
Abstract
Neurotropic viruses affecting the central nervous system cause significant short- and long-term morbidity and mortality. Sequelae are extremely prevalent, with up to 50% of survivors experiencing neurological, neuropsychiatric, and behavioral issues. However, the immunopathological and virological mechanisms and factors influencing these outcomes remain poorly understood. In this review, we outline the available knowledge on the long-term outcomes of brain infections of several widespread viruses and highlight key current research gaps.
Similar content being viewed by others
Introduction
A diverse group of viruses is neurotropic in humans and can affect the central nervous system (CNS) to cause disease. This includes encephalitis (inflammatory disease of the brain), as well as meningitis (inflammation of the meninges), and myelitis (inflammation of the spinal cord). While meningitis and myelitis are important causes of acute and chronic neurological disability in their own right, and sometimes share pathogenic mechanisms with encephalitis1,2, this review places primary emphasis on encephalitis and the short‑ to medium-term neurological sequelae that follow brain infection.
Acute viral encephalitis can impair physical, cognitive, and behavioral functions, and, in many cases, proves fatal. The mortality rate varies between viruses, and it has decreased over the past three decades, but fatality estimates remain at ~6% for encephalitis overall3. In addition, even in individuals who survive the acute disease, the viral brain infection can be followed by severely debilitating neurological sequelae in up to 50% of patients. These can include cognitive, language, and sensory impairments, motor deficits, seizures, depression, anxiety, personality changes, and fatigue, and overall can drastically impair the quality of life of the patient4. Numerous brain regions have been reported to be involved in the different forms of viral encephalitis, including the temporal lobe, basal ganglia, cerebellum, and thalamus, reflecting the wide range of possible sequelae5. When including all forms of encephalitis, the estimated disability-adjusted life years worldwide were 4.8 million in 20193, and thus, it is a major public health concern around the globe.
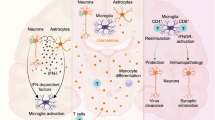
Yet, to date, there has been relatively limited research on these post-infectious brain conditions and the mechanisms underlying their development and persistence. Within the last decade, progress in the understanding of virology, brain physiology, and immunology, combined with methodological advancements, has begun to address some of these questions. Here, we outline several viruses associated with post-encephalitic neurological complications and the evidence as to the underlying mechanisms of action that have been obtained to date (overview in Fig. 1). It is important to note that several viruses that are not classically neurotropic, such as SARS-CoV-2, human immunodeficiency virus (HIV), and hepatitis C, can affect the brain during and following primary infection6,7,8. Furthermore, viruses have been implicated as contributors to several neurodegenerative disorders9,10,11, including herpes simplex virus (HSV) type 1 in Alzheimer’s disease12 and Epstein-Barr virus (EBV) in multiple sclerosis13. However, systemic viruses and the potential links of viruses to neurodegenerative disease fall outside the scope of this review, which is centered on post-infectious brain conditions that, while sometimes persistent, are distinct from classical neurodegenerative disorders.

Viruses can enter the CNS via brain–blood barrier disruption, transcellular, paracellular, or transsynaptic spread, or by infecting peripheral immune cells, which then cross into the CNS. Viral infection of the CNS can trigger neuronal death, disruption of neurogenesis, and dysregulation of the immune system. These disturbances in homeostasis can result in long-term neurological complications, such as cognitive deficits, seizures, headache, fatigue, mood disorders, and changes in personality and behavior. Figure created with BioRender.
Flaviviruses
Flaviviruses are a diverse group of positive-sense, single-stranded RNA viruses, primarily transmitted by mosquitoes and ticks. This genus consists of over 70 viruses, including many that induce human disease14. The burden of flaviviruses worldwide is substantial, with an estimated 400 million people infected annually by dengue alone15. Despite the enormous global impact, no specific antivirals exist for the treatment of flaviviral infections16. Flaviviruses can cause systemic diseases such as dengue and yellow fever, while others, including West Nile virus and Japanese encephalitis virus, can induce severe neurological complications14. In this review, we have focused on several common flaviviruses that cause neurological disease with the most data available.
West Nile virus
West Nile virus (WNV) is a mosquito-borne flavivirus, with birds serving as natural amplification hosts17. It was first isolated from the blood of a febrile patient in the West Nile district of Uganda in 193718. Today, it is one of the most widespread causes of arboviral disease worldwide, and has been detected in every continent except Antarctica19. Currently, there are no human vaccines available20.
An estimated 75% of human WNV infections are asymptomatic21,22,23. Most symptomatic cases manifest as a flu-like illness known as West Nile fever. However, in <1% of infections, the individual develops West Nile neuroinvasive disease (WNND), which may manifest as encephalitis, meningitis, acute flaccid paralysis, or a combination thereof, depending on the spread of the virus19,21,24. A diagnosis of WNND is usually obtained by testing of serum or CSF for anti-WNV immunoglobulin M (IgM) antibodies, which are usually detectable 3–8 days after illness onset, and can persist for 90 days25. Several risk factors have been identified for the development of neuroinvasive disease, including advanced age, immunosuppression, and comorbidities such as diabetes, hypertension, and cardiovascular disease26,27,28. Host genetic factors also influence susceptibility to West Nile virus infection, most notably the C–C chemokine receptor type 5 Δ32 (CCR5-Δ32) mutation29,30.
The fatality rate of WNND is ~10%31,32,33,34, and amongst the survivors, 40–60% will experience neurological sequelae, which may persist for a few months or for up to several years35,36,37,38. These complications include muscle weakness, fatigue, depression, headache, memory loss, and confusion39. However, the mechanisms remain poorly understood, and while it has been reported that a clinical presentation of encephalitis is associated with the highest risk of sequelae development, long-term complications have also been reported in those with only febrile illness, indicating that the occurrence of WNND does not reliably predict sequelae35,37,40.
Following a mosquito bite, the incubation time for WNV is typically 3–14 days41. The virus initially replicates in the keratinocytes and Langerhans cells of the skin, before progressing to the local draining lymph nodes, where it is amplified. The virus then spreads to the kidney, spleen, and other organs, and following systemic infection, it can invade the CNS around 5 days post-infection24,42. WNV has been proposed to enter the brain through multiple mechanisms, including disruption of the blood–brain barrier (BBB), transsynaptic or transcellular spread, and by infection of peripheral immune cells, which subsequently enter the CNS, also known as a ‘Trojan horse’ mechanism43,44,45,46,47,48. However, the relative contribution of these mechanisms across different species and viral strains remains unclear.
Within the CNS, WNV primarily replicates within neurons, but there is also evidence that it may be able to infect glial cells, such as astrocytes49,50,51. Viral particles have been detected postmortem in multiple brain regions, including the brainstem, cerebellum, and thalamus52,53. Furthermore, in individuals with a history of WNV infection and ongoing neurological issues, magnetic resonance imaging (MRI) showed regional atrophy in those same brain regions, and cortical thinning in both hemispheres compared to controls54. This diversity of infection and pathology may explain the wide range of clinical sequelae associated with the disease.
WNV can directly induce neuronal injury and apoptosis through several mechanisms, such as caspase 3-dependent apoptosis, accumulation of ubiquitinated proteins, and activation of the unfolded protein response55,56,57. Recognition of the virus through pattern recognition receptors, including toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I), is crucial for the initiation of the innate immune response to WNV in the CNS58,59. However, the immune response might also be harmful, as the release of inflammatory cytokines and chemokines from glia or infected cells can indirectly induce neuronal damage and death51,60,61.
Impairments in cognition and memory are among the most frequent sequelae of WNND39. In a mouse model using an attenuated WNV strain, mice displayed impaired spatial learning and memory in the Barnes maze despite no loss of hippocampal neurons. However, there was a loss of hippocampal CA3 presynaptic terminals, which was also observed in postmortem tissue from patients with WNND62. This was attributed to microglial complement activation, as C1Qa (the initiating factor of the classical complement pathway) was upregulated and localized to infected neurons and microglia in the mice. This finding was supported by the absence of synaptic terminal loss in mice lacking complement C3 or its receptor C3aR162.
The signaling of T cells to microglia is also important in the context of learning deficits following WNND. Specifically, antiviral T cells were observed to persist in the hippocampus after infection in mice, and interferon-gamma (IFN-γ) released from these CD8+ T cells drove microglial activation and impairments in spatial learning. Similar mechanisms were observed after Zika virus infection, another flavivirus with distinct neuropathological outcomes63. Furthermore, CCR2 signaling modulates the phenotype of persisting CD8 resident memory T cells (TRM) in the hippocampus and regulates their expression of IFN-γ, which was implicated in the prevention of memory impairment in the novel object recognition test64. The CXCL16/CXCR6 chemokine signaling pathway also supports both the maintenance of these cells post-infection and their expression of TRM markers in the forebrain65.
In addition, hippocampal neurogenesis is disrupted following WNND, which may explain why the hippocampus is unable to restore learning even though adult neurogenesis occurs within the dentate gyrus and subventricular zone (SVZ). Using the same attenuated WNV mouse model as in the studies described above, transcriptional alterations in genes regulating adult neurogenesis, such as interleukin-1 (IL-1), were observed in the hippocampus. Mice that recovered from WNND showed increased astrogenesis compared to mocks, and ex vivo, these cells were determined to be the primary source of IL-1β, suggesting that astrocytes function via IL-1 to interfere with neurogenesis and memory function after viral infection. Mice deficient in the IL-1 receptor IL-1R1 did not show the impairment of neurogenesis and memory observed in their wild-type counterparts, highlighting the role of this cytokine in post-infectious cognitive dysfunction66. Figure 2 summarizes some of the current mechanistic understanding of impairment of cognition and memory after WNND.

a CD8+ T cells release IFN-γ, which activates microglial cells (green). Microglia upregulate C1Qa, leading to complement-dependent loss of CA3 pre-synaptic terminals in the hippocampus. b Increased astrogenesis leads to the secretion of IL-1, which inhibits neurogenesis in the dentate gyrus. c Both mechanisms outlined in panels a and b contribute to deficits in learning and memory of mice, assessed in the Barnes maze. See main text for references. Figure created with BioRender.
Depression is highly prevalent following WNV infection, affecting between 20% and 40% of survivors36,39,67,68. Despite the frequency of this condition, there are no animal models of WNV-induced depression or anxiety at chronic time points. One patient study investigated the associations between blood levels of 15 cytokines and the onset of depression following WNV infection. Univariate analyses suggested that elevated chemokine (C–C motif) ligand 2 (CCL2) and decreased tumor necrosis factor-alpha (TNF-α) were associated with depression, but this was not found to be significant after adjusting for multiple comparisons69.
Persistent inflammation has, however, been linked to other long-term symptoms after WNV infection. Increased serum levels of pro-inflammatory and antiviral cytokines such as IFN-γ, interferon gamma-induced protein 10 (IP-10/CXCL10), interleukin-6 (IL-6), and granulocyte-macrophage colony-stimulating factor (GM-CSF) were reported in WNND patients suffering from fatigue post-infection, compared to those who did not have fatigue70. Elevated TNF-α in serum was also reported in patients suffering from persistent post-infectious symptoms71. Levels of the inflammatory mediators interleukin-4 (IL-4), interleukin-8 (IL-8), interleukin-10 (IL-10), and CCL2 also remained elevated one month post-infection, although this was not directly linked to sequelae risk72.
