
Abstract
Identifying plasma-based biomarkers that can accurately differentiate Lewy body disease (LBD) from Alzheimer’s disease (AD) remains a major challenge. Extracellular vesicles (EVs), which carry molecular cargo from their parent cells and can cross the blood-brain barrier, offer a new path forward. We developed the multiplexed Track-Etch magnetic NanoPOre (mTENPO) platform, a highly parallelized microfluidic technology for cell-specific EV isolation, and demonstrated independent enrichment of GluR2+ (neuron-derived) and GLAST+ (astrocyte-derived) EVs from the antemortem plasma of 137 autopsy-confirmed LBD, AD, mixed pathology, and control subjects. By integrating miRNA sequencing of GluR2+ and GLAST + EV cargo with plasma measurements of Aβ40, Aβ42, tau, p-Tau181, and p-Tau231, we identified a multimodal 15-feature panel that more comprehensively reflects brain pathology than conventional biomarkers. Using tenfold cross-validation to mitigate overfitting, the panel achieved an accuracy of 0.95 and an area under the curve of 0.96 for distinguishing LBD versus AD.
Similar content being viewed by others

Introduction
The clinical differentiation between Lewy body disease (LBD) and Alzheimer’s disease (AD), the two most prevalent histopathologies underlying neurodegenerative dementia1, remains a major challenge that affects patient outcomes and hinders therapeutic development. LBD and AD share overlapping clinical manifestations, despite distinct pathophysiological mechanisms2. LBD presents clinically as dementia with Lewy bodies (DLB), Parkinson’s disease (PD), or Parkinson’s disease dementia (PDD), and is histopathologically characterized by α-synuclein (α-syn) aggregates1, whereas AD is marked by amyloid β (Aβ) plaques3. While efforts have yielded promising biomarkers for diagnosing AD4,5,6, non-invasive biomarkers that are capable of accurately diagnosing LBD or differentiating between LBD and AD remain lacking1,7,8,9. Clinical diagnostic accuracies for LBD remain low, ranging from 34-89.5% when compared to associated gold-standard postmortem pathological diagnoses10,11,12,13, a challenge accentuated by the approximately 50% of LBD and AD patients who exhibit mixed pathology at death1,3,8.
The incorrect classification of neurodegenerative dementia can have a substantial impact on patient outcomes, such as adverse side effects in patients with LBD who receive anticholinergic and conventional antipsychotic medications2,11. Recently approved AD therapeutics, such as lecanemab and donanemab, are also more effective when administered early in the disease course14. Moreover, improved LBD and AD biomarkers can lower the cost and accelerate the pace of clinical trials by enriching the patient population for those most likely to benefit, which is critical given the current absence of disease-modifying therapies for LBD1.
The current standard of care for differentiating LBD versus AD primarily relies on (1) clinical assessment, which lacks specificity; (2) neuroimaging, which is not always accessible and has limited sensitivity/specificity8,15,16; or (3) cerebrospinal fluid (CSF) analysis, which requires invasive lumbar punctures5,17,18. By contrast, blood-based biomarkers are minimally invasive and more accessible. Numerous plasma biomarkers have shown potential in differentiating neurodegenerative dementias6,19,20,21,22. However, (1) prior studies rely on clinical diagnosis rather than postmortem pathology as the ground truth, limiting their accuracy19,21,22; (2) biomarkers that have shown efficacy in diagnosing dementia from controls show poor performance in classifying between different dementias8,9,23; and (3) many circulating biomarkers degrade rapidly in blood, and can be non-specifically released from non-neurological sources or comorbidities23.
Extracellular vesicles (EVs) offer a promising opportunity to address the limitations of conventional biomarkers for neurodegenerative disease. EVs not only carry surface proteins and nucleic acid cargoes reflecting their cells of origin24 but also are able to cross the blood-brain barrier (BBB)25, therefore representing a source for peripherally circulating brain-derived biomarkers whose changes reflect the molecular underpinnings of neurodegenerative disease. As mediators of cell-to-cell signaling, EVs are central to central nervous system (CNS) homeostasis and neural plasticity, which they maintain through the transfer of biomolecules, including proteins and miRNA25. In the context of neurodegenerative disease, aberrant signaling networks within parental cells can cause changes in EV-associated miRNA expression, whose transfer to recipient cells result in dysregulated post-transcriptional modification of gene expression24. Multiple studies have interrogated EV cargoes for LBD and AD diagnosis26; despite early promise, these studies have been limited by the absence of sufficiently specific enrichment for subpopulations of brain-specific EVs from the large background in blood (1010–1012 EVs/mL)27, relatively small cohort sizes (N < 50 total subjects), or a lack of postmortem pathology7,28,29,30.
We address these challenges through multimodal analysis of brain-derived EV miRNA cargo—separately profiling nanomagnetically isolated neuron- and astrocyte-derived EVs, together with conventional AD protein biomarkers (Aβ40, Aβ42, tau, p-Tau181, p-Tau231)6 from the antemortem plasma of 137 subjects with postmortem-confirmed LBD, AD, or mixed pathology, and controls. From this analysis, we identified a 15-feature multimodal biomarker panel that more comprehensively reflects brain pathology than conventional biomarkers and robustly distinguishes LBD from AD. A key feature of our approach is the ability to sensitively and specifically isolate multiple brain-derived EV subpopulations directly from plasma using our multiplexed Track-Etch magnetic NanoPOre (mTENPO) microfluidic EV sorting platform. mTENPO can achieve high-throughput, cell-specific EV isolation by quantitatively sorting EVs at the single-particle level based on immunomagnetic tagging of target surface proteins. In the presence of an applied magnetic field, magnetic traps are formed along the edges of nanopores within the mTENPO device’s ion track-etched permalloy-coated membranes. By massively parallelizing the magnetic sorting of individual EVs (through 107 nanopores/chip)27, mTENPO achieves clog-resistant high capture efficiency and throughput directly from complex biofluids with greater specificity than conventional EV immunoisolation approaches. We have previously demonstrated the device’s utility in classifying brain injury using a single EV subpopulation of neuron-derived EVs31,32,33,34, as well as reproducibility of our isolation technique within various cancers27,35,36. Here, we expand upon this prior work to isolate two EV subpopulations from each plasma sample, using GluR2 surface marker expression to enrich for neuron-derived EVs and GLAST surface marker expression to enrich for astrocyte-derived EVs, where GluR237,38,39,40,41 and GLAST42,43,44,45,46 expression within EVs derived from these two cell types have been extensively validated in the literature. Using a multiplexed analysis allows us to capture a more comprehensive state of the brain than possible with a single pulldown. This builds upon our recent study demonstrating this dual-pulldown approach in breast cancer diagnostics36, and represents the first use of precision nanoscale microfluidics to evaluate EV miRNAs from multiple brain cell types from the plasma of pathologically-confirmed neurodegenerative disease patients.
