
Abstract
The transmissible locus of stress tolerance (tLST) confers enhanced resilience to stressors, including chlorine-based disinfectants and heat. A systematic examination of 48,183 complete bacterial genomes representing 7,190 unique species identified tLST, including six novel variants, in 2,128 genomes spanning 46 bacterial species across 10 families. Among the analysed sequences, tLST was most common in ESKAPE-E bacteria, including highly drug-resistant pandemic lineages of Pseudomonas aeruginosa ST111 and Klebsiella pneumoniae ST20. The tLST1_AW1.7 variant was predominantly associated with F plasmids and was the most common variant in Enterobacter and Klebsiella, whereas tLST1 was near exclusive to E. coli, and tLST3a was dominant in Pseudomonas. This comprehensive mapping of tLST distribution highlights its potential significance in bacterial adaptation and persistence across diverse ecological and anthropogenically impacted niches, and has implications for infection control and eradication, waste and drinking water management, food animal and fresh produce production, and food safety.
Similar content being viewed by others

Introduction
ESKAPE-E pathogens (Enterobacter spp., Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterococcus faecium and Escherichia coli) are the leading cause of healthcare-associated infections globally1,2,3. ESKAPE-E pathogens are notorious for their capacity to acquire resistance to multiple antibiotics, metals and biocides by horizontal gene transfer. Multidrug-resistant (MDR) infections significantly increase healthcare costs, morbidity and mortality rates4 with annual economic impacts exceeding $20 billion in the US alone5. ESKAPE-E pathogens primarily colonise the human and animal gastrointestinal tract as colonising opportunistic pathogens (COPs), but their impact extends beyond host organisms. Their capacity to form biofilms enables persistent colonisation of water systems and survival on fomites, facilitating transmission within healthcare settings and more generally within drinking water infrastructure6,7. Wastewater treatment plants (WWTPs) are recognised as important reservoirs for antimicrobial resistance (AMR) within taxa that include ESKAPE-E pathogens8,9. WWTPs and the biosolids generated from waste treatment harbour sufficient nutrients and high microbial and bacteriophage densities, creating favourable conditions for the growth of environmental bacteria and for horizontal gene transfer10,11,12. These environments also expose microbes to a wide array of selective agents—including pharmaceuticals, biocides, metals, and oxidising agents—that drive horizontal gene acquisition and adaptive evolution, potentially enriching resistance determinants and virulence traits. Although ESKAPE-E bacteria have been isolated from diverse environmental sources, their survival mechanisms in the built environment remain poorly understood— representing a critical gap in the One Health framework12,13. Chlorine-based disinfectants serve as a cornerstone of global sanitation practices14,15,16, with widespread applications in wastewater treatment, drinking water production, and food safety17. Hence, the emergence of chlorine-resistant bacteria represents a potential public health concern. Studies have identified several important resistance mechanisms, including chlorine-sensitive transcription factors (hypT, rclR, and nemR)18,19,20, an RpoS-regulated general stress response, and several oxidative stress regulons (oxyR and soxR) and heat-shock proteins21,22,23,24. Of particular concern is the discovery of the transmissible locus of stress tolerance (tLST)—a 15–19 kb mobile genetic element (MGE) encoding resistance to not only chlorine-based oxidising agents, but also to extreme heat and pressure25,26,27. The acquisition of the tLST, also known as the locus of heat resistance (LHR) and the transmissible locus of protein quality control (tLPQC)28,29 has enabled resistance to many antimicrobial interventions, including heat (up to 70 °C), sodium chlorite (NaClO) (32 mM; 5 min), hydrogen peroxide (H2O2; 120 mM; 5 min), and peroxyacetic acid (105 mg/L; 5 min)28,30,31. This sophisticated genetic system is found on both chromosomes and plasmids 32,33). Bacteria carrying tLST have been recovered from meat processing plants34, beef35,36,37,38 and dairy cattle39,40, raw milk cheese39, in municipal wastewater41,42 and in wildlife adapted to anthropogenic sites43. The detection of bacteria carrying tLST in food animals, on meat surfaces and in processed food more broadly poses a significant food safety risk, as these organisms can both survive standard sanitisation procedures and potentially transfer this resistance element to other bacteria within the human gut microbiome.
A recent study of 18,959 E. coli genomes revealed four variants of tLST (tLST1, tLST2, tLST3a and tLST3b) with tLST1 being the most prevalent33. These variants were predominately found in commensal phylogroup A isolates, with much lower frequencies in phylogroup B1 and C, and were notably absent from virulent phylogroups B2, D and G33, which are responsible for most extraintestinal pathogenic E. coli (ExPEC) infections (i.e. UTI, pyelonephritis and blood stream infections). Although tLST was primarily found in traditionally commensal E. coli phylogroups A and B1, these groups include emerging pathogenic lineages such as ST1044,45, ST21646 and ST5847. The tLST has been identified in other clinically important Gram-negative bacteria48, including Cronobacter species49, Salmonella enterica Serovar Senftenberg50, Klebsiella species27, Pseudomonas aeruginosa28, and Enterobacter cloacae32. However, our understanding of tLST distribution and any correlations with antimicrobial resistance genes (ARGs) and virulence factors in organisms other than E. coli remains largely unexplored. In this study, we analysed 48,183 completed genome sequences from public repositories to investigate tLST carriage across all bacterial species available. Our analysis identified the presence of tLST across 10 bacterial families encompassing 46 species, including members of Enterobacteriaceae, Pseudomonadaceae, Yersiniaceae, and Geobacteraceae. Notably, we identified tLST in high-risk MDR ESKAPE-E lineages. These findings underscore the urgent need for innovative solutions to combat these emerging threats to public health.
Results
tLST variant screening and distribution
The original dataset comprised 48,183 complete genomes, sourced from humans (n = 16,803), the environment (n = 11,123), animals (n = 8910), and wastewater (n = 738), with no source information available for 10,609 genomes. Taxonomic analysis revealed Escherichia (n = 4411), Klebsiella (n = 3032), Staphylococcus (n = 2451), Salmonella (n = 2203), and Pseudomonas (n = 1988) as the most abundant genera, among a total of 1956 identified genera. Geographical distribution showed that most prevalent countries were USA (n = 9014), China (n = 8159), South Korea (n = 3002), Japan (n = 1930) and UK (n = 1773).
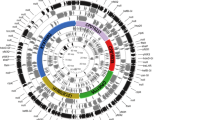
Our initial analysis, using the four tLST variants described by Zhang & Yang, 2022, identified 4569 tLST-positive genomes (~0.5% of all analysed genomes). After applying stringent quality filters requiring ≥90% sequence identity, 2128 genomes remained for detailed investigation. Among these, we observed varying degrees of tLST coverage: 968 genomes contained partial sequences spanning 20–30% of the full-length variant, while 559 genomes showed minimal coverage of <20%, and 385 genomes had coverage >30% but <80%. For our definitive analysis, we focused on 216 genomes that demonstrated both high sequence identity (≥90%) and substantial coverage (≥80%) of tLST variants. tLST3a emerged as the predominant form, present in 118 genomes, followed by tLST1 with 82 occurrences. tLST2 and tLST3b appeared in only seven (E. coli n = 4, S. enterica n = 2, E. ludwigii n = 1) and nine (P. aeruginosa n = 8, E. coli n = 1) genomes respectively. tLST1 occurring almost exclusively in E. coli, with a single instance detected in Raoutella terrigena. In contrast, tLST3a exhibited broad distribution across multiple bacterial families, including Pseudomonadaceae, Burkholderiaceae, Yersiniaceae, and Enterobacteriaceae (Fig. 1b).