IgM antibodies were detected in 23% of patients 8 years following WNV infection, suggesting ongoing low-level antigenic stimulation or a chronic immune response73. WNV RNA was also detected in the urine of 20% of patients years after primary infection74. However, this was a relatively small study, and while 4 out of the 5 patients with detectable RNA had chronic symptoms, it did not assess if the presence of WNV RNA increased the risk of any particular sequela.
Although many individuals infected with WNV are asymptomatic, those who develop WNND face a substantial risk of death or disabling long-term neurological sequelae. Despite the increasing geographical range and growing public health threat of the virus, no human vaccines or targeted antivirals are currently available20,42. Further studies are required to assess the cellular mechanisms underpinning the progression of the disease and the development of post-infectious complications in order to aid therapeutic development.
Japanese encephalitis virus
Japanese encephalitis virus (JEV) is a mosquito-borne flavivirus in the Asia-Pacific region, with over 3 billion people across 24 countries at risk of infection75. Although effective vaccines have been commercially available for decades76,77, incomplete vaccination coverage and the continued circulation of the virus in nature78,79,80 mean that Japanese encephalitis (JE) remains a significant public health concern. While the majority of cases are asymptomatic or have mild symptoms, an estimated 1% of infected individuals will develop encephalitis, which particularly affects children, as most adults from endemic regions have developed immunity due to prior JEV exposures81,82,83. Diagnosis is usually confirmed by testing serum or CSF for virus-specific IgM antibodies, which are typically detectable ~3–8 days after illness onset84. This disease is associated with a substantial disease burden; recent estimates indicate that although fatality rates are falling, the current mortality remains at ~14%85. Furthermore, nearly half of survivors will suffer from long-term neuropsychiatric sequelae, including effects on memory, language, and emotional and behavioral disturbances85,86,87,88.
As the first symptoms of disease generally manifest 5–15 days post-exposure41, the mechanisms active during the early phases of infection remain poorly characterized. It is proposed that the virus initially replicates in the skin surrounding the mosquito bite, including keratinocytes, fibroblasts, endothelial cells, and various immune cells89,90,91,92. After infecting the nearby lymph nodes, it can enter the bloodstream and cross the BBB by several proposed mechanisms, including cytokine-mediated BBB disruption, transcellular transport, or using infected immune cells in the ‘Trojan horse’ strategy93,94,95,96,97. Once in the CNS, acute JE manifests with neurological symptoms such as altered consciousness, fever, seizures, and headache, sometimes accompanied by non-neurological features such as abnormal breathing patterns and pulmonary edema98,99. Postmortem, viral antigen has been detected in the hippocampus, thalamus, substantia nigra, and medulla oblongata100. While JEV is highly neurotropic, there is evidence that it can occasionally infect microglia, astrocytes, and perivascular macrophages101,102.
Several risk factors influence JEV transmission, including climate, with higher temperatures supporting mosquito development, and land use, as activities like rice paddy cultivation and pig farming increase exposure to JEV amplifying hosts such as wading birds and pigs103,104,105,106. Another risk factor is host genetic polymorphisms in immunity genes, such as toll-like receptor 3 (TLR3)107, IL-1β108, and TNF-α109,110, which also modulate susceptibility to JEV infection.
The nature of the immune response likely contributes to long-term outcomes following JEV exposure. Infection of the CNS triggers pronounced activation of microglia and astrocytes, infiltration of dendritic cells and T cells, and widespread neuronal injury111,112,113,114. While the initial acute inflammatory response is critical in controlling the viral infection, a prolonged inflammatory response can lead to long-term undesirable consequences. Proteomic profiling of CSF defined two subtypes of JE (JE1 and JE2)115. The JE2 subtype was associated with a higher rate of cognitive impairment, alongside downregulation of proteins involved in myeloid cell-mediated immunity and leukocyte/neutrophil degranulation, and significant upregulation of several complement pathway components, indicating roles for these pathways in the prognoses of JE patients. An increased neutrophil-to-lymphocyte ratio was also observed to increase the risk of neurological sequelae in patients following JE, as was the quality of the T cell response115. Poor outcome and sequelae were associated with a pro-inflammatory TNF-α CD4+ response, whereas complete recovery was linked to a polyfunctional CD4+ response116. Furthermore, levels of IFN-γ, commonly produced by T cells, were found to be inversely proportional to the severity of sequelae117. The cytokines IL-1β and IL-10 also play roles, as polymorphisms of these genes were associated with an altered risk of neurological sequelae108. Collectively, these data indicate an altered inflammatory response and potential immune dysregulation post-JE that may contribute to greater neuronal injury and impaired recovery within the central nervous system.
Following CNS injury, effective neuronal repair is critical, with neurogenesis serving as a key mechanism in restoring homeostatic balance and promoting a return to functional health118. JEV has been observed to significantly impair neurogenesis by depleting neural progenitor cells from the SVZ. By use of a combination of mouse models and neurosphere cultures, this was ascertained to occur not only due to direct viral cytolysis, but also through inhibition of cell cycle progression at the G1 to S transition by upregulation of checkpoint proteins119. Moreover, the differentiation potential of neural stem/progenitor cells (NSPCs) was affected following JEV infection, with neuronal differentiation observed to be more adversely impacted than astrocytic differentiation120. A proteomic study in human neural stem cells revealed endoplasmic reticulum (ER) stress post-JEV infection, a feature that was also observed in the SVZ of JEV-infected mice, indicating a mechanism of JEV-induced apoptosis of neural stem cells121. Treatment of JEV-infected mice with minocycline, a broad-spectrum antibiotic, has, in addition to its anti-inflammatory and neuroprotective effects, been observed to restore neurogenesis in the SVZ, indicating a potential therapeutic avenue122,123. However, minocycline did not significantly improve outcome in a trial of acute encephalitis syndrome, although the number of JE patients was too small for a separate analysis124. At present, although no direct causal link between impairment of neurogenesis and JE sequelae has been documented, it is likely to be a contributing factor and should be explored in more detail.
Despite widespread vaccination regimes, JE continues to exert significant mortality and long-term disability in endemic regions, underscoring the requirement for new treatments. Furthermore, follow-up studies of JEV encephalitis are relatively scarce, which is partly due to the difficulty of following up with patients, as many live in rural areas with limited healthcare access125,126. Progress will require integrated research strategies combining long-term, community-based cohort follow-ups with mechanistic studies in both human and model systems to clarify immune and cellular drivers of persistent deficits and evaluate targeted interventions that could prevent or mitigate neurological sequelae.
Tick-borne encephalitis virus
Tick-borne encephalitis virus (TBEV) is an arbovirus that causes tick-borne encephalitis (TBE), a major health concern across Europe and Asia. Humans usually become infected through a bite from an infected Ixodes tick, though transmission can also occur due to the ingestion of unpasteurized dairy products from infected animals127,128. The incubation period for TBE in humans can range from 4 to 28 days following a tick bite, but is typically shorter with foodborne infections128,129,130.
Following a tick bite, TBEV initially replicates in Langerhans cells and neutrophils in the skin before spreading to the regional lymph nodes and then entering the bloodstream. In some patients, it then crosses the BBB via transcellular, paracellular, or ‘Trojan horse’ mechanisms127,131. A recent study mapping the three-dimensional distribution of TBEV in the mouse brain demonstrated its strong neuronal tropism, primarily infecting neurons within the hippocampus, lateral ventricles, and cerebral cortex132.
Infection with TBEV can be asymptomatic or present as a mild monophasic illness with influenza-like symptoms such as fever, fatigue, headache, and myalgia. However, in over half of symptomatic patients, a brief period of remission is followed by a second neurological stage, with persistent fever, insomnia, confusion, and muscle pain and weakness. This neurological phase can be due to meningitis, encephalitis, myelitis, or combinations thereof127,128,133. Diagnosis of TBE is usually accomplished via detection of specific antiviral IgM or immunoglobulin G (IgG) antibodies in the CSF or serum, which typically appear 0-6 days after disease onset and may persist for up to 10 months in some cases134.
While efficacious vaccines are available135, awareness and uptake are often heterogeneous, with many regions reporting low coverage136,137. In recent years, an increase in both the number of cases and the geographical range of the virus has been observed, with over 3500 cases reported by the EU in 2022138.
The fatality rate of TBE varies dramatically between the subtypes of TBEV, from less than 2% for the European subtype, up to 8% for the Siberian subtype, and as high as 40% for the Far-Eastern subtype128,139,140. Host factors also influence the propensity to severe disease, including age, immunosuppression, comorbidities, and vaccination status, in addition to polymorphisms in immune-related genes such as TLR3 and CCR5141,142,143,144,145,146.
TBE can result in an array of neurological and neuropsychiatric sequelae such as headaches, fatigue, anxiety, and issues with memory, concentration, balance, and sleep. These long-term effects are more common in adults than in children, with up to half of adults suffering from persistent symptoms147,148,149,150,151,152. The rate of various sequelae changes over time, but deficits that persist over a year are frequently permanent and can have a large impact on a person’s quality of life127,153. Indeed, the post-encephalitic symptoms of TBE have been reported to have a higher burden in terms of disability-adjusted life years than the acute phase of the disease154.
Several factors have been reported that affect the risk of sequelae development after TBE in adults, including older age, comorbidities, abnormal MRI findings, elevated CSF protein concentration, CSF pleocytosis, and acute disease severity130,143,152,155,156,157. However, acute features that can predict incomplete recovery in children following TBE are scarce. One study detected higher acute CSF levels of the cytokines IFN-γ, IL-4, IL-6, and IL-8 in children who subsequently developed sequelae158. In adults, no correlation was detected between acute levels of 24 different cytokines or chemokines in the CSF and long-term outcome in one study159, but in another, the CSF levels of several cytokines and mediators were observed to associate with the risk of sequelae, including IL-1, IL-8, CCL2, and matrix metalloproteinase-8160. Although it has been suggested that the more favorable outcome in pediatric patients may be due to a less mature immune system and, consequently, a milder inflammatory response158, these findings indicate that the inflammatory responses in the CNS may influence prognosis across all ages.
Indeed, the immune response appears to be critical in the long-term prognosis of TBE. Acute CNS inflammation driven by activated microglia, macrophages, and T cells, amongst others, disrupts neural homeostasis and damages brain tissue, which can have long-term consequences161. Several immune factors have been directly linked to such sequelae. By the time the neurological features of TBE manifest, the virus is typically no longer detectable in the blood, and therefore, the diagnosis of TBE is largely based on clinical symptoms and the presence of specific IgM and IgG antibodies in the serum162. Lower levels of IgG antibodies to TBEV in the acute stage of the illness are associated with both a more severe acute disease course and an increased risk of developing sequelae163. Furthermore, in TBE patients presenting with meningitis or meningoencephalitis, a favorable long-term prognosis was associated with patients who had strong specific IFN-γ-producing T cell responses to the TBEV structural protein E and the non-structural proteins NS1 and NS5. However, this effect was not observed in those who presented with meningoencephalomyelitis, a very severe clinical manifestation of the disease, or if the analyses were carried out without stratification by clinical presentation164. At follow-ups of patients 2–7 years after TBE, individuals with poorer outcomes had higher serum levels of most mediators associated with innate, Th1, or B cell responses, but lower levels of mediators associated with Th17 responses159. Neutrophils may also contribute: while they are common in acute CSF, they usually disappear within two weeks. However, persistence of neutrophils in the CSF at the two-week follow-up was associated with neurological deficits165. Furthermore, there was also a trend between acute blood neutrophil count and sequelae development, although this did not reach significance165. These data collectively suggest that inappropriate or prolonged immune responses may affect the clinical outcome of the disease.