In this work, we combine mTENPO with next-generation miRNA sequencing and gold-standard digital enzyme-linked immunosorbent assay (ELISA) to: (1) identify differentially expressed biomarkers between disease cohorts across multiple modalities; (2) assess the degree to which each compartment of biomarkers (GluR2+ EVs, GLAST+ EVs, and proteins) yields complementary diagnostic information; (3) demonstrate associations between EV miRNA expression and neurodegenerative processes via pathway analysis; and (4) validate the hypothesis that a multimodal biomarker panel can improve dementia diagnosis, using cross-validation to mitigate overfitting. This work demonstrates a proof-of-concept approach toward developing non-invasive, accessible diagnostic tools for differentiating LBD and AD.
Results
Study design and isolation of neuron- and astrocyte-derived EVs
We profiled neuron- and astrocyte-derived EV miRNAs and circulating plasma proteins from 137 patients across five pathologically confirmed disease cohorts: LBD, AD, mixed AD and LBD (AD/LBD), AD with amygdala Lewy bodies (AD/ALB), and controls (Fig. 1A). Patient numbers per cohort and average ages for symptom onset, sample collection, and death, as well as additional demographic, clinical, and neuropathologic characteristics are summarized in Table 1. Histograms showing the distribution of ages for symptom onset, sample collection, and death are presented in Supplementary Fig. 1 for each of the five disease cohorts.

A Outline of study time-course and sample processing for EV isolation. Antemortem plasma samples with postmortem pathological confirmation of neurological diagnoses were processed to isolate cell-specific EVs using our mTENPO microfluidic platform, alongside plasma protein biomarkers using commercial digital ELISA, for patients with LBD (n = 30), AD (n = 31), AD/LBD (n = 30), AD/ALB (n = 19), and controls (n = 27). B The mTENPO platform, illustrating the external magnet, inlet reservoir, outlet ports, and tubing connections to syringe pumps. Syringes are connected to the waste outlet for blocking, washing, and sample addition steps, and then replaced and switched to the lysate outlet before captured EVs are lysed on-chip. The inset shows a photo of the mTENPO chip with a quarter for scale. C Schematic of operation of the mTENPO platform for cell-specific EV isolation using antibody-labeled magnetic nanoparticles (MNPs) for GluR2+ (top) and GLAST+ (bottom) EV pulldowns. D Scanning electron microscopy (SEM) images of GluR2+ EVs immobilized on the edges of pores of the mTENPO device’s surface. E SEM images of GLAST+ EVs immobilized on the edges of pores of the mTENPO device’s surface. F Representative cropped western blot images showing protein expression of GluR2, GLAST, and EV-associated marker TSG101 using mTENPO-isolated GluR2+ or GLAST + EV lysates from n = 2 human plasma samples. Full-length western blot images are shown in Supplementary Fig. 7.
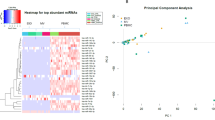
We compared demographic factors and clinical characteristics between the cohorts and did not observe statistically significant (P value < 0.05 from Chi-square test) differences between each of the five groups in education level or race (Supplementary Table 1). We did note statistically significant differences in the sex ratios of the groups, driven by the higher proportion of males (90%) in the LBD cohort and AD/ALB cohort (84.2%) as compared to the AD, AD/LBD, and control groups (Supplementary Table 1, Supplementary Table 2); however, LBD is shown in the literature to be more common in men47. Statistical comparison of differences using one-way analysis of variance (ANOVA) in mean age of symptom onset, mean age at time of sample collection, and mean age at death yielded no significant P value < 0.05 differences between the five groups (Supplementary Table 3). Principal component analyses (PCAs) were performed to further interrogate age and sex as confounders for miRNA expression data from all five cohorts for GluR2+ (Supplementary Fig. 2A) or GLAST+ (Supplementary Fig. 2B) EVs, isolated using the mTENPO platform as described below. This analysis did not reveal associations with either age at sample collection, as evaluated using Pearson correlation (GluR2 PC1 versus age, r = -0.042, P value = 0.629; GluR2 PC2 versus age, r = 0.083, P value = 0.332; GLAST PC1 versus age, r = –0.051, P value = 0.554; GLAST PC2 versus age, r = 0.092, P value = 0.284), or sex, as evaluated using the Mann–Whitney U test (GluR2 PC1 versus sex, P value = 0.424; GluR2 PC2 versus sex, P value = 0.850; GLAST PC1 versus sex, P value = 0.228; GLAST PC2 versus sex, P value = 0.200).
For the enrichment of neuron- and astrocyte-derived EVs from patient plasma, we developed and performed an mTENPO EV isolation protocol using GluR2 and GLAST antibodies. The mTENPO platform consists of a chip that houses three layers of track-etched film with d = 3 μm pores, coated with permalloy (200 nm Ni80Fe20) and a 30 nm Au passivation layer (Fig. 1B)27. The chip sits in a uniform magnetic field (B0) provided by a small external NdFeB magnet (d = 1.5 inches, h = 0.75 inches)35. As plasma is pulled through the mTENPO device, only EVs that are sufficiently magnetized (due to high degree of GluR2 or GLAST surface expression) are pulled to the edges of pores and retained on-chip, as these EVs have sufficient magnetophoretic force (Fm) from bound magnetic nanoparticles (MNPs) to counteract the strong drag forces (Fd) from fluid flow (Fig. 1C)27. Non-specific EV binding to mTENPO surfaces is mitigated through (1) the design of the chip, which minimizes the possibility of clogs and dead volume trapping non-target-EV background; (2) the flow rates used in our protocol, which maximize capture of strongly-tagged EVs and minimize capture of weakly-labeled EVs; and (3) the pre-blocking of mTENPO chips with Pluronic F-127 to minimize non-specific adsorption. Optimization of these parameters and characterization of EV enrichment were reported in Lin, A.A. et al., Sci. Rep., 202327.
To characterize these mTENPO-captured EVs, we performed scanning electron microscopy (SEM) on EV-MNP complexes immobilized on-chip; we also performed SEM, helium ion microscopy (HIM), and cryo-transmission electron microscopy (cryo-TEM) for EV-MNP complexes eluted off-chip to confirm that our immunomagnetic labeling was binding EVs to MNPs. For SEM, we identified particles with consistent size and morphology as EVs within pores of our mTENPO devices (Fig. 1D, E, Supplementary Fig. 3). We also identified eluted EVs on SEM, HIM, and cryo-TEM bound to MNPs approximately 50 nm in size (Supplementary Fig. 4, Supplementary Fig. 5). Eluted EVs were within the typical EV size range, although EVs appeared larger when imaged by cryo-TEM compared to SEM/HIM, which is consistent with the literature48. To compare the GluR2 and GLAST pulldowns, we performed nanoparticle tracking analysis (NTA) on eluted EVs from mTENPO and found no differences in EV concentration or size distribution for either pulldown (Supplementary Fig. 6).