a Structural comparison of tLST variants showing open reading frames (ORFs) colour-coded by function: protein homeostasis/heat resistance (red), cell envelope stress response (yellow), oxidative stress response (blue), variable ORFs (pink), and insertion elements (green). b Midpoint rooted phylogenetic tree, generated by mashtree software, of tLST-positive species with corresponding variant distribution. The heatmap indicates variant presence (darker red indicating higher frequency) and shows the percentage of tLST-positive genomes within each species (rightmost column). Species are colour-coded by family affiliation. Novel variants are highlighted by bars above the distribution matrix. c Table showing the genomic context (plasmid versus chromosomal) and source origin of each tLST variant.
Under further scrutiny, it was noted that several genomes displayed consistent hit patterns matching known tLST variants, despite not meeting our initial stringent criteria for inclusion. By mapping these sequences back to reference tLST variants (Supplementary Figs. 1–4), we identified structural variations that our initial screening parameters had not captured. This reassessment led to the identification of seven additional tLST variants (Fig. 1a). We incorporated these newly identified variants into our sequence alignment and screening protocols, resulting in the detection of 213 additional tLST-positive genomes. This discovery nearly doubled our total count of tLST-positive genomes from 216 to 429.
Detailed structural analysis of these newly identified variants revealed distinct modifications in key genetic elements. The tLST1 derivatives, designated as tLST1a and tLST1b, exhibited more extensive deletions in ftsH compared to the original variant (Fig. 1a). The tLST2 family showed more complex structural variations. Three new variants—tLST2a, tLST2b, and tLST2c—demonstrated modifications from the original tLST2, including minor deletions in ftsH and a DNA duplication between the kefB and orfE genes. The tLST2c variant displayed additional distinguishing features, notably the absence of orfC and orfD genes, along with an ISPa85 insertion positioned between the trx and kefB genes. Among all tLST2 variants, tLST2d showed the most substantial structural changes, characterised by complete deletion of ftsH, orfA, orfC, and orfD genes. Although tLST2d appeared structurally more similar to tLST1 (Fig. 1a), sequence analysis confirmed its closer relationship to tLST2, with 98% sequence identity at 100% coverage compared to only 85% identity, 91% coverage with tLST1.
tLST distribution across bacterial species and families (including new variants)
Our analysis revealed significant variation in tLST prevalence across 46 out of 7190 bacterial species screened, with notably high frequencies in several clinically relevant pathogens. tLST was detected in ESKAPE-E pathogens such as Enterobacter hormaechei (8.3%, n = 34), P. aeruginosa (7.7%, n = 69), K. pneumoniae (3.1%, n = 80) and E. coli (2.6%, n = 111) (Fig. 1). We also observed distinct patterns in tLST variant distribution (Fig. 1b). P. aeruginosa showed the most concentrated distribution, with tLST3a dominating (88%) and tLST3b representing the remainder (12%). E. coli displayed greater variant diversity, with tLST1 as the predominant form (73%), followed by tLST1_AW1.7 (8%), tLST2c (6.3%), tLST3a (5.4%), tLST2 (3.6%), tLST2b (2.7%), and tLST3b (0.9%). E. hormaechei harboured primarily tLST1_AW1.7 (62%) and tLST1a (38%), while K. pneumoniae showed a strong preference for tLST1_AW1.7 (77.5%), with lower frequencies of tLST2b (17.5%), tLST1b (2.5%), and tLST3a (2.5%). At the family level, we observed a clear phylogenetic signal in tLST variant distribution. Enterobacteriaceae members predominantly carried tLST1 and its variants, while non-Enterobacteriaceae families, particularly Pseudomonadaceae, primarily harboured tLST3a. This family-specific distribution pattern suggests selective pressures, horizontal gene transfer constraints or fitness impost have shaped the evolution and dissemination of tLST variants across bacterial taxa.
Source distribution of tLST variants
We categorised genome sources into broad ecological niches based on available metadata (Fig. 1c). Despite some limitations in source attribution for certain genomes, distribution patterns emerged across different environmental contexts. tLST3a (associated with P. aeruginosa) demonstrated a strong bias toward clinical settings, with 76.9% (83/108) of isolates originating from human sources, while environmental (n = 15) and wastewater (n = 10) isolates accounted for the remainder. In contrast, tLST1 (E. coli associated) exhibited a more balanced distribution: animal (39.4%, n = 26), human (27.3%, n = 18), wastewater (18.2%, n = 12), and environmental sources (15.2%, n = 10). For tLST1-AW1.7, 53.9% (n = 82) of isolates were from human sources and 36.8% (n = 56) from wastewater, while animal and environmental sources each contributed only 5.3% (n = 8) and 3.9% (n = 6) of isolates. tLST1a followed a similar pattern, predominantly occurring in wastewater (59.5%, n = 25) and human samples (33.3%, n = 14), with minimal representation in environmental (4.8%, n = 2) and animal (2.4%, n = 1) sources. For tLST2b 86.4% (n = 19) of isolates were from human sources and the remaining 13.6% (n = 3) from wastewater. However, given the inherent sampling biases in public genome databases, we caution interpreting these patterns as prevalence differences and instead report them to contextualise where tLST variants have been identified to date. For example, the apparent clinical bias of tLST3a likely reflects the overrepresentation of clinical P. aeruginosa isolates in sequence databases rather than a true ecological preference. Conversely, the broader source distribution of tLST1 may reflect the more extensive sampling of E. coli across multiple niches due to its key role in OneHealth AMR surveillance, driving sampling from clinical, animal, food, and environmental sources.
Plasmid-mediated distribution of tLST1_AW1.7
Among all tLST variants, tLST1_AW1.7 displayed a distinctive pattern of plasmid association (Fig. 1c). This variant demonstrated a confined distribution within the Enterobacteriaceae family, primarily occurring in Enterobacter (47/145, 32.4%) and Klebsiella (84/145, 57.9%) species, with occasional presence in E. coli (9/145, 6.2%). Plasmid typing revealed that 91.7% of tLST1_AW1.7 was carried on IncF plasmids (133/145). BLAST analysis of these plasmids revealed limited sequence conservation beyond individual strains, with similarity dropping rapidly to 60% or 20% coverage, suggesting substantial plasmid diversity despite shared replicon types.