In addition to immune-mediated mechanisms, direct viral effects on neural regeneration may contribute to the chronic neurological burden of TBE. Experimental findings suggest a possible mechanistic link between acute neuronal infection and long-term deficits. In a mouse model utilizing a TurboGFP-TBEV reporter virus, pronounced viral localization was observed within immature neurogenic cells of the SVZ132. Infection of these progenitor cell populations may impair neurogenesis, potentially contributing to the persistent cognitive and neuropsychiatric sequelae observed after TBE. Further understanding of the mechanisms underlying the neuropathology of TBE will be essential in order to address this growing public health challenge and improve patient outcomes.
Herpesviruses
Several members of the Herpesviridae family can cause severe neurological disease and long-term sequelae. This is a family of double-stranded DNA viruses, classified into three subfamilies: α, β, and γ viruses. With regard to human herpesviruses, the α-herpesvirus subfamily most commonly induces serious CNS disease in otherwise healthy individuals: this subfamily consists of HSV-1, HSV-2, and varicella zoster virus (VZV). The β-herpesvirus subfamily includes human cytomegalovirus and human herpesvirus (HHV) type 6, while the γ-herpesvirus subfamily comprises Epstein-Barr virus and HHV-8166. For the sake of relevance and clarity, we will focus on α-herpesviruses in this review, as they are highly neurotropic, establish life-long latency in sensory ganglia, and can be periodically reactivated throughout life166.
Herpes simplex virus type 1
HSV-1 is highly prevalent worldwide, infecting over 65% of people under the age of 50167. It is mainly transmitted through oral secretions, and during primary infection, the virus replicates in mucosal epithelial cells before establishing latency in the sensory neurons of peripheral ganglia. These reservoirs of latent virus are crucial to the survival of the virus, as they can persist throughout an individual’s lifetime and can be periodically reactivated168.
Many infected individuals are asymptomatic, and the dominant symptom the virus is best recognized for is small blisters or ‘cold sores’ around the mouth. In addition to this typical manifestation, both HSV-1 and HSV-2 can cause devastating disease in neonates. Infection may occur intrapartum via exposure to the virus in the genital tract during delivery, or through intrauterine or postnatal routes169. Neonatal infections can present as localized skin, eye, and mouth (SEM) disease, disseminated multi-organ disease, or localized CNS disease. The mortality rate is ~40% for disseminated disease, even with antiviral treatment, and nearly half of infants with CNS disease still exhibit neurological abnormalities at 24 months of age170. While neonatal disease is rare, affecting less than 5 cases per 10,000 births in the US171, this highlights the capacity of HSV to induce profound and lasting pathology.
On rare occasions, in both children and adults, the virus can invade the brain and cause herpes simplex encephalitis (HSE)172, the most common cause of sporadic infectious encephalitis in humans4,173. HSV-2 may also cause this disease, but the vast majority of instances are attributable to HSV-1173,174. Primary infection of HSV-1 is estimated to cause only 30% of encephalitis cases, with reactivation of the virus responsible for the other two-thirds172,175. The gold standard for HSE diagnosis is CSF HSV polymerase chain reaction (PCR), which should be performed as early as possible within the first two weeks of symptom onset, in conjunction with MRI and clinical signs176. Neurons are the primary target in the CNS for the virus, but it can also infect astrocytes and, importantly, microglia, which undergo an abortive infection and launch a strong immune response177,178,179. The virus can induce extensive necrosis and edema, most predominantly in the medial temporal lobes, disrupting critical limbic circuits180,181.
HSE typically presents with fever, disorientation, behavioral changes, and seizures182,183,184. Prior to the introduction of acyclovir in the treatment regimen of HSE, the fatality rate was ~70%, but with early antiviral therapy, it has dropped dramatically to <20%185. Despite these therapeutic improvements, the prevalence of sequelae following HSE remains high, affecting an estimated 30–70% of patients, significantly impacting their quality of life4,186,187,188. These sequelae encompass a wide range of conditions, such as seizures, motor disabilities, cognitive issues, and behavioral and personality changes, as well as psychological effects like depression and anxiety189,190,191,192. These complications can last for months to years; one study of 43 patients with HSE showed that only 14% of patients had fully recovered 3 years later190.
Numerous factors have been identified that are associated with increased morbidity and mortality in patients with HSV-1 CNS disease. These include age, pre-existing conditions, immunocompromisation, and delay of initiation of acyclovir treatment193. Additionally, several inborn errors in innate immune signaling pathways that increase HSV-1 susceptibility have been ascertained, including those in the TLR3 signaling pathway and the Interferon Alpha/Beta Receptor (IFNAR) pathway194. MRI detects brain abnormalities in 80–100% of HSE cases, such as T2 and FLAIR hyperintensities in the medial temporal lobes, the insular cortex, and the frontobasal cortex195. Early, extensive brain involvement observed on MRI has been linked to poor outcomes183,196. Cerebrospinal fluid levels of cytokines have also been suggested as a prognostic tool, with higher initial IFN-γ and maximal IL-6 levels associated with unfavorable outcomes197.
The development of autoimmune encephalitis (AE), primarily N-methyl-D-aspartate receptor (NMDAR) encephalitis, occurs in up to a quarter of HSE patients198,199. These cases may be mistaken for ‘relapsing HSE’. The detection of neuronal antibodies within 3 weeks following HSE has been reported as a risk factor for developing autoimmune encephalitis. None of these patients tested positive for these antibodies at the onset of HSE, indicating that the virus triggered the immune response198. Interestingly, the development of NMDAR antibodies was associated with cognitive issues199. AE phenotype varies with age, with young children associated with a worse outcome and a higher risk of suffering from choreoathetosis, decreased consciousness, and seizures, whereas older children and adults are more likely to present with behavioral and cognitive issues, such as irritability, memory deficits, and confusion198,200. The underlying mechanisms as to why HSV infection may trigger the development of autoimmune encephalitis are not fully understood. The effect of HSV-1 on brain tissue may cause the release of neuronal proteins that induce autoimmunity activation, but specific maladaptive immune responses induced by HSV-1 or molecular mimicry have also been suggested as contributing factors201.
Besides the temporal lobe and frontal lobe, the hippocampus appears to be particularly vulnerable to HSV-1 infection202. The hippocampus expresses HSV-1 entry receptors at a higher level than other brain regions203, and one study suggests a lower antiviral cytokine response in the hippocampus in mice compared to other regions204. Amongst the cells in the hippocampus, the cells of the dentate gyrus, a site of adult neurogenesis, are particularly vulnerable205. The accumulation of amyloid-β protein, which is strongly implicated in Alzheimer’s disease pathology, has been reported in the neural stem cells in this region, which may impair neurogenesis206. Given the key role the hippocampus plays in memory and learning, these effects of HSV-1 on the hippocampus likely contribute to the cognitive sequelae observed, although they have not been directly linked to behavioral effects thus far.
Persistence of inflammatory mechanisms has been linked to neuronal damage and behavioral impairments following HSV-1 infection. In a non-lethal intracranial mouse model using a neural-attenuated HSV-1 strain, there were persistent neuroinflammatory responses and a dramatic increase in microglial numbers. This resulted in neuronal loss in the cerebral cortex near the injection site and decreased spontaneous motor activity in the open field test207. In addition to increases in microglia, astrogliosis and lymphocytic infiltration have also been reported chronically, with subsequent neuronal loss, decreased brain volume, and spatial memory deficits208. The cytokine IL-1β may be crucial to these outcomes, as repeated HSV-1 reactivations induced increases in IL-1β, which impaired cognitive performances, affected hippocampal synaptic plasticity, and reduced expression of pre- and post-synaptic proteins. Furthermore, pharmacological blockade of the IL-1β receptor ameliorated effects on synaptic plasticity and rescued cognitive impairments, although this latter effect was delayed209. Interestingly, IL-1 has also been implicated in the pathogenesis of anti-NMDAR encephalitis, which, as previously mentioned, may develop post-HSE210,211,212. In HSV-1-infected mouse microglia, NLR family pyrin domain-containing 3 (NLRP3) inflammasome activation leads to IL-1β release213. This inflammasome has been implicated in the development of cognitive impairment214,215,216,217 and of neurodegenerative diseases that have cognitive components, such as Alzheimer’s and Parkinson’s disease218,219,220.
It is important to note that HSV-1 has been detected in the brains of individuals who died of non-neurological causes221,222, potentially causing neuroinflammation and neuroimmune responses even without symptoms. In mice, asymptomatic HSV infection was associated with sex-specific changes in inflammatory and senescence markers and increased permeability of the BBB223,224,225, and asymptomatic reactivation of the virus from latency induced upregulation of neuroinflammatory and early neurodegenerative markers226. Furthermore, a single low-dose intranasal HSV infection in neonatal mice did not induce acute signs of infection but led to cognitive and memory impairments months later in adulthood227, suggesting that subclinical infections can impact brain homeostasis and induce long-term impairments. Most research regarding asymptomatic HSV-1 infections has investigated the link to Alzheimer’s disease, wherein there is evidence that the risk of developing Alzheimer’s disease is increased in seropositive individuals12,219,228,229,230. However, overall, very little is known about the effects of asymptomatic HSV-1 CNS infections in humans, as the presence of the virus in the brain of patients is only confirmed postmortem, making it challenging to investigate. The broader long-term consequences of both asymptomatic and symptomatic HSV-1 infections warrant further investigation.
Varicella zoster virus
VZV predominantly causes chickenpox (varicella) during primary infection, and shingles (herpes zoster) upon reactivation. Infection is typically acquired via close contact transmission, after which VZV replicates in the epithelial cells of the respiratory tract, followed by dissemination to the lymph nodes. Infected T cells can deliver the virus to the skin, which, after an incubation period of 10-21 days, results in the characteristic itchy skin lesions of chickenpox231.
Following primary infection, VZV establishes latency in cranial nerve, dorsal root, and autonomic ganglia, where latency is largely controlled by cell-mediated immunity232. With increasing age or immunocompromisation, a decline in this VZV-specific immunity occurs, permitting the virus to reactivate and replicate in neurons, which causes shingles, associated with painful blister eruptions232. The virus may also progress to the CNS, infecting neurons and astrocytes233,234,235, and causing severe complications. Diagnosis relies on CSF analysis for anti-VZV antibodies, PCR detection of VZV DNA in CSF, and evaluation of any recent rash, although the optimal timing for these tests remains poorly defined236,237.
The CNS pathology of VZV is diffuse, with lesions reported in many sites, including frontal, temporal, parietal, and occipital lobes, in addition to the basal ganglia, brainstem, and cerebellum238,239. While cerebellitis and encephalitis can occur in children with varicella, VZV-induced neurological diseases are much more commonly associated with reactivation infections than primary infections, and include meningitis and encephalitis240,241,242. Although live-attenuated VZV vaccines have markedly reduced the incidence of disease243,244, the vaccine itself can, in extremely rare cases, cause delayed-onset meningitis245,246.