We have previously shown that western blotting on GluR2 pulldown protein lysates from mouse plasma displays positive expression of GluR2 and EV markers TSG101, Alix, and CD931. To build upon this work and further validate the specificity of our GluR2 and GLAST EV isolations, western blotting was used to confirm protein expression of GluR2, GLAST, and EV marker TSG101 within EV protein lysates from mTENPO pulldowns for n = 4 human plasma samples, with representative images for n = 2 samples depicted in Fig. 1F. Full-length images of the blots presented in Fig. 1F are pictured in Supplementary Fig. 7A–C. GluR2 mTENPO EV lysates showed a substantial increase in GluR2 western blot signal relative to GLAST (Supplementary Fig. 7D); likewise, GLAST mTENPO EV lysates showed increased GLAST signal relative to GluR2 (Supplementary Fig. 7E). Both pulldowns showed positivity for extracellular vesicle marker TSG101 (Supplementary Fig. 7C).
We confirmed the specificity of the GluR2+ EV and GLAST + EV-derived miRNA signals by finding ΔCq values > 0 in the expression of multiple brain-relevant miRNAs, when comparing EVs isolated from plasma using GluR2 or GLAST versus isotype controls (Supplementary Fig. 8). A commercially available panel of CSF markers (Qiagen) was used for this assay, as it provided a readily accessible measure for CNS-specific RNA cargo. The mean Cq across replicates was taken for each individual panel marker, and the resulting values were compared across antibody and isotype pulldowns using a one-tailed two-sample Mann–Whitney U test, showing a statistically significant difference across the panel for both GluR2 versus isotype (P value = 0.0027) and GLAST versus isotype (P value = 0.0022).
Lastly, to validate that the GluR2 pulldown isolates neuronal EV signals and the GLAST pulldown isolates astrocytic EV signals, we performed mTENPO isolation on 500 µL of conditioned media from mixed neuron cell cultures (neuron and astrocyte co-culture) or astrocyte-only cell cultures derived from rat neocortical tissue. Within these cultures, it was necessary to co-culture neurons and astrocytes for our mixed neuron cell culture model to improve neuronal viability. The conditioned media used within these experiments contain EVs from well-defined originating cell types, and after input into mTENPO, resulted in selective enrichment of EVs from the corresponding cell type for each antibody. NTA was performed on EVs eluted off-chip, showing additional EV-sized particles isolated using GluR2 mTENPO from mixed neuron media that were not present when performing GLAST mTENPO on the same sample (Supplementary Fig. 9A). Similarly, for astrocyte-only media, NTA showed additional EV-sized particles for GLAST mTENPO that were not present for GluR2 mTENPO (Supplementary Fig. 9B). Furthermore, NTA for GLAST mTENPO on mixed neuron media and GluR2 mTENPO on astrocyte-only media showed similar size distributions to bead-only NTA measurements (Supplementary Fig. 9C), suggesting that the particles detected for these two conditions may be primarily from beads that passed into the eluate, with a rightward shift of the NTA peak for the former, presumably due to the presence of astrocytes within the mixed cultures.
Neuron- and astrocyte-derived EV miRNAs and plasma proteins are differentially expressed between LBD and AD
The mTENPO-isolated neuron and astrocyte EV miRNAs alongside the plasma protein measurements defined a biomarker expression profile for each patient in this study. In a comparison of subjects with LBD (n = 30) versus those with AD (n = 31), in which each subject did not exhibit any degree of mixed postmortem pathology of the two diseases, we observed multiple nucleic acid and protein biomarkers with significant differential expression after Benjamini-Hochberg false discovery rate (FDR) correction (FDR-corrected P value < 0.1). To visualize the spread of miRNA and protein expression across our LBD and AD cohorts, we performed hierarchical clustering of the z-score of log2(expression) data between patients (Fig. 2A). We identified 16 differentially expressed miRNAs from GluR2+ EVs (11 upregulated, five downregulated), eight differentially expressed miRNAs from GLAST+ EVs (seven upregulated, one downregulated), and four differentially expressed plasma protein biomarkers, including Aβ42/Aβ40 ratio (one upregulated, three downregulated) (Fig. 2B).

A Heatmap of z-score of log2(expression) for biomarkers with Benjamini-Hochberg FDR-corrected P value < 0.1. Subjects (columns) are hierarchically clustered within cohort and biomarkers within each compartment (rows) are sorted by descending fold-change. B Volcano plot demonstrating differential expression of GluR2+ EV miRNAs, GLAST + EV miRNAs, and plasma proteins. C Venn diagram showing overlap in FDR P value significant miRNAs (P value < 0.1) between GluR2+ EVs and GLAST+ EVs. D Top 30 biomarkers in all compartments ranked by descending area under the curve (AUC). Error bars represent standard error from bootstrapping 10x.
To assess the extent to which the GluR2 versus GLAST compartments yielded complementary biological information, making them suitable for integration into a biomarker panel35,36, we identified the overlap between differentially expressed miRNA markers from each pulldown. There were six overlapping markers, each of which exhibited consistent upregulation or downregulation within both compartments: miR-144-3p, miR-144-5p, miR-4732-5p, miR-486-5p, and miR-451a were upregulated in both compartments; and miR-1299 was downregulated in both compartments (Fig. 2C). We also identified a combined 12 differentially expressed markers (10 in GluR2, two in GLAST) unique to a single pulldown, highlighting both shared and distinct molecular signatures among the different EV subtypes.
We assessed the capability of each individual nucleic acid and protein marker to classify LBD versus AD by performing a receiver operating characteristic (ROC) analysis and reporting the area under the curve (AUC); the AUCs for the top 30 biomarkers are shown in Fig. 2D. A total of five markers in the top 30 markers ranked by AUC was both statistically significant for differential expression after FDR correction and had a significant AUC compared to a non-predictive (AUC = 0.5) marker using Delong’s test. Of these markers, GLAST + EV miR-203a-3p had the greatest AUC at 0.76 (Delong’s P value = 0.0094), followed by p-Tau231 (AUC = 0.73, Delong’s P value = 0.0048), GluR2+ EV miR-144-5p (AUC = 0.72, Delong’s P value = 0.026), GluR2+ EV miR-144-3p (AUC = 0.71, Delong’s P value = 0.045), and p-Tau181 (AUC = 0.69, Delong’s P value = 0.039).
