
Abstract
Environmental dissemination of antimicrobial-resistant Escherichia coli is a major One Health challenge in Bangladesh. This study assessed the occurrence of third-generation cephalosporin and carbapenem-resistant E. coli across key environmental interfaces in Mymensingh. In May 2022, 28 water samples were collected from hospital wastewater, livestock effluents, aquaculture ponds, and the Khiro River. Samples were processed via the WHO Tricycle protocol, with 26 isolates undergoing whole-genome sequencing. Third-generation cephalosporin-resistant E. coli were detected in 86% of samples, while carbapenem-resistant isolates (18%) were found exclusively in hospital and river samples. Highest ESBL-producing E. coli concentrations occurred in hospital and poultry wastewater (mean 6.9–7.1 log10 CFU/ml). Sequencing identified 93 resistance genes, dominated by blaCTX-M-15 (79%), tet(A) (75%), aadA1 (54%), qnrS1 (50%), and mph(A) (50%). Carbapenem resistance was associated with blaNDM-5 in hospital and river isolates. While most isolates showed niche-specific clustering in our Phylogenetic analysis, highly conserved core-genome profiles (0 SNPs) between hospital and downstream river isolates (ST2363/ST410) provided strong genomic evidence consistent with effluent-mediated dissemination. These findings highlight the role of wastewater and livestock systems in AMR transmission, underscoring the urgent need for integrated One Health surveillance and improved waste management in Bangladesh.
Similar content being viewed by others


Introduction
Antimicrobial resistance (AMR) is a global public health threat with significant implications for human and animal health1. Of particular concern is the emergence of resistance to critically important antibiotics, including third-generation cephalosporins and carbapenems, which limits treatment options for severe infections.2. These antibiotics are often used as last-resort treatments for serious infections, and the development of resistance severely limits therapeutic options, leading to increased risk of treatment failure and mortality3,4.
Environmental compartments are increasingly recognized as key reservoirs and transmission pathways for antimicrobial resistance (AMR). Resistant bacteria and antibiotic resistance genes (ARGs) can be released into the environment through various pathways, including wastewater discharge from hospitals, farms, and households, as well as through the improper disposal of animal waste and agricultural runoff5. Once released into soil, water, or sediments, resistant bacteria can persist and accumulate in the environment, while their resistance genes can disseminate through microbial communities via horizontal gene transfer. Contaminated environmental reservoirs may subsequently contribute to human and animal exposures, amplifying the AMR burden across ecosystems6.
Bangladesh faces multiple AMR risk factors, including high population density, intensive livestock production, widespread antimicrobial use (AMU) in human and animal health, and inadequate wastewater treatment7,8. A 2019 Lancet report estimated that approximately 98,800 deaths in Bangladesh were associated with AMR9. The close interactions between humans, livestock, and aquatic environments, coupled with inadequate waste management and sanitation infrastructure, create an ideal setting for the emergence and spread of resistant bacteria. Several studies have detected multidrug-resistant E. coli across rivers, ponds, tap water, and household water, underscoring extensive environmental contamination10,11. Furthermore, a study revealed that 26% of municipal tap water samples in Dhaka harbored ESBL-producing E. coli12. Evidence of strain sharing and dissemination is also emerging, with studies tracking the environmental spread of New Delhi metallo-β-lactamase-1 (NDM-1)-producing multidrug-resistant bacteria in Dhaka13 and investigating the genomic diversity of ESBL-producing and carbapenem-resistant E. coli to understand transmission in urban and rural communities14. However, most studies examine isolated environmental niches, and few adopt an integrated One Health design that simultaneously assesses hospital, livestock, aquaculture, and river systems. Therefore, this study aimed to (i) quantify third-generation cephalosporin- and carbapenem-resistant E. coli across linked environmental sources, and (ii) characterize their resistance genes and phylogenetic relationships to explore potential dissemination pathways.
Results
Antimicrobial resistance in E. coli Isolates
All 28 environmental samples yielded E. coli growth on TBX agar. Overall, 24/28 samples (86%) were positive for third-generation cephalosporin-resistant E. coli, while 5/28 samples (18%) contained carbapenem-resistant E. coli (CRE). CRE were detected only in hospital wastewater (4/6 isolates) and river samples (one downstream isolate). Sampling sites, locations, and types are summarized in Table 1 and Fig. 1.

