitis C virus: implications for direct-acting antiviral use. Drugs 77, 1043–1055 (2017).
Simonsen, L. et al. The genesis and spread of reassortment human influenza A/H3N2 viruses conferring adamantane resistance. Mol. Biol. Evol. 24, 1811–1820 (2007).
Nelson, M. I., Simonsen, L., Viboud, C., Miller, M. A. & Holmes, E. C. The origin and global emergence of adamantane resistant A/H3N2 influenza viruses. Virology 388, 270–278 (2009).
Yang, J.-R. Reassortment and mutations associated with emergence and spread of oseltamivir-resistant seasonal influenza A/H1N1 viruses in 2005–2009. PLoS ONE 6, e18177 (2011).
Nora, T. et al. Contribution of recombination to the evolution of human immunodeficiency viruses expressing resistance to antiretroviral treatment. J. Virol. 81, 7620–7628 (2007).
Dalakas, M. C. et al. Mitochondrial myopathy caused by long-term zidovudine therapy. N. Engl. J. Med. 322, 1098–1105 (1990).
Arnaudo, E. et al. Depletion of muscle mitochondrial DNA in AIDS patients with zidovudine-induced myopathy. Lancet 337, 508–510 (1991).
Peters, B. S., Winer, J., Landon, D. N., Stotter, A. & Pinching, A. J. Mitochondrial myopathy associated with chronic zidovudine therapy in AIDS. QJM Int. J. Med. https://doi.org/10.1093/oxfordjournals.qjmed.a068738 (1993).
Tisdale, M., Kemp, S. D., Parry, N. R. & Larder, B. A. Rapid in vitro selection of human immunodeficiency virus type 1 resistant to 3’-thiacytidine inhibitors due to a mutation in the YMDD region of reverse transcriptase. Proc. Natl. Acad. Sci. USA. 90, 5653–5656 (1993).
Cooper, D. A. et al. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N. Engl. J. Med. 359, 355–365 (2008).
Fransen, S. et al. Loss of raltegravir susceptibility by human immunodeficiency virus type 1 is conferred via multiple nonoverlapping genetic pathways. J. Virol. 83, 11440–11446 (2009).
Margot, N. et al. Phenotypic resistance to lenacapavir and monotherapy efficacy in a proof-of-concept clinical study. J. Antimicrob. Chemother. 77, 989–995 (2022).
Wirden, M. et al. Ultra-rapid selection of the N74D capsid inhibitor resistance mutation after 3 weeks on lenacapavir. J. Antimicrob. Chemother. 79, 1706–1707 (2024).
Kandeel, M. The impact of the M184V resistance mutation on treatment outcomes in patients with HIV infection: a systematic review and meta-analysis. AIDS Rev. 26, 136–144 (2023).
Smith, I. L. et al. High-level resistance of cytomegalovirus to ganciclovir is associated with alterations in both the UL97 and DNA polymerase genes. J. Infect. Dis. 176, 69–77 (1997).
Feng, M. et al. Doravirine suppresses common nonnucleoside reverse transcriptase inhibitor-associated mutants at clinically relevant concentrations. Antimicrob. Agents Chemother. 60, 2241–2247 (2016).
Reddy, N., Papathanasopoulos, M., Steegen, K. & Basson, A. E. K. 103N, V106M and Y188L significantly reduce HIV-1 subtype C phenotypic susceptibility to doravirine. Viruses 16, 1493 (2024).
Fujii, H. et al. Application of next-generation sequencing to detect acyclovir-resistant herpes simplex virus type 1 variants at low frequency in thymidine kinase gene of the isolates recovered from patients with hematopoietic stem cell transplantation. J. Virol. Methods 251, 123–128 (2018).
Schalkwijk, H. H. et al. Heterogeneity and viral replication fitness of HSV-1 clinical isolates with mutations in the thymidine kinase and DNA polymerase. J. Antimicrob. Chemother. 77, 3153–3162 (2022).
Smith-Palmer, J., Cerri, K. & Valentine, W. Achieving sustained virologic response in hepatitis C: a systematic review of the clinical, economic and quality of life benefits. BMC Infect. Dis. 15, 19 (2015).
Lahser, F. et al. Interim analysis of a 3-year follow-up study of NS5A and NS3 resistance-associated substitutions after treatment with grazoprevir-containing regimens in participants with chronic HCV infection. Antivir. Ther. 23, 593–603 (2018).
Popping, S. et al. Transmission of NS5A-inhibitor resistance-associated substitutions among men who have sex with men recently infected with hepatitis C virus genotype 1a. Clin. Infect. Dis. 71, e215–e217 (2020).
Niman, H. Emergence and fixing of antiviral resistance in influenza A via recombination and hitch hiking. Nat. Preced. https://doi.org/10.1038/npre.2009.2832.1 (2009).
Bright, R. A. Adamantane resistance among influenza A viruses isolated early during the 2005-2006 influenza season in the United States. JAMA 295, 891 (2006).
Salloum, S., Kluge, S. F., Kim, A. Y., Roggendorf, M. & Timm, J. The resistance mutation R155K in the NS3/4A protease of hepatitis C virus also leads the virus to escape from HLA-A*68-restricted CD8 T cells. Antiviral Res. 87, 272–275 (2010).
Roy, S., Sukla, S., De, A. & Biswas, S. Non-cytopathic herpes simplex virus type-1 isolated from acyclovir-treated patients with recurrent infections. Sci. Rep. 12, 1345 (2022).
Powdrill, M. H. et al. Contribution of a mutational bias in hepatitis C virus replication to the genetic barrier in the development of drug resistance. Proc. Natl. Acad. Sci. USA 108, 20509–20513 (2011).
Domingo, E. Quasispecies dynamics in disease prevention and control. in Virus as Populations 263–297 (Elsevier, 2016).
Strasfeld, L. & Chou, S. Antiviral drug resistance: mechanisms and clinical implications. Infect. Dis. Clin. North Am. 24, 413–437 (2010).
Ghany, M. G. & Doo, E. C. Antiviral resistance and hepatitis B therapy. Hepatology 49, S174–S184 (2009).
Chen, R. et al. Antiviral drug delivery system for enhanced bioactivity, better metabolism and pharmacokinetic characteristics. Int. J. Nanomedicine 16, 4959–4984 (2021).
Fox, Z. et al. Viral resuppression and detection of drug resistance following interruption of a suppressive non-nucleoside reverse transcriptase inhibitor-based regimen. AIDS 22, 2279–2289 (2008).
