
Abstract
This study developed a new approach to produce sterile, hyperpolarized [13C,15N2]urea as a novel molecular imaging probe and applied it for first-ever healthy brain volunteer studies. Hyperpolarized [13C,15N2]urea, as a small, metabolically inert molecule, offers significant advantages for perfusion imaging due to its endogenous nature and excellent safety profile. The developed methods achieved a hyperpolarized [13C,15N2]urea solution (132 ± 6 mM) with 27.4 ± 5.6% polarization and a T1 = 50.4 ± 0.2 s. In healthy brain volunteer studies, high-resolution 13C imaging captured blood flow with a spatial resolution of 7.76 × 7.76 × 15 (or 10) mm3 over ~1 min following hyperpolarized [13C,15N2]urea injection, visualizing detailed vascular structures. Time-to-peak and centroid analyses showed consistent arterial and venous signal patterns across subjects. Findings suggest hyperpolarized [13C,15N2]urea may have applications beyond brain imaging, including the non-invasive perfusion assessment in various organs, cancer microenvironment, and renal function, paving the way for clinical translation.
Similar content being viewed by others


Introduction
Over the past decade, hyperpolarized (HP) carbon-13 MRI, utilizing dissolution dynamic nuclear polarization (dDNP)1, has become a powerful clinical-research molecular imaging approach2,3,4. This stable-isotope (non-radioactive) technology enables real-time imaging of cellular metabolic processes by tracking the uptake and enzymatic conversions of injected HP substrates, most commonly [1-13C]pyruvate, to provide unique insights into abnormal metabolism in disease states5,6,7,8,9. While HP 13C MRI has primarily been employed to investigate metabolic pathways, its ability to capture dynamic blood flow measurements has also positioned it as a powerful tool for perfusion imaging, particularly in the brain10.
Perfusion imaging is essential for diagnosing and monitoring neurological conditions like stroke, brain tumors, and neurodegenerative diseases, offering critical insights into blood flow and tissue health. Among HP 13C agents explored for this purpose, 13C-urea has gained significant attention in preclinical studies due to its unique properties as an endogenous, metabolically inert molecule that directly reflects blood flow and tissue perfusion11,12,13. Compared to Gadolinium (Gd)-based MR contrast agents, urea’s lower molecular weight—approximately 15 times smaller—allows for rapid distribution into extracellular, interstitial spaces and microvasculature, making it uniquely effective in characterizing tissue perfusion12,13. In addition, urea offers a direct proportionality of signal-to-probe concentration and an inherently high contrast-to-noise ratio due to the absence of background signal14, whereas Gd contrast agents in 1H DCE-MRI often suffer from non-specific uptake, non-linear relationship between signal intensity and concentration, and reduced sensitivity in non-enhancing tumors15.
The dual-labeled [13C,15N2]urea provides much longer relaxation times (T2 increase by 250-fold15) than using natural abundance 14N and, therefore, greatly benefits higher usable polarizations and longer lifetimes for HP 13C MRI16,17. Preclinical research has validated the use of HP [13C,15N2]urea for safe and effective investigation of conditions such as tumor hypoxia18,19, cardiomyopathy20, renal ischemia21,22,23, and acute kidney injury24,25 by observing decreased urea perfusion with high SNR. While it perfuses into most tissues, urea typically does not cross the blood–brain barrier (BBB)26 and, in fact, has been used clinically as a hyperosmolar agent to decrease brain swelling27. Also, ex vivo brain tissue studies from patients with Alzheimer’s, Huntington’s, Parkinson’s disease, Lewy Body, and vascular dementia demonstrated elevated cerebral urea28,29.
For human research, HP [13C,15N2]urea was first translated as part of a dual-agent probe with [1-13C]pyruvate, enabling simultaneous measurement of pyruvate metabolism and urea tissue perfusion30,31. In a first-in-human study involving a prostate cancer patient, the co-administration of 150 mM HP [1-13C]pyruvate and 35 mM [13C,15N2]urea revealed a mismatch between metabolism and tissue-perfusion that is characteristic of high-grade prostate cancer, where decreased urea tissue perfusion aligned with increased pyruvate metabolism32. This finding underscored the potential of HP [13C,15N2]urea to provide unique insights into tumor perfusion and hypoxia in aggressive pathologies. Furthermore, this may eventually serve as a potential method to investigate blood–brain barrier integrity in conditions such as dementia, trauma, other neurodegenerative disorders, and aging.
In the dual-agent probe, the pyruvate-to-urea concentration ratio was 4:1, resulting in a urea probe concentration of ~35 mM30. Compared to the coarser resolution achieved in the dual-agent study for tissue perfusion and tumor hypoxia, this project developed a single-agent urea probe at a 4-fold higher concentration, enabling higher resolution imaging of brain vasculature and perfusion. To achieve this, we designed a standard operating procedure (SOP) for producing sterile HP single-agent [13C,15N2]urea and obtained IRB & FDA-IND approvals. This protocol was validated for clinical translation and feasibility studies of urea perfusion, regional vasculature, and its distribution in the healthy human brain.