Sites include hospitals, poultry and cattle farms, aquaculture ponds, and the Khiro River system.
Total E. coli levels varied significantly across environmental compartments (p = 0.002), with the highest concentrations observed in poultry wastewater and hospital effluent (mean 7.7 and 7.9 log₁₀ CFU/100 mL, respectively), as shown in Fig. 2. Cattle farms showed moderately elevated levels (7.0 log₁₀ CFU/100 mL), while aquaculture ponds exhibited substantially lower counts (3.3 log₁₀ CFU/100 mL). River samples showed intermediate contamination (5.6 log₁₀ CFU/100 mL). Concentrations of ESBL-producing E. coli showed highly significant variation between sources (p < 0.001). Hospital and poultry wastewater had the highest ESBL counts (mean 6.9–7.1 log₁₀ CFU/100 mL), followed by cattle farm effluents (mean 5.5 log₁₀ CFU/100 mL). River samples had moderate counts (4.6 log₁₀ CFU/100 mL), while aquaculture ponds had the lowest levels (1.2 log₁₀ CFU/100 mL). Although carbapenem-resistant E. coli were less frequent, their distribution across sites was statistically significant (p = 0.007), with detection limited to hospital and river samples. The highest levels were found in hospital wastewater (3.7 log₁₀ CFU/100 mL), with low-level detection in downstream river water (0.5 log₁₀ CFU/100 mL). CRE were absent from all livestock and aquaculture samples. Kruskal–Wallis tests confirmed significant differences across sampling sources for total E. coli (p = 0.002) and ESBL-producing E. coli (p = 0.0003). Significant variation was also observed for carbapenem-resistant E. coli (p = 0.0074), driven by their detection in hospital and river samples.

Data were presented as mean log10 CFU/100 mL. Error bars represent standard deviation.
Phenotypic resistance patterns among the ESBL-producing E. coli isolates (n = 28) are summarized in Table 2. Near universal 100% resistance was observed to third-generation cephalosporins and the monobactam aztreonam. High levels of resistance were also observed for tetracycline, ranging from 67 to 100% across most environments, while azithromycin resistance was most pronounced in cattle farm wastewater (83%). Fluoroquinolone resistance (ciprofloxacin and nalidixic acid) was detected in 50–67% of isolates from hospital, cattle, and poultry wastewater. Trimethoprim–sulfamethoxazole resistance was particularly high in poultry wastewater (83%). No resistance to nitrofurantoin was detected in any ESBL-producing isolates. Gentamicin resistance was observed mainly in hospital (33%), poultry (17%), and cattle (17%) samples, whereas Cefoxitin resistance was restricted to the clinical sector, with half (50%) of the hospital isolates exhibiting a resistant phenotype. The two aquaculture pond isolates exhibited noticeably lower resistance across most antibiotic classes compared with isolates from other environmental sources.
Among the five CRE isolates identified, the hospital wastewater isolates (n = 4) demonstrated 100% resistance to gentamicin, nalidixic acid, meropenem, azithromycin, and ceftriaxone, and 75% (3/4) were also resistant to ciprofloxacin. The single downstream river CRE isolate showed resistance to most antibiotics, including ampicillin, ceftriaxone, ciprofloxacin, nalidixic acid, azithromycin, trimethoprim–sulfamethoxazole, and chloramphenicol. However, this isolate was sensitive to gentamicin and nitrofurantoin. All five CRE isolates exhibited reduced susceptibility or resistance to both imipenem and meropenem, consistent with their carbapenem-resistant phenotype.
Whole-genome sequencing analysis
Of the 26 sequenced isolates, 19 were ESBL-E. coli, five were CRE, and two (isolates 205C1 and 206C1 from aquaculture ponds) were non-ESBL-E. coli isolates. This selection ensured representation of all sample types and resistance phenotypes observed in the study.