Sethi, A. K., Celentano, D. D., Gange, S. J., Moore, R. D. & Gallant, J. E. Association between adherence to antiretroviral therapy and human immunodeficiency virus drug resistance. Clin. Infect. Dis. 37, 1112–1118 (2003).
Dayyeh, B. K. A. et al. IL28B alleles exert an additive dose effect when applied to HCV-HIV coinfected persons undergoing peginterferon and ribavirin therapy. PLoS ONE 6, e25753 (2011).
Mallal, S. et al. HLA-B*5701 screening for hypersensitivity to abacavir. N. Engl. J. Med. 358, 568–579 (2008).
DeLaurentis, C. E., Kiser, J. & Zucker, J. New perspectives on antimicrobial agents: tecovirimat for treatment of human monkeypox virus. Antimicrob. Agents Chemother. 66, e01226–22 (2022).
Almehmadi, M. et al. A glance at the development and patent literature of tecovirimat: the first-in-class therapy for emerging monkeypox outbreak. Viruses 14, 1870 (2022).
Karan, A. et al. Surveillance of complicated Mpox cases unresponsive to oral tecovirimat in Los Angeles County, 2022. J. Infect. Dis. 229, S249–S254 (2024).
Martinez, A. et al. Successful outcome after treatment with cidofovir, vaccinia, and extended course of tecovirimat in a newly-diagnosed HIV patient with severe Mpox: a case report. Vaccines 11, 650 (2023).
Brown, L. et al. Severe ocular Mpox in person living with advanced HIV treated with extended course of tecovirimat. HIV Med. 25, 1086–1090 (2024).
Griffith, D. C. et al. Mpox recurrence and tecovirimat resistance in a patient with advanced human immunodeficiency virus disease. Open Forum Infect. Dis. 11, ofae549 (2024).
Roosenhoff, R. et al. Viral kinetics and resistance development in children treated with neuraminidase inhibitors: the Influenza Resistance Information Study (IRIS). Clin. Infect. Dis. 71, 1186–1194 (2020).
Nguyen Thi Thu, P., Ngo Thi Quynh, M., Pham Van, L., Nguyen Van, H. & Nguyen Thanh, H. Determination of risk factors associated with the failure of 12 weeks of direct-acting antiviral therapy in patients with hepatitis C: a prospective study. BioMed Res. Int. 2022, 6054677 (2022).
Sutherland, K. A. et al. HIV-1 subtype influences susceptibility and response to monotherapy with the protease inhibitor lopinavir/ritonavir. J. Antimicrob. Chemother. 70, 243–248 (2015).
Kai, Y. et al. Baseline quasispecies selection and novel mutations contribute to emerging resistance-associated substitutions in hepatitis C virus after direct-acting antiviral treatment. Sci. Rep. 7, 41660 (2017).
Palanisamy, N. et al. Worldwide prevalence of baseline resistance-associated polymorphisms and resistance mutations in HCV against current direct-acting antivirals. Antivir. Ther. 23, 485–493 (2018).
Faiz, S. et al. Study of drug resistance-associated genetic mutations, and phylo-genetic analysis of HCV in the Province of Sindh, Pakistan. Sci. Rep. 13, 12213 (2023).
Nakamoto, S., Kanda, T., Wu, S., Shirasawa, H. & Yokosuka, O. Hepatitis C virus NS5A inhibitors and drug resistance mutations. World J. Gastroenterol. 20, 2902–2912 (2014).
Paolucci, S. et al. Baseline and breakthrough resistance mutations in HCV patients failing DAAs. Sci. Rep. 7, 16017 (2017).
Götte, M. The distinct contributions of fitness and genetic barrier to the development of antiviral drug resistance. Curr. Opin. Virol. 2, 644–650 (2012).
Grimes, S. L. et al. A mutation in the coronavirus nsp13-helicase impairs enzymatic activity and confers partial remdesivir resistance. mBio 14, e0106023 (2023).
Stannard, H. L. et al. Assessing the fitness of a dual-antiviral drug resistant human influenza virus in the ferret model. Commun. Biol. 5, 1026 (2022).
Back, N. K. et al. Reduced replication of 3TC-resistant HIV-1 variants in primary cells due to a processivity defect of the reverse transcriptase enzyme. EMBO J. 15, 4040–4049 (1996).
Hung, M. et al. Elucidating molecular interactions of L-nucleotides with HIV-1 reverse transcriptase and mechanism of M184V-caused drug resistance. Commun. Biol. 2, 469 (2019).
Bacon, T. H., Levin, M. J., Leary, J. J., Sarisky, R. T. & Sutton, D. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin. Microbiol. Rev. 16, 114–128 (2003).
Pottage, J. C. & Kessler, H. A. Herpes simplex virus resistance to acyclovir: clinical relevance. Infect. Agents Dis. 4, 115–124 (1995).
Piret, J. & Boivin, G. Resistance of herpes simplex viruses to nucleoside analogueues: mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 55, 459–472 (2011).
Hickerson, B. T. et al. Impact of baloxavir resistance-associated substitutions on influenza virus growth and drug susceptibility. J. Virol. 97, e0015423 (2023).
Bloom, J. D., Gong, L. I. & Baltimore, D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 328, 1272–1275 (2010).
Ono, S. K. et al. The polymerase L528M mutation cooperates with nucleotide binding-site mutations, increasing hepatitis B virus replication and drug resistance. J. Clin. Invest. 107, 449–455 (2001).
Xu, H.-T. et al. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J. Virol. 85, 11300–11308 (2011).
Xu, H.-T. et al. Role of the K101E substitution in HIV-1 reverse transcriptase in resistance to rilpivirine and other nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 57, 5649–5657 (2013).
Delaney, W. E. et al. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J. Virol. 77, 11833–11841 (2003).
Goldhill, D. H. et al. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. USA. 115, 11613–11618 (2018).
Delang, L. et al. Mutations in the chikungunya virus non-structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral. J. Antimicrob. Chemother. 69, 2770–2784 (2014).
Abdelnabi, R. et al. Understanding the mechanism of the broad-spectrum antiviral activity of favipiravir (T-705): key role of the F1 motif of the viral polymerase. J. Virol. 91, e00487-17 (2017).
Müller, J. A. et al. Reduced susceptibility to VIRIP-based HIV-1 entry inhibitors has a high genetic barrier and severe fitness costs. J. Virol. 92, e00733-18 (2018).
Sulkowski, M. S. et al. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N. Engl. J. Med. 370, 211–221 (2014).