Results
Standard operating procedure (SOP) development for sterile HP urea production
In this project, we successfully developed and optimized a new SOP for the on-site production of sterile hyperpolarized (HP) urea, following Good Manufacturing Practice (GMP) guidelines as outlined in the U.S. Code of Federal Regulation Title 21, Part 212. To dissolve solid urea and achieve a DNP-compatible sample with ~9 M urea concentration, lactic acid was used as a solvent due to its similar physical properties to pyruvic acid (i.e., a self-glassing agent) as previously demonstrated33. The urea/lactic acid/electron paramagnetic agent (EPA) mixture, sterile water for injection (SWFI) for dissolution, and buffer for neutralization were prepared as described in the Methods section and loaded into a GE Pharmacy Kit. The composition of the Pharmacy Kit preparation for the HP [13C,15N2]urea probe is summarized in Table 1.
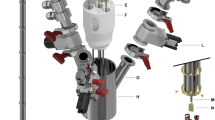
Figure 1a illustrates the components of the GE Pharmacy Kit, including a cryovial with the urea/lactic acid/EPA solution (i), transfer tubing (j, m), a dissolution valve (k), a dissolution syringe filled with 41 ml SWFI (l), a size-exclusion EPA filter (n), a receiver bag containing SWFI and NaOH-Tris buffer (o), a terminal sterilization filter (p), a power-injector syringe (Medrad, Bayer Germany) (q), and a QC syringe (r).
![Fig. 1: Fluid path workflow and quality control tests for preparing hyperpolarized [13C,15N2]urea.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs44303-025-00073-3/MediaObjects/44303_2025_73_Fig1_HTML.png?as=webp)
a Fluid path (Pharmacy Kit) components and steps for pre-release quality control tests: (i) Cryovial in the SPINlab system at 0.8 K with microwave irradiation; (j) Dual-lumen dissolution tubing; (k) Dissolution syringe valve; (l) Dissolution syringe; (m) Single-lumen transfer tubing; (n) electron paramagnetic agent (EPA) filter; (o) Receiver bag; (p) sterility filter; (q) MedRad® syringe for patient administration; (r) quality control (QC) syringe; (1) Solid-state 13C NMR measurements performed on the frozen urea sample to monitor polarization build-up (2) Visual inspection of injection product volume (3) pH test of the sample collected in the QC syringe (4) EPA concentration measurements by UV spectrophotometry (5) Bubble point test to ensure the sterility filter integrity. b First timepoint HP 13C NMR spectrum of the injection product acquired on a 1.4 T benchtop NMR spectrometer.
In addition, Fig. 1a outlines the quality control (QC) measurements required before releasing the HP urea probe, which are also summarized in Table S1. Firstly, solid-state 13C NMR signals from the sample in a cryovial were monitored during microwave irradiation to confirm a more than 100-fold increase in signal intensity, ensuring that there would be sufficient hyperpolarization before dissolution (Fig. 1a(1)). During the dissolution process in the SPINlab DNP polarizer, the HP urea was rapidly dissolved by superheated SWFI from the dissolution syringe and transferred through tubing to a receiver bag, passing through a size-exclusion filter that removed EPA. In the receiver bag, the dissolved product was neutralized with Tris-NaOH, resulting in an HP urea solution at physiological temperature and pH, referred to as the “injection product.” The injection product then passed through a terminal sterilization filter before reaching the administering syringe and QC syringe. Upon completion of the dissolution process, the injection product’s volume was visually inspected to confirm that more than 38 mL was collected in the Medrad syringe (Fig. 1a(2)). The Medrad syringe was then prepared for patient administration. Meanwhile, 3–4 mL of the injection product in the QC syringe underwent pH testing (acceptable range: 5.0–9.0) using a pH strip and EPA concentration testing (less than 7 μM) using a UV spectrophotometer (Fig. 1a(3), (4)). Finally, a bubble point test was performed to ensure the sterility filter’s integrity (Fig. 1a(5)). Once all five QC tests were successfully passed, the pharmacist released the injection product for human administration. Post-dissolution QC tests, including pH electrode measurement, endotoxin, and sterility testing, were also conducted. The SOP was validated through three consecutive successful Process Qualification trials (Table S2).