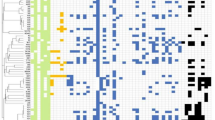
Across all 26 genomes, 93 antimicrobial resistance genes were detected. A wide range of antibiotic resistance genes were identified across various environments (Fig. 3). Hospitals harbored a high diversity of resistance genes, including the ESBL blaCTX-M-15; the AmpC beta-lactamases blaCMY and blaDHA-1; carbapenemases such as blaNDM-5 and blaOXA-181; and other beta-lactamase variants including blaTEM, blaOXA-1, blaOXA-10, and blaOXA-9. Other environments, however, primarily contained blaCTX-M-15, along with occasional occurrences of blaTEM, blaCMY, or blaNDM-5. Notably, the blaCTX-M family, particularly blaCTX-M-15, was the most prevalent, detected in all sampling locations. Conversely, blaCMY was prevalent in hospitals and rivers, while blaTEM was found in all environments except rivers. The concerning blaNDM-5 gene, known for conferring broad resistance, was identified in three hospital E. coli isolates and one river isolate. Virulence genes associated with adhesion (e.g., fyuA, papC) and toxin production (e.g., astA, ltcA) were also identified in some isolates. Further details of the genomes are provided in Supplementary Tables 1–4.

Genes are arranged in descending order of prevalence across the 26 sequenced isolates.
Heatmap analysis
Heatmap analysis showed distinct clustering patterns according to source category. Hospital-derived isolates exhibited the highest density and diversity of AMR genes, forming a tightly grouped cluster. Poultry and cattle isolates formed separate clusters dominated by blaCTX-M, tetracycline resistance genes, and various aminoglycoside resistance determinants. River isolates were heterogeneous. The downstream river isolate clustered closely with several hospital wastewater isolates due to the presence of blaNDM-5, multiple blaCMY variants, and shared plasmid-associated resistance genes. Upstream river isolates showed fewer resistance determinants. Virulence gene clustering demonstrated broader distribution across sample types but was most enriched in cattle and hospital isolates (Supplementary Table 4). Heatmap results are presented in Fig. 4.

Hierarchical clustering illustrates the grouping of isolates based on their genetic profiles.
Phylogenetic analysis
The core-genome SNP phylogeny (Fig. 5) revealed distinct genomic clustering primarily associated with sampling sources. Most poultry isolates (e.g., 201K1 and 204K2) grouped together, as did the cattle isolates belonging to ST4981 (e.g., 401K1 and 405K2), indicating genetic similarity within specific agricultural production systems. Hospital wastewater isolates exhibited greater genetic diversity, forming multiple clusters across different phylogroups. Importantly, a high-resolution linkage was identified between hospital wastewater isolate 102K1 (ST2363) and the downstream river isolate 501K1 (ST410). Through plasmid analysis, it was found that both isolate carrying the blaNDM-5 carbapenemase gene; however, while the hospital isolate (102K1) harbored an IncX3 replicon, the river isolate (501K1) carried a multireplicon profile including IncF-type and IncI1 plasmids. Aquaculture isolates (e.g., 301K1) formed a distinct branch, consistent with their lower AMR gene burden and phenotypic resistance levels.

The maximum-likelihood tree was constructed using a core SNP alignment of all 26 sequenced isolates against the E. coli K-12 MG1655 reference genome. Branch support was assessed with 1000 bootstrap replicates. Isolates are labeled by their unique ID (e.g., 102K1 and 501K1) and colored by sampling source: Hospital (Red), poultry (Green), cattle (Blue), river (Purple), and fisheries (Gold).
Core-genome SNP analysis identified a high-resolution linkage between hospital wastewater isolates (102K1, 106K1) and the downstream river isolate (501K1). While assigned to closely related sequence types (ST2363 and ST410, respectively) due to a single allelic variation in the recA gene, these isolates exhibited 0-SNP divergence across the shared core-genome alignment. This provides strong genomic evidence consistent with clonal dissemination along the hospital-river axis.