The miRNA and protein profiles revealed complex patterns of expression, with outliers and heterogeneity within disease cohorts, even for strongly predictive markers (Supplementary Fig. 10). For upregulated markers, subclusters of patients exhibited high expression for both GluR2 and GLAST pulldowns (Fig. 2A). Some downregulated GluR2 markers (miR-490-3p, miR-34a-5p, miR-1299, miR-9-3p) appeared to be influenced by outlier expression profiles in a few patients, resulting in correspondingly low AUCs (AUCs of 0.58, 0.55, 0.54 and 0.53, respectively) (Fig. 2D, Supplementary Fig. 10A). A single outlier patient within the AD cohort had high expression in the Aβ42/Aβ40 ratio, driving overall downregulation of Aβ42/Aβ40 for LBD versus AD, and yielding a nonsignificant AUC (AUC = 0.68, Delong’s P value = 0.12). ROC curves for the Delong’s-significant differentially expressed markers are shown in Supplementary Fig. 11A-C. We also performed a comparison of the AUCs for the top 30 miRNA markers between the GluR2 and GLAST pulldowns to assess their degree of correlation, identifying that the pulldowns did not yield significant correlation for these miRNAs (Supplementary Fig. 11D).
Dementia and control subjects exhibit distinct miRNA and protein profiles
We compared the biomarker profiles of patients with pathologically defined dementia to controls using three comparisons: (1) a comparison of combined dementias (n = 30 LBD, n = 31 AD) versus n = 27 controls, (2) a comparison of n = 31 AD patients versus n = 27 controls, and (3) a comparison of n = 30 LBD patients versus n = 27 controls. As performed previously for LBD versus AD, we generated hierarchically clustered heatmaps of the z-score of log2(expression) between patients for all three comparisons (Supplementary Fig. 12).
For LBD and AD versus control, we identified seven differentially expressed miRNAs (two GluR2, five GLAST, with one overlapping miRNA between the pulldowns) and two differentially expressed protein biomarkers (Aβ42 and Aβ42/Aβ40). Of these biomarkers, three GLAST + EV miRNAs and one protein were upregulated in the combined dementia cohort, while the remaining biomarkers were upregulated in the control cohort (Supplementary Fig. 13A–D). By performing an ROC analysis on each biomarker with a statistically significant fold-change (Supplementary Fig. 13E, F), we identified one GLAST + EV miRNA (miR-204-5p, AUC = 0.67, Delong’s P value = 0.062) and two protein biomarkers (Aβ42/Aβ40, AUC = 0.67, Delong’s P value = 0.051; Aβ42, AUC = 0.66, Delong’s P value = 0.045) with a significant AUC as compared to a non-predictive (AUC = 0.5) marker using Delong’s test, for which Aβ42/Aβ40 featured the highest classifying AUC (Supplementary Fig. 13G). miR-204-5p has been previously identified as a dementia biomarker in CSF; however, it was downregulated in AD patients versus controls49, whereas it is upregulated in the combined LBD and AD cohort in this study.
In our comparison of AD versus control, we identified 13 miRNAs (11 GluR2, two GLAST, with one overlapping miRNA between the pulldowns) and three proteins (p-Tau181, Aβ42, and Aβ42/Aβ40) with statistically significant differences between AD versus control. Within these biomarkers, eight miRNAs (seven from GluR2, one from GLAST), Aβ42/Aβ40, and p-Tau181 were upregulated in the AD cohort; the remaining biomarkers were upregulated in the control cohort (Supplementary Fig. 14A–D). ROC analysis of these biomarkers yielded four biomarkers (GluR2+ EV miR-132-3p alongside proteins p-Tau181, Aβ42, and Aβ42/Aβ40) with statistically significant AUCs per Delong’s test (Delong’s P value = 0.0039, 0.027, 0.017, and 0.018, respectively), with miR-132-3p having the highest AUC of the group at 0.76 (Supplementary Fig. 14E–G). Aβ42 was upregulated in controls in a manner consistent with the literature19,50. p-Tau181 was also upregulated in AD versus controls, similar to what has been previously reported in the literature4. While overall Aβ42/Aβ40 expression increased for both the combined dementia versus control and AD versus control comparisons, which runs contrary to the literature19,50, this was largely driven by one outlier, as median Aβ42/Aβ40 expression for the combined dementia and AD cohort were lower than for the control group.
Our comparison of LBD versus control yielded only two differentially expressed biomarkers (p-Tau231 and GLAST + EV miR-200b-3p) (Supplementary Fig. 15A–C), and only p-Tau231 had a statistically significant AUC at 0.69 (Delong’s P value = 0.020) (Supplementary Fig. 15D, E). The limited number of differentially expressed markers between LBD versus control is consistent with the documented lack of diagnostic biomarkers for LBD to date.
EV miRNA enrichment analysis shows associations to neurodegenerative processes
To determine whether the identified biomarkers relate to known pathways in the biology of LBD and AD, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses on the union of differentially expressed miRNAs from each individual pulldown. We pinpointed the top 10 terms per GO category [biological process (BP), cellular component (CC), and molecular function (MF)] and top 10 KEGG pathways by the number of target genes affected by each set of GluR2+ (Fig. 3A, B) or GLAST+ (Fig. 3C, D) EV miRNAs with FDR P value < 0.05. Additional significantly enriched (FDR P value < 0.05) GO BP terms associated with brain-related processes and neuroinflammation are shown in Supplementary Table 4 and significantly enriched (FDR P value < 0.05) KEGG pathways associated with the nervous system, neuron development, and brain-specific diseases are shown in Supplementary Table 5. Within the GluR2 pulldown, a maximum of 14 miRNAs were involved in identified GO terms or KEGG pathways. Within the GLAST pulldown, our GO and KEGG analyses identified terms involving a maximum of seven miRNAs.

GO and KEGG pathway analyses were performed on differentially expressed miRNAs using DIANA miRPath v4.0 using the TarBase v8.0 database. FDR P values for identified GO terms and KEGG pathways were calculated using a one-sided Fisher’s exact test and considered significant at P value < 0.05. The top 10 (ranked by number of target genes) terms within each of the three GO categories (BP, CC, MF) and top 10 (ranked by number of target genes) KEGG pathways were identified for each pulldown. A Top 10 terms within each GO category for GluR2+ EV miRNAs. B Top 10 KEGG pathways for GluR2+ EV miRNAs. C Top 10 terms within each GO category for GLAST + EV miRNAs. D Top 10 KEGG pathways for GLAST + EV miRNAs. In all panels, each bar is labeled to the right with the number of differentially expressed miRNAs associated with the given GO term or KEGG pathway.
The GO enrichment analysis identified terms such as “cell cycle” and “regulation of transcription” (GO BP) that are shown to be dysregulated in studies concerning general aging or other age-related pathologies such as cancer51. Of note, “negative regulation of apoptotic process” only appeared as a BP term within the GLAST pulldown. While LBD and AD are marked by neuronal cell death52, they are also associated with upregulation in both astrocytic pro-inflammatory or neuroprotective, proliferative states53.