Imaging probe characterization
After 3.5 h of polarization, the solid-state 13C polarization of [13C,15N2]urea reached ~90% of the maximum achievable polarization determined from the fitting curve, with a build-up time constant of 5480 s (Fig. S1). Following rapid dissolution, a HP [13C,15N2]urea solution with a neutral pH of 7–8 was successfully prepared. A representative HP 13C NMR spectrum of the injection product displays the urea signal as a triplet, attributed to J-coupling between 13C and 15N spins (20.2 Hz) at 164 ppm, along with a lactate signal at natural abundance at 183.7 ppm (Fig. 1b). In addition, two minor peaks observed upfield from the lactate signal at 176.1 and 178.8 ppm were identified as lactate dimers. No other impurities were detected in the spectrum.
Thermal 13C NMR spectrum analysis revealed that the urea concentration was 132 ± 6 mM, nearly four times higher than the urea concentrations used in the prior co-polarized pyruvate and urea experiments (~35 mM30), while the natural-abundance lactate concentration was 81 ± 9 mM. The liquid-state 13C polarization of urea at the time of dissolution was calculated to be 27.4 ± 5.6%. The average concentration of the residual EPA was measured to satisfy specifications at 4.1 ± 2.2 μM, with a pH of 8.2 ± 0.2. The T1 of the HP 13C urea was 43.3 ± 2.6 s at 1.4 T on the benchtop NMR and 50.4 ± 0.2 s at 3 T MRI.
First-in-human brain imaging with HP [13C,15N2]urea
The time course of HP [13C,15N2]urea signals from healthy brain volunteers is illustrated in Fig. 2a and Supporting Information Figs. S2 and S3. The spatial resolution achieved was 7.76 × 7.76 × 15 mm3, comparable to the highest reported resolution of HP [1-13C]pyruvate acquisition (7.5 × 7.5 × 15 mm3)34. Urea images clearly delineated the cerebrovascular system, including the anterior circulation (internal carotid and middle cerebral arteries), posterior circulation (vertebral and basilar arteries), and dural venous sinuses (transverse/superior sagittal sinus, sigmoid sinus). These images captured dynamic signal intensity changes corresponding to the in-flow and washout of HP [13C,15N2]urea compounded with T1 relaxations of HP magnetization.
![Fig. 2: Representative hyperpolarized [13C,15N2]urea images from a healthy brain volunteer.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs44303-025-00073-3/MediaObjects/44303_2025_73_Fig2_HTML.png?as=webp)
a Corresponding 1H anatomical images and the dynamic HP [13C,15N2]urea images (7.76 × 7.76 × 15 mm3) overlaid on the 1H anatomical images are displayed. A total of 20 slices and 32 timeframes were acquired, and this figure displays the middle 6 slices and the first 16 timeframes (1.5 s temporal resolution). White arrows indicate the signal from the urea phantom. b Total signal (area-under-curve, AUC) images of urea from the same subject. The urea images overlaid on the corresponding 1H anatomical images are shown.
Initially, the urea bolus signal appeared in the internal carotid and middle cerebral arteries, followed by the distal arterial branches, and eventually in the venous system, specifically well seen in the transverse and sagittal sinuses (Fig. 2a). Asymmetric flow of urea into the left and right transverse sinuses was observed in most volunteer datasets, consistent with normal anatomic variation35. The peak dynamic SNR in this dataset was 380, observed within the confluence of sinuses at the 8th timepoint (10.5 s after the saline injection ended). An average peak SNR of 253 ± 136 was measured in the superior sagittal sinus across all volunteer datasets (n = 8) with one exception where the peak signal was observed in the superficial temporal artery. Area-under-curve (AUC) images from a representative subject, shown in Fig. 2b, highlighted signals from the blood vessels, but no urea signal was observed in the brain parenchyma, consistent with urea not crossing the blood–brain barrier (BBB).
Figure 3b displays dynamic urea images from selected slices of another volunteer. The time course of urea signals from regions of interest (ROIs) placed on arteries and veins across different axial slices revealed that arterial signal peaks occurred 4.5 to 9 s earlier than venous signals. As expected, larger differences were observed in the inferior slices (ex. slice 8 vs slice 12). With a temporal resolution of 1.5 s, we detected a slight difference in peak times between the urea signals in the internal carotid (1) and basilar arteries (2), with a delay of one timeframe. Across four datasets with a temporal resolution of 1.5 s, the average peak time difference between urea signals in the internal carotid artery and venous sinuses on the same axial slice was 6.1 ± 1.4 s (n = 4).
![Fig. 3: Time-course analysis of hyperpolarized [13C,15N2]urea signals in a healthy brain volunteer.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs44303-025-00073-3/MediaObjects/44303_2025_73_Fig3_HTML.png?as=webp)
a Dynamic HP [13C,15N2]urea images of selected slices from a volunteer in grayscale. Every other timeframe between 1.5 and 22.5 s is shown. Eleven ROIs are indicated by numbers (1–9) and letters (used for the same anatomical structures but on different slices) on the 1H images, and by arrows on the 13C images (1: internal carotid artery; 2: basilar artery; 3: sigmoid sinus/internal jugular vein; 4: transverse sinus; 5: confluence of sinuses; 6: M2 middle cerebral artery; 7: vein of Galen; 8: superior sagittal sinus; 9: urea phantom). b Time-course plots of urea signals from the ROIs, with time-to-peak points indicated by dotted vertical lines.