Patients with VZV meningitis generally show more favorable long-term outcomes compared to encephalitis patients, with 70–100% of meningitis patients making a full recovery, compared to less than half of patients with encephalitis193. The predominant acute symptoms of VZV encephalitis include an altered mental state, headaches, and occasionally seizures247, whereas sequelae comprise attention and memory deficits, language issues, and emotional and behavioral changes248,249,250. Herpes zoster can cause complications with a CNS component even in the absence of encephalitis, including vasculopathies leading to stroke, myelitis, and postherpetic neuralgia251.
Numerous host factors have been determined that predispose to severe VZV infection, reactivation, and CNS infection, such as older age, chronic medical comorbidities, and immunosuppression242,252. This includes primary immunodeficiencies affecting T cells, NK cells, and B cells, and secondary immunosuppression such as cancer, transplantation, HIV/AIDS, and immunosuppressive medications253. Defects in the DNA sensor RNA polymerase III (POL III) have also been associated with increased risk of severe VZV-induced CNS disease254. However, thus far, our understanding regarding the risk factors for long-term persistence of symptoms or sequelae following VZV infection is limited.
The most frequent chronic complication of herpes zoster is postherpetic neuralgia (PHN)251, which is difficult to treat with current therapies. Encephalitis is not necessary to induce this condition, but it has a complex pathogenesis with changes in both peripheral and central somatosensory processing255. PHN is a chronic neuropathic pain condition lasting over 3 months following a herpes zoster outbreak256,257 and is also associated with concurrent depression, anxiety, and insomnia258,259,260. Peripherally, it is the activation of stress nociceptors in tissues via inflammation-driven mechanisms that causes the pain. Centrally, several pathological mechanisms contribute to the disorder, including inflammation, alterations in brain structure and function, and changes in central pain modulation255. MRI studies showed that these abnormalities were mainly located in ‘pain matrix’ brain regions, including the thalamus, insula, amygdala, and dorsolateral prefrontal cortex. However, other regions outside this system, such as the precentral gyrus and cerebellum, which play crucial roles in motor control, have also been reported to be affected261.
Productive VSV infection of cerebral vessels can cause vasculopathy, which may lead to transient ischemic attacks and ischemic or hemorrhagic stroke, amongst various other complications262. The average duration between the herpes zoster rash and the presentation of neurological symptoms related to vasculopathies is approximately 4 months263. While the risk for developing stroke is highest in the month following herpes zoster, it still remains elevated years later, with younger people at higher risk264,265,266. If the stroke affects key cerebral arteries, this could result in brain damage and thus impairment of cognitive functions, a common sequela of VZV CNS disease.
CSF concentrations of various biomarkers have been used in an attempt to assess brain pathophysiology following VZV infections. Increased CSF levels of neurofilament light chain were detected several months post-VZV CNS infection, which were highest in patients with encephalitis. Alterations were also detected in the levels of S100B, primarily found in astrocytes, but no markers were found to correlate with long-term outcome267. In another study, in patients with long-term cognitive impairments followed up between 1.5 and 7 years after VZV encephalitis, there were no alterations in CSF levels of neurofilament light chain, S100B, or GFAP, indicating that ongoing degeneration is not occurring despite the neurological deficits248.
Overall, the pathogenesis and neuropathogenesis of VZV are poorly understood to date, as it is a highly human-specific virus and thus challenging to investigate in animal model systems. Simian varicella virus (SVV) infection in non-human primates has provided valuable insights into the pathogenesis, as it results in varicella in the animals, and as the virus has a similar genome in terms of size, structure, and content to VZV268,269. However, these non-human primate studies are highly resource-demanding; therefore, the development of severe combined immunodeficiency (SCID) xenograft transplant mouse models was a major advance in the field231. Dorsal root ganglia (DRG) xenografts are utilized to investigate the neurological facets of the disease, while skin and thymus-liver xenografts are used to study the skin and T cell aspects, respectively270,271. The use of DRG xenograft models has already provided insights into the neuropathology of VZV. Glycoprotein I, both alone and in its interactions with glycoprotein E, plays a crucial role in the virulence of VZV272,273. The DRG neuronal subtype also appears to influence neurovirulence, as productive VZV replication is restricted in mechanoreceptive neurons compared to nociceptive neurons. Interestingly, at later stages, mechanoreceptive neurons undergo selective depletion despite their reduced permissiveness274. The continuing use of these models will hopefully enlighten us further on VZV pathological mechanisms and how they contribute to long-term neurological complications, although carrying out behavioral tests may be challenging due to the severe immunodeficiency of these animals.
Conclusion
Viral infection of the central nervous system can result in significant acute morbidity, but its impact often extends far beyond the initial illness, manifesting as long-term neurological, cognitive, and psychiatric sequelae. Despite advances in antiviral therapies and vaccination programs, the treatment of post-infectious sequelae remains largely supportive as the underlying causes are still not fully understood.
Evidence points to persistent inflammation, immune dysregulation, and impaired neurogenesis as key contributors to the pathogenesis of CNS injury following viral infection. Only a few factors have been identified so far, such as age or immune status, which may predispose a patient to an adverse outcome. A deeper understanding of these processes and recognition of the shared mechanisms underlying CNS damage across different viral families could pave the way for targeted interventions aimed at preventing or mitigating post-viral neurological damage.
Data availability
No datasets were generated or analysed during the current study.
References
Blackburn, K. M. & Wang, C. Post-infectious neurological disorders. Ther. Adv. Neurol. Disord. 13, 1756286420952901 (2020).
Swanson, P. A. & McGavern, D. B. Viral diseases of the central nervous system. Curr. Opin. Virol. 11, 44–54 (2015).
Wang, H. et al. Global magnitude of encephalitis burden and its evolving pattern over the past 30 years. J. Infect. 84, 777–787 (2022).
Kvam, K. A. et al. Outcome and Sequelae of Infectious Encephalitis. J. Clin. Neurol. 20, 23–36 (2024).
Tyler, K. L. Acute viral encephalitis. N. Engl. J. Med. 379, 557–566 (2018).
Heaton, R. K. et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy. Neurology 75, 2087–2096 (2010).
Matschke, J. et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. 19, 919–929 (2020).
Senzolo, M. et al. Neuropsychological alterations in hepatitis C infection: the role of inflammation. World J. Gastroenterol. 17, 3369–3374 (2011).
Leblanc, P. & Vorberg, I. M. Viruses in neurodegenerative diseases: more than just suspects in crimes. PLoS Pathog. 18, e1010670 (2022).
Levine, K. S. et al. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 111, 1086–1093.e2 (2023).
Shouman, S., Hesham, N. & Salem, T. Z. Viruses and neurodegeneration: a growing concern. J. Transl. Med. 23, 46 (2025).
Itzhaki, R. F. Overwhelming evidence for a major role for herpes simplex virus type 1 (HSV1) in Alzheimer’s disease (AD); underwhelming evidence against. Vaccines 9, 679 (2021).
Bjornevik, K., Münz, C., Cohen, J. I. & Ascherio, A. Epstein–Barr virus as a leading cause of multiple sclerosis: mechanisms and implications. Nat. Rev. Neurol. 19, 160–171 (2023).
van Leur, S. W., Heunis, T., Munnur, D. & Sanyal, S. Pathogenesis and virulence of flavivirus infections. Virulence 12, 2814–2838 (2021).
Bhatt, S. et al. The global distribution and burden of dengue. Nature 496, 504–507 (2013).
Bifani, A. M. et al. Therapeutics for flaviviral infections. Antiviral Res. 210, 105517 (2023).
Colpitts, T. M., Conway, M. J., Montgomery, R. R. & Fikrig, E. West Nile virus: biology, transmission, and human infection. Clin. Microbiol. Rev. 25, 635–648 (2012).
Smithburn, K. C., Hughes, T. P., Burke, A. W. & Paul, J. H. A Neurotropic Virus Isolated from the Blood of a Native of Uganda https://doi.org/10.4269/ajtmh.1940.s1-20.471 (1940).
Chancey, C., Grinev, A., Volkova, E. & Rios, M. The global ecology and epidemiology of West Nile virus. BioMed. Res. Int. 2015, 376230 (2015).
Kocabiyik, D. Z., Álvarez, L. F., Durigon, E. L. & Wrenger, C. West Nile virus - a re-emerging global threat: recent advances in vaccines and drug discovery. Front. Cell. Infect. Microbiol. 15, 1568031 (2025).
Mostashari, F. et al. Epidemic West Nile encephalitis, New York, 1999: results of a household-based seroepidemiological survey. Lancet Lond. Engl. 358, 261–264 (2001).
Petersen, L. R. et al. Estimated cumulative incidence of West Nile virus infection in US adults, 1999–2010. Epidemiol. Infect. 141, 591–595 (2013).
Zou, S., Foster, G. A., Dodd, R. Y., Petersen, L. R. & Stramer, S. L. West Nile fever characteristics among viremic persons identified through blood donor screening. J. Infect. Dis. 202, 1354–1361 (2010).
Fulton, C. D. M., Beasley, D. W. C., Bente, D. A. & Dineley, K. T. Long-term, West Nile virus-induced neurological changes: a comparison of patients and rodent models. Brain Behav. Immun. - Health 7, 100105 (2020).
CDC. Clinical Testing and Diagnosis for West Nile Virus Disease https://www.cdc.gov/west-nile-virus/hcp/diagnosis-testing/index.html (2024).
Jean, C. M., Honarmand, S., Louie, J. K. & Glaser, C. A. Risk factors for West Nile Virus neuroinvasive disease, California, 2005. Emerg. Infect. Dis. 13, 1918–1920 (2007).
Lindsey, N. P., Staples, J. E., Lehman, J. A. & Fischer, M. Medical risk factors for severe West Nile Virus disease, United States, 2008–2010. Am. J. Trop. Med. Hyg. 87, 179–184 (2012).
Murray, K. et al. Risk factors for encephalitis and death from West Nile virus infection. Epidemiol. Infect. 134, 1325–1332 (2006).
Glass, W. G. et al. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 203, 35–40 (2006).
Lim, J. K. et al. CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J. Infect. Dis. 201, 178–185 (2010).
Danis, K. et al. Outbreak of West Nile virus infection in Greece, 2010. Emerg. Infect. Dis. 17, 1868–1872 (2011).
Kopel, E. et al. Surveillance of West Nile virus disease, Tel Aviv district, Israel, 2005 to 2010. Euro Surveill. Bull. Eur. Sur. Mal. Transm. Eur. Commun. Dis. Bull. 16, 19894 (2011).
Philpott, D. C. E. et al. Acute and delayed deaths after West Nile virus infection, Texas, USA, 2002–2012. Emerg. Infect. Dis. 25, 256–264 (2019).
Riccetti, N., Ferraccioli, F., Fasano, A. & Stilianakis, N. I. Demographic characteristics associated with West Nile virus neuroinvasive disease—a retrospective study on the wider European area 2006–2021. PLoS ONE 18, e0292187 (2023).
Cook, R. L. et al. Demographic and clinical factors associated with persistent symptoms after West Nile virus infection. Am. J. Trop. Med. Hyg. 83, 1133–1136 (2010).
Klee, A. L. et al. Long-term prognosis for clinical West Nile virus infection. Emerg. Infect. Dis. 10, 1405–1411 (2004).