Costa, V. D. et al. Treatment of chronic HCV infection with DAAs in Rio de Janeiro/Brazil: SVR rates and baseline resistance analyses in NS5A and NS5B genes. PLoS ONE 14, e0216327 (2019).
Wahid, B. Successful treatment of HBV, HCV & HEV, with 12-week long use of tenofovir, sofosbuvir, daclatasvir, and ribavirin: a case report. J. Infect. Public Health 13, 149–150 (2020).
Wagoner, J. et al. Combinations of host- and virus-targeting antiviral drugs confer synergistic suppression of SARS-CoV-2. Microbiol. Spectr. 10, e0333122 (2022).
Schreiber, A. et al. The MEK1/2 inhibitor ATR-002 (zapnometinib) synergistically potentiates the antiviral effect of direct-acting anti-SARS-CoV-2 drugs. Pharmaceutics 14, 1776 (2022).
Franco, E. J., Pires De Mello, C. P. & Brown, A. N. Antiviral evaluation of UV-4B and interferon-alpha combination regimens against dengue virus. Viruses 13, 771 (2021).
Nitta, S. et al. Impact of novel NS5A resistance-associated substitutions of hepatitis C virus detected in treatment-experienced patients. Sci. Rep. 9, 5722 (2019).
Wensing, A. M. et al. 2022 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 30, 559–574 (2022).
Paton, N. I. et al. Dolutegravir or darunavir in combination with zidovudine or tenofovir to treat HIV. N. Engl. J. Med. 385, 330–341 (2021).
Soul-Lawton, J. et al. Absolute bioavailability and metabolic disposition of valaciclovir, the L-valyl ester of acyclovir, following oral administration to humans. Antimicrob. Agents Chemother. 39, 2759–2764 (1995).
Fife, K. H., Barbarash, R. A., Rudolph, T., Degregorio, B. & Roth, R. Valaciclovir versus acyclovir in the treatment of first-episode genital herpes infection. Results of an international, multicenter, double-blind, randomized clinical trial. The Valaciclovir International Herpes Simplex Virus Study Group. Sex. Transm. Dis. 24, 481–486 (1997).
Erice, A. et al. Ganciclovir treatment of cytomegalovirus disease in transplant recipients and other immunocompromised hosts. JAMA 257, 3082–3087 (1987).
Anderson, R. D. et al. Ganciclovir absolute bioavailability and steady-state pharmacokinetics after oral administration of two 3000-mg/d dosing regimens in human immunodeficiency virus- and cytomegalovirus-seropositive patients. Clin. Ther. 17, 425–432 (1995).
Boyd, M. R., Bacon, T. H., Sutton, D. & Cole, M. Antiherpesvirus activity of 9-(4-hydroxy-3-hydroxy-methylbut-1-yl)guanine (BRL 39123) in cell culture. Antimicrob. Agents Chemother. 31, 1238–1242 (1987).
Tyring, S. et al. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia. A randomized, double-blind, placebo-controlled trial. Collaborative Famciclovir Herpes Zoster Study Group. Ann. Intern. Med. 123, 89–96 (1995).
Hull, M. W. & Montaner, J. S. G. Ritonavir-boosted protease inhibitors in HIV therapy. Ann. Med. 43, 375–388 (2011).
Scott, J. D. Simplifying the treatment of HIV infection with ritonavir-boosted protease inhibitors in antiretroviral-experienced patients. Am. J. Health. Syst. Pharm. 62, 809–815 (2005).
Youle, M. Overview of boosted protease inhibitors in treatment-experienced HIV-infected patients. J. Antimicrob. Chemother. 60, 1195–1205 (2007).
Raedler, L. A. Viekira Pak (Ombitasvir, Paritaprevir, and Ritonavir Tablets; Dasabuvir Tablets): all-oral fixed combination approved for genotype 1 chronic hepatitis C infection. Am. Health Drug Benefits 8, 142–147 (2015).
Owen, D. R. et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021).
Alshammari, M. K. et al. A systematic review of clinical pharmacokinetics of inhaled antiviral. Medicina 59, 642 (2023).
Sanna, V., Satta, S., Hsiai, T. & Sechi, M. Development of targeted nanoparticles loaded with antiviral drugs for SARS-CoV-2 inhibition. Eur. J. Med. Chem. 231, 114121 (2022).
Žigrayová, D., Mikušová, V. & Mikuš, P. Advances in antiviral delivery systems and chitosan-based polymeric and nanoparticulate antivirals and antiviral carriers. Viruses 15, 647 (2023).
Holmes, E. C. Error thresholds and the constraints to RNA virus evolution. Trends Microbiol. 11, 543–546 (2003).
Vignuzzi, M., Stone, J. K., Arnold, J. J., Cameron, C. E. & Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439, 344–348 (2006).
Fitzsimmons, W. J. et al. A speed-fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. 16, e2006459 (2018).
Mattenberger, F., Vila-Nistal, M. & Geller, R. Increased RNA virus population diversity improves adaptability. Sci. Rep. 11, 6824 (2021).
Bouvet, M. et al. RNA 3’-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Natl. Acad. Sci. USA. 109, 9372–9377 (2012).
Gribble, J. et al. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 17, e1009226 (2021).
Ferron, F. et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. USA. 115, E162–E171 (2018).
Shannon, A. et al. Favipiravir strikes the SARS-CoV-2 at its Achilles heel, the RNA polymerase. bioRxiv https://doi.org/10.1101/2020.05.15.098731 (2020).
Peng, Q. et al. Structural basis of SARS-CoV-2 polymerase inhibition by favipiravir. Innovation 2, 100080 (2021).
Kabinger, F. et al. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 28, 740–746 (2021).
Pfeiffer, J. K. & Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogues via increased fidelity. Proc. Natl. Acad. Sci. USA. 100, 7289–7294 (2003).
Mejer, N. et al. Mutations identified in the hepatitis C virus (HCV) polymerase of patients with chronic HCV treated with ribavirin cause resistance and affect viral replication fidelity. Antimicrob. Agents Chemother. 64, e01417-20 (2020).
Sierra, M. et al. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: implications for error catastrophe. J. Virol. 81, 2012–2024 (2007).
Sanjuán, R., Cuevas, J. M., Furió, V., Holmes, E. C. & Moya, A. Selection for robustness in mutagenized RNA viruses. PLoS Genet. 3, e93 (2007).
Graci, J. D. et al. Mutational robustness of an RNA virus influences sensitivity to lethal mutagenesis. J. Virol. 86, 2869–2873 (2012).