The time course centroid maps represent the center of mass of the time course36, providing insights into the distribution of urea signals over time across the brain. As shown in Fig. 4, the centroid maps from two representative subjects demonstrate a symmetric distribution of urea signals between the left and right hemispheres. Signals from the arteries appear more “blue”, indicating an earlier centroid, while venous signals are more “red” in these images, reflecting a delayed centroid. Gaussian distribution analysis revealed two distinct time regimes within the centroid maps: for the dataset in Fig. 4a, the mean and standard deviation (μ, σ) were (8.8, 1.2) seconds and (10.4, 2.6) seconds, while for the dataset in Fig. 4b, they were (7.5, 0.9) seconds and (9.4, 1.4) seconds (Fig. S4). The distributions with smaller mean values likely represent arterial signals, while those with larger mean values correspond to venous signals, indicating a difference of 1.6 and 1.9 s between the two regimes. Across four datasets collected with the same temporal resolution (1.5 s), an average difference of 2.0 ± 0.3 s was observed between arterial and venous portions of the signals. This consistency suggests the reproducibility of the measurement across individuals.
![Fig. 4: Hyperpolarized [13C,15N2]urea area-under-curve (AUC) and time-course centroid maps from two subjects.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs44303-025-00073-3/MediaObjects/44303_2025_73_Fig4_HTML.png?as=webp)
1H anatomical images (first column), urea AUC images (second column), and time course centroid maps third column) acquired from two subjects (a, b). The HP urea AUC and time course centroid maps were masked for voxels with SNR greater than 10 in the hyperpolarized urea AUC images.
In a separate study, a slice thickness of 10 mm was used while maintaining the same in-plane resolution. Representative dynamic urea images are displayed in Fig. 5, featuring axial, coronal, and sagittal planes. In the axial images, clear signals from the sigmoid sinus and the confluence of sinuses were visible. The coronal images depicted the urea bolus flowing through the internal carotid artery, while sagittal images showed the urea signal first appearing in the basilar artery, then moving through the superior sagittal sinus to the confluence of sinuses.
![Fig. 5: Dynamic hyperpolarized [13C,15N2]urea images in multiple planes.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs44303-025-00073-3/MediaObjects/44303_2025_73_Fig5_HTML.png?as=webp)
Dynamic HP [13C,15N2]urea images (7.76 × 7.76 × 10 mm3) and corresponding AUC images (Σ) acquired from a volunteer are displayed in axial (top), coronal (middle), and sagittal (bottom) planes.
Discussion
This study was the first to successfully prepare sterile, single agent hyperpolarized (HP) [13C,15N2]urea and demonstrate its feasibility and safety for human use. None of the four volunteers experienced any adverse effects during or after the urea injections, including when administered twice in a single exam. The urea concentration used in this imaging study was sub-physiological, as urea naturally occurs in the body at millimolar concentrations, reaching over a hundred millimolar in the renal medulla. The administered dose of 5.6 mg/kg was ~2–5 times lower than normal blood urea levels (naturally produced as a waste product of metabolism; 10.5–30 mg/kg) and 150 to 300 times lower than the urea doses clinically used for conditions like glaucoma37, cerebral edema38,39, and hyponatremia40, which range from 1 to 1.5 g/kg. In addition, a previous study investigating the toxicity of [13C,15N2]urea in Sprague Dawley rats, co-administered with hyperpolarized [1-13C]pyruvate, found no adverse effects on vital signs, laboratory results, or the gross pathology of major organs30. Based on the successful process qualification tests performed using the SOP developed in this study, our HP [13C,15N2]urea protocol received both FDA IND (under IND# 109956) and UCSF IRB approval for studies in healthy volunteers.
For this project, lactic acid was selected as the solvent and glassing agent for dissolving solid urea due to its self-glassing properties at low temperatures, as demonstrated in a previous study where neat lactic acid was successfully hyperpolarized without additional glassing agents33. As a naturally occurring metabolite, lactate minimizes the risk of adverse reactions, and its lower reactivity compared to pyruvate reduces the chance of side reactions with urea, as confirmed by NMR analyses. Its endogenous nature ensures biological compatibility for clinical applications. Lactic acid also offers practical advantages: It is less viscous than glycerol, which has been used as a glassing agent for urea in preclinical studies12,13,14, facilitating easier handling. In this study, food-grade lactic acid (88%), compliant with United States Pharmacopeia (USP) standards for purity and safety for consumption, was used. The 12% water aided the dissolution of urea to ~9 M, ensuring a uniform and concentrated solution. This material is also inexpensive and readily available, further supporting its practical use.