Murray, K. O. et al. Survival analysis, long-term outcomes, and percentage of recovery up to 8 years post-infection among the Houston West Nile Virus Cohort. PLoS ONE 9, e102953 (2014).
Nikolić, N. et al. Long-term follow-up of patients with West Nile neuroinvasive disease. Viruses 17, 878 (2025).
Patel, H., Sander, B. & Nelder, M. P. Long-term sequelae of West Nile virus-related illness: a systematic review. Lancet Infect. Dis. 15, 951–959 (2015).
Weatherhead, J. E. et al. Long-term neurological outcomes in West Nile virus-infected patients: an observational study. Am. J. Trop. Med. Hyg. 92, 1006–1012 (2015).
Rudolph, K. E., Lessler, J., Moloney, R. M., Kmush, B. & Cummings, D. A. T. Incubation periods of mosquito-borne viral infections: a systematic review. Am. J. Trop. Med. Hyg. 90, 882–891 (2014).
Winkelmann, E. R., Luo, H. & Wang, T. West Nile Virus infection in the central nervous system. F1000Research 5, F1000 Faculty Rev-105 (2016).
Hasebe, R. et al. Transcellular transport of West Nile virus-like particles across human endothelial cells depends on residues 156 and 159 of envelope protein. BMC Microbiol. 10, 165 (2010).
Paul, A. M. et al. Osteopontin facilitates West Nile virus neuroinvasion via neutrophil ‘Trojan horse’ transport. Sci. Rep. 7, 4722 (2017).
Roe, K. et al. West Nile virus-induced disruption of the blood–brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J. Gen. Virol. 93, 1193–1203 (2012).
Samuel, M. A., Wang, H., Siddharthan, V., Morrey, J. D. & Diamond, M. S. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl Acad. Sci. USA 104, 17140–17145 (2007).
Verma, S. et al. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: transmigration across the in vitro blood–brain barrier. Virology 385, 425–433 (2009).
Wang, P. et al. Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain. J. Virol. 82, 8978–8985 (2008).
Cheeran, M. C.-J. et al. Differential responses of human brain cells to West Nile virus infection. J. Neurovirol. 11, 512–524 (2005).
Diniz, J. A. P. et al. West Nile virus infection of primary mouse neuronal and neuroglial cells: the role of astrocytes in chronic infection. Am. J. Trop. Med. Hyg. 75, 691–696 (2006).
van Marle, G. et al. West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J. Virol. 81, 10933–10949 (2007).
Armah, H. B. et al. Systemic distribution of West Nile Virus infection: postmortem immunohistochemical study of six cases. Brain Pathol. 17, 354–362 (2007).
Guarner, J. et al. Clinicopathologic study and laboratory diagnosis of 23 cases with West Nile virus encephalomyelitis. Hum. Pathol. 35, 983–990 (2004).
Murray, K. O. et al. The neurocognitive and MRI outcomes of West Nile Virus infection: preliminary analysis using an External Control Group. Front. Neurol. 9, 111 (2018).
Kobayashi, S., Orba, Y., Yamaguchi, H., Kimura, T. & Sawa, H. Accumulation of ubiquitinated proteins is related to West Nile virus-induced neuronal apoptosis. Neuropathology 32, 398–405 (2012).
Medigeshi, G. R. et al. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 81, 10849–10860 (2007).
Samuel, M. A., Morrey, J. D. & Diamond, M. S. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile Virus Encephalitis. J. Virol. 81, 2614–2623 (2007).
Errett, J. S., Suthar, M. S., McMillan, A., Diamond, M. S. & Gale, M. The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J. Virol. 87, 11416–11425 (2013).
Sabouri, A. H. et al. TLR signaling controls lethal encephalitis in WNV-infected brain. Brain Res. 1574, 84–95 (2014).
Kumar, M., Verma, S. & Nerurkar, V. R. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J. Neuroinflamm. 7, 73 (2010).
Quick, E. D., Seitz, S., Clarke, P. & Tyler, K. L. Minocycline has anti-inflammatory effects and reduces cytotoxicity in an ex vivo spinal cord slice culture model of West Nile virus infection. J. Virol. 91, e00569–17 (2017).
Vasek, M. J. et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543 (2016).
Garber, C. et al. T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat. Neurosci. 22, 1276–1288 (2019).
Ai, S. et al. CCR2 restricts IFN-γ production by hippocampal CD8 TRM cells that impair learning and memory during recovery from WNV encephalitis. J. Neuroinflamm. 21, 330 (2024).
Rosen, S. F. et al. Single-cell RNA transcriptome analysis of CNS immune cells reveals CXCL16/CXCR6 as maintenance factors for tissue-resident T cells that drive synapse elimination. Genome Med. 14, 108 (2022).
Garber, C. et al. Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via IL-1. Nat. Immunol. 19, 151–161 (2018).
Murray, K. O., Resnick, M. & Miller, V. Depression after Infection with West Nile Virus1. Emerg. Infect. Dis. 13, 479–481 (2007).
Nolan, M. S., Hause, A. M. & Murray, K. O. Findings of long-term depression up to 8 years post infection from West Nile Virus. J. Clin. Psychol. 68, 801–808 (2012).
Lino, A., Erickson, T. A., Nolan, M. S., Murray, K. O. & Ronca, S. E. A preliminary study of proinflammatory cytokines and depression following West Nile virus infection. Pathogens 11, 650 (2022).
Garcia, M. N. et al. Evaluation of prolonged fatigue post-West Nile virus infection and association of fatigue with elevated antiviral and proinflammatory cytokines. Viral Immunol. 27, 327–333 (2014).
Leis, A. A. et al. Tumor necrosis factor-alpha signaling may contribute to chronic west Nile Virus post-infectious proinflammatory state. Front. Med. 7, 164 (2020).
Constant, O., Barthelemy, J., Nagy, A., Salinas, S. & Simonin, Y. West Nile virus neuroinfection in humans: peripheral biomarkers of neuroinflammation and neuronal damage. Viruses 14, 756 (2022).
Murray, K. O., Garcia, M. N., Yan, C. & Gorchakov, R. Persistence of detectable immunoglobulin M antibodies up to 8 years after infection with West Nile Virus. Am. J. Trop. Med. Hyg. 89, 996–1000 (2013).
Murray, K. et al. Persistent infection with West Nile virus years after initial infection. J. Infect. Dis. 201, 2–4 (2010).
WHO. Japanese Encephalitis https://www.who.int/news-room/fact-sheets/detail/japanese-encephalitis (2015).
Wilder-Smith, A. & Halstead, S. B. Japanese encephalitis: update on vaccines and vaccine recommendations. Curr. Opin. Infect. Dis. 23, 426 (2010).
Yun, S.-I. & Lee, Y.-M. Japanese encephalitis: the virus and vaccines. Hum. Vaccines Immunother. 10, 263–279 (2014).
Letson, G. W., Marfin, A. A., Mooney, J., Minh, H. V. & Hills, S. L. Impact of vaccination against Japanese encephalitis in endemic countries. PLoS Negl. Trop. Dis. 18, e0012390 (2024).
Konishi, E., Kitai, Y., Tabei, Y., Nishimura, K. & Harada, S. Natural Japanese encephalitis virus infection among humans in west and east Japan shows the need to continue a vaccination program. Vaccine 28, 2664–2670 (2010).
Yin, Q. et al. Spatiotemporal distribution and host–vector dynamics of Japanese encephalitis virus. Viruses 17, 815 (2025).
Campbell, G. L. et al. Estimated global incidence of Japanese encephalitis: a systematic review. Bull. World Health Organ. 89, 766–774E (2011).
Filgueira, L. & Lannes, N. Review of emerging Japanese encephalitis virus: new aspects and concepts about entry into the brain and inter-cellular spreading. Pathogens 8, 111 (2019).
Hills, S. L., Netravathi, M. & Solomon, T. Japanese Encephalitis among adults: a review. Am. J. Trop. Med. Hyg. 108, 860–864 (2023).
CDC. Clinical features and diagnosis of Japanese Encephalitis. Japanese Encephalitis Virus https://www.cdc.gov/japanese-encephalitis/hcp/clinical-diagnosis/index.html (2024).
Cheng, Y., Minh, N. T., Minh, Q. T., Khandelwal, S. & Clapham, H. E. Estimates of Japanese Encephalitis mortality and morbidity: a systematic review and modeling analysis. PLoS Negl. Trop. Dis. 16, e0010361 (2022).
Sarkari, N. B. S. et al. Japanese encephalitis (JE) part II: 14 years’ follow-up of survivors. J. Neurol. 259, 58–69 (2012).
Wang, X. et al. Long-term neurological sequelae and disease burden of Japanese Encephalitis in Gansu Province, China. Ann. Glob. Health 87, 103 (2021).
Yin, Z. et al. Neurological sequelae of hospitalized Japanese Encephalitis cases in Gansu Province, China. Am. J. Trop. Med. Hyg. 92, 1125–1129 (2015).
García-Nicolás, O., Lewandowska, M., Ricklin, M. E. & Summerfield, A. Monocyte-derived dendritic cells as model to evaluate species tropism of mosquito-borne flaviviruses. Front. Cell. Infect. Microbiol. 9, (2019).
Kumar, G., Date, O. S., Kim, K. S. & Manjunath, R. Infection of human amniotic and endothelial cells by Japanese Encephalitis virus: Increased expression of HLA-F. Virology 471–473, 29–37 (2014).
Sharma, K. B., Vrati, S. & Kalia, M. Pathobiology of Japanese encephalitis virus infection. Mol. Aspects Med. 81, 100994 (2021).
Shwetank, Date, O. S., Kim, K. S. & Manjunath, R. Infection of human endothelial cells by Japanese Encephalitis virus: increased expression and release of soluble HLA-E. PLoS ONE 8, e79197 (2013).
Dutta, K., Mishra, M. K., Nazmi, A., Kumawat, K. L. & Basu, A. Minocycline differentially modulates macrophage mediated peripheral immune response following Japanese encephalitis virus infection. Immunobiology 215, 884–893 (2010).
Frank, J. C., Song, B.-H. & Lee, Y.-M. Mice as an animal model for Japanese Encephalitis virus research: mouse susceptibility, infection route, and viral pathogenesis. Pathogens 12, 715 (2023).
Hsieh, J. T. & John, A. L. S. Japanese encephalitis virus and its mechanisms of neuroinvasion. PLoS Pathog. 16, e1008260 (2020).
Liou, M.-L. & Hsu, C.-Y. Japanese encephalitis virus is transported across the cerebral blood vessels by endocytosis in mouse brain. Cell Tissue Res. 293, 389–394 (1998).
Mathur, A., Khanna, N. & Chaturvedi, U. C. Breakdown of blood–brain barrier by virus-induced cytokine during Japanese encephalitis virus infection. Int. J. Exp. Pathol. 73, 603–611 (1992).
Lin, F.-H., Chou, Y.-C., Hsieh, C.-J. & Yu, C.-P. Epidemiological features, clinical symptoms, and environmental risk factors for notifiable Japanese Encephalitis in Taiwan From 2008 to 2020: retrospective study. JMIR Public Health Surveill. 11, e63053 (2025).
Sarkari, N. B. S. et al. Japanese encephalitis (JE). Part I: clinical profile of 1,282 adult acute cases of four epidemics. J. Neurol. 259, 47–57 (2012).