The KEGG analysis revealed significant enrichment in multiple neurodegenerative pathways and diseases. The MAPK signaling and PI3K-Akt pathways, which appeared for both pulldowns, are known regulators of synaptic plasticity and neuronal survival54,55. Additionally, “endocytosis”, which appeared as a GluR2 KEGG-enriched pathway, is consistent with our understanding of EV uptake and spread of pathological proteins in neurodegeneration24,25. Lastly, “regulation of actin cytoskeleton”, enriched within the GLAST pulldown, maintains normal synaptic function and is impaired in both LBD and AD56.
miRNAs control gene expression by binding to target mRNAs; therefore, the examination of gene targets influenced by differential miRNA expression can demonstrate the functional implications of miRNA dysregulation in driving disease states. The top 10 enriched KEGG pathways showed the greatest interaction between differentially expressed GluR2+ EV miRNAs with target genes IGF1R (inhibition of which is linked to Aβ accumulation and tau phosphorylation)57, KIF5B (inhibition of which causes memory deficits in mice)58, and MAPK1 (which influences a wide network of genes affected in LBD and AD)59 (Supplementary Fig. 16A). For differentially expressed GLAST + EV miRNAs, KEGG pathways showed the greatest interaction with apoptosis regulators BCL260 and MYC61, along with PTK2, which controls tau-induced neurotoxicity62 (Supplementary Fig. 16B).
Feature selection identifies multimodal panels for differentiating LBD from AD
We used least absolute shrinkage and selection operator (LASSO) feature selection to identify candidate miRNAs and proteins which were most informative for classifying LBD versus AD as combined panels. For input into LASSO, we used all neuron- and astrocyte-derived EV miRNAs which cleared the expression threshold for the Qiagen RNA sequencing analysis pathway to generate an FDR-corrected P value. Similarly, we included all plasma proteins that generated an FDR-corrected P value.
In sweeping the number of selected biomarkers from two to 20, we generated a composite list of biomarkers including 14 GluR2+ EV miRNAs, four GLAST + EV miRNAs, p-Tau231, and p-Tau181 (Table 2), with many of the LASSO-selected markers having been identified by previous literature concerning neurological conditions (Supplementary Table 6). As with our previous analyses, we generated a heatmap of the z-score of log2(expression) of LASSO-selected biomarkers with hierarchically clustered patients (Fig. 4A). We also performed a pairwise Kendall tau correlation analysis between each of the biomarkers (Fig. 4B), identifying only limited correlation. While several biomarkers did not feature statistically significant differential expression between LBD versus AD, the top 14 biomarkers ranked by AUC, in addition to GluR2+ EV hsa-miR-4748, were all found to have AUCs significantly above 0.5 per Delong’s test.

A Heatmap of z-score of log2(expression) for LASSO-selected biomarkers. Subjects (columns) are hierarchically clustered within cohort and biomarkers within each compartment (rows) are sorted by descending AUC. B Kendall correlation staircase plots identifying the extent to which biomarker information was correlated between the LASSO-selected GluR2+ EV, GLAST + EV, and protein biomarkers. Biomarkers are sorted within compartments by AUC. The inset shows the correlation distribution of Kendall’s τ, where the dotted line represents the median count. C LASSO panel accuracy versus panel size for classifying LBD versus AD, shown in blue; accuracy is assessed through tenfold cross-validation, with error bars representing standard error from 5 repeats of panel training on the LBD vs AD patient groups. Average accuracy and standard error for control experiments performed by scrambling patient labels 10x are shown in orange. D LASSO panel AUC versus panel size for classifying LBD versus AD, shown in blue with error bars as described in (C). Average AUC and standard error for the same control experiments described in (C) are shown in orange. E AUCs for the 15-marker LASSO panel and individual LASSO biomarkers, sorted by descending AUC. Error bars represent 95% confidence intervals, calculated from 5x repeats of tenfold cross-validation for the 15-marker panel or from bootstrapping 10x for individual markers.
To assess the combined performance of biomarkers within each panel, we used an ensemble model on the entire cohort of LBD versus AD patients with tenfold cross-validation repeated 5x to mitigate overfitting35,63, displaying a plateau in panel accuracy for a panel size of 15 (accuracy = 0.95) (Fig. 4C), at which the panel AUC = 0.96 (Fig. 4D). The ROC curve for the combined 15-marker panel shows that the panel greatly outperformed each individual LASSO-selected biomarker (Supplementary Fig. 17), showing an improvement in AUC of 0.21 over the best performing individual LASSO biomarker, and non-overlapping 95% confidence intervals between the 15-marker panel and any of the individual LASSO biomarkers (Fig. 4E).
To confirm whether we had included a sufficient number of patients necessary to train our model, we then generated a learning curve by evaluating the model’s performance using different sizes of training data35 with 100 bootstraps64,65,66 per size (Supplementary Fig. 18). The learning curve stabilized and showed a plateau in AUC and accuracy near the maximum possible size of the training set, corresponding to the entire combined LBD and AD cohorts. This suggests that our cohort size was sufficient for discrimination but also indicates that inclusion of the full cohort was necessary to reach optimal model performance.
Features within the 15-marker panel were assessed for contributions to model discrimination using (1) permutation feature importance (Supplementary Fig. 19), which quantifies how perturbing each of the features independently degrades model AUC and accuracy64; and (2) Shapley Additive exPlanations (SHAP) analyses (Supplementary Fig. 20), which measures the contribution of each feature to model predictions67. We found good agreement between permutation feature importance and SHAP value rankings; the highest ranked markers were GluR2+ EV miR-4748, GLAST + EV miR-148b-5p, and p-Tau231 in all three feature importance rankings: permutation feature importance by change in AUC (Supplementary Fig. 19A) and accuracy (Supplementary Fig. 19B), as well as SHAP value (Supplementary Fig. 20A).
Notably, although the order of LASSO selection closely followed univariate AUC rankings, feature importance analyses found that the features with the highest impacts on model performance and predictions within the multivariate model were not always those with the highest individual predictive power. Moreover, perturbing individual features within the 15-marker panel led to relatively small changes in performance, with means ranging from –0.0006 to 0.059 for decrease in AUC and 0.0087 to 0.085 for accuracy. This indicated that no single feature disproportionally drove model performance, which is supported by the SHAP analysis. Only one feature showed a negative decrease in AUC when perturbed (GluR2+ EV miR-200c-3p, decrease in AUC = –0.0006) (Supplementary Fig. 19A) although the magnitude of this change was small and indicative of only a marginal improvement in performance.
Feature stability was then examined by generating a Spearman’s rank correlation matrix68,69 of SHAP-based feature rankings (Supplementary Fig. 20B). Stability of ranking order was assessed using SHAP feature rankings rather than permutation feature importance rankings, as SHAP values measure feature contribution rather than performance drop. The resulting distribution of pairwise rank correlations exhibited a median Spearman’s correlation coefficient of 0.57, indicating moderate agreement in relative feature importance across folds and repeats of the 5x repeated tenfold cross-validation framework. This suggests that, though there was some expected variability in feature rankings due to fold splits, top-ranked features consistently and meaningfully contributed to model predictions across folds and repeats in a manner above chance.