However, lactic acid’s lower acidity (pKa = 3.86) compared to pyruvic acid (pKa = 2.50) decreased the efficiency of radical removal via size-exclusion filters, as the EPA precipitates more effectively in more acidic environments. To counter this, we reduced the initial EPA concentration to 12 mM (down from the typical 15 mM used in pyruvate studies) and adjusted the release criterion for EPA concentration from 5 μM to 7 μM. These modifications improved the success rate in meeting the quality control standards by consistently lowering the final EPA concentrations to below 7 μM. While the reduced EPA concentration led to a slight increase in the polarization build-up time constant to ~5500 s (~1.3 times longer than with 15 mM EPA), it remained within a practical range for clinical applications. Future improvements may include the use of C18 cartridges for more effective EPA removal, which could support higher EPA concentrations and shorter polarization build-up times while maintaining residual EPA under the specified limit.
The significant advantages of [13C,15N2]urea over [13C]urea with natural abundance 14N for clinical translation have been described in the preclinical literature16,20, including its much longer T1 and T2 relaxation times, which provide higher in vivo signals for perfusion imaging particularly when using bSSFP-based acquisition. The longer T1 relaxation time of [13C,15N2]urea also makes it less sensitive to low magnetic fields encountered during clinical quality-control tests and sample transfer, preserving hyperpolarization, which begins to decay immediately after dissolution due to T1 relaxation. As demonstrated in preclinical studies, replacing the current dissolution solvent (H2O) with deuterated water (D2O) is expected to further increase both T1 and T2 relaxation times41, potentially enhancing SNR in future applications.
With the current SOP, our imaging results demonstrated that single-agent HP [13C,15N2]urea MRI can dynamically assess cerebral perfusion with high sensitivity, offering novel flow and perfusion information in addition to what gadolinium-based contrast agents provide. Although urea does not normally cross the blood–brain barrier, this characteristic makes it a potentially valuable tool for evaluating blood–brain barrier integrity in conditions such as dementia, multiple sclerosis, traumatic brain injury, and brain tumors that show increased permeability due to blood–brain barrier leakage42. Compared to conventional MRI techniques for measuring cerebral blood flow such as Arterial Spin Labeling (ASL)43, HP [13C,15N2]urea MRI offers the benefit of positive contrast with no background signal and much longer T1 relaxation times, providing more sensitive detection of perfusion abnormalities from arteries to capillaries to veins. Beyond brain applications, HP [13C,15N2]urea MRI may be beneficial for non-invasive assessment of tissue perfusion, cancer aggressiveness, cardiac perfusion, urea transporters, kidney function, liver function, and other perfusion-related diseases.
In this study, we developed and optimized a new approach for producing sterile, hyperpolarized [13C,15N2]urea as a new MR molecular imaging agent, and successfully demonstrated its feasibility and safety in imaging the cerebral vasculature and blood flow in healthy volunteers. The results demonstrated that HP [13C,15N2]urea could be a viable and safe MR imaging probe, with significant potential for perfusion imaging in the brain. HP urea effectively captures blood flow by detecting signals from both arterial and venous circulation, making it a promising tool for assessing changes in blood–brain barrier integrity in neurological disorders, as well as evaluating perfusion characteristics in various other organs. This study provides a foundation for the clinical translation of HP urea, offering new possibilities for non-invasive perfusion imaging across multiple organ systems.
Methods
Preparation of HP [13C,15N2]urea probe for clinical studies
To produce a single dose of HP urea probe, 810 mg of [13C,15N2]urea (GMP grade; ISOTEC, MilliporeSigma, Miamisburg, OH) was dissolved with 1.39 g of natural abundance (not C-13 enriched) lactic acid (Food grade 88%; Lab Alley, Spicewood, TX), and 28.5 mg of AH111501 (GE Healthcare; an electron paramagnetic agent (EPA)), was added to 1.905 g of the [13C,15N2]urea/lactic acid mixture. This resulted in a solution of ~9.0 M [13C,15N2]urea with 12 mM EPA.
For hyperpolarization, 1.52 g of the prepared [13C,15N2]urea solution was loaded into a cryovial and polarized on a 5 T SPINlab system (GE Healthcare) operating at 0.8 K. The sample was polarized for 3–4 h with microwaves at a frequency of 139.88 GHz, which was 0.01 GHz lower than the microwave frequency used for polarizing pyruvate. Following polarization, the frozen urea sample was rapidly dissolved with 41 g of super-heated (130 °C), pressurized sterile water for injection (SWFI) and subsequently neutralized with 29.85 g of 600 mM sodium hydroxide (NaOH) and 333 mM Tris(hydroxyethyl)aminomethane (Tris) buffer.