Zhou, S. et al. β-d-N4-hydroxycytidine inhibits SARS-CoV-2 through lethal mutagenesis but is also mutagenic to mammalian cells. J. Infect. Dis. 224, 415–419 (2021).
Brandsma, I. et al. Genotoxicity assessment of potentially mutagenic nucleoside analogueues using ToxTracker®. Toxicol. Lett. 362, 50–58 (2022).
Waters, M. D., Warren, S., Hughes, C., Lewis, P. & Zhang, F. Human genetic risk of treatment with antiviral nucleoside analogue drugs that induce lethal mutagenesis: the special case of molnupiravir. Environ. Mol. Mutagen. 63, 37–63 (2022).
Pauly, M. D. & Lauring, A. S. Effective lethal mutagenesis of influenza virus by three nucleoside analogues. J. Virol. 89, 3584–3597 (2015).
Goldhill, D. H. et al. Favipiravir-resistant influenza A virus shows potential for transmission. PLoS Pathog. 17, e1008937 (2021).
Sanderson, T. et al. A molnupiravir-associated mutational signature in global SARS-CoV-2 genomes. Nature 623, 594–600 (2023).
Hansel, T. T., Kropshofer, H., Singer, T., Mitchell, J. A. & George, A. J. T. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 9, 325–338 (2010).
Köhler, G. & Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975).
Ulrich, S. & Ebel, F. Monoclonal antibodies as tools to combat fungal infections. J. Fungi 6, 22 (2020).
Mitra, S. & Tomar, P. C. Hybridoma technology; advancements, clinical significance, and future aspects. J. Genet. Eng. Biotechnol. 19, 159 (2021).
Chen, Z. et al. Overview of tumor immunotherapy based on approved drugs. Life Sci 340, 122419 (2024).
Cummings, J., Osse, A. M. L., Cammann, D., Powell, J. & Chen, J. Anti-amyloid monoclonal antibodies for the treatment of Alzheimer’s disease. BioDrugs 38, 5–22 (2024).
Jorgensen, S. C. J. Nirsevimab: review of pharmacology, antiviral activity and emerging clinical experience for respiratory syncytial virus infection in infants. J. Antimicrob. Chemother. 78, 1143–1149 (2023).
Bierle, D. M., Ganesh, R. & Razonable, R. R. Breakthrough COVID-19 and casirivimab-imdevimab treatment during a SARS-CoV-2 B1.617.2 (Delta) surge. J. Clin. Virol. 145, 105026 (2021).
Herman, G. A. et al. Efficacy and safety of a single dose of casirivimab and imdevimab for the prevention of COVID-19 over an 8-month period: a randomised, double-blind, placebo-controlled trial. Lancet Infect. Dis. 22, 1444–1454 (2022).
Pochtovyi, A. A. et al. In vitro efficacy of antivirals and monoclonal antibodies against SARS-CoV-2 omicron lineages XBB.1.9.1, XBB.1.9.3, XBB.1.5, XBB.1.16, XBB.2.4, BQ.1.1.45, CH.1.1, and CL.1. Vaccines 11, 1533 (2023).
Hagihara, M. et al. Clinical efficacy of imdevimab/casirivimab for persistent omicron SARS-CoV-2 infection in patients with hematological malignancies. Intern. Med. 63, 2283–2287 (2024).
Arora, P. et al. Omicron sublineage BQ.1.1 resistance to monoclonal antibodies. Lancet Infect. Dis. 23, 22–23 (2023).
Jensen, B. et al. Emergence of the E484K mutation in SARS-COV-2-infected immunocompromised patients treated with bamlanivimab in Germany. Lancet Reg. Health Eur. 8, 100164 (2021).
Halfmann, P. J. et al. Evolution of a globally unique SARS-CoV-2 Spike E484T monoclonal antibody escape mutation in a persistently infected, immunocompromised individual. Virus Evol. 9, veac104 (2023).
Huygens, S. et al. Clinical and virological outcome of monoclonal antibody therapies across SARS-CoV-2 variants in 245 immunocompromised patients: a multicenter prospective cohort study. Clin. Infect. Dis. 78, 1514–1521 (2024).
Paul, M.-K. et al. Impact of SARS-CoV-2 resistance to antiviral monoclonal antibody therapy on neutralizing antibody response. Pathog. Immun. 9, 79–93 (2024).
Boonkrai, C. et al. Efficacy of the combination of monoclonal antibodies against the SARS-CoV-2 Beta and Delta variants. PLoS ONE 18, e0284173 (2023).
Rayaprolu, V. et al. Structure of the Inmazeb cocktail and resistance to Ebola virus escape. Cell Host Microbe 31, 260–272.e7 (2023).
Liu, G. et al. A pan-ebolavirus monoclonal antibody cocktail provides protection against ebola and sudan viruses. J. Infect. Dis. 228, S691–S700 (2023).
Thakur, A., Huang, M. & Lum, L. G. Bispecific antibody based therapeutics: strengths and challenges. Blood Rev. 32, 339–347 (2018).
Klein, C., Brinkmann, U., Reichert, J. M. & Kontermann, R. E. The present and future of bispecific antibodies for cancer therapy. Nat. Rev. Drug Discov. 23, 301–319 (2024).
Nyakatura, E. K., Soare, A. Y. & Lai, J. R. Bispecific antibodies for viral immunotherapy. Hum. Vaccines Immunother 13, 836–842 (2017).
Wagner, E. K. & Maynard, J. A. Engineering therapeutic antibodies to combat infectious diseases. Curr. Opin. Chem. Eng. 19, 131–141 (2018).
Struble, E. B., Rawson, J. M. O., Stantchev, T., Scott, D. & Shapiro, M. A. Uses and challenges of antiviral polyclonal and monoclonal antibody therapies. Pharmaceutics 15, 1538 (2023).
Chang, Z. et al. Bispecific antibodies targeting two glycoproteins on SFTSV exhibit synergistic neutralization and protection in a mouse model. Proc. Natl. Acad. Sci. USA 121, e2400163121 (2024).
Moirangthem, R. et al. Dual neutralization of influenza virus hemagglutinin and neuraminidase by a bispecific antibody leads to improved antiviral activity. Mol. Ther. 32, 3712–3728 (2024).
Lee, J. H. et al. A dual-targeting approach using a human bispecific antibody against the receptor-binding domain of the Middle East Respiratory Syndrome Coronavirus. Virus Res. 345, 199383 (2024).