Genetic characterisation of tLST1_AW1.7-carrying plasmids revealed selective carriage of virulence determinants. The mrkABCDFJ operon, associated with biofilm formation and enhanced biocide tolerance51, was present in 31.7% (46/145) of plasmids. In contrast, other virulence factors were rare, with only astA detected in only two plasmids. ARGs were similarly uncommon, with only 11 completed plasmids carrying resistance determinants. Nevertheless, many of these tLST-positive plasmids had extensive AMR profiles. For example, IncHI2:ST1 plasmid OW849321.1, originating from a clinical Enterobacter cloacae isolate from Spain in 2018, carried 18 ARGs conferring in silico resistance to carbapenem (blaVIM-1), colistin (mcr-9), ESBL (blaCTX-M-9), rifampicin (arr-3), macrolides (mphA), fluoroquinolones (qnrA1, aac(6′)-lb-cr6), chloramphenicol (catB3), aminoglycosides (aac(6′)-lb9, ant(2′′)-Ia, ant(3′′)-Ila), trimethoprim (dfrA16, dfrB1), beta-lactams (blaOXA-1), sulphonamides (sul1) and tetracyclines (tet(A)). Another eight tLST-positive IncF plasmids, all from Enterobacter species (E. asburiae, E. roggenkampii, E. kobei), carried colistin resistance gene mcr10.1.
tLST sequence type and lineage associations
Within the study collection, the tLST was found to be most prevalent in specific high-risk lineages (Supplementary Data 1E), notably Enterobacter hormaechei ST108 (n = 11) (phylogeny provided in Supplementary Fig. 5), Klebsiella pneumoniae ST20 (n = 10) and Pseudomonas aeruginosa ST111 (n = 10), as well as emerging pathogenic lineages such as E. coli ST399 (n = 11). Complete genotyping, including AMR, VAG and metal resistance was performed for all isolates (Supplementary Data 2–5). This association between tLST and these STs warranted detailed examination given their established and emerging clinical significance, thus draft genomes for each ST were analysed in addition to the original completed genome dataset.
tLST association in Klebsiella pneumoniae ST20
Hypervirulent and MDR K. pneumoniae ST20 has been identified in Europe (Spain and Greece), North America (Canada), South America (Brazil), Asia (China, South Korea) and New Zealand52. Among the 220 K. pneumoniae ST20 isolates analysed here, from 31 countries, more than half (n = 124, 56.4%) harboured tLST. The plasmid-associated variant, tLST1_AW1.7, predominated, accounting for 98.4% (n = 122) of all tLST-positive isolates. The source distribution of ST20 isolates primarily reflected human origins (n = 184, 84%), with smaller numbers from environmental (n = 9, 4.1%), animal (n = 2, 0.9%), and wastewater sources (n = 2, 0.9%). This pattern was similarly reflected in the tLST-positive isolates: human (n = 103, 91.2%), environmental (n = 8, 7.1%), animal (n = 1, 0.9%), and wastewater (n = 1, 0.9%). Analysis of closely related clades revealed variation in tLST presence, indicating that tLST acquisition likely occurred through horizontal gene transfer rather than strictly following vertical inheritance (Fig. 2).

Leaf labels in red indicate complete genomes included in the earlier part of study. From inner to outer, the coloured rings show tLST variant, source, and country of origin. IncF RST shown as determined by pMLST. The blue heatmap represents yersiniabactin high pathogenicity island (ICEKp) associated genes, followed by intI1 gene length detected. The red heatmap summarises antimicrobial-resistance classes of detected ARGs; with specific colours for ESBLs (light green: blaCTX-M, dark green: others) and carbapenemases (dark brown: blaKPC, light brown: others).
K. pneumoniae ST20 isolates exhibited highly conserved intrinsic AMR profiles, with fosfomycin resistance genes (fosA, fosA10) detected universally across all 220 isolates. Phenicol/quinolone resistance genes (oqxA, oqxB32) were also highly prevalent (n = 183, 83.2%). Non-ESBL/non-carbapenemase beta-lactamases (blaSHV-1,11,14,187) were found in 95.5% (n = 210). Conversely, acquired resistance determinants displayed more variability—ESBLs (blaCTX-M-3,12,14,15,154, blaSHV-2,5,7,12) and carbapenemases (blaIMP-1,4,26, blaVIM-1,4,24, blaNDM-1,5,7, blaOXA-48,181,232, blaKPC-2,3) were each detected in 27.8% (n = 61) of isolates while other non-ESBL/non-carbapenemase beta-lactamases (blaOXA-1,2,9, blaLAP-2, blaTEM-1, blaSCO-1) were found in 35.1% (n = 79). Additional resistance determinants included sulphonamide resistance genes (sul1,2,3; 35.5%, n = 78), quinolone resistance genes (qnrA1, qnrB1,2,4,6,7,19,91, qnrS1; 33.6%, n = 74), streptomycin resistance genes (aadA1,2,4,5,16, aph(3′′)-Ib, aph(6)-Id; 33.6%, n = 74), and trimethoprim resistance genes (dfrA1,5,7,8,12,14,15,17,19,25,27; 30%, n = 66). Statistical comparison of AMR profiles between tLST-positive and tLST-negative isolates revealed important observation. The tLST-positive isolates demonstrated significantly lower ESBL carriage (17% vs. 42%, p = 0.002, chi-squared test with Bonferroni correction). No statistically significant differences were observed for other resistance determinants. The class 1 integron integrase gene (intI1) was present in 34.1% (n = 75) of all ST20 isolates, with 24% (n = 30) of tLST-positive isolates harbouring this element. Notably, tLST-positive isolates were significantly associated with copper (pco; p = 0.002), arsenic (ars; p = 0.001) and silver (sil; p = 0.00006) resistance genes.
A distinct lineage of 14 isolates from Singapore was identified, with a high prevalence of tLST (78.6%, n = 11). These isolates differed by an average of 4 SNPs (range: 0–9 core SNPs), carried IncF K5:A-:B- plasmids and demonstrated a consistent multidrug resistance profile, harbouring acquired genes conferring resistance to multiple antimicrobial classes: blaOXA-1, aadA16, sul1, dfrA27, tet(A), qnrB6, aac(6′)-Ib, catB3, blaKPC-2, aac(3)-IIe, and arr-3. Despite being highly clonal, these isolates from Singapore were collected from at least three hospitals, from different pathologies and sources (e.g. blood, liver abscess, rectal swabs) and different years (2012–2015).
tLST associations in Pseudomonas aeruginosa ST111
Our analysis encompassed 380 P. aeruginosa ST111 isolates, predominantly of human origin (n = 274, 72%). The ST111 lineage exhibited close genetic relatedness, with pairwise core SNP distances ranging from 0 to 3635 (mean = 119 core SNPs). The isolates demonstrated a global distribution, with representatives from diverse geographical regions including Germany, Lebanon, Russia, Colombia, and other countries. The tLST (either tLST3a or tLST3b), present in 141 isolates (37.1%), showed restricted distribution within the ST111 population, being primarily concentrated in one specific sublineage (Fig. 3, blue highlighted clade). This sublineage comprised 149 isolates, with 77.2% (n = 115) derived from human sources, including isolates from blood, sputum, bronchoalveolar lavage, bone, skin and soft tissue, and urine (Supplementary Data 3A and 4.0% (n = 6) from environmental sources. Within this sublineage, tLST was highly prevalent, present in 81.2% (n = 121) of isolates (all tLST3b). There was a strong association of Class 1 integrons (intI1) carriage across the entire ST111 collection (n = 300, 78.9%) and this association was also evident in the lineage carrying tLST3b (100%; all 1014 bp). The ST111 lineage exhibited extensive AMR. We observed universal resistance to kanamycin and fosfomycin (100%), with near-complete resistance to quinolones (98.9%) and chloramphenicol (95.8%). Resistance to sulphonamides was also common (83%) (Supplementary Data 3B). Most strikingly, tLST-positive isolates showed a strong association with MBL carbapenemase genes, such as blaIMP-1, blaIMP-18, blaVIM-2, and blaVIM-9 (91.5%, n = 129) compared to tLST-negative isolates (34.3%, n = 83; p = 3.48 × 10⁻26). All isolates carrying tLST3b carried blaVIM-2, with six additionally carrying either blaVIM-9 (n = 3) or blaIMP-18 (n = 3). Conversely, tLST-negative isolates exhibited significantly higher prevalence of aminoglycoside resistance genes conferring resistance to gentamicin (48.1%, n = 115 vs. 16.3%, n = 23; p = 2.18 × 10−8) and streptomycin (39.3%, n = 95 vs. 12.1%, n = 17; p = 4.84 × 10−7).