Nuclear magnetic resonance (NMR) spectroscopy
To characterize T1 and polarization of the HP urea imaging probe, HP 13C NMR spectra were acquired on a 1.4 T benchtop NMR system (Magritek) with the following parameters: 1° RF flip angle, 5 s temporal resolution for a total of 128 repetitions. After the HP 13C magnetization was fully relaxed, the signal intensity at thermal equilibrium was measured by applying 1% v/v Gd-DTPA (Magnevist®, Bayer, Whippany, NJ) and using parameters including a 10 s temporal resolution, a 90° flip angle, and 512 averages. The T1 for [13C,15N2]urea was determined from an exponential fit to the dynamic HP signal intensities after correcting for the signal loss caused by RF pulses. Liquid-state polarization of the HP urea was calculated by comparing the urea signal from the first HP spectrum to the thermal-equilibrium spectrum, then back-calculating to the time of dissolution using T1 measured from the HP NMR data. A 9.4 T NMR instrument (Bruker Avance III) was used to determine the urea concentration of the dose for injection.
HP 13C MRI
The HP 13C urea data of the brain collected in this study were acquired from four healthy volunteers (Median age 37, range 26–64, 3 males, 1 female) using a 3D balanced steady-state free precession (bSSFP) sequence with a stack-of-spiral acquisition31 following the injection of HP [13C,15N2]urea. The 3D-bSSFP pulse sequence consisted of a series of excitation pulses (50°) with alternating phases, employing 6 interleaves and 20 (or 30) stacks for kz encoding, with 8-ms TR and a field-of-view (FOV) of 45.0 × 45.0 × 30.0 cm3 with a matrix size of 58 × 58 × 20 (or 30). At the beginning of each timeframe, four catalyzation pulses with increasing amplitudes (i.e., 6°, 19°, 34.5°, and 46.5°) were applied, and the reversed order of catalyzation pulses was used at the end of the timeframe to restore the magnetization back to the z-axis. A spoiler gradient was added to crush residual transverse magnetization. A non-selective 1-ms RF pulse was used for excitation. The acquisition achieved a spatial resolution of 7.76 × 7.76 × 15 mm3 with a temporal resolution of 1.5 s (N = 4) or 3 s (N = 2) or a spatial resolution of 7.76 × 7.76 × 10 mm3 with a temporal resolution of 1.5 s (N = 2) over thirty-two timeframes. For anatomic reference, proton T2-weighted and 3D T1-weighted images were acquired. All experiments were performed on a 3 T clinical MR scanner (Premier, GE Healthcare, Waukesha, WI). A 1H/13C dual-frequency transmit/receive radiofrequency (RF) coil with volume 13C transmit and multichannel 8 1H & 24 13C channel receive (Rapid Biomedical, Rimper, Germany) was used in this study. Transmit RF power was calibrated on a head-shaped phantom containing unenriched ethylene glycol (HOCH2CH2OH, anhydrous, 99.8%, Sigma Aldrich, St. Louis, MO) prior to the volunteer study. All human studies were conducted with informed consent following a protocol approved by both the Institutional Review Board (IRB) and the Food and Drug Administration for an investigational new drug (FDA IND).
HP 13C data processing and analysis
HP 13C urea data were reconstructed by first gridding the k-space data using the Kaiser-Bessel gridding method44, followed by an inverse Fourier transform to generate the reconstructed images. The resulting images were then pre-whitened, coil-combined45, and zero-padded to match the resolution of the proton anatomical images. For time-to-peak analysis, regions of interest (ROIs) were drawn over different branches of arteries and veins, and the signal intensities within the ROIs were averaged to plot the dynamic curves. The centroid of the urea dynamic curve was calculated as the time-weighted average of signal intensity, obtained by dividing the sum of time-weighted signal intensities by the total area under the curve36. In addition, two Gaussian functions were fitted to histograms for analyzing the time course centroids. All processing and analysis were performed using MATLAB.
Data availability
Deidentified data will be made available upon request to the Authors, after the initiation of formal data sharing agreement as required by the data sharing policy at our institution (UCSF).
References
Ardenkjær-Larsen, J. H. et al. Increase in signal-to-noise ratio of >10,000 times in liquid-state NMR. Proc. Natl Acad. Sci. USA 100, 10158–10163 (2003).
Wang, Z. J. et al. Hyperpolarized 13C MRI: state of the art and future directions. Radiology 291, 273–284 (2019).
Kurhanewicz, J. et al. Hyperpolarized 13C MRI: path to clinical translation in oncology. Neoplasia 21, 1–16 (2019).
Larson, P. E. Z. et al. Current methods for hyperpolarized [1‐13C]pyruvate MRI human studies. Magn. Reson. Med. 91, 2204–2228 (2024).
Nelson, S. J. et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate. Sci. Transl. Med. 5, 198ra108–198ra108 (2013).