Li, M. et al. Bispecific antibodies provide broad neutralization of emerging beta-coronaviruses by targeting ACE2 and viral spikes. Emerg. Microbes Infect. 13, 2404166 (2024).
Cox, M. et al. SARS-CoV-2 variant evasion of monoclonal antibodies based on in vitro studies. Nat. Rev. Microbiol. 21, 112–124 (2023).
Liu, C. et al. Emerging variants develop total escape from potent monoclonal antibodies induced by BA.4/5 infection. Nat. Commun. 15, 3284 (2024).
Hannon, G. J. RNA interference. Nature 418, 244–251 (2002).
Agrawal, N. et al. RNA interference: biology, mechanism, and applications. Microbiol. Mol. Biol. Rev. 67, 657–685 (2003).
Svoboda, P. Key mechanistic principles and considerations concerning RNA interference. Front. Plant Sci. 11, 1237 (2020).
Ghildiyal, M. & Zamore, P. D. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 10, 94–108 (2009).
Marques, J. T., Meignin, C. & Imler, J.-L. An evolutionary perspective to innate antiviral immunity in animals. Cell Rep. 43, 114678 (2024).
Kang, H. et al. Small interfering RNA (siRNA)-based therapeutic applications against viruses: principles, potential, and challenges. J. Biomed. Sci. 30, 88 (2023).
Qureshi, A., Tantray, V. G., Kirmani, A. R. & Ahangar, A. G. A review on current status of antiviral siRNA. Rev. Med. Virol. 28, e1976 (2018).
Takahashi, T., Heaton, S. M. & Parrish, N. F. Mammalian antiviral systems directed by small RNA. PLoS Pathog 17, e1010091 (2021).
Kalke, K. et al. Herpes simplex virus type 1 clinical isolates respond to UL29-targeted siRNA swarm treatment independent of their acyclovir sensitivity. Viruses 12, 1434 (2020).
Lasanen, T. et al. Single therapeutic dose of an antiviral UL29 siRNA swarm diminishes symptoms and viral load of mice infected intranasally with HSV‐1. Smart Med. 2, e20230009 (2023).
Idris, A. et al. An intranasally delivered ultra-conserved siRNA prophylactically represses SARS-CoV-2 infection in the lung and nasal cavity. Antiviral Res. 222, 105815 (2024).
Pei, J. et al. A novel recombinant ORF7-siRNA delivered by flexible nano-liposomes inhibits varicella zoster virus infection. Cell Biosci. 13, 167 (2023).
Jiang, H.-F. et al. ORF7 of varicella-zoster virus is required for viral cytoplasmic envelopment in differentiated neuronal cells. J. Virol. 91, e00127-17 (2017).
Zhan, S., Wang, Y. & Chen, X. RNA virus-encoded microRNAs: biogenesis, functions and perspectives on application. ExRNA 2, 15 (2020).
Jafarzadeh, A. et al. MicroRNA-155 and antiviral immune responses. Int. Immunopharmacol. 101, 108188 (2021).
Pandey, N. & Singh, S. K. MicroRNA-155 triggers a cellular antiviral immune response against Chandipura virus in human microglial cells. Microbes Infect. 25, 105173 (2023).
Bivalkar-Mehla, S. et al. Viral RNA silencing suppressors (RSS): Novel strategy of viruses to ablate the host RNA interference (RNAi) defense system. Virus Res. 155, 1–9 (2011).
Wang, J. & Li, Y. Current advances in antiviral RNA interference in mammals. FEBS J. 291, 208–216 (2024).
Ding, S.-W. Transgene silencing, RNA interference, and the antiviral defense mechanism directed by small interfering RNAs. Phytopathology® 113, 616–625 (2023).
Li, W.-X. & Ding, S.-W. Mammalian viral suppressors of RNA interference. Trends Biochem. Sci. 47, 978–988 (2022).
Chen, X. et al. The subgenomic flaviviral RNA suppresses RNA interference through competing with siRNAs for binding RISC components. J. Virol. 98, e01954-23 (2024).
Li, Y. et al. Induction and suppression of antiviral RNA interference by influenza A virus in mammalian cells. Nat. Microbiol. 2, 16250 (2016).
Muñoz-Moreno, R. et al. Viral fitness landscapes in diverse host species reveal multiple evolutionary lines for the NS1 gene of influenza A viruses. Cell Rep. 29, 3997–4009.e5 (2019).
Lezcano, O. M. et al. Parallel evolution and enhanced virulence upon in vivo passage of an RNA virus in Drosophila melanogaster. Virus Evol. 9, vead074 (2023).
Samuel, G. H. et al. RNA interference is essential to modulating the pathogenesis of mosquito-borne viruses in the yellow fever mosquito Aedes aegypti. Proc. Natl. Acad. Sci. USA 120, e2213701120 (2023).
Ziegler, C. M. & Botten, J. W. Defective interfering particles of negative-strand RNA viruses. Trends Microbiol. 28, 554–565 (2020).
Jones, J. E., Le Sage, V. & Lakdawala, S. S. Viral and host heterogeneity and their effects on the viral life cycle. Nat. Rev. Microbiol. 19, 272–282 (2021).
Singh, K., Mehta, D., Dumka, S., Chauhan, A. S. & Kumar, S. Quasispecies nature of RNA viruses: lessons from the past. Vaccines 11, 308 (2023).
Vignuzzi, M. & López, C. B. Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol. 4, 1075–1087 (2019).
Magnus, P. V. Studies on interference in experimental influenza. I. Biological observations. Ark. Kemi Mineral. Och Geol. 24 B, 1–6 (1949).
Frensing, T., Pflugmacher, A., Bachmann, M., Peschel, B. & Reichl, U. Impact of defective interfering particles on virus replication and antiviral host response in cell culture-based influenza vaccine production. Appl. Microbiol. Biotechnol. 98, 8999–9008 (2014).
Kupke, S. Y., Riedel, D., Frensing, T., Zmora, P. & Reichl, U. A novel type of influenza A virus-derived defective interfering particle with nucleotide substitutions in its genome. J. Virol. 93, e01786–18 (2019).
Li, D. et al. Defective interfering viral particles in acute dengue infections. PLoS ONE 6, e19447 (2011).
Genoyer, E. & López, C. B. Defective viral genomes alter how sendai virus interacts with cellular trafficking machinery, leading to heterogeneity in the production of viral particles among infected cells. J. Virol. 93, e01579–18 (2019).
Girgis, S. et al. Evolution of naturally arising SARS-CoV-2 defective interfering particles. Commun. Biol. 5, 1140 (2022).