Desai, A., Shankar, S. K., Ravi, V., Chandramuki, A. & Gourie-Devi, M. Japanese encephalitis virus antigen in the human brain and its topographic distribution. Acta Neuropathol. 89, 368–373 (1995).
Myint, K. S. A. et al. Neuropathogenesis of Japanese encephalitis in a primate model. PLoS Negl. Trop. Dis. 8, e2980 (2014).
Li, F. et al. Viral infection of the central nervous system and neuroinflammation precede blood-brain barrier disruption during Japanese Encephalitis virus infection. J. Virol. 89, 5602–5614 (2015).
Hsu, S. M., Yen, A. M. F. & Chen, T. H. H. The impact of climate on Japanese Encephalitis. Epidemiol. Infect. 136, 980–987 (2008).
Le Flohic, G., Porphyre, V., Barbazan, P. & Gonzalez, J.-P. Review of climate, landscape, and viral genetics as drivers of the Japanese Encephalitis virus ecology. PLoS Negl. Trop. Dis. 7, e2208 (2013).
Liu, W. et al. Risk factors for Japanese encephalitis: a case-control study. Epidemiol. Infect. 138, 1292–1297 (2010).
Walsh, M. G. et al. High-risk landscapes of Japanese encephalitis virus outbreaks in India converge on wetlands, rain-fed agriculture, wild Ardeidae, and domestic pigs and chickens. Int. J. Epidemiol. 51, 1408–1418 (2022).
Biyani, S. et al. Toll-like receptor-3 gene polymorphism in patients with Japanese encephalitis. J. Neuroimmunol. 286, 71–76 (2015).
Ghildiyal, S. et al. Pro-inflammatory and anti-inflamatory cytokine genes polymorphisms and susceptibility to Japanese encephalitis disease in the North Indian population. Cytokine 149, 155716 (2022).
Deval, H. et al. Association of single nucleotide polymorphisms in the CD209, MMP9, TNFA and IFNG genes with susceptibility to Japanese encephalitis in children from North India. Gene 808, 145962 (2022).
Pujhari, S. K., Ratho, R. K., Prabhakar, S., Mishra, B. & Modi, M. TNF-α promoter polymorphism: a factor contributing to the different immunological and clinical phenotypes in Japanese encephalitis. BMC Infect. Dis. 12, 23 (2012).
Ashraf, U. et al. Pathogenicity and virulence of Japanese encephalitis virus: neuroinflammation and neuronal cell damage. Virulence 12, 968–980 (2021).
Ghoshal, A. et al. Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 55, 483–496 (2007).
Soni, N. & Rameshwari, R. Silent messengers of chaos: unveiling the dual threat of immune infiltrates in Japanese encephalitis virus neuroinflammatory storm. Virol. J. 22, 173 (2025).
Zhang, F. et al. PD1+CCR2+CD8+ T cells infiltrate the central nervous system during acute Japanese encephalitis virus infection. Virol. Sin. 34, 538–548 (2019).
Yin, R. et al. Proteomic landscape subtype and clinical prognosis of patients with the cognitive impairment by Japanese encephalitis infection. J. Neuroinflamm. 19, 77 (2022).
Turtle, L. et al. Human T cell responses to Japanese encephalitis virus in health and disease. J. Exp. Med. 213, 1331–1352 (2016).
Kumar, P. et al. Impaired T helper 1 function of nonstructural protein 3-specific T cells in Japanese patients with encephalitis with neurological sequelae. J. Infect. Dis. 189, 880–891 (2004).
Arzate, D. M. & Covarrubias, L. Adult neurogenesis in the context of brain repair and functional relevance. Stem Cells Dev. 29, 544–554 (2020).
Das, S. & Basu, A. Japanese encephalitis virus infects neural progenitor cells and decreases their proliferation. J. Neurochem. 106, 1624–1636 (2008).
Ariff, I. M., Thounaojam, M. C., Das, S. & Basu, A. Japanese encephalitis virus infection alters both neuronal and astrocytic differentiation of neural stem/progenitor cells. J. Neuroimmune Pharmacol. 8, 664–676 (2013).
Mukherjee, S. et al. Japanese encephalitis virus induces human neural stem/progenitor cell death by elevating GRP78, PHB and hnRNPC through ER stress. Cell Death Dis. 8, e2556 (2018).
Das, S. et al. Abrogated inflammatory response promotes neurogenesis in a murine model of Japanese Encephalitis. PLoS ONE 6, e17225 (2011).
Mishra, M. K. & Basu, A. Minocycline neuroprotects, reduces microglial activation, inhibits caspase 3 induction, and viral replication following Japanese encephalitis. J. Neurochem. 105, 1582–1595 (2008).
Kumar, R. et al. Role of oral Minocycline in acute encephalitis syndrome in India—a randomized controlled trial. BMC Infect. Dis. 16, 67 (2016).
Chow, C. & Dehority, W. Long-term outcomes in children surviving tropical arboviral encephalitis: a systematic review. J. Trop. Pediatr. 67, fmab028 (2021).
Mayxay, M. et al. Outcome of Japanese Encephalitis Virus (JEV) Infection in Pediatric and Adult Patients at Mahosot Hospital, Vientiane, Lao PDR https://doi.org/10.4269/ajtmh.20-0581 (2021).
Chiffi, G., Grandgirard, D., Leib, S. L., Chrdle, A. & Růžek, D. Tick-borne encephalitis: a comprehensive review of the epidemiology, virology, and clinical picture. Rev. Med. Virol. 33, e2470 (2023).
Kwasnik, M., Rola, J. & Rozek, W. Tick-borne Encephalitis—review of the current status. J. Clin. Med. 12, 6603 (2023).
Elbaz, M. et al. Systematic review and meta-analysis of foodborne tick-borne encephalitis, Europe, 1980–2021. Emerg. Infect. Dis. 28, 1945–1954 (2022).
Kaiser, R. The clinical and epidemiological profile of tick-borne encephalitis in southern Germany 1994–98: a prospective study of 656 patients. Brain J. Neurol. 122, 2067–2078 (1999).
Mustafá, Y. M., Meuren, L. M., Coelho, S. V. A. & de Arruda, L. B. Pathways exploited by flaviviruses to counteract the blood–brain barrier and invade the central nervous system. Front. Microbiol. 10, 525 (2019).
Berankova, M. et al. Three-dimensional mapping of tick-borne encephalitis virus distribution in the mouse brain using a newly engineered TurboGFP reporter virus. Emerg. Microbes Infect. https://www.tandfonline.com/doi/abs/10.1080/22221751.2025.2542246 (2025).
Kohlmaier, B. et al. Clinical characteristics of patients with Tick-Borne Encephalitis (TBE): a European Multicentre study from 2010 to 2017. Microorganisms 9, 1420 (2021).
European Centre for Disease Prevention and Control. Factsheet About Tick-borne Encephalitis (TBE) https://www.ecdc.europa.eu/en/tick-borne-encephalitis/facts/factsheet (2024).
Miazga, W. et al. The long-term efficacy of tick-borne encephalitis vaccines available in Europe—a systematic review. BMC Infect. Dis. 23, 621 (2023).
Erber, W. & Schmitt, H.-J. Self-reported tick-borne encephalitis (TBE) vaccination coverage in Europe: results from a cross-sectional study. Ticks Tick-Borne Dis. 9, 768–777 (2018).
Pilz, A., Erber, W. & Schmitt, H.-J. Vaccine uptake in 20 countries in Europe 2020: focus on tick-borne encephalitis (TBE). Ticks Tick-Borne Dis. 14, 102059 (2023).
European Centre for Disease Prevention and Control. Tick-borne encephalitis—Annual Epidemiological Report for 2022. https://www.ecdc.europa.eu/en/publications-data/tick-borne-encephalitis-annual-epidemiological-report-2022 (2024).
Bogovic, P. & Strle, F. Tick-borne encephalitis: a review of epidemiology, clinical characteristics, and management. World J. Clin. Cases WJCC 3, 430–441 (2015).
European Centre for Disease Prevention and Control. Tick-borne Encephalitis https://www.ecdc.europa.eu/en/tick-borne-encephalitis (2010).
Bartholdsson, S. et al. Clinical characteristics of tick-borne Encephalitis in adult patients: a 10-year retrospective study in Stockholm, Sweden. J. Infect. Dis. 231, e195–e205 (2024).
Kindberg, E. et al. A functional toll-like receptor 3 gene (TLR3) may be a risk factor for tick-borne encephalitis virus (TBEV) infection. J. Infect. Dis. 203, 523–528 (2011).
Mickiene, A. et al. Tickborne encephalitis in an area of high endemicity in lithuania: disease severity and long-term prognosis. Clin. Infect. Dis. 35, 650–658 (2002).
Mickienė, A. et al. Polymorphisms in chemokine receptor 5 and toll-like receptor 3 genes are risk factors for clinical tick-borne Encephalitis in the Lithuanian Population. PLoS ONE 9, e106798 (2014).
Nygren, T. M. et al. Tick-borne encephalitis: acute clinical manifestations and severity in 581 cases from Germany, 2018–2020. J. Infect. 86, 369–375 (2023).
Radzišauskienė, D. et al. The epidemiology, clinical presentation, and predictors of severe Tick-borne encephalitis in Lithuania, a highly endemic country: a retrospective study of 1040 patients. PLoS ONE 15, e0241587 (2020).
Haglund, M., Forsgren, M., Lindh, G. & Lindquist, L. A 10-year follow-up study of tick-borne encephalitis in the Stockholm area and a review of the literature: need for a vaccination strategy. Scand. J. Infect. Dis. 28, 217–224 (1996).
Haglund, M. & Günther, G. Tick-borne encephalitis—pathogenesis, clinical course and long-term follow-up. Vaccine 21, S11–S18 (2003).
Halsby, K. et al. Clinical spectrum and dynamics of sequelae following tick-borne encephalitis virus infection: a systematic literature review. Open Forum Infect. Dis. 12, ofaf317 (2025).
Halsby, K. et al. Clinical manifestations and outcomes of Tick-borne encephalitis: a systematic literature review. Ticks Tick-Borne Dis. 15, 102407 (2024).
Krawczuk, K., Czupryna, P., Pancewicz, S., Ołdak, E. & Moniuszko-Malinowska, A. Comparison of tick-borne encephalitis between children and adults—analysis of 669 patients. J. Neurovirol. 26, 565–571 (2020).
Nygren, T. M. et al. Recovery and sequelae in 523 adults and children with tick-borne encephalitis in Germany. Infection 51, 1503–1511 (2023).
Ruzek, D. et al. Tick-borne encephalitis in Europe and Russia: review of pathogenesis, clinical features, therapy, and vaccines. Antiviral Res. 164, 23–51 (2019).
Šmit, R. & Postma, M. J. The burden of tick-borne encephalitis in Disability-Adjusted Life Years (DALYs) for Slovenia. PLoS ONE 10, e0144988 (2015).
Bogovič, P. et al. The long-term outcome of tick-borne encephalitis in Central Europe. Ticks Tick-Borne Dis. 9, 369–378 (2018).
Czupryna, P. et al. Sequelae of tick-borne encephalitis in retrospective analysis of 1072 patients. Epidemiol. Infect. 146, 1663–1670 (2018).