Gallagher, F. A. et al. Imaging breast cancer using hyperpolarized carbon-13 MRI. Proc. Natl Acad. Sci. USA 117, 2092–2098 (2020).
Hackett, E. P. et al. Imaging acute metabolic changes in patients with mild traumatic brain injury using hyperpolarized [1-13C]pyruvate. iScience 23, 101885 (2020).
Gordon, J. W. et al. Hyperpolarized 13C metabolic MRI of patients with pancreatic ductal adenocarcinoma. Magn. Reson. Imaging 60, 741–749 (2024).
Autry, A. W. et al. Advanced hyperpolarized 13C metabolic imaging protocol for patients with gliomas: a comprehensive multimodal MRI approach. Cancers 16, 354 (2024).
Hu, J. Y. et al. Investigating cerebral perfusion with high resolution hyperpolarized [1‐13C]pyruvate MRI. Magn. Reson. Med. 90, 2233–2241 (2023).
Golman, K., Ardenkjær-Larsen, J. H., Petersson, J. S., Månsson, S. & Leunbach, I. Molecular imaging with endogenous substances. Proc. Natl Acad. Sci. USA 100, 10435–10439 (2003).
von Morze, C. et al. Imaging of blood flow using hyperpolarized [13C]Urea in preclinical cancer models. J. Magn. Reson. Imaging 33, 692–697 (2011).
Von Morze, C. et al. Simultaneous multiagent hyperpolarized 13C perfusion imaging. Magn. Reson. Med. 72, 1599–1609 (2014).
Von Morze, C. et al. Investigating tumor perfusion and metabolism using multiple hyperpolarized 13C compounds: HP001, pyruvate and urea. Magn. Reson. Imaging 30, 305–311 (2012).
Falk Delgado, A. et al. Diagnostic value of alternative techniques to gadolinium-based contrast agents in MR neuroimaging—a comprehensive overview. Insights Imaging 10, 84 (2019).
Reed, G. D. et al. High resolution 13C MRI with hyperpolarized urea: in vivo T2 mapping and 15N labeling effects. IEEE Trans. Med. Imaging 33, 362–371 (2014).
Milshteyn, E. et al. Simultaneous T1 and T2 mapping of hyperpolarized 13C compounds using the bSSFP sequence. J. Magn. Reson. 312, 106691 (2020).
Bok, R. et al. The role of lactate metabolism in prostate cancer progression and metastases revealed by dual-agent hyperpolarized 13C MRSI. Cancers 11, 257 (2019).
Chen, H.-Y. et al. Assessing prostate cancer aggressiveness with hyperpolarized dual-agent 3D dynamic imaging of metabolism and perfusion. Cancer Res. 77, 3207–3216 (2017).
Lau, A. Z., Miller, J. J., Robson, M. D. & Tyler, D. J. Simultaneous assessment of cardiac metabolism and perfusion using copolarized [1‐13C]pyruvate and 13C‐urea. Magn. Reson. Med. 77, 151–158 (2017).
Nielsen, P. M. et al. Renal ischemia and reperfusion assessment with three-dimensional hyperpolarized [13C,15N2]-urea. Magn. Reson. Med. 76, 1524–1530 (2016).
Kjærgaard, U. et al. Assessment of focal renal ischemia–reperfusion injury in a porcine model using hyperpolarized [1‐13C]pyruvate MRI. Magn. Reson. Med. 90, 655–663 (2023).
Mariager, C. Ø., Nielsen, P. M., Qi, H., Ringgaard, S. & Laustsen, C. Hyperpolarized [13C,15N2]-urea T2 relaxation changes in acute kidney injury. Magn. Reson. Med. 80, 696–702 (2018).
Grist, J. T., Mariager, C. Ø., Qi, H., Nielsen, P. M. & Laustsen, C. Detection of acute kidney injury with hyperpolarized [13C,15N2]urea and multiexponential relaxation modeling. Magn. Reson. Med. 84, 943–949 (2020).
Lindhardt, J. L. et al. The hemodynamic and metabolic effects of spironolactone treatment in acute kidney injury assessed by hyperpolarized MRI. NMR Biomed. 33, e4371 (2020).
Lee, S. J., Park, I., Talbott, J. F. & Gordon, J. Investigating the feasibility of in vivo perfusion imaging methods for spinal cord using hyperpolarized [13C]t-butanol and [13C,15N2]urea. Mol. Imaging Biol. 24, 371–376 (2022).
Otvos, B., Kshettry, V. R. & Benzel, E. C. The history of urea as a hyperosmolar agent to decrease brain swelling. FOC 36, E3 (2014).
Scholefield, M. et al. Severe and regionally widespread increases in tissue urea in the human brain represent a novel finding of pathogenic potential in Parkinson’s disease dementia. Front. Mol. Neurosci. 14, 711396 (2021).