Plasmid profiling and resistance gene mobility
Plasmid profiling identified a diverse array of replicons (IncF, IncI1, IncN, and IncX) across all sectors (Supplementary Table 5). Notably, blaNDM-5 was physically linked to IncX3 in hospital isolates 102K1 (NODE_43) and 106K1 (NODE_40). Conversely, the downstream river isolate 501K1 harbored blaNDM-5 on a different IncF/IncI1 multireplicon platform (NODE_25; 81.8 kb).
blaCTX-M-15 was frequently co-located with IncF-type replicons across the interface. Source-specific associations included IncN, IncHI2, and IncFII in cattle (ST4981), IncI1 and IncFIB in poultry, and IncFII/IncFIB variants in aquaculture isolates.
Discussion
This study provides an integrated assessment of third-generation cephalosporin- and carbapenem-resistant E. coli across hospitals, livestock farms, aquaculture ponds, and a river system within a shared landscape in Bangladesh. The detection of ESBL-producing E. coli in 86% of samples and carbapenem-resistant E. coli in hospital and downstream river water demonstrates substantial environmental contamination with resistant organisms across multiple sectors. The consistently high ESBL-E. coli counts in hospital, poultry, and cattle wastewater reflect the marked bacterial loads and the widespread use of antimicrobials in these settings. In contrast, aquaculture ponds showed low levels of both total E. coli and resistant strains, consistent with lower antimicrobial inputs in these systems15.
In contrast, carbapenem resistance was less frequent, confined to hospital wastewater and river water. This aligns with the limited use of these antibiotics in livestock and aquatic farms in Bangladesh16,17. Genomic analysis revealed blaNDM as the sole carbapenemase gene, detected in 60% of carbapenem-resistant isolates, while blaIMP, blaVIM, blaKPC, and blaOXA-48 were absent. The detection of blaNDM in both hospital and river isolates suggests dissemination from healthcare facilities into the wider environment, raising concern given the clinical importance of NDM producers and their potential to limit last-resort treatment options.
Whole-genome sequencing confirmed a diverse array of AMR genes, including blaCTX-M-15, tet(A), aadA1, qnrS1, and mph(A). The frequent detection of these genes aligns closely with known antimicrobial resistance patterns and use patterns in Bangladesh8,18,19. In hospitals, third-generation cephalosporins are heavily prescribed, which corresponds with the prevalence of blaCTX-M-15. Similarly, the common use of macrolides and fluoroquinolones in both hospitals and poultry production likely contributes to the presence of mph(A) and qnrS1, respectively. This confirms the direct link between antimicrobial use in specific sectors and the genetic profiles observed in the environment. The results also align with other studies in Bangladesh and other LMIC countries20,21,22.
Phylogenetic analysis demonstrated source-specific clustering, suggesting localized persistence of strains. However, the detection of blaNDM-5 across sectors, carried by diverse plasmid replicons such as IncX3 (hospital) and IncF/IncI1 (river), underscores the complexity of environmental AMR transmission. The near-identity of isolates across different environmental compartments is highlighted by the 0-SNP distance between hospital and riverine E. coli. Although these isolates were partitioned into ST2363 and ST410 based on a single SNP in the recA locus, their overall genomic backbone suggests a very recent common ancestry. This supports the positioning of hospital effluent as a significant point source for AMR contamination in the local aquatic ecosystem, aligning with local studies, though we frame this as a high-resolution snapshot rather than a definitive transmission chain23,24. Furthermore, the distinct plasmid profiles in livestock, specifically the prevalence of IncN in cattle, highlight a separate agricultural resistome. These findings suggest that while clonal spread drives clinical-aquatic dissemination, horizontal gene transfer mediated by broad-host-range plasmids facilitates persistence in agricultural niches. This dual mechanism underscores the complexity of AMR transmission across the “One Health” interface in Bangladesh.
Furthermore, by linking AMR in hospitals, farms, and a river, our study provides a clear basis for cross-sectoral policy. The high AMR levels in hospital effluent emphasize the need for upgraded wastewater treatment protocols. In the animal health sector, our findings support the Bangladesh government’s existing efforts to reduce antibiotic use in food-producing animals. Moreover, the presence of AMR in river water highlights the risk to both human and animal populations who rely on this water source.