We compared the model’s performance to control experiments in which LASSO was implemented with randomly scrambled patient labels. The resulting panels were assessed for diagnostic utility in categorizing patients based on their true labels. This was repeated 10 times with the average accuracy and AUC falling below 0.6 for all panel sizes (Fig. 4C, D). At all panel sizes, the cross-validated panel AUCs were also significantly higher than for their associated scrambled controls (P value < 0.001 using the Mann–Whitney U test).
We then examined the relative expression levels of LASSO-selected biomarkers in mixed-pathology patients with histopathologic diagnoses of AD/LBD (n = 30) or AD/ALB (n = 19) as compared to LBD and AD patients (Supplementary Fig. 21). We observed that the AD/LBD and AD/ALB patient cohorts generally featured median biomarker expression levels in between the LBD and AD groups, which is consistent with the status of AD/LBD and AD/ALB as mixed pathologies. For most miRNA biomarkers we examined, we did not observe large differences between the AD/LBD and AD/ALB cohorts; however, in both LASSO-selected protein biomarkers p-Tau181 and p-Tau231, we observed higher expression of these proteins in AD/ALB versus AD/LBD patients.
Finally, we performed an analysis of confidence interval precision to determine the necessary sample size for a future independent validation cohort using a method proposed by Grolleau, F. et al., AMIA Jt. Summits Transl. Sci. Proc., 202570. Assuming a more modest model performance within the validation cohort of AUC = 0.9 and an event prevalence of 0.5, we determined that at least an additional n = 158 subjects within our independent validation set would be necessary to achieve a 95% confidence interval width of ≤0.1 (Supplementary Fig. 22).
Discussion
While clarifying the role of EV cargoes within LBD and AD remains an ongoing process, this study identifies potential biomarkers that support the hypothesis that mTENPO enables enrichment of brain-derived EVs which are linked to signaling changes associated with neurodegeneration. Recent studies have supported the pathogenic involvement of EVs in LBD and AD, finding that β-amyloid plaques within the AD brain are interdigitated by EVs71, suggesting that they participate in plaque formation, and EVs may propagate neurotoxic Aβ, tau, and α-syn between astrocytes and neurons, leading to increased neuronal death and proximal microglial activation25,26. Astrocyte-derived EVs that cross the BBB can also increase neuroinflammation through peripheral leukocytic recruitment into the brain25. However, EVs may still retain some role in neuroprotection within the context of dementia; in the AD brain, EV-bound PrPc preferentially binds Aβ and promotes its fibrillization and decreased toxicity25. In our study, neuron- and astrocyte-derived EV miRNAs revealed unique associations with GO terms and KEGG pathways, indicating that altered signaling networks may be cell-specific. There is a complex interplay between cell types within the pathological brain; as the molecular content of EVs is influenced by their originating cell’s pathological state24, our ability to capture signal from these two specific cell types using mTENPO allows us to restrict our view to biomarkers that are specific to the disease and may be involved these pathways, minimizing background noise that may be present from other EVs in the blood.
The heterogeneity in individual miRNAs and proteins across our patient cohorts underscores the difficulty of relying on a single marker for differential diagnosis, as shown by the modest AUCs for individual miRNAs. In contrast, the integration of a minimally correlated multi-marker panel selected by LASSO from all three compartments reveals an identifiable pattern of differential expression, alongside clinically relevant (AUC > 0.8) AUCs for all panel sizes of at least three biomarkers and high AUCs (AUC > 0.9) for panel sizes >10. To date, even emerging AD biomarkers such as p-tau217 have only achieved an AUC of 0.84 in distinguishing AD for patients with early-onset/atypical dementia16. Similarly, α-syn seed amplification assays (SAAs), which have been used for LBD diagnosis, have primarily achieved success in CSF, brain tissue homogenates, and skin17,72. While preliminary SAA results from the serum of non-autopsy-confirmed subjects showed good performance for PD versus controls (AUC = 0.96 in the initial cohort; AUC = 0.86 in a blinded independent cohort), 4 of 25 AD subjects were notably SAA-positive73.
This study utilizes exceptionally valuable biospecimens which can only be obtained postmortem; to mitigate this constraint, the AUCs and accuracies in our approach were generated using a robust cross-validation strategy. Because of the rarity of these specific samples, reserving a portion of the cohort as a held-out test set would have left too few instances for both model training and evaluation, resulting in less reliable and statistically meaningful performance estimates. Although the absence of a separate validation set is a limitation of this study, and a clear next step in future work, repeated tenfold cross-validation has been shown to be a low bias74 estimator of performance that can approximate independent validation75,76. By combining this method with LASSO, we were able to perform an internal assessment of model performance while minimizing overfitting and maximizing our available sample size for biomarker discovery.
We also noted minimal impact of perturbing any individual LASSO-selected marker on overall panel AUC and found positive ranking stability of features across resampled data sets, demonstrating that our panel is not being purely driven by noise. In the presence of repeated cross-validation, these results provide evidence that feature contributions are reproducible rather than overfitting to specific splits of the data. Despite this, we recognize the need for future validation experiments with an independent pathology-confirmed cohort to ensure the robustness and generalizability of our panel. The size of our panel may also help facilitate reproducibility across larger cohorts, mitigating the variability inherent in dementia populations. Although panels of n > 10 markers may seem considerable, the ability to measure large biomarker panels has become more accessible with the advancement of technologies enabling multiplexed nucleic acid and protein quantification, which may be employed in future studies.
To date, histopathology remains the gold standard for the diagnosis of LBD, AD, and mixed-pathologies3,77. The inability to reliably detect non-AD or mixed pathologies prior to death remains an obstacle in designing effective clinical trials, particularly given that postmortem studies indicate 35–50% of PD patients and >70% of DLB patients exhibit AD co-pathology77. Clinically, AD/LBD and AD/ALB bear close symptomatic resemblance to pure AD patients in advanced stages52. Existing single LBD and AD biomarkers, like SAAs and p-tau217, may not be effective in detecting these mixed pathology cases; SAAs may still produce a positive result for some patients with Lewy bodies that do not meet neuropathological criteria for LBD17,18 and p-Tau217 is unable to predict presence or absence of LBD co-pathology in AD patients4. Likewise, imaging biomarkers such as 18F-fluorodeoxyglucose-positron emission tomography (18F-FDG PET) have shown limited performance in distinguishing between AD with concomitant amygdala-predominant versus limbic/neocortical Lewy bodies78.