Manzoni, T. B. & López, C. B. Defective (interfering) viral genomes re-explored: impact on antiviral immunity and virus persistence. Future Virol. 13, 493–503 (2018).
Brennan, J. W. & Sun, Y. Defective viral genomes: advances in understanding their generation, function, and impact on infection outcomes. mBio 15, e0069224 (2024).
Sun, Y. et al. Immunostimulatory defective viral genomes from respiratory syncytial virus promote a strong innate antiviral response during infection in mice and humans. PLoS Pathog. 11, e1005122 (2015).
Pelz, L. et al. Semi-continuous propagation of influenza A virus and its defective interfering particles: analyzing the dynamic competition to select candidates for antiviral therapy. J. Virol. 95, e01174-21 (2021).
Pitchai, F. N. N. et al. Engineered deletions of HIV replicate conditionally to reduce disease in nonhuman primates. Science 385, eadn5866 (2024).
Dethoff, E. A. et al. Pervasive tertiary structure in the dengue virus RNA genome. Proc. Natl. Acad. Sci. USA 115, 11513–11518 (2018).
Tavares, R. et al. The global and local distribution of RNA structure throughout the SARS-CoV-2 genome. J. Virol. 95, e02190-20 (2021).
Lan, T. C. T. et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nat. Commun. 13, 1128 (2022).
Yan, S., Zhu, Q., Hohl, J., Dong, A. & Schlick, T. Evolution of coronavirus frameshifting elements: competing stem networks explain conservation and variability. Proc. Natl. Acad. Sci. USA 120, e2221324120 (2023).
Das, N. K. et al. Crystal structure of a highly conserved enteroviral 5’ cloverleaf RNA replication element. Nat. Commun. 14, 1955 (2023).
Akiyama, B. M. et al. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 354, 1148–1152 (2016).
Chapman, E. G. et al. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science 344, 307–310 (2014).
Dilweg, I. W. et al. All genera of Flaviviridae host a conserved Xrn1-resistant RNA motif. RNA Biol. 18, 2321–2329 (2021).
Tomezsko, P. J. et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 582, 438–442 (2020).
Baranovskaya, I. et al. Changes in RNA secondary structure affect NS1 protein expression during early stage influenza virus infection. Virol. J. 16, 162 (2019).
Cagliani, R., Forni, D., Clerici, M. & Sironi, M. Coding potential and sequence conservation of SARS-CoV-2 and related animal viruses. Infect. Genet. Evol. 83, 104353 (2020).
Hagey, R. J. et al. Programmable antivirals targeting critical conserved viral RNA secondary structures from influenza A virus and SARS-CoV-2. Nat. Med. 28, 1944–1955 (2022).
Yang, R. et al. Mapping of the influenza A virus genome RNA structure and interactions reveals essential elements of viral replication. Cell Rep. 43, 113833 (2024).
Qassem, S., Breier, D., Naidu, G. S., Hazan-Halevy, I. & Peer, D. Unlocking the therapeutic potential of locked nucleic acids through lipid nanoparticle delivery. Mol. Ther. Nucleic Acids 35, 102224 (2024).
Herkt, M. & Thum, T. Pharmacokinetics and proceedings in clinical application of nucleic acid therapeutics. Mol. Ther. 29, 521–539 (2021).
Okamoto, S. et al. Antiviral efficacy of RNase H-dependent gapmer antisense oligonucleotides against Japanese Encephalitis Virus. Int. J. Mol. Sci. 24, 14846 (2023).
Tuplin, A., Struthers, M., Cook, J., Bentley, K. & Evans, D. J. Inhibition of HCV translation by disrupting the structure and interactions of the viral CRE and 3′ X-tail. Nucleic Acids Res. 43, 2914–2926 (2015).
Zhu, C. et al. An intranasal ASO therapeutic targeting SARS-CoV-2. Nat. Commun. 13, 4503 (2022).
Huston, N. C. et al. Comprehensive in vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms. Mol. Cell 81, 584–598.e5 (2021).
Triebel, S. et al. Comprehensive survey of conserved RNA secondary structures in full-genome alignment of Hepatitis C virus. Sci. Rep. 14, 15145 (2024).
Prosser, O., Stonehouse, N. J. & Tuplin, A. Inhibition of Chikungunya virus genome replication by targeting essential RNA structures within the virus genome. Antiviral Res. 211, 105523 (2023).
Burton, T. D. & Eyre, N. S. Applications of deep mutational scanning in virology. Viruses 13, 1020 (2021).
Wang, S. et al. Deep mutational scanning of influenza A virus neuraminidase facilitates the identification of drug resistance mutations in vivo. mSystems 8, e00670-23 (2023).
Dadonaite, B. et al. Deep mutational scanning of H5 hemagglutinin to inform influenza virus surveillance. PLoS Biol. 22, e3002916 (2024).
Carr, C. R. et al. Deep mutational scanning reveals functional constraints and antibody-escape potential of Lassa virus glycoprotein complex. Immunity 57, 2061–2076.e11 (2024).
Johnson, M. S., Reddy, G. & Desai, M. M. Epistasis and evolution: recent advances and an outlook for prediction. BMC Biol. 21, 1–12 (2023).
Bank, C. Epistasis and adaptation on fitness landscapes. Annu. Rev. Ecol. Evol. Syst. 53, 457–479 (2022).
Rajaram, S. et al. The impact of candidate influenza virus and egg-based manufacture on vaccine effectiveness: literature review and expert consensus. Vaccine 38, 6047–6056 (2020).
Wu, N. C. et al. Preventing an antigenically disruptive mutation in egg-based H3N2 seasonal influenza vaccines by mutational incompatibility. Cell Host Microbe 25, 836–844.e5 (2019).
Liang, W. et al. Egg-adaptive mutations of human influenza H3N2 virus are contingent on natural evolution. PLoS Pathog. 18, e1010875 (2022).
Lei, R. et al. Epistasis mediates the evolution of the receptor binding mode in recent human H3N2 hemagglutinin. Nat. Commun. 15, 5175 (2024).
Duwe, S. C. et al. Increase of synergistic secondary antiviral mutations in the evolution of A(H1N1)pdm09 influenza virus neuraminidases. Viruses 16, 1109 (2024).
Ghosh, N., Nandi, S. & Saha, I. A review on evolution of emerging SARS-CoV-2 variants based on spike glycoprotein. Int. Immunopharmacol. 105, 108565 (2022).