This study adds important environmental data to the broader understanding of AMR circulation in Bangladesh. By sampling multiple interconnected compartments using a One Health framework, we demonstrate how antimicrobial use and wastewater management practices shape the environmental distribution of resistant bacteria and genes. A comparison of our findings with data from the Mymensingh Medical College Hospital confirms that resistance levels in our hospital wastewater samples for certain drugs were higher than those in clinical isolates25. These findings highlight the need for improved wastewater treatment in healthcare facilities, strengthened antimicrobial stewardship in livestock production, and expanded environmental AMR surveillance to monitor dissemination pathways. Beyond technical interventions, there is a clear necessity for targeted awareness programs to bridge existing knowledge gaps among key stakeholders, including doctors, nurses, and farmers, concerning the environmental dimensions of resistance and the critical role of prudent antimicrobial stewardship26.
Several limitations should be noted. The study used a purposive sampling design with a relatively small sample size, which may limit generalizability. Sampling was restricted to a single month and did not capture potential seasonal variation. Only E. coli was investigated, and other relevant organisms may exhibit different patterns. Antibiotic concentrations in environmental samples were not measured, preventing linkage of resistance profiles to specific antimicrobial residues. Despite these limitations, this study represents a high-resolution genomic snapshot of the environmental distribution of resistant E. coli. While it identifies clear potential pathways, further longitudinal analysis is required to characterize the long-term transmission dynamics at these human–animal–environment interfaces.
In conclusion, this study demonstrates widespread environmental contamination with ESBL-producing E. coli and carbapenem-resistant E. coli across clinical, aquatic, and livestock sectors in Mymensingh, Bangladesh. While the detection of blaNDM-5 on diverse plasmid replicons, such as IncX3 in hospital wastewater and IncF/IncI1 in river water, underscores the complexity of environmental AMR transmission, core-genome SNP analysis revealed highly conserved genomic profiles (0-SNP divergence) between hospital and downstream river isolates (ST2363/ST410). This high-resolution linkage provides strong genomic evidence consistent with clonal, effluent-associated dissemination within the local ecosystem. These findings emphasize the urgent need for strengthened wastewater treatment in healthcare facilities, prudent antimicrobial use in livestock, and integrated One Health surveillance to contain the spread of “superbugs.” Such measures are essential to support the implementation of Bangladesh’s National Strategy and Action Plan on AMR (2023–2028).
Methods
Study design and study site
We conducted a cross-sectional environmental sampling study in May 2022 in Bhaluka Upazila, Mymensingh District, Bangladesh. This semi-urban area (~308,758 population; 444.05 km²) was selected because it contains clustered hospitals, commercial poultry and cattle farms, aquaculture ponds, and a river system within the same ecological landscape, enabling an integrated One Health assessment. The proximity of these sites to an AMR sentinel hospital also facilitated comparison with human AMR data. Hospitals were selected due to their high antibiotic consumption and potential to discharge resistant organisms into wastewater. Poultry, cattle, and aquaculture farms were chosen as major food production systems with known antibiotic use. Two points along the Khiro River (upstream and downstream of hospital discharge points) were sampled to assess potential dissemination into the aquatic environment.
Sampling size, sample site, and sample collection
A purposive sampling approach was used to collect 28 environmental water samples from selected sites in Bhaluka Upazila, based on logistical feasibility and findings from an initial pilot survey that informed site selection. The study included three hospitals, three poultry farms, three cattle farms, and three aquaculture farms, each of which was sampled twice, 2 weeks apart, with approval from local authorities and farm owners. In addition, the Khiro River was sampled at two locations, 1.6 km apart, upstream and downstream, to capture the potential influence of hospital effluent entering the river system, and both sites were sampled twice. All samples were collected using sterile procedures, transported in insulated cool boxes, and processed within 6 h of collection. GPS coordinates, sampling time, and basic water characteristics (appearance, odor, and turbidity) were recorded for each sampling event. Table 1 summarizes the sample types collected, and Fig. 1 illustrates the geographic distribution of all sampling sites.
Bacterial isolation and identification
Samples were processed following the WHO Tricycle ESBL Surveillance Protocol (WHO, 2021). For each sample, three 10 mL subsamples were vacuum-filtered through 0.45 μm cellulose nitrate membranes (Millipore). Filters were placed onto (i) Tryptone Bile X-glucuronide (TBX) agar for total E. coli counts, (ii) CHROMagar™ ESBL (CHROMagar, France) for ESBL-producing E. coli, and (iii) CHROMagar™ mSuperCARBA for carbapenem-resistant E. coli.