In our pathology-confirmed patient cohort, select miRNAs showed identifiable trends in median expression level associated with degree of Lewy pathology. At the same time, we also observed some overlap in individual LASSO-selected biomarker profiles between mixed and pure-pathology cohorts. These findings, alongside the lack of reliable single biomarkers for detection of mixed pathology thus far, underscore the benefits of using larger multimodal panels to increase the likelihood of stratifying varying Lewy body and AD co-pathology. This remains a critical challenge, as not knowing the true state of enrolled participants would affect the design of clinical trials in two ways: (1) in clinical trials aimed at targeting a single disease, the estimated rates of decline and survival times would be inappropriate for patients with hidden co-pathology, impacting measures of trial success79 and (2) because the anticipated mechanisms of decline would be improperly characterized, there is the potential for drug inefficacy or toxicity induced by off-target activity80. Conversely, an attempt to exclude mixed pathology patients could result in the development of therapeutics that may not work on the general population. Future studies involving patients along the mixed AD and LBD pathological spectrum will need to identify means of understanding the biology driving the disease process to better characterize the “ground truth” for determining differentially expressed biomarkers between these populations.
There are several limitations of our study and additional opportunities for future work. First, it is important to note that our biomarker measurements are collected at a time point separated by years from the postmortem pathology. While detectable cognitive changes were present in our patient populations, we cannot determine the precise point at which each patient was located within the course of their disease. This uncertainty could have affected subjects with mixed pathology (AD/LBD and AD/ALB), as patients may not have had both diseases at sample draw or may have lacked sufficient core clinical features to detect one or both diseases. Each patient had a recorded clinical diagnosis at the time of visit; however, given the number of subjects with a diagnosis of mild cognitive impairment (MCI), the potential for non-AD diagnoses including MCI to evolve into AD, and the lack of knowledge of when patient MCI evolved into frank dementia, we did not attempt to directly correlate accuracy of clinical diagnosis versus histopathologic diagnosis. Despite this limitation, we observed promising predictive performance within our biomarker panels, motivating future longitudinal studies aimed at capturing EV-derived signals across multiple stages of disease progression to better define the exact point of disease onset. This would also enable evaluation of the generalizability of our panel to subjects with early-onset disease, who may present with atypical symptoms and rates of decline81,82,83.
Second, given the sample type (plasma collected years before death for patients with pathology-proven subtypes of dementia), our sample size, subject diversity, and sample volumes were limited. Because our plasma samples were collected as part of the Mayo Clinic Alzheimer’s Disease Research Center (ADRC) study or Mayo Clinic Study of Aging (MCSA), our subjects reflect the predominantly white population in the upper Midwest. Likewise, we had a high percentage of males within both LBD and AD/ALB cohorts; while this is consistent with the literature for LBD47,84, for AD/ALB, this discrepancy may have due to the limited cohort size. Studies have shown that both race85 and sex differences47,84 may play important roles in disease progression, clinical presentation, and survival of LBD, AD, and mixed pathology patients; thus, further studies with greater diversity and balanced sex from multiple collection centers are needed to validate these markers within the general population. Additionally, since our study focuses on detecting biomarkers within a complex aging population, many of the subjects had concomitant non-LBD and non-AD pathologies, including vascular dementia, primary age-related tauopathy, and age-related degeneration, that may have also driven miRNA and protein expression.
Finally, there are limitations to the definition of any cell-specific EV marker. Although GluR2 and GLAST are well-defined EV surface proteins that are predominantly associated with neuron- and astrocyte-derived EVs, we acknowledge that cellular origin cannot be strictly attributed to expression of a single surface protein86. Characterization of brain-derived EVs is ongoing, and brain-derived EVs may exhibit some degree of co-expression of these proteins87. Furthermore, GluR2 and GLAST can be expressed in cells outside the brain; GluR2 has been found in pancreatic beta cells88, in the inner ear89, and within soft tissue tumors90, whereas GLAST has been found on osteocytes91, within dorsal root ganglia and the sciatic nerve92, and in epithelial cells of peripheral organs93. Nonetheless, our methodology focuses on the enrichment of brain-derived signals given GluR2 and GLAST’s highly elevated expression profile within the brain, acknowledging that this may not entirely exclude some lower-level peripheral signals94 and that even with rigorous efforts to minimize non-specific binding27, residual background noise is unavoidable in any EV isolation technique86,94,95. Subsequent studies employing multi-marker tagging approaches for greater specificity may help clarify cellular origin of isolated EV subpopulations with better precision.
Future work aims to address these limitations by using the set of biomarkers identified in this study in multi-omic and multi-center clinical studies to survey neurodegenerative dementia across more diverse patient populations. This study establishes a multiplexed EV-based diagnostic framework enabled by mTENPO’s high throughput and specificity. By doing so, we also identify a set of candidate biomarkers which could be interrogated in cellular models of neurons and astrocytes to further the mechanistic understanding of the biology of LBD and AD, and can be adapted to include additional cell types. While in this work we primarily focused on miRNA cargo within GluR2+ and GLAST+ EVs, we have previously demonstrated mTENPO’s ability to be used for measurement of cell-specific EV protein cargo when combined with Quanterix SIMOA32. Looking ahead, the use of mTENPO for neurodegenerative disease diagnostics can be expanded beyond miRNA EV signatures to include these additional molecular cargoes, which can be used to quantify seeding-competent or cytotoxic proteins and provide a more comprehensive characterization of disease impact on EV content.
The mTENPO platform is being commercialized, and through a startup company, a fluid handling system was built to automate mTENPO isolation and minimize user variability (Supplementary Fig. 23). This automation is designed to enable clinical implementation with improved reproducibility. Combining this automated platform with our predictive biomarker panel offers a scalable solution that can be integrated into standard diagnostic workflows. Given the accelerating pace of the development of therapeutics for neurodegenerative dementia, this work articulates a multimodal and biomarker-driven approach that aims to provide greater diagnostic clarity for clinicians and patients and further the understanding of these complex diseases.
Methods
Study population and sample collection
Participants were recruited as part of the ADRC or MCSA between 2004 and 2019. Subjects selected for this study were chosen based upon postmortem pathology for LBD (n = 30)11, AD (n = 31)96,97, AD/LBD (n = 30), AD/ALB (n = 19)98, or absence of AD and Lewy body pathology (controls) (n = 27). Blood samples were collected during multiple visits for most patients. The sample selected for this study was (1) defined as early in the course of the disease (preferably within the initial 1–2 years of participation), (2) one for which sufficient plasma was available, and (3) taken from a patient for which autopsy was performed. In all cases, and regardless of age, plasma samples were selected for patients along their clinical progression early in their course when they were in the mild cognitive impairment [global Clinical Dementia Rating (CDR)99 = 0.5] or mild dementia (global CDR = 1) stage. Age at symptom onset was consistent with previous studies on incidence of cognitive impairment and dementia for LBD and AD81,100,101. For controls, samples were selected using the same criteria with preference being within the initial 1–2 years of participation. For selected visits, ethylenediamine tetraacetic acid (EDTA) plasma samples were collected following an overnight fast. Samples were centrifuged, and plasma was aliquoted into polypropylene tubes and stored at −80 °C until mTENPO EV isolation. During the same visit as sample collection, detailed clinical data was also recorded, including demographic information and clinical diagnoses.