Leaf labels in red indicate complete genomes included in the earlier part of study. Blue highlighted area indicates major tLST presence. From inner to outer, the coloured rings show tLST variant, source and country of origin. These are followed by the length of intI1 gene if present, and serotype. Red heatmap summarises antimicrobial-resistance classes of detected ARGs with specific colours for ESBL (green) and carbapenemases (dark brown: blaKPC, light brown: others).
tLST associations in Escherichia coli clonal complex (CC) 399
E. coli ST399 and ST635 had the most instances of tLST in the completed genome dataset and both STs belong to CC399. For more comprehensive tLST screening, and to elucidate any AMR and virulence correlations, ST399 (n = 514) and ST635 (n = 317) genomes were sourced from Enterobase and analysed. In ST399, 196 isolates (38%) carried at least one tLST variant, while ST635 showed an even higher proportion with 198 isolates (62%) being tLST-positive. The distribution pattern of tLST variants showed both similarities and distinctions between the sequence types. In ST399, tLST1 was predominant (n = 139), followed by tLST2b (n = 40), tLST1_AW1.7 (n = 21), tLST2c (n = 19), tLST3a (n = 19), and tLST2 (n = 5). Carriage of more than one tLST variant was notable in 51 ST399 genomes, with tLST1/tLST2b (n = 29) and tLST1/tLST2c (n = 16) being the most frequent combinations. Similarly, ST635 isolates primarily carried tLST1 (n = 167), followed by tLST2c (n = 89) and tLST3a (n = 3), with 61 isolates harbouring two tLST variants simultaneously.
Both sequence types generally maintained limited AMR profiles (Figs. 4 and 5). However, an outbreak of a clonal lineage of ST399 carrying the Yersiniabactin high pathogenicity island (HPI) and plasmid encoded blaOXA-48 engendering resistance to carbapenems, occurred on a vascular surgery ward housing predominantly diabetic patients with foot ulcers and compromised vasculature in a newly built hospital53. These outbreak ST399 isolates carried either tLST2b (n = 6) or tLST2b and tLST1 (n = 14). A second, predominantly environment-sourced (hospital sinks) E. coli ST635 subclade of 85 isolates (Fig. 5) also revealed a distinct AMR profile, of which 76% carried tLST (65/85), and 77 (91%) harboured class 1 integrons (intI1). The same isolates harboured consistent ARGs, including β-lactamases (blaTEM-1 or blaOXA-1), sulphonamide resistance determinants (sul1, sul2), aminoglycoside resistance genes (aadA1, aph(3′′)-Ib, aph(6)-Id), trimethoprim resistance (dfrA14 or dfrA19), and aminoglycoside-modifying enzymes conferring gentamicin resistance (aac(3)-IIe, aac(3)-IIg). ESBL genes (blaCTX-M-15, blaSHV-12) were detected in 76 isolates (89.4%) and colistin resistance (mcr-9) in 64 isolates (75.3%). Most (91%) carried F100:A-:B- plasmids and the subclade as a whole demonstrated high genetic relatedness with a mean SNP distance of 27 (range: 0–115 core SNPs). These observations reinforce the concept that lineages harbouring tLST can carry an impressive arsenal of antibiotic resistance genes, expanding on their ability to resist heat and a broad array of chlorine-based oxidising agents.

Leaf labels in red indicate complete genomes included in the earlier part of study. The inner coloured rings show tLST variants, source, and country of origin. These are followed by serotype and IncF RST as text labels. The blue heatmap represents yersinabactin high pathogenicity island (HPI) associated genes, which are followed by the length of intI1 gene detected and the outer red heatmap summarises antimicrobial-resistance classes of detected ARGs with specific colours for ESBL (light green = blaCTX-M; dark green = other) and carbapenemases (dark brown: blaKPC, light brown: others).

Leaf labels in red indicate complete genomes included in the earlier part of study. The inner coloured rings show tLST variant, source, and country of origin, followed by serotype and IncF RST. Blue heatmap represent yersiniabactin high pathogenicity island (HPI) associated genes, followed by the length of intI1 gene detected and outer red heatmap summarises antimicrobial-resistance classes of detected ARGs with specific colours for ESBL (light green: blaCTX-M; dark green: other) and carbapenemases (brown: blaKPC, light brown: others).
Discussion
Bacterial survival and adaptation across diverse environments represent fundamental drivers of evolutionary success, enabling microorganisms to persist in the face of environmental challenges and selective pressures. The tLST, an MGE that confers enhanced stress resilience, exemplifies how bacteria acquire adaptive traits through horizontal gene transfer. Our comprehensive genomic analysis of 48,183 bacterial genomes revealed the distribution of tLST across 46 bacterial species spanning multiple genera, including the identification of six previously undescribed tLST variants. We demonstrated that tLST1_AW1.7, which we found predominantly associated with F plasmids, dominates in Enterobacter and Klebsiella species, while tLST1 was almost exclusive to E. coli, and tLST3a prevails in Pseudomonas. This taxonomic partitioning extended to the family level, with Enterobacteriaceae members predominantly harbouring tLST1 and its variants, while non-Enterobacteriaceae families, particularly Pseudomonadaceae, primarily carried tLST3a. Characterisation of tLST1_AW1.7-carrying plasmids revealed selective carriage of virulence factors, notably the mrkABCDFJ operon present in 31.7% of plasmids, conferring enhanced biofilm formation capability and biocide tolerance. Although ARGs were generally uncommon on these plasmids, those that did contain ARGs often displayed extensive in silico resistance profiles, including resistance to last-resort antibiotics such as colistin and carbapenems. Importantly, we identified notable prevalence of tLST in specific high-risk ESKAPE-E lineages. Heat, chlorine and chlorine-based oxidising agents have been instrumental in controlling microbial loads in healthcare environments including cooling towers, WWTPs, recreational water systems, agricultural operations and food production. As such, the concentration of tLST in specific high-risk ESKAPE-E lineages represents one of the most concerning aspects of our findings because these successful pandemic lineages appear unencumbered by fitness imposts that normally accompany the accumulation and maintenance of the diversity of laterally acquired genetic cargo required to encode virulence and resistance to diverse and clinically relevant antibiotics, chlorine-based oxidising agents, heat, biocides and metals. The remarkably high prevalence in K. pneumoniae ST20 and P. aeruginosa ST111, suggests that stress tolerance provides additional selective advantages to successful pathogenic lineages.