For the samples having a high number of colonies, a tenfold serial dilution was done using sterile distilled water to get a countable number of colonies in a culture plate. Plates were incubated at 37 °C for 18–24 h. Blue/blue-green colonies on TBX and pink, mauve or pink to reddish colonies on chromogenic media were counted. From each plate type per sample, 1–2 morphologically distinct colonies presumed to be E. coli were sub-cultured and confirmed biochemically using a commercially available API-20E kit (bioMerieux, France), as described previously27. Confirmed isolates were stored at −70 °C in glycerol stocks.
Antimicrobial susceptibility testing
We determined the antibiotic susceptibility profile of E. coli isolates (one isolate from each type of plate) against 21 clinically important antibiotics from 11 classes of antibiotics following CLSI guidelines28. The antibiotics used in this study were ampicillin (10 mg), mecillinam (25 mg), piperacillin-tazobactam (110 mg), cefepime (30 mg), cefotaxime (30 mg), ceftriaxone (30 mg), cefoxitin (30 mg), ceftazidime (30 mg), cefixime (5 mg), aztreonam (30 mg), ertepenem (10 mg), imipenem (10 mg), meropenem (10 mg), gentamicin (10 mg), tetracycline (30 mg), ciprofloxacin (5 mg), nalidixic acid (30 mg), trimethoprim–sulfamethoxazole (25 mg), chloramphenicol (30 mg), nitrofurantoin (F300 mg). Zone diameters were interpreted using CLSI breakpoints. E. coli ATCC 25922 served as the quality control strain.
Whole-genome sequencing and bioinformatic analysis
A total of 26 isolates were selected for whole-genome sequencing to represent the diversity of sample types and phenotypes (19 ESBL-Ec, five CRE, and two non-ESBL aquatic isolates: 205C1 and 206C1). Genomic DNA was extracted using the Maxwell culture DNA extraction kit and Maxwell automated nucleic acid extraction system following the manufacturer’s instructions, and sequencing libraries were prepared with the Nextera XT kit. Paired-end sequencing (2 × 150 bp) was performed on the Illumina NextSeq500 platform. Raw sequencing reads were assessed for quality using FastQC v0.11.4. High-quality reads were assembled de novo using SPAdes v3.13.0 using default parameters29. AMR genes were detected using ABRicate v1.0.1 with the ResFinder v4.1 database, applying thresholds of ≥90% identity and ≥90% coverage30,31,32. Plasmid replicons were characterized using PlasmidFinder 2.1 (Enterobacterales database).
To visualize the distribution of antimicrobial resistance genes (ARGs) across different sampling sectors, a heatmap was generated using the pheatmap (v1.0.12) package in R (v4.1.3)33. The presence (1) or absence (0) of ARGs, as identified by ResFinder, was used to create a binary matrix. A core-genome single-nucleotide polymorphism (SNP) phylogeny was constructed to investigate the genetic relatedness and potential clonality of the E. coli isolates. Whole-genome sequences were aligned to a reference E. coli genome (e.g., E. coli K-12 MG1655) using Snippy v4.6.0 to identify high-quality variants34. A pairwise SNP distance matrix was generated using snp-dists (v0.8.2). A core SNP alignment was generated using snippy-core, and low-quality or closely spaced SNPs were filtered to reduce noise. A maximum-likelihood phylogenetic tree was then inferred from the core SNP alignment using IQ-TREE v2.1.2 with 1000 bootstrap replicates to assess branch support35. All 26 sequenced isolates, including those from aquaculture and river systems, were taxonomically confirmed as Escherichia coli using Kraken2 and ribosomal MLST (rMLST).
Statistical data analysis
Descriptive statistics were computed in STATA v17. CFU counts were log₁₀-transformed and summarized by mean and standard deviation. Proportions of ESBL-Ecoli and CRE positivity were calculated per sample type. Differences in CFU levels between environmental sources were assessed using non-parametric tests (Kruskal–Wallis). Significance was defined as a p value <0.05.
Ethical considerations
No human or animal subjects were involved in this study; only environmental samples were collected. Administrative approval was obtained from the relevant government authorities.