Scholefield, M. et al. Human dementia with Lewy bodies brain shows widespread urea elevations. Parkinsonism Relat. Disord. 124, 107017 (2024).
Qin, H. et al. Clinical translation of hyperpolarized 13C pyruvate and urea MRI for simultaneous metabolic and perfusion imaging. Magn. Reson. Med. 87, 138–149 (2022).
Liu, X. et al. Development of specialized magnetic resonance acquisition techniques for human hyperpolarized [13C,15N2]urea + [1‐13C]pyruvate simultaneous perfusion and metabolic imaging. Magn. Reson. Med. 88, 1039–1054 (2022).
De Kouchkovsky, I. et al. Dual hyperpolarized [1-13C]pyruvate and [13C]urea magnetic resonance imaging of prostate cancer. J. Magn. Reson. Open 21, 100165 (2024).
Chen, A. P., Lau, J. Y. C., Alvares, R. D. A. & Cunningham, C. H. Using [1-13C]lactic acid for hyperpolarized 13C MR cardiac studies: lactic acid for 13C MR cardiac studies. Magn. Reson. Med. 73, 2087–2093 (2015).
Gordon, J. W. et al. A variable resolution approach for improved acquisition of hyperpolarized 13C metabolic MRI. Magn. Reson Med. 84, 2943–2952 (2020).
Ayanzen, R. H. et al. Cerebral MR venography: normal anatomy and potential diagnostic pitfalls. AJNR Am. J. Neuroradiol. 21, 74–78 (2000).
Larson, P. E. Z. et al. Investigation of tumor hyperpolarized [1-13C]-pyruvate dynamics using time-resolved multiband RF excitation echo-planar MRSI: dynamic C13 MRSI of the TRAMP mouse. Magn. Reson. Med. 63, 582–591 (2010).
Keith, C. G. Intravenous urea in glaucoma. Br. J. Ophthalmol. 45, 307–311 (1961).
Javid, M. Effect of urea on cerebrospinal fluid pressure in human subjects: preliminary report. JAMA 160, 943 (1956).
Levin, A. B., Duff, T. A. & Javid, M. J. Treatment of increased intracranial pressure: a comparison of different hyperosmotic agents and the use of thiopental. Neurosurgery 5, 570–575 (1979).
Reeder, R. F. & Harbaugh, R. E. Administration of intravenous urea and normal saline for the treatment of hyponatremia in neurosurgical patients. J. Neurosurg. 70, 201–206 (1989).
Cho, A., Eskandari, R., Miloushev, V. Z. & Keshari, K. R. A non-synthetic approach to extending the lifetime of hyperpolarized molecules using D2O solvation. J. Magn. Reson. 295, 57–62 (2018).
Sweeney, M. D., Sagare, A. P. & Zlokovic, B. V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150 (2018).
Williams, D. S., Detre, J. A., Leigh, J. S. & Koretsky, A. P. Magnetic resonance imaging of perfusion using spin inversion of arterial water. Proc. Natl Acad. Sci. USA 89, 212–216 (1992).
Jackson, J. I., Meyer, C. H., Nishimura, D. G. & Macovski, A. Selection of a convolution function for Fourier inversion using gridding (computerised tomography application). IEEE Trans. Med. Imaging 10, 473–478 (1991).
Zhu, Z. et al. Coil combination methods for multi-channel hyperpolarized 13C imaging data from human studies. J. Magn. Reson. 301, 73–79 (2019).
Acknowledgements
This work was supported by NIH grants P41EB013598, P01CA118816, and S10OD025077.
Author information
Authors and Affiliations
Contributions
D.V. contributed to the conceptualization, experiment design, resources, and editing of the manuscript; H.C. contributed to experimental design and sequence development; T.N. and I.S. contributed to performing hyperpolarized 13C-urea MRI experiments; D.D. and J.S. contributed to developing SOP for the human hyperpolarized 13C-urea MRI experiments; C.W. and J.G. contributed to data analysis and manuscript editing; C.T.T., C.S., and S.M. contributed to developing the GMP grade [13C,15N2]urea; A.G. and R.C. contributed to developing urea sample formulation method for DNP and experiment; J.V.M., D.X., P.E.Z.L., J.K., R.B., and S.C. contributed to conceptualization and experiment design; Y.K. contributed to conceptualization, experiment design, experiment, analysis, and manuscript writing. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, Y., Chen, HY., Nickles, T. et al. Translation of hyperpolarized [13C,15N2]urea MRI for novel human brain perfusion studies. npj Imaging 3, 11 (2025). https://doi.org/10.1038/s44303-025-00073-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44303-025-00073-3
This article is cited by
-
The state of imaging glycolytic metabolism in cancer with magnetic resonance
npj Imaging (2026)