Kaiser, R. & Holzmann, H. Laboratory findings in tick-borne encephalitis—correlation with clinical outcome. Infection 28, 78–84 (2000).
Fowler, Å, Ygberg, S., Bogdanovic, G. & Wickström, R. Biomarkers in cerebrospinal fluid of children with tick-borne encephalitis: association with long-term outcome. Pediatr. Infect. Dis. J. 35, 961 (2016).
Bogovič, P. et al. Inflammatory immune responses in patients with tick-borne encephalitis: dynamics and association with the outcome of the disease. Microorganisms 7, 514 (2019).
Czupryna, P. et al. The assessment of usefulness of cytokines and other soluble mediators as the predictors of sequalae development in various forms of tick-borne encephalitis (TBE). Cytokine 184, 156767 (2024).
Blom, K. et al. Cell-mediated immune responses and immunopathogenesis of human tick-borne encephalitis virus-infection. Front. Immunol. 9, 2174 (2018).
Reusken, C. et al. An evaluation of serological methods to diagnose tick-borne encephalitis from serum and cerebrospinal fluid. J. Clin. Virol. 120, 78–83 (2019).
Bogovič, P. et al. Low virus-specific IgG antibodies in adverse clinical course and outcome of tick-borne encephalitis. Microorganisms 9, 332 (2021).
Aregay, A. et al. Poor virus-specific T-cell responses early after tick-borne encephalitis virus infection correlate with disease severity. Emerg. Microbes Infect. 13, 2317909 (2024).
Grygorczuk, S. et al. The intrathecal expression and pathogenetic role of Th17 cytokines and CXCR2-binding chemokines in tick-borne encephalitis. J. Neuroinflamm. 15, 115 (2018).
Ludlow, M. et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 131, 159–184 (2016).
Looker, K. J. et al. Global and regional estimates of prevalent and incident Herpes Simplex Virus Type 1 infections in 2012. PLoS ONE 10, e0140765 (2015).
Nicoll, M. P., Proença, J. T. & Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 36, 684 (2012).
Rose, D. U. D. et al. Neonatal herpes simplex virus infection: from the maternal infection to the child outcome. J. Med. Virol. 95, e29024 (2023).
Melvin, A. J. et al. Neonatal Herpes Simplex virus infection: epidemiology and outcomes in the modern era. J. Pediatr. Infect. Dis. Soc. 11, 94–101 (2022).
Mahant, S. et al. Neonatal Herpes Simplex virus infection among medicaid-enrolled children: 2009–2015. Pediatrics 143, e20183233 (2019).
Gnann, J. W. Jr. & Whitley, R. J. Herpes Simplex Encephalitis: an update. Curr. Infect. Dis. Rep. 19, 13 (2017).
Rohani, H. et al. The worldwide prevalence of herpes simplex virus encephalitis and meningitis: a systematic review and meta-analysis. Turk. Arch. Pediatr. 58, 580–587 (2023).
Dennett, C., Cleator, G. M. & Klapper, P. E. HSV-1 and HSV-2 in herpes simplex encephalitis: a study of sixty-four cases in the United Kingdom. J. Med. Virol. 53, 1–3 (1997).
Steiner, I. Herpes simplex virus encephalitis: new infection or reactivation?. Curr. Opin. Neurol. 24, 268 (2011).
Ak, A. K., Bhutta, B. S. & Mendez, M. D. Herpes Simplex Encephalitis (StatPearls Publishing, 2024).
Reinert, L. S. et al. Sensing of HSV-1 by the cGAS–STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 7, 13348 (2016).
Patrycy, M., Chodkowski, M. & Krzyzowska, M. Role of microglia in herpesvirus-related neuroinflammation and neurodegeneration. Pathogens 11, 809 (2022).
Aravalli, R. N., Hu, S., Rowen, T. N., Gekker, G. & Lokensgard, J. R. Differential apoptotic signaling in primary glial cells infected with herpes simplex virus 1. J. Neurovirol. 12, 501–510 (2006).
Defres, S. et al. A feasibility study of quantifying longitudinal brain changes in herpes simplex virus (HSV) encephalitis using magnetic resonance imaging (MRI) and stereology. PLoS ONE 12, e0170215 (2017).
Sili, U., Kaya, A. & Mert, A. Herpes simplex virus encephalitis: clinical manifestations, diagnosis and outcome in 106 adult patients. J. Clin. Virol. 60, 112–118 (2014).
Riera-Mestre, A., Gubieras, L., Martínez-Yelamos, S., Cabellos, C. & Fernández-Viladrich, P. Adult herpes simplex encephalitis: fifteen years’ experience. Enferm. Infecc. Microbiol. Clín. 27, 143–147 (2009).
Sili, U., Kaya, A. & Mert, A. Herpes simplex virus encephalitis: Clinical manifestations, diagnosis and outcome in 106 adult patients. J. Clin. Virol. 60, 112 (2014).
Stahl, J. P. & Mailles, A. Herpes simplex virus encephalitis: an update. Mediterr. J. Infect. Microbes Antimicrob. 7, 24 (2018).
Bradshaw, M. J. & Venkatesan, A. Herpes Simplex Virus-1 encephalitis in adults: pathophysiology, diagnosis, and management. Neurotherapeutics 13, 493–508 (2016).
Raschilas, F. et al. Outcome of and prognostic factors for herpes simplex encephalitis in adult patients: results of a Multicenter Study. Clin. Infect. Dis. 35, 254–260 (2002).
Riancho, J., Delgado-Alvarado, M., Sedano, M. J., Polo, J. M. & Berciano, J. Herpes simplex encephalitis: clinical presentation, neurological sequelae and new prognostic factors. Ten years of experience. Neurol. Sci. 34, 1879–1881 (2013).
Stahl, J. P., Mailles, A., Broucker, T. D. & Group, the S. C. and I. Herpes simplex encephalitis and management of acyclovir in encephalitis patients in France. Epidemiol. Infect. 140, 372–381 (2012).
Harris, L. et al. Neuropsychological and psychiatric outcomes in encephalitis: a multi-centre case-control study. PLoS ONE 15, e0230436 (2020).
Mailles, A. et al. Long-term Outcome of patients presenting with acute infectious encephalitis of various causes in France. Clin. Infect. Dis. 54, 1455–1464 (2012).
McGrath, N., Anderson, N. E., Croxson, M. C. & Powell, K. F. Herpes simplex encephalitis treated with acyclovir: diagnosis and long term outcome. J. Neurol. Neurosurg. Psychiatry 63, 321–326 (1997).
Rocha, N. D., de Moura, S. K., da Silva, G. A. B., Mattiello, R. & Sato, D. K. Neurological sequelae after encephalitis associated with herpes simplex virus in children: systematic review and meta-analysis. BMC Infect. Dis. 23, 55 (2023).
Abbuehl, L. S., Hofmann, E., Hakim, A. & Dietmann, A. Can we forecast poor outcome in herpes simplex and varicella zoster encephalitis? A narrative review. Front. Neurol. 14, 1130090 (2023).
Skouboe, M. K., Werner, M. & Mogensen, T. H. Inborn errors of immunity predisposing to herpes simplex virus infections of the central nervous system. Pathogens 12, 310 (2023).
Bertrand, A. et al. MR imaging of adult acute infectious encephalitis. Méd. Mal. Infect. 47, 195–205 (2017).
Sarton, B. et al. Assessment of magnetic resonance imaging changes and functional outcomes among adults with severe herpes simplex encephalitis. JAMA Netw. Open 4, e2114328 (2021).
Kamei, S. et al. Prognostic value of cerebrospinal fluid cytokine changes in herpes simplex virus encephalitis. Cytokine 46, 187–193 (2009).
Armangue, T. et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol. 17, 760–772 (2018).
Westman, G. et al. N-methyl-d-aspartate receptor autoimmunity affects cognitive performance in herpes simplex encephalitis. Clin. Microbiol. Infect. 22, 934–940 (2016).
Armangue, T. et al. Autoimmune post-herpes simplex encephalitis of adults and teenagers. Neurology 85, 1736–1743 (2015).
Esposito, S., Autore, G., Argentiero, A., Ramundo, G. & Principi, N. Autoimmune encephalitis after herpes simplex encephalitis: a still undefined condition. Autoimmun. Rev. 21, 103187 (2022).
Yong, S. J. et al. The hippocampal vulnerability to herpes simplex virus type I infection: relevance to Alzheimer’s disease and memory impairment. Front. Cell. Neurosci. 15, 695738 (2021).
Lathe, R. & Haas, J. G. Distribution of cellular HSV-1 receptor expression in human brain. J. Neurovirol. 23, 376–384 (2017).
Aniszewska, A. et al. The expression of interleukin-6 and its receptor in various brain regions and their roles in exploratory behavior and stress responses. J. Neuroimmunol. 284, 1–9 (2015).
Ando, Y., Kitayama, H., Kawaguchi, Y. & Koyanagi, Y. Primary target cells of herpes simplex virus type 1 in the hippocampus. Microbes Infect. 10, 1514–1523 (2008).
Li Puma, D. D. et al. Herpes simplex virus type-1 infection impairs adult hippocampal neurogenesis via amyloid-β protein accumulation. Stem Cells 37, 1467–1480 (2019).
Wang et al. Persistent inflammation and neuronal loss in the mouse brain induced by a modified form of attenuated herpes simplex virus type I. Virol. Sin. 38, 108–118 (2022).
Armien, A. G. et al. Chronic cortical and subcortical pathology with associated neurological deficits ensuing experimental herpes encephalitis. Brain Pathol. 20, 738–750 (2009).
Li Puma, D. D. et al. Interleukin 1β triggers synaptic and memory deficits in Herpes simplex virus type-1-infected mice by downregulating the expression of synaptic plasticity-related genes via the epigenetic MeCP2/HDAC4 complex. Cell. Mol. Life Sci. 80, 172 (2023).
Peng, Y. et al. Higher CSF levels of NLRP3 inflammasome is associated with poor prognosis of anti-N-methyl-D-aspartate receptor encephalitis. Front. Immunol. 10, 905 (2019).
Taraschenko, O. et al. Seizures and memory impairment induced by patient-derived anti-NMDA receptor antibodies in mice are attenuated by anakinra, an interleukin-1 receptor antagonist. Epilepsia 62, 671–682 (2021).
Yang, J. et al. Elevated serum levels of the NLRP3 inflammasome are associated with the severity of anti-NMDAR encephalitis in children. Clin. Chim. Acta Int. J. Clin. Chem. 551, 117587 (2023).
Hu, X. et al. Herpes simplex virus 1 induces microglia gasdermin D-dependent pyroptosis through activating the NLR family pyrin domain containing 3 inflammasome. Front. Microbiol. 13, 838808 (2022).
Hou, B. et al. Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis. 11, 377 (2020).
Niu, L. et al. The critical role of the hippocampal NLRP3 inflammasome in social isolation-induced cognitive impairment in male mice. Neurobiol. Learn. Mem. 175, 107301 (2020).
Xu, Y. et al. NLRP3 inflammasome in cognitive impairment and pharmacological properties of its inhibitors. Transl. Neurodegener. 12, 49 (2023).
Zhu, W. et al. NLRP3 inflammasome activation contributes to long-term behavioral alterations in mice injected with lipopolysaccharide. Neuroscience 343, 77–84 (2017).