Data availability
All genome sequence data are available under BioProject PRJNA1372464.
References
Walsh, T. R., Gales, A. C., Laxminarayan, R. & Dodd, P. C. Antimicrobial resistance: addressing a global threat to humanity. PLoS Med. 20, e1004264 (2023).
Naghavi, M. et al. Global burden of bacterial antimicrobial resistance 1990–2021: a systematic analysis with forecasts to 2050. Lancet 404, 1199–1226 (2024).
Stewardson, A. J. et al. Effect of carbapenem resistance on outcomes of bloodstream infection caused by Enterobacteriaceae in low-income and middle-income countries (PANORAMA): a multinational prospective cohort study. Lancet Infect. Dis. 19, 601–610 (2019).
Chen, J. et al. Extended-spectrum β-lactamase-producing Escherichia coli and Klebsiella pneumoniae: risk factors and economic burden among patients with bloodstream infections. Risk Manag. Healthc. Policy 17, 375–385 (2024).
Pal, C., Bengtsson-Palme, J., Kristiansson, E. & Larsson, D. J. The structure and diversity of human, animal and environmental resistomes. Microbiome. 4, 54 (2016).
Read, D. S. et al. Dissemination and persistence of antimicrobial resistance (AMR) along the wastewater-river continuum. Water Res. 264, 122204 (2024).
Chowdhury, S. et al. Antibiotic usage and resistance in food animal production: what have we learned from Bangladesh?. Antibiotics 10, 1032 (2021).
Rahman, A. et al. Patterns of antimicrobial resistance among major bacterial pathogens isolated from clinical samples in Bangladesh (2017–2020): a nationwide cross-sectional study. Microbiol. Res. 16, 122 (2025).
Murray, C. J. et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
Islam, M. M., Islam, M. A., Neela, F. A. & Hasanuzzaman, A. F. M. Occurrence of antibiotic-resistant bacteria in urban surface water sources in Bangladesh. Curr. Microbiol. 82, 96 (2025).
Asaduzzaman, M. et al. Spatiotemporal distribution of antimicrobial resistant organisms in different water environments in urban and rural settings of Bangladesh. Sci. Total Environ. 831, 154890 (2022).
Talukdar, P. K. et al. Antimicrobial resistance, virulence factors and genetic diversity of Escherichia coli isolates from household water supply in Dhaka, Bangladesh. PLoS ONE 8, e61090 (2013).
Islam, M. A. et al. Environmental spread of New Delhi metallo-β-lactamase-1-producing multidrug-resistant bacteria in Dhaka, Bangladesh. Appl. Environ. Microbiol. 83, e00793-17 (2017).
Flatgard, B. M. et al. Tracking antimicrobial resistance transmission in urban and rural communities in Bangladesh: a One Health study of genomic diversity of ESBL-producing and carbapenem-resistant Escherichia coli. Microbiol. Spectr. 12, e03956-23 (2024).
Rheman, S. et al. Antimicrobial and other aquamedicine use for disease treatment in pond-based commercial tilapia farms in Bangladesh. Aquaculture 618, 743814 (2026).
Hasan, M. M. et al. Patterns of antimicrobial usage (AMU) in small and mediumscale commercial chicken farms in Bangladesh. Int. J. Infect. Dis. 134, S20–S21 (2023).
Amin, M. B. et al. Prevalence and antimicrobial resistance of major foodborne pathogens isolated from pangas and tilapia fish sold in retail markets of Dhaka city, Bangladesh. Int. J. Food Microbiol. 418, 110717 (2024).
Ibrahim, N. et al. Antimicrobial resistance in Escherichia coli and its correlation with antimicrobial use on commercial poultry farms in Bangladesh. Antibiotics 12, 1361 (2023).
Rashid, M. M. et al. Pattern of antibiotic use among hospitalized patients according to WHO access, watch, reserve (AWaRe) classification: findings from a point prevalence survey in Bangladesh. Antibiotics 11, 810 (2022).