K. pneumoniae ST20 is a pandemic lineage known for its association with MDR and frequent hospital outbreaks, particularly within neonatal units54,55,56,57. Of the 220 K. pneumoniae ST20 genomes investigated here, 124 (56%) carried tLST, with the plasmid-associated variant tLST1_AW1.7 dominating 98.4% of tLST-positive isolates. This finding aligns with earlier studies on the ClpK protein (also referred to as ClpG, confers remarkable heat resistance and now known to be part of the tLST). Bojer et al.58, found ClpK in 30% of K. pneumoniae clinical isolates within an intensive care unit and demonstrated its correlation with increased thermotolerance, suggesting this mechanism could facilitate strain persistence in healthcare environments. This was subsequently confirmed by Jørgensen et al.59, who isolated a ClpK-producing K. pneumoniae strain from an endoscope biofilm that survived standard chemothermal disinfection during an outbreak investigation. Like most K. pneumoniae ST20 isolates, those harbouring tLST were predominantly MDR. A striking example of this convergence of MDR and stress tolerance was observed in a highly clonal cluster of K. pneumoniae ST20 tLST-positive isolates from Singapore, where despite minimal genetic differences (mean 4 SNPs), isolates persisted across multiple hospitals, pathologies, and years while maintaining extensive MDR profiles including carbapenem resistance (blaKPC-2). The plasmid-mediated co-localisation of stress tolerance, biofilm formation capabilities, and extensive AMR in K. pneumoniae ST20 represents a significant challenge for infection control, as these strains can potentially survive both antimicrobial therapy and standard disinfection procedures.
P. aeruginosa ST111 represents one of the most clinically significant high-risk P. aeruginosa clones with global distribution, second only to ST235 in its widespread prevalence60. Our genomic analysis of 380 ST111 isolates was comprised of strains from diverse geographical regions yet a close genetic relatedness was observed (mean 119 SNPs). Most notably, our investigation revealed a ST111 sublineage characterised by a high prevalence of tLST (81.2%). Interestingly, the tLST3b variant dominated ST111, contrasting with the broader Pseudomonas population where tLST3a was more prevalent. The difference between these two variants is the presence of orfB in tLST3b, a gene that encodes for a diguanylate cyclase, known to control exopolysaccharide-associated biofilm development61. This variant-specific distribution suggests a potential selective advantage or co-evolutionary relationship within the ST111 lineage. We observed a significant co-occurrence of tLST and MBL carbapenemase genes in the ST111 population (91.5% of tLST-positive isolates versus 34.3% in tLST-negative isolates, p = 3.48 × 10−26). Indeed, all tLST3b-positive isolates carried blaVIM-2. Furthermore, the universal carriage of Class 1 integrons in the tLST-positive sublineage aligns with findings by Rath et al.62), who identified qacE/qacEΔ1 genes (part of a typical class 1 integron structure) conferring quaternary ammonium compound resistance in ST111 outbreak isolates. This convergence of disinfectant and AMR mechanisms highlights how genetic environmental persistence elements like tLST may contribute to the success of high-risk clones in healthcare settings, where both disinfectant and antibiotic selection pressures are substantial.
Our analysis of E. coli ST399 and ST635 isolates provides critical insights that challenge previous assumptions about tLST-positive E. coli lineages and their relationship with AMR and virulence factors. The high prevalence of tLST variants within these sequence types (38% in ST399 and 62% in ST635) supports their association with environmental adaptation, particularly in engineered environments63,64. However, our examination of specific subclades reveals a more complex evolutionary narrative. The tLST-positive ST399 outbreak strain in the UK carried both the carbapenemase gene blaOXA-48 and the Yersiniabactin high pathogenicity island, which contradicts earlier characterisations of tLST-positive strains as typically lacking virulence determinants42,65. The Yersiniabactin High Pathogenicity Island encoding Yersiniabactin, a key iron acquisition siderophore, is highly associated with death in a mouse sepsis model ranking higher than other important iron acquisition operons strongly implicated in invasive ExPEC disease including aerobactin and the sitABCD operon66. Similarly, the predominantly water faucet-derived ST635 subclade combining tLST positivity (76%) with extensive AMR profiles aligns with observations that a significant proportion (87.5%) of ST635 isolates from hospital wastewater were carbapenemase producers67. These findings suggest that environmental persistence conferred by tLST provides certain lineages with an evolutionary platform upon which they can accumulate clinically relevant MGEs, particularly in healthcare settings where selective pressures are intense64. This is especially concerning given that hospital wastewater contributes disproportionately to the environmental burden of resistance genes, potentially creating a cyclical relationship where environmentally persistent strains acquire clinical resistance determinants and subsequently establish resilient reservoirs in hospital infrastructure68. The evolutionary convergence of environmental persistence with AMR in these lineages underscores the public health significance of monitoring tLST-positive E. coli in healthcare environments, particularly as they may contribute to ongoing transmission chains and persistent outbreaks64.
Our study, while comprehensive in its genomic analysis of tLST variants, faced several limitations. As a purely WGS study using genomes from public databases, our analysis was constrained by the available bacterial genomes and their associated metadata, which may not fully represent global and source distributions, particularly from regions with limited sequencing infrastructure, and is biased towards clinical isolates. Without accompanying clinical or environmental metadata in many cases, we were unable to systematically assess clinical outcomes or environmental prevalence. Without access to corresponding isolates, we could not experimentally verify phenotypic traits associated with identified tLST variants, or test disinfection efficacies against tLST-positive strains, limiting our ability to provide evidence-based infection control recommendations. Nonetheless, extensive studies have been undertaken to evaluate the function(s) of specific genes carried across most tLST variants29,48. While all identified variants retain the core genes necessary for heat and chlorine resistance27, their structural variations may reflect evolutionary optimisation for different ecological contexts, with gene duplications potentially enhancing specific resistance mechanisms while deletions may reduce metabolic burden without sacrificing essential protective functions. Despite identifying novel tLST variants, our understanding of their evolutionary origins remains incomplete due to the nature of public database sampling. Methodologically, our reliance on high sequence identity thresholds may have excluded divergent variants.
The distribution of tLST across diverse bacterial pathogens represents an observation that warrants further investigation regarding its potential clinical and One Health implications. The presence of these elements in certain high-risk lineages, often in association with extensive AMR and enhanced environmental persistence capabilities, suggests these strains possess multiple mechanisms for withstanding both therapeutic interventions and infection control measures. Moving forward, further research into the functional characteristics of different tLST variants and their interactions with host bacterial genetics will enhance our understanding of their adaptive significance. Our findings highlight the value of comprehensive genomic surveillance incorporating both clinical and environmental sampling to monitor the distribution and evolution of these concerning elements. Increased awareness about tLST in clinical microbiology, industry and public health contexts would be beneficial based on our analysis. Such approaches will help us better understand the role of sanitation-tolerant, multidrug-resistant pathogens, including pandemic lineages, in healthcare settings and prevent their further dissemination through environmental reservoirs.