Chatterjee, S., Bhattacharya, M., Nag, S., Dhama, K. & Chakraborty, C. A detailed overview of SARS-CoV-2 omicron: its sub-variants, mutations and pathophysiology, clinical characteristics, immunological landscape, immune escape, and therapies. Viruses 15, 167 (2023).
Taylor, A. L. & Starr, T. N. Deep mutational scans of XBB.1.5 and BQ.1.1 reveal ongoing epistatic drift during SARS-CoV-2 evolution. PLoS Pathog. 19, e1011901 (2023).
Dultz, G. et al. Epistatic interactions promote persistence of NS3-Q80K in HCV infection by compensating for protein folding instability. J. Biol. Chem. 297, 101031 (2021).
Zhang, H., Quadeer, A. A. & McKay, M. R. Direct-acting antiviral resistance of Hepatitis C virus is promoted by epistasis. Nat. Commun. 14, 7457 (2023).
Lukačišinová, M., Fernando, B. & Bollenbach, T. Highly parallel lab evolution reveals that epistasis can curb the evolution of antibiotic resistance. Nat. Commun. 11, 3105 (2020).
Dewachter, L. et al. Deep mutational scanning of essential bacterial proteins can guide antibiotic development. Nat. Commun. 14, 241 (2023).
Rouleau, F. D. Deep mutational scanning of Pneumocystis jirovecii dihydrofolate reductase reveals allosteric mechanism of resistance to an antifolate. PLoS Genet. 20, 1–29 (2024).
Wu, X. et al. Mutational profiling of SARS-CoV-2 papain-like protease reveals requirements for function, structure, and drug escape. Nat. Commun. 15, 6219 (2024).
Chen, D. et al. High throughput profiling identified PA-L106R amino acid substitution in A(H1N1)pdm09 influenza virus that confers reduced susceptibility to baloxavir in vitro. Antiviral Res. 229, 105961 (2024).
Goodacre, N., Devkota, P., Bae, E., Wuchty, S. & Uetz, P. Protein-protein interactions of human viruses. Semin. Cell Dev. Biol. 99, 31–39 (2020).
Tanushree, Sharma, A., Monika, Singh, R. P. & Jhawat, V. Human immunodeficiency virus infection challenges: Current therapeutic limitations and strategies for improved management through long‐acting injectable formulation. Rev. Med. Virol. 34, e2563 (2024).
Farooq, Q. U. A., Shaukat, Z., Aiman, S. & Li, C.-H. Protein-protein interactions: methods, databases, and applications in virus-host study. World J. Virol. 10, 288–300 (2021).
González-Avendaño, M., López, J., Vergara-Jaque, A. & Cerda, O. The power of computational proteomics platforms to decipher protein-protein interactions. Curr. Opin. Struct. Biol. 88, 102882 (2024).
Hajjo, R., Sabbah, D. A., Abusara, O. H., Kharmah, R. & Bardaweel, S. Targeting human proteins for antiviral drug discovery and repurposing efforts: a focus on protein kinases. Viruses 15, 568 (2023).
Dey, S. & Mondal, A. Unveiling the role of host kinases at different steps of influenza A virus life cycle. J. Virol. 98, e01192–23 (2024).
Haas, K. M. et al. Proteomic and genetic analyses of influenza A viruses identify pan-viral host targets. Nat. Commun. 14, 6030 (2023).
Bouhaddou, M. et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182, 685–712.e19 (2020).
Stukalov, A. et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 594, 246–252 (2021).
Lin, D. et al. Proteomic and phosphoproteomic analysis of responses to enterovirus A71 infection reveals novel targets for antiviral and viral replication. Antiviral Res. 220, 105761 (2023).
Gordon, D. E. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020).
Álvarez-Machancoses, Ó. & Fernández-Martínez, J. L. Using artificial intelligence methods to speed up drug discovery. Expert Opin. Drug Discov. 14, 769–777 (2019).
Popova, M., Isayev, O. & Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 4, eaap7885 (2018).
Matevosyan, M. et al. Design of new chemical entities targeting both native and H275Y mutant influenza a virus by deep reinforcement learning. J. Biomol. Struct. Dyn. 41, 10798–10812 (2023).
Izmailyan, R. et al. Discovery of new antiviral agents through artificial intelligence: In vitro and in vivo results. Antiviral Res. 222, 105818 (2024).
Hao, Y., Cai, M. & Li, L. Drug repositioning via matrix completion with multi-view side information. IET Syst. Biol. 13, 267–275 (2019).
Mongia, A., Saha, S. K., Chouzenoux, E. & Majumdar, A. A computational approach to aid clinicians in selecting anti-viral drugs for COVID-19 trials. Sci. Rep. 11, 9047 (2021).
Hie, B., Zhong, E. D., Berger, B. & Bryson, B. Learning the language of viral evolution and escape. Science 371, 284–288 (2021).
Bloom, J. D. & Neher, R. A. Fitness effects of mutations to SARS-CoV-2 proteins. Virus Evol. 9, vead055 (2024).
Flynn, J. M. et al. Comprehensive fitness landscape of SARS-CoV-2 Mpro reveals insights into viral resistance mechanisms. eLife 11, e77433 (2022).
Acevedo, A., Brodsky, L. & Andino, R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 505, 686–690 (2014).
Quadeer, A. A., Barton, J. P., Chakraborty, A. K. & McKay, M. R. Deconvolving mutational patterns of poliovirus outbreaks reveals its intrinsic fitness landscape. Nat. Commun. 11, 377 (2020).
Zanini, F., Puller, V., Brodin, J., Albert, J. & Neher, R. A. In vivo mutation rates and the landscape of fitness costs of HIV-1. Virus Evol. 3, vex003 (2017).
Dolan, P. T. et al. Principles of dengue virus evolvability derived from genotype-fitness maps in human and mosquito cells. eLife 10, e61921 (2021).
Łuksza, M. & Lässig, M. A predictive fitness model for influenza. Nature 507, 57–61 (2014).
Tisthammer, K. H. et al. Assessing in vivo mutation frequencies and creating a high-resolution genome-wide map of fitness costs of Hepatitis C virus. PLoS Genet. 18, e1010179 (2022).
Wu, N. C. et al. Major antigenic site B of human influenza H3N2 viruses has an evolving local fitness landscape. Nat. Commun. 11, 1233 (2020).
Perfetto, L. et al. The IMEx coronavirus interactome: an evolving map of Coronaviridae–host molecular interactions. Database 2020, baaa096 (2020).
Thadani, N. N. et al. Learning from prepandemic data to forecast viral escape. Nature 622, 818–825 (2023).