Ayzanoa, B. et al. Genomic surveillance of multidrug-resistant E. Coli and Klebsiella in clinical and wastewater isolates from a pediatric hospital in Lima, Peru. Access Microbiol. 8, 001006-v3 (2026)
Mbanga, J., Abia, A. L., Amoako, D. G. & Essack, S. Y. Longitudinal surveillance of antibiotic resistance in Escherichia coli and Enterococcus spp. from a wastewater treatment plant and its associated waters in KwaZulu-Natal, South Africa. Microb. Drug Resist. 27, 904–918 (2021).
Davies, A. R. et al. Genomic characterisation of Escherichia coli isolated from poultry at retail through sink surveillance in Dhaka, Bangladesh reveals high levels of multi-drug resistance. Front. Microbiol. 15, 1418476 (2024).
Nashwan, A. J., Shah, H. H., Hussain, T., Rauf, S. A. & Ahmed, S. K. Environmental drivers of antimicrobial resistance in low and middle-income countries: the impacts of a changing world. Environ. Health Insights 18, 11786302241246420 (2024).
Islam, M. S., Rahman, A. T., Hassan, J. & Rahman, M. T. Extended-spectrum beta-lactamase in Escherichia coli isolated from humans, animals, and environments in Bangladesh: a One Health perspective systematic review and meta-analysis. One Health 16, 100526 (2023).
Roy, S. et al. Trends and patterns of antimicrobial resistance in a tertiary care hospital in Bangladesh. Microbiol. Res. 16, 30 (2025).
Rahman, A., Jannat, H., Deb, P. K. & Rizvi, S. S. Knowledge, attitudes, and practices of antimicrobial stewardship among healthcare professionals in Bangladesh: a cross-sectional survey. Bangladesh J. Infect. Dis. 12, 18–26 (2025).
Amin, M. B. et al. Identifying the sources of intestinal colonization with extended-spectrum β-lactamase-producing Escherichia coli in healthy infants in the community. Front. Microbiol. 13, 803043 (2022).
Humphries, R., Bobenchik, A. M., Hindler, J. A. & Schuetz, A. N. Overview of changes to the clinical and laboratory standards institute performance standards for antimicrobial susceptibility testing, M100. J. Clin. Microbiol. 59, 10–128 (2021).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Clausen, P., Aarestrup, F. M. & Lund, O. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics 19, 307 (2018).
Bortolaia, V. et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500 (2020).
Zankari, E. et al. PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 72, 2764–2768 (2017).
Kolde R. pheatmap: Pretty Heatmaps. R package version 1.0.13. https://github.com/raivokolde/pheatmap (2025).
Seemann T. snippy: fast bacterial variant calling from NGS reads. https://github.com/tseemann/snippy (2015).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Acknowledgements
We thank the Fleming Fund for their financial support through the Fleming Fellowship, and icddr,b, for providing sample collection and laboratory testing facilities. This study was conducted as part of the collaborative activities under the Fleming Fellowship for Bangladesh, Phase 1. Funding to support sample collection and laboratory testing was provided through the Fleming Fellows’ activity budget. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Open access funding provided by CGIAR Science Programme “Sustainable Animal and Aquatic Foods (SAAF)” which is supported by contributors to the CGIAR Trust Fund.
Author information
Authors and Affiliations
Contributions
All authors combinedly designed the study. A.R., S.R., B.A., N.A., M.G.S.A., M.S., A.K., F.T., B.A., S.Y., R.H., and S.A. coordinated field activities and supervised the sample collection. AR and AM drafted the manuscript. A.R.L., L.M.C., J.L., J.D.-D., A.M., and A.P. finalized laboratory methodology. K.I.H., M.B.A., S.R., B.A., M.G.S.A., M.S., A.K., F.T., B.A., and S.A. (icddr,b) supported phenotypic tests, whole-genome sequencing, and bioinformatic analyses. A.R. conducted, and A.M., M.G.S.A., M.S., K.I.H., and M.B.A. supported the data analysis. All authors critically reviewed the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rahman, A., Roy, S., Afreen, N. et al. Environmental dissemination of multidrug-resistant Escherichia coli across one health interfaces in Mymensingh, Bangladesh. npj Antimicrob Resist 4, 27 (2026). https://doi.org/10.1038/s44259-026-00202-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44259-026-00202-x