Methods
Screening for tLST
A total of 48,183 completed bacterial genomes and their corresponding metadata, representing 7190 unique species, were downloaded on February 5, 2024 (all completed genomes at the time), from the National Center for Biotechnology Information (NCBI) using NCBI Datasets69. To identify the presence of tLST variants, we screened all genomes using abricate v0.9.8 (github.com/tseemann/abricate) against four known reference sequences: tLST1 (NZ_CP025739), tLST2 (NZ_CP061749), tLST3a (NZ_CP010237), and tLST3b (NZ_CP061755). Genomes were classified as tLST-positive if they demonstrated sequence alignment with ≥90% identity and ≥80% coverage length to any of the reference variants. Further analysis focused on isolates showing multiple tLST hits with ≥90% identity and <80% but ≥20% coverage length. These sequences were mapped against complete tLST reference sequences, leading to the identification of six previously unreported tLST variants. Additionally, we confirmed the presence of two previously reported variants: AW1.7 (LDYJ01000141) and FAM21805 (KY416992). New variants were named according to their closest matching tLST version, with alphabetical designations for differentiation. The workflow used to screen for tLST is illustrated in Fig. 6. We selected one representative genome from each species containing any tLST variant and constructed a phylogenetic tree of diverse bacterial species using mashtree70, confidence values were estimated via 250 bootstrap replicates and visualised the relationships using the Interactive Tree of Life (iTOL)71. Structural comparisons of all tLST variants were visualised using EasyFig v2.2.572.

Bioinformatic workflow for identifying and analysing transmissible locus of stress tolerance (tLST) variants across bacterial genomes.
The data were then systematically classified into six distinct categories: Human, Animal, Environment, Wastewater, Laboratory and Unknown. For automated classification, the ChatGPT model ‘GPT-4o-mini’ was employed with the following query: ‘Classify the following sample isolation source into one of the categories: human, animal, environment, wastewater, laboratory. If the description is vague or unclear, and you cannot confidently classify it, respond with ‘unknown’. Answer with just one term. Sample: ‘$Sample’.’ All model-generated classifications were subsequently subjected to manual curation to ensure accuracy and consistency. Metadata was processed in R to extract key attributes, including host source and associated details.
Genotyping and phylogenetic analyses of tLST positive genomes
For sequence type (ST) determination, we analysed tLST-positive assemblies using mlst (github.com/tseemann/mlst) with default parameters, which require perfect hits for alleles (100% coverage and identity). We conducted a detailed investigation of E. coli, P. aeruginosa, K. pneumoniae and E. hormachei STs showing the highest tLST prevalence by retrieving all publicly available corresponding ST genomes from EnteroBase73, pubMLST74 and/or NCBI. Phylogenetic analysis of K. pneumoniae ST20, P. aeruginosa ST111 and E. coli ST399 and ST635 proceeded in two steps: first, we generated core genome sequence alignments (Block Mapping and Gathering with Entropy [BMGE] filtered) using panaroo v1.5.275 with -a core --remove-invalid-genes --aligner MAFFT options, followed by maximum likelihood phylogenetic tree construction using IQ-TREE 2 v2.2.076 with ModelFinderPlus (-m MFP) to determine best-fit model and single branch testing using ultrafast bootstrapping (1000 replicates) (--B 1000). The resulting phylogenetic relationships were visualised using iTOL71. Because recombination was not explicitly masked, branch lengths/supports may be influenced by homologous exchange; we therefore avoided micro-scale transmission inferences. To characterise these isolates comprehensively, we enriched the phylogenetic visualisation with metadata and molecular features on iTOL figures. We screened for ARGs, metal resistance genes, insertion sequences, plasmid replicons, and virulence factors using Abricate v1.0.1, accessing the CARD77, ISfinder78, PlasmidFinder79, and VFDB80 databases, respectively. AbritAMR81 was used to classify ARGs according to their corresponding antibiotic classes. Additionally, we conducted F family plasmid replicon sequence typing using pmlst79, serotype detection was done using ECTyper82 for E. coli and via PAst83 for P. aeruginosa. K. pneumoniae ST20 isolates were additionally analysed using Kleborate v3.2.484 (default parameters) to separate intrinsic Klebsiella AMR and virulence determinants from acquired ones. All genotyping data and metadata are available in Supplementary Data 1.
Data availability
To support the reproducibility of this work we have made the R scripts used to process, analyse and visualise this data available at github.com/Tu6ka/tLST_supdata_2025.
Code availability
To support the reproducibility of this work we have made the R scripts used to process, analyse and visualise this data available at github.com/Tu6ka/tLST_supdata_2025.
References
Rice, L. B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 197, 1079–1081 (2008).
Boucher, H. W. et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12 (2009).
Pendleton, J. N., Gorman, S. P. & Gilmore, B. F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti Infect. Ther. 11, 297–308 (2013).
Zhen, X., Lundborg, C. S., Sun, X., Hu, X. & Dong, H. Economic burden of antibiotic resistance in ESKAPE organisms: a systematic review. Antimicrob. Resist. Infect. Control 8, 1–23 (2019).
Centers for Disease Control, U. Antibiotic Resistance Threats in the United States, 2019. https://doi.org/10.15620/cdc:82532. (2019)
Ly, Y. T., Leuko, S. & Moeller, R. An overview of the bacterial microbiome of public transportation systems—risks, detection, and countermeasures. Front. Public Health 12, 1367324 (2024).
Gholipour, S., Shamsizadeh, Z., Gwenzi, W. & Nikaeen, M. The bacterial biofilm resistome in drinking water distribution systems: a systematic review. Chemosphere 329, 138642 (2023).
Li, S., Ondon, B. S., Ho, S. H., Jiang, J. & Li, F. Antibiotic resistant bacteria and genes in wastewater treatment plants: from occurrence to treatment strategies. Sci. Total Environ. 838, 156544 (2022).
Marutescu, L. G. et al. Wastewater treatment plants, an ‘escape gate’ for ESCAPE pathogens. Front. Microbiol. 14, 1193907 (2023).
Harrison, J. C. et al. Determinants of antimicrobial resistance in biosolids: a systematic review, database, and meta-analysis. Sci. Total Environ. 957, 177455 (2024).
Law, A. et al. Biosolids as a source of antibiotic resistance plasmids for commensal and pathogenic bacteria. Front. Microbiol. 12, 606409 (2021).
Kim, C. et al. Examining antimicrobial resistance in Escherichia coli: a case study in Central Virginia’s environment. Antibiotics 13, 223 (2024).
Djordjevic, S. P. et al. Genomic surveillance for antimicrobial resistance — a One Health perspective. Nat. Rev. Genet. 25, 142–157 (2023).
Ding, W. et al. Ozone disinfection of chlorine-resistant bacteria in drinking water. Water Res. 160, 339–349 (2019).
Simões, L. C., Simões, M. & Vieira, M. J. Influence of the diversity of bacterial isolates from drinking water on resistance of biofilms to disinfection. Appl. Environ. Microbiol. 76, 6673 (2010).
Luo, L. W. et al. Evaluating method and potential risks of chlorine-resistant bacteria (CRB): a review. Water Res. 188, 116474 (2021).
Malka, S. K. & Park, M. H. Fresh produce safety and quality: chlorine dioxide’s role. Front. Plant Sci. 12, 775629 (2022).