Codolo, G. et al. Triggering of inflammasome by aggregated α–synuclein, an inflammatory response in synucleinopathies. PLoS ONE 8, e55375 (2013).
Wang, Z. et al. Herpes simplex virus 1 accelerates the progression of Alzheimer’s disease by modulating microglial phagocytosis and activating NLRP3 pathway. J. Neuroinflamm. 21, 176 (2024).
Zhou, K., Shi, L., Wang, Y., Chen, S. & Zhang, J. Recent advances of the NLRP3 inflammasome in central nervous system disorders. J. Immunol. Res. 2016, 9238290 (2016).
Baringer, J. R. & Pisani, P. Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann. Neurol. 36, 823–829 (1994).
Wozniak, M. A., Shipley, S. J., Combrinck, M., Wilcock, G. K. & Itzhaki, R. F. Productive herpes simplex virus in brain of elderly normal subjects and Alzheimer’s disease patients. J. Med. Virol. 75, 300–306 (2005).
Duarte, L. F. et al. Asymptomatic herpes simplex virus brain infection elicits cellular senescence phenotypes in the central nervous system of mice suffering multiple sclerosis-like disease. Commun. Biol. 7, 811 (2024).
Duarte, L. F. et al. Asymptomatic herpes simplex virus type 1 infection causes an earlier onset and more severe experimental autoimmune encephalomyelitis. Front. Immunol. 12, 635257 (2021).
Sivasubramanian, M. K. et al. Herpes simplex virus type 1 preferentially enhances neuro-inflammation and senescence in brainstem of female mice. J. Virol. 96, e01081–22 (2022).
Martin, C. et al. Inflammatory and neurodegeneration markers during asymptomatic HSV-1 reactivation. J. Alzheimers Dis. 39, 849–859 (2014).
Dutton, A. J. et al. Asymptomatic neonatal herpes simplex virus infection in mice leads to persistent CNS infection and long-term cognitive impairment. PLoS Pathog. 21, e1012935 (2025).
Itzhaki, R. F. et al. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 349, 241–244 (1997).
Letenneur, L. et al. Seropositivity to herpes simplex virus antibodies and risk of Alzheimer’s disease: a Population-Based Cohort Study. PLoS ONE 3, e3637 (2008).
Liu, Y., Johnston, C., Jarousse, N., Fletcher, S. P. & Iqbal, S. Association between Herpes Simplex Virus Type 1 and the Risk of Alzheimer’s Disease: A Retrospective Case-control Study https://doi.org/10.1136/bmjopen-2024-093946 (2025).
Zerboni, L., Sen, N., Oliver, S. L. & Arvin, A. M. Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Microbiol. 12, 197–210 (2014).
Patil, A., Goldust, M. & Wollina, U. Herpes zoster: a review of clinical manifestations and management. Viruses 14, 192 (2022).
Baiker, A. et al. Varicella-zoster virus infection of human neural cells in vivo. Proc. Natl Acad. Sci. USA 101, 10792–10797 (2004).
Bubak, A. N., Como, C. N., Blackmon, A. M., Jones, D. & Nagel, M. A. Varicella zoster virus differentially alters morphology and suppresses proinflammatory cytokines in primary human spinal cord and hippocampal astrocytes. J. Neuroinflamm. 15, 318 (2018).
Carpenter, J. E. et al. Defensive perimeter in the central nervous system: predominance of astrocytes and astrogliosis during recovery from varicella-zoster virus encephalitis. J. Virol. 90, 379–391 (2015).
Nagel, M. A., Niemeyer, C. S. & Bubak, A. N. Central nervous system infections produced by varicella zoster virus. Curr. Opin. Infect. Dis. 33, 273 (2020).
Skripuletz, T. et al. Varicella zoster virus infections in neurological patients: a clinical study. BMC Infect. Dis. 18, 238 (2018).
Mirouse, A. et al. Neurologic outcome of VZV encephalitis one year after ICU admission: a multicenter cohort study. Ann. Intensive Care 12, 32 (2022).
Kleinschmidt-Demasters, B. K., Amlie-Lefond, C. & Gilden, D. H. The patterns of varicella zoster virus encephalitis. Hum. Pathol. 27, 927–938 (1996).
Bozzola, E. et al. Neurological complications of varicella in childhood: case series and a systematic review of the literature. Vaccine 30, 5785–5790 (2012).
Nagel, M. A. & Gilden, D. Neurological complications of VZV reactivation. Curr. Opin. Neurol. 27, 356–360 (2014).
Tyrberg, T., Hagberg, L., Nilsson, S. & Grahn, A. Incidence and risk factors for varicella-zoster virus-associated central nervous system infections: a Nationwide Swedish Retrospective Case-Control Study. J. Med. Virol. 97, e70166 (2025).
Wang, L., Zhu, L. & Zhu, H. Efficacy of varicella (VZV) vaccination: an update for the clinician. Ther. Adv. Vaccines https://doi.org/10.1177/2051013616655980 (2016).
Shapiro, E. D. & Marin, M. The effectiveness of varicella vaccine: 25 years of postlicensure experience in the United States. J. Infect. Dis. 226, S425–S430 (2022).
Grose, C. & Bonthius, D. J. Meningitis caused by the varicella vaccine virus in 17 immunized children and adolescents from the United States, Europe, and Japan. Ann. Child Neurol. Soc. 1, 96–101 (2023).
Ramachandran, P. S. et al. Meningitis caused by the live varicella vaccine virus: metagenomic next generation sequencing, immunology exome sequencing and cytokine multiplex profiling. Viruses 13, 2286 (2021).
Herlin, L. K. et al. Varicella zoster virus encephalitis in Denmark from 2015 to 2019—a Nationwide Prospective Cohort Study. Clin. Infect. Dis. 72, 1192–1199 (2021).
Eckerström, M., Nilsson, S., Zetterberg, H., Blennow, K. & Grahn, A. Cognitive impairment without altered levels of cerebrospinal fluid biomarkers in patients with encephalitis caused by varicella-zoster virus: a pilot study. Sci. Rep. 10, 22400 (2020).
Grahn, A., Nilsson, S., Nordlund, A., Lindén, T. & Studahl, M. Cognitive impairment 3 years after neurological Varicella-zoster virus infection: a long-term case control study. J. Neurol. 260, 2761–2769 (2013).
Hokkanen, L. et al. Subcortical type cognitive impairment in herpes zoster encephalitis. J. Neurol. 244, 239–245 (1997).
Giannelos, N. et al. The incidence of herpes zoster complications: a systematic literature review. Infect. Dis. Ther. 13, 1461–1486 (2024).
Marra, F., Parhar, K., Huang, B. & Vadlamudi, N. Risk factors for herpes zoster infection: a meta-analysis. Open Forum Infect. Dis. 7, ofaa005 (2020).
Kennedy, P. G. & Mogensen, T. H. Determinants of neurological syndromes caused by varicella zoster virus (VZV). J. Neurovirol. 26, 482–495 (2020).
Carter-Timofte, M. E., Paludan, S. R. & Mogensen, T. H. RNA polymerase III as a gatekeeper to prevent severe VZV infections. Trends Mol. Med. 24, 904–915 (2018).
Liu, Q., Han, J. & Zhang, X. Peripheral and central pathogenesis of postherpetic neuralgia. Skin Res. Technol. 30, e13867 (2024).
Ding, S. et al. Association of the incidence of postherpetic neuralgia with early treatment intervention of herpes zoster and patient baseline characteristics: a systematic review and meta-analysis of cohort studies. Int. J. Infect. Dis. 147, 107181 (2024).
Zhou, H., Wang, Z., Jin, H., Chen, X. & Lei, L. A systematic review and meta-analysis of independent risk factors for postherpetic neuralgia. Ann. Palliat. Med. 10, 12181–12189 (2021).
Drolet, M. et al. The impact of herpes zoster and postherpetic neuralgia on health-related quality of life: a prospective study. Can. Med. Assoc. J. J. Assoc. Medicale Can. 182, 1731–1736 (2010).
Du, J. et al. Prevalence and risk factors of anxiety and depression in patients with postherpetic neuralgia: a Retrospective Study. Dermatology 237, 891–895 (2021).
Lee, D. H. et al. Factors associated with increased risk for clinical insomnia in patients with postherpetic neuralgia: a retrospective cross-sectional study. Pain Med. 17, 1917–1922 (2016).
Tang, Y. et al. Structural and functional brain abnormalities in postherpetic neuralgia: a systematic review of neuroimaging studies. Brain Res. 1752, 147219 (2021).
Nagel, M. A. & Gilden, D. Update on varicella zoster virus vasculopathy. Curr. Infect. Dis. Rep. 16, 407 (2014).
Nagel, M. A. et al. The varicella zoster virus vasculopathies. Neurology 70, 853–860 (2008).
Kwon, S. U. et al. Risk of stroke and transient ischaemic attack after herpes zoster. Clin. Microbiol. Infect. 22, 542–548 (2016).
Langan, S. M., Minassian, C., Smeeth, L. & Thomas, S. L. Risk of stroke following herpes zoster: a self-controlled case-series study. Clin. Infect. Dis. 58, 1497–1503 (2014).
Sreenivasan, N. et al. The short- and long-term risk of stroke after herpes zoster—A Nationwide Population-Based Cohort Study. PLoS ONE 8, e69156 (2013).
Grahn, A. et al. Cerebrospinal fluid biomarkers in patients with varicella-zoster virus CNS infections. J. Neurol. 260, 1813–1821 (2013).
Gray, W. L., Starnes, B., White, M. W. & Mahalingam, R. The DNA sequence of the simian varicella virus genome. Virology 284, 123–130 (2001).
White, T. M., Gilden, D. H. & Mahalingam, R. An animal model of varicella virus infection. Brain Pathol. 11, 475–479 (2006).
Moffat, J. F., Stein, M. D., Kaneshima, H. & Arvin, A. M. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J. Virol. 69, 5236–5242 (1995).
Zerboni, L., Ku, C.-C., Jones, C. D., Zehnder, J. L. & Arvin, A. M. Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc. Natl Acad. Sci. USA 102, 6490–6495 (2005).
Zerboni, L. et al. Varicella-zoster virus glycoprotein E is a critical determinant of virulence in the SCID mouse-human model of neuropathogenesis. J. Virol. 85, 98–111 (2011).
Zerboni, L. et al. Aberrant infection and persistence of varicella-zoster virus in human dorsal root ganglia in vivo in the absence of glycoprotein I. Proc. Natl Acad. Sci. USA 104, 14086–14091 (2007).
Zerboni, L. & Arvin, A. Neuronal subtype and satellite cell tropism are determinants of varicella-zoster virus virulence in human dorsal root ganglia xenografts in vivo. PLoS Pathog. 11, e1004989 (2015).
Acknowledgements
The work was supported by the Lundbeck Foundation (R359-2020-2287) and the Danish National Research Foundation (DNRF164).
Author information
Authors and Affiliations
Contributions
R.K. and S.R.P. conceptualized the review. R.K. performed the literature search, wrote the original draft, and prepared the figures. L.S.R. and S.R.P. contributed to writing and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kelly, R., Reinert, L.S. & Paludan, S.R. Sequelae of viral CNS infections including outcomes, mechanisms, and knowledge gaps. npj Viruses 3, 79 (2025). https://doi.org/10.1038/s44298-025-00160-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44298-025-00160-7