Biswas, S. et al. Pharmacokinetics-pharmacodynamics of the helicase-primase inhibitor pritelivir following treatment of wild-type or pritelivir-resistant virus infection in a murine herpes simplex virus 1 infection model. Antimicrob. Agents Chemother. 58, 3843–3852 (2014).
Bernstein, D. I. et al. Intermittent therapy with helicase-primase inhibitor IM-250 efficiently controls recurrent herpes disease and reduces reactivation of latent HSV. Antiviral Res. 219, 105733 (2023).
Gege, C. et al. A helicase-primase drug candidate with sufficient target tissue exposure affects latent neural herpes simplex virus infections. Sci. Transl. Med. 13, eabf8668 (2021).
Quenelle, D. C. et al. Efficacy of pritelivir and acyclovir in the treatment of herpes simplex virus infections in a mouse model of herpes simplex encephalitis. Antiviral Res. 149, 1–6 (2018).
Hussein, I. T. M., Brooks, J. & Bowlin, T. L. The discovery and development of filociclovir for the prevention and treatment of human cytomegalovirus-related disease. Antiviral Res. 176, 104710 (2020).
Diamond, T. L. et al. No antagonism or cross-resistance and a high barrier to the emergence of resistance in vitro for the combination of islatravir and lenacapavir. Antimicrob. Agents Chemother. 68, e00334-24 (2024).
Liu, X. et al. SARS‐CoV‐2–host proteome interactions for antiviral drug discovery. Mol. Syst. Biol. 17, e10396 (2021).
Yang, S. L. et al. Comprehensive mapping of SARS-CoV-2 interactions in vivo reveals functional virus-host interactions. Nat. Commun. 12, 5113 (2021).
Li, G. et al. Atlas of interactions between SARS-CoV-2 macromolecules and host proteins. Cell Insight 2, 100068 (2023).
Muccini, C., Canetti, D., Castagna, A. & Spagnuolo, V. Efficacy and safety profile of fostemsavir for the treatment of people with human immunodeficiency virus-1 (HIV-1): current evidence and place in therapy. Drug Des. Dev. Ther. 16, 297–304 (2022).
Matthews, T. et al. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 3, 215–225 (2004).
Li, Y.-Y. et al. A small molecule compound targeting hemagglutinin inhibits influenza A virus and exhibits broad-spectrum antiviral activity. Acta Pharmacol. Sin. 45, 2380–2393 (2024).
De Clercq, E. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 5, 1015–1025 (2006).
Pevear, D. C., Tull, T. M., Seipel, M. E. & Groarke, J. M. Activity of pleconaril against enteroviruses. Antimicrob. Agents Chemother. 43, 2109–2115 (1999).
Elion, G. B. Mechanism of action and selectivity of acyclovir. Am. J. Med. 73, 7–13 (1982).
Bhatia, H. K., Singh, H., Grewal, N. & Natt, N. K. Sofosbuvir: a novel treatment option for chronic hepatitis C infection. J. Pharmacol. Pharmacother. 5, 278–284 (2014).
Wassner, C., Bradley, N. & Lee, Y. A review and clinical understanding of tenofovir: tenofovir disoproxil fumarate versus tenofovir alafenamide. J. Int. Assoc. Provid. AIDS Care 19, 2325958220919231 (2020).
Hadj Hassine, I., Ben M’hadheb, M. & Menéndez-Arias, L. Lethal mutagenesis of RNA viruses and approved drugs with antiviral mutagenic activity. Viruses 14, 841 (2022).
Sluis-Cremer, N. & Tachedjian, G. Mechanisms of inhibition of HIV replication by non-nucleoside reverse transcriptase inhibitors. Virus Res. 134, 147–156 (2008).
Smith, S. J. et al. Integrase strand transfer inhibitors are effective anti-HIV drugs. Viruses 13, 205 (2021).
Lv, Z., Chu, Y. & Wang, Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV AIDS 7, 95–104 (2015).
de Leuw, P. & Stephan, C. Protease inhibitors for the treatment of hepatitis C virus infection. GMS Infect. Dis. 5, Doc08 (2017).
Bege, M. & Borbás, A. The design, synthesis and mechanism of action of paxlovid, a protease inhibitor drug combination for the treatment of COVID-19. Pharmaceutics 16, 217 (2024).
Pillai, S. et al. A novel viral assembly inhibitor blocks SARS-CoV-2 replication in airway epithelial cells. Res. Sq. rs.3.rs-2887435 (2023)
Davies, B. E. Pharmacokinetics of oseltamivir: an oral antiviral for the treatment and prophylaxis of influenza in diverse populations. J. Antimicrob. Chemother. 65, ii5–ii10 (2010).
Rossi, E., Meuser, M. E., Cunanan, C. J. & Cocklin, S. Structure, function, and interactions of the HIV-1 capsid protein. Life 11, 100 (2021).
Dubois, E. A. & Cohen, A. F. Maraviroc and raltegravir. Br. J. Clin. Pharmacol. 68, 651–652 (2009).
Sadler, A. J. & Williams, B. R. G. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8, 559–568 (2008).
Fu, H., Zhang, Z., Dai, Y., Liu, S. & Fu, E. Brequinar inhibits enterovirus replication by targeting biosynthesis pathway of pyrimidines. Am. J. Transl. Res. 12, 8247–8255 (2020).
Wu, M. et al. Mycophenolate mofetil exerts broad-spectrum antiviral activity against coronaviruses including SARS-CoV-2. Virol. J. 22, 56 (2025).
Saha, P. et al. Unveiling the antiviral potential of minocycline: modulation of nuclear export of viral ribonuclear proteins during influenza virus infection. Viruses 16, 1317 (2024).
Acknowledgements
We would like to thank the members of Vignuzzi lab for helpful discussion and reviewing.
Author information
Authors and Affiliations
Contributions
D.Z.H.A. and D.X.G.Z. contributed equally in the literature research and primary writing of the review. M.V. was primary editor and supervisor of the review.
Corresponding author
Ethics declarations
Competing interests
All three authors have current ongoing collaborations with DeNovo Biosciences and Recursion, two AI antiviral drug discovery companies. There was no bias towards any of the work directly performed by these companies, and is not covered in this review.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Aw, D.Z.H., Zhang, D.X. & Vignuzzi, M. Strategies and efforts in circumventing the emergence of antiviral resistance against conventional antivirals. npj Antimicrob Resist 3, 54 (2025). https://doi.org/10.1038/s44259-025-00125-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44259-025-00125-z