Drazic, A. et al. Role of cysteines in the stability and DNA-binding activity of the hypochlorite-specific transcription factor HypT. PLoS ONE 8, e75683 (2013).
Parker, B. W., Schwessinger, E. A., Jakob, U. & Gray, M. J. The RclR protein is a reactive chlorine-specific transcription factor in Escherichia coli. J. Biol. Chem. 288, 32574–32584 (2013).
Gray, M. J., Wholey, W.-Y., Parker, B. W., Kim, M. & Jakob, U. NemR is a bleach-sensing transcription factor. J. Biol. Chem. 288, 13789–13798 (2013).
Winter, J., Linke, K., Jatzek, A. & Jakob, U. Severe oxidative stress causes inactivation of DnaK and activation of the redox-regulated chaperone Hsp33. Mol. Cell 17, 381–392 (2005).
Du, Z., Nandakumar, R., Nickerson, K. W. & Li, X. Proteomic adaptations to starvation prepare Escherichia coli for disinfection tolerance. Water Res. 69, 110–119 (2015).
Cabiscol, E., Tamarit, J. & Ros, J. Oxidative stress in bacteria and protein damage by reactive oxygen species. Int. Microbiol. 3, 3–8 (2000).
Hillion, M. & Antelmann, H. Thiol-based redox switches in prokaryotes. Biol. Chem. 396, 415 (2015).
Li, H. et al. Heat and pressure resistance in Escherichia coli relates to protein folding and aggregation. Front. Microbiol. 11, 485630 (2020).
Wang, Z. et al. The locus of heat resistance confers resistance to chlorine and other oxidizing chemicals in Escherichia coli. Appl. Environ. Microbiol. 86, e02123-19 (2020).
Wang, Z. et al. Ecology and function of the transmissible locus of stress tolerance in Escherichia coli and plant-associated enterobacteriaceae. mSystems 6, e0037821 (2021).
Lee, C. et al. A novel protein quality control mechanism contributes to heat shock resistance of worldwide-distributed Pseudomonas aeruginosa clone C strains. Environ. Microbiol. 17, 4511–4526 (2015).
Mercer, R., Nguyen, O., Ou, Q., McMullen, L. & Gänzle, M. G. Functional analysis of genes comprising the locus of heat resistance in Escherichia coli. Appl. Environ. Microbiol. 83, e01400–e01417 (2017).
Gajdosova, J. et al. Analysis of the DNA region mediating increased thermotolerance at 58°C in Cronobacter sp. and other enterobacterial strains. Antonie Van Leeuwenhoek 100, 279–289 (2011).
Li, H. & Gänzle, M. Some like it hot: heat resistance of Escherichia coli in food. Front. Microbiol. 7, 1763 (2016).
Mercer, R. G., Walker, B. D., Yang, X., McMullen, L. M. & Gänzle, M. G. The locus of heat resistance (LHR) mediates heat resistance in Salmonella enterica, Escherichia coli and Enterobacter cloacae. Food Microbiol. 64, 96–103 (2017).
Zhang, P. & Yang, X. Genetic characteristics of the transmissible locus of stress tolerance (tLST) and tLST harboring Escherichia coli as revealed by large-scale genomic analysis. Appl. Environ. Microbiol. 88, e02185–21 (2022).
Dlusskaya, E. A., McMullen, L. M. & Gänzle, M. G. Characterization of an extremely heat-resistant Escherichia coli obtained from a beef processing facility. J. Appl. Microbiol. 110, 840–849 (2011).
Guragain, M., Brichta-Harhay, D. M., Bono, J. L. & Bosilevac, J. M. Locus of heat resistance (LHR) in meat-borne Escherichia coli: screening and genetic characterization. Appl. Environ. Microbiol. 87, 1–13 (2021).
Fang, Y., Tran, F., Stanford, K. & Yang, X. Stress Resistance and virulence gene profiles associated with phylogeny and phenotypes of Escherichia coli from cattle. J. Food Prot. 86, 100122 (2023).
Machado, M. A. M. et al. Genomic features and heat resistance profiles of Escherichia coli isolated from Brazilian beef. J. Appl. Microbiol. 134, lxac027 (2023).
Zhang, P., Tran, F., Stanford, K. & Yang, X. Are Antimicrobial interventions associated with heat-resistant Escherichia coli on meat? Appl. Environ. Microbiol. 86, e00512–e00520 (2020).
Marti, R. et al. Short communication: heat-resistant Escherichia coli as potential persistent reservoir of extended-spectrum β-lactamases and Shiga toxin-encoding phages in dairy. J. Dairy Sci. 99, 8622–8632 (2016).
Boll, E. J. et al. Turn up the heat-food and clinical Escherichia coli isolates feature two transferrable loci of heat resistance. Front. Microbiol. 8, 579 (2017).
Jarocki, V. M. et al. Comparative genomic analysis of ESBL-selected and non-selected Escherichia coli in Australian wastewater: elucidating differences in diversity, antimicrobial resistance, and virulence profiles. Sci. Total Environ. 949, 175079 (2024).
Zhi, S. et al. Evidence of naturalized stress-tolerant strains of Escherichia coli in municipal wastewater treatment plants. Appl. Environ. Microbiol. 82, 5505 (2016).
Wyrsch, E. R. et al. The faecal microbiome of the Australian silver gull contains phylogenetically diverse ExPEC, aEPEC and Escherichia coli carrying the transmissible locus of stress tolerance. Sci. Total Environ. 919, 170815 (2024).
Manges, A. R. et al. Global extraintestinal pathogenic Escherichia coli (ExPEC) lineages. Clin. Microbiol. Rev. 32, e00135-18 (2019).
Reid, C. J., De Maere, M. Z. & Djordjevic, S. P. Australian porcine clonal complex 10 (CC10) Escherichia coli belong to multiple sublineages of a highly diverse global CC10 phylogeny. Microb. Genom. 5, e000225 (2019).
Tarabai, H., Wyrsch, E. R., Bitar, I., Dolejska, M. & Djordjevic, S. P. Epidemic HI2 plasmids mobilising the carbapenemase gene blaIMP-4 in Australian clinical samples identified in multiple sublineages of Escherichia coli ST216 colonising silver gulls. Microorganisms 9, 1–21 (2021).
Reid, C. J. et al. A role for ColV plasmids in the evolution of pathogenic Escherichia coli ST58. Nat. Commun. 13, 1–15 (2022).
Kamal, S. M., Simpson, D. J., Wang, Z., Gänzle, M. & Römling, U. Horizontal transmission of stress resistance genes shape the ecology of beta- and gamma-proteobacteria. Front. Microbiol. 12, 696522 (2021).
Liu, H. et al. Diverse genotypes of Cronobacter spp. Associated with dairy farm systems in Jiangsu and Shandong Provinces in China. Foods 13, 871 (2024).
Nguyen, S. V., Harhay, G. P., Bono, J. L., Smith, T. P. L. & Harhay, D. M. Genome sequence of the thermotolerant foodborne pathogen Salmonella enterica Serovar Senftenberg ATCC 43845 and phylogenetic analysis of loci encoding increased protein quality control mechanisms. mSystems 2, e00190–16 (2017).
Gual-de-Torrella, A. et al. Prevalence of the fimbrial operon mrkABCD, mrkA expression, biofilm formation and effect of biocides on biofilm formation in carbapenemase-producing Klebsiella pneumoniae isolates belonging or not belonging to high-risk clones. Int. J. Antimicrob. Agents 60, 106663 (2022).
Sydow, K. et al. Geno- and phenotypic characteristics of a Klebsiella pneumoniae ST20 isolate with unusual colony morphology. Microorganisms 10, 2063 (2022).
Ledda A, et al. Hospital outbreak of carbapenem-resistant Enterobacterales associated with a blaOXA-48 plasmid carried mostly by Escherichia coli ST399. Microb Genom 8, 000675 (2022).
Thuy, D. B. et al. Colonization with Staphylococcus aureus and Klebsiella pneumoniae causes infections in a Vietnamese intensive care unit. Microb. Genom. 7, 1–14 (2021).
Jin, Y. et al. Outbreak of multidrug resistant NDM-1-producing Klebsiella pneumoniae from a neonatal unit in Shandong Province, China. PLoS ONE 10, e0119571 (2015).
Mavroidi, A. et al. Successful control of a neonatal outbreak caused mainly by ST20 multidrug-resistant SHV-5-producing Klebsiella pneumoniae, Greece. BMC Pediatr. 14, 1–8 (2014).
Pei, N. et al. Large-scale genomic epidemiology of klebsiella pneumoniae identified clone divergence with hypervirulent plus antimicrobial-resistant characteristics causing within-ward strain transmissions. Microbiol. Spectr.10, e0269821 (2022).
Bojer, M. S. et al. Concurrent emergence of multidrug resistance and heat resistance by CTX-M-15-encoding conjugative plasmids in Klebsiella pneumoniae. APMIS 120, 699–705 (2012).
Jørgensen, S. B. et al. Heat-resistant, extended-spectrum β-lactamase-producing Klebsiella pneumoniae in endoscope-mediated outbreak. J. Hosp. Infect. 93, 57–62 (2016).
del Barrio-Tofiño, E., López-Causapé, C. & Oliver, A. Pseudomonas aeruginosa epidemic high-risk clones and their association with horizontally-acquired β-lactamases: 2020 update. Int. J. Antimicrob. Agents 56, 106196 (2020).
Whiteley, C. G. & Lee, D. J. Bacterial diguanylate cyclases: structure, function and mechanism in exopolysaccharide biofilm development. Biotechnol. Adv. 33, 124–141 (2015).
Rath, A. et al. Whole-genome sequencing reveals two prolonged simultaneous outbreaks involving Pseudomonas aeruginosa high-risk strains ST111 and ST235 with resistance to quaternary ammonium compounds. J. Hosp. Infect. 145, 155–164 (2024).
Yu, D., Stothard, P. & Neumann, N. F. Emergence of potentially disinfection-resistant, naturalized Escherichia coli populations across food- and water-associated engineered environments. Sci. Rep. 14, 1–14 (2024).
Constantinides, B. et al. Genomic surveillance of Escherichia coli and Klebsiella spp. in hospital sink drains and patients. Microb. Genom. 6, 4–16 (2020).
Behruznia, M. & Gordon, D. M. Molecular and metabolic characteristics of wastewater associated Escherichia coli strains. Environ. Microbiol. Rep. 14, 646–654 (2022).
Royer, G. et al. Epistatic interactions between the high pathogenicity island and other iron uptake systems shape Escherichia coli extra-intestinal virulence. Nat. Commun. 14, 1–15 (2023).
Carlsen, L. et al. High burden and diversity of carbapenemase-producing Enterobacterales observed in wastewater of a tertiary care hospital in Germany. Int. J. Hyg. Environ. Health 242, 113968 (2022).
Sib, E. et al. Bacteria isolated from hospital, municipal and slaughterhouse wastewaters show characteristic, different resistance profiles. Sci. Total Environ. 746, 140894 (2020).
O’Leary, N. A. et al. Exploring and retrieving sequence and metadata for species across the tree of life with NCBI datasets. Sci. Data 11, 1–10 (2024).
Katz, L. S. et al. Mashtree: a rapid comparison of whole genome sequence files. J. Open Source Softw. 4, 1762 (2019).
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82 (2024).
Sullivan, M. J., Petty, N. K. & Beatson, S. A. Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010 (2011).
Dyer, N. P. et al. EnteroBase in 2025: exploring the genomic epidemiology of bacterial pathogens. Nucleic Acids Res. 53, D757–D762 (2025).
Jolley, K. A., Bray, J. E. & Maiden, M. C. J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3, 124 (2018).
Tonkin-Hill, G. et al. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 21, 1–21 (2020).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Alcock, B. P. et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 51, D690–D699 (2023).
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J. & Chandler, M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32-6 (2006).
Carattoli, A. & Hasman, H. PlasmidFinder and in silico pMLST: identification and typing of plasmid replicons in whole-genome sequencing (WGS). Methods Mol. Biol. 2075, 285–294 (2020).
Liu, B., Zheng, D., Zhou, S., Chen, L. & Yang, J. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917 (2022).
Sherry, N. L. et al. An ISO-certified genomics workflow for identification and surveillance of antimicrobial resistance. Nat Commun 14, https://doi.org/10.1038/S41467-022-35713-4 (2023).
Bessonov, K. et al. ECTyper: in silico Escherichia coli serotype and species prediction from raw and assembled whole-genome sequence data. Microb. Genom. 7, 000728 (2021).
Thrane, S. W., Taylor, V. L., Lund, O., Lam, J. S. & Jelsbak, L. Application of whole-genome sequencing data for O-specific antigen analysis and in silico serotyping of Pseudomonas aeruginosa isolates. J. Clin. Microbiol. 54, 1782–1788 (2016).
Lam, M. M. C. et al. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat. Commun. 12, 1–16 (2021).
Acknowledgements
This work was partially funding by The Joint Programming Initiative on Antimicrobial Resistance (JPIAMR), Towards next-generation AMR surveillance: Assessment of novel technologies with high-throughput and multiplexing potential (TEXAS). JPIAMR is a global collaborative organisation and platform, engaging 29 nations to curb AMR with a One Health approach. This work has also been supported by the Solving Antimicrobial Resistance in Agribusiness, Food, and Environments Cooperative Research Centre whose activities are funded by the Australian Government’s Cooperative Research Centre Programme. This is SAAFE CRC Publication VJ001_PB001.
Author information
Authors and Affiliations
Contributions
D.L.: Methodology, formal analysis, data curation, visualisation, writing—review and editing; V.M.J.: Investigation, supervision, writing—original draft, writing—review and editing. E.R.W.: Formal analysis, writing—review and editing. M.L.C.: Methodology, formal analysis, writing—review and editing. S.P.D.: Conceptualisation, supervision, project administration, writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, D., Jarocki, V.M., Wyrsch, E.R. et al. Next generation ESKAPE-E superbugs: identifying transmissible locus of stress tolerance and antibiotic resistance in pandemic bacterial lineages. npj Antimicrob Resist 3, 91 (2025). https://doi.org/10.1038/s44259-025-00162-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44259-025-00162-8