Postmortem brain tissue was collected, processed, and analyzed through standard procedures as described in Lowe, V.J. et al., Alzheimers Dement., 2024102. Neuropathologic diagnoses were determined as described above using the National Alzheimer’s Coordinating Center (NACC) Neuropathology Data Form.
All participants or their proxies provided informed consent for antemortem sample collection and postmortem analysis. All methods were conducted in accordance with appropriate clinical guidelines and recommendations, and were approved by the Mayo Clinic Institutional Review Board (IRB number 712-98 for ADRC; 14-004401 for MCSA). Patients were anonymized during data analysis and confidentiality of patient information was maintained throughout the study.
Isolation of EVs using mTENPO
mTENPO chips were assembled using a protocol specified in our published work27. Briefly, 500 μL of patient plasma was incubated for 20 min at a concentration of 1 μg/mL with either biotinylated GluR2 capture antibody (GluR1 + GluR2 polyclonal antibody, Bioss bs-10042R-Biotin) for neuron-derived EVs per our previous work32 or biotinylated GLAST capture antibody [GLAST (ACSA-1) antibody, anti-human/mouse/rat Biotin, Miltenyi Biotec, 130-118-984] for astrocyte-derived EVs, where the use of GLAST as a protein target for astrocyte EV isolation has been previously reported30,42,43,44,45,46,103. The sample was then incubated with 50 μL of 50 nm MNPs at manufacturer stock concentration (Anti-Biotin MicroBeads UltraPure, Miltenyi Biotec, 130-090-485) for another 20 min. During incubation, the mTENPO devices were pre-blocked with 700 μL 1% Pluronic F-127 before a 1 mL phosphate-buffered saline (PBS) wash at 15 mL/hr using a syringe pump (Braintree Scientific). Following off-chip sample incubation and device blocking/washing, sample was added on-chip while on magnet and flowed through at 1 mL/hr. 100 μL PBS was added once the entire sample had passed through the chip. The device was then washed three times with 700 μL PBS at 15 mL/hr to remove non-specific background.
RNA isolation from EVs
To isolate RNA from the EVs captured on-chip, 700 μL of Qiazol (Qiagen) was added to the device and incubated for 2 min on-chip for lysis and release of RNA cargo. Lysate was then collected at 15 mL/hr and frozen at –80 °C until RNA isolation. RNA isolation was conducted using the miRNeasy Mini Kit (Qiagen) per manufacturer instructions; isolated RNA (20 μL elution volume) was aliquoted into three aliquots and frozen at –80 °C until sequencing preparation.
Next-generation miRNA sequencing and pathway analysis
Isolated RNA was prepared into libraries for miRNA sequencing using the Qiagen QIAseq miRNA library kit (96) following manufacturer instructions. To prepare 274 miRNA samples (two samples for each of the 137 patients—one per EV pulldown), library preparation was conducted in four rounds. Libraries were then barcoded using the QIAseq miRNA 96 Index IL (96). The concentration of each library was assessed using a Qubit fluorometer (dsDNA HS Kit, Invitrogen), and library size distribution for selected libraries was assessed using a Bioanalyzer HS DNA Analysis kit (Agilent). Libraries were sequenced on the Novaseq 6000 at the Next-Generation Sequencing Core at the University of Pennsylvania using the Novaseq S1 kit (100 cycles) (Illumina) per manufacturer instructions. Libraries and 1% PhiX spike-in (Illumina) were pooled based on Qubit readings and diluted to 2 nM in Trizma (Sigma-Aldrich). 100 μL of diluted library alongside 1 μL diluted PhiX were denatured with 25 μL of 0.2 N NaOH before neutralization with 25 μL of 400 mM Tris-HCl and loading into the sequencer for single-read sequencing per Qiagen instructions. For all sequencing runs following the first run, overlapping libraries from previous batches were re-run to check for batch-to-batch variation. Comparison of the libraries run on different batch days showed average Kendall correlation > 0.9 for the sequencing reads from the same patient sample run on two different batch days (Supplementary Fig. 24), as well as close rank-order matching of individual patient samples between sequencing runs (Supplementary Fig. 25).
RNA sequencing data was demultiplexed using Illumina BCL Convert before alignment, counting, and counts-per-million (CPM) normalization using the Qiagen RNA-seq Analysis Portal. GO and KEGG analyses were conducted using DIANA miRPath v4.0 using the TarBase v8.0 database (accessed September 2024)104.
Plasma protein analysis
Analysis of protein in patient plasma was conducted using the Neuro3Plex (Aβ40, Aβ42, tau), p-Tau181, and p-Tau231 SIMOA assays from Quanterix. All SIMOA assays were run on an HD-X instrument per manufacturer instructions at the Human Immunology Core at the University of Pennsylvania. Samples were run in triplicate and fitted to a standard curve to yield protein concentrations. For samples outside of the dynamic range, protein concentrations were set to either the standard curve’s minimum fitted concentration on-plate corresponding to 0 pg/mL (if below dynamic range) or upper assay range as reported by Quanterix (if above dynamic range). The Aβ42/Aβ40 ratio for each patient was calculated using their Aβ42 and Aβ40 concentrations and incorporated into downstream differential analysis.
NTA of mTENPO-isolated EVs
Plasma from eight different patients (62.5 µL per patient) was pooled to create a 500 µL composite patient sample for each of the five disease cohorts investigated in this study. EVs were eluted off-chip using a previously reported protocol27. NTA was performed at the Extracellular Vesicle Core at the University of Pennsylvania using the Particle Metrix ZetaView instrument. All dilutions were performed on-site in deionized water.
Isotype control validation of GluR2 and GLAST antibodies
To confirm the specificity of the GluR2 and GLAST antibodies, quantitative polymerase chain reaction (qPCR) was performed on miRNA from EVs captured by mTENPO using either 0.5 µg GluR2 or GLAST capture antibody, alongside the appropriate isotype control antibodies at the same concentrations (rabbit IgG isotype control antibody, BioLegend, 910801 for GluR2 or mouse IgG2a isotype control antibody, Miltenyi Biotec, 130-106-546 for GLAST, biotinylated using the One-Step Antibody Biotinylation Kit, Miltenyi Biotec, 130-093-385) on 500 µL of pooled healthy human plasma (BioIVT, HUMANPLK2-0101481). To demonstrate specificity of the antibodies for EV miRNAs associated with the CNS, we used a commercially available plate containing probes for 87 miRNAs known to be associated with EVs in CSF (miRCURY LNA miRNA Focus PCR Panel, CSF Exosome, Qiagen, YAHS-124Y, 384-well format). qPCR was performed using the miRCURY LNA SYBR Green PCR Kit (Qiagen, 339347) per manufacturer instructions. Antibody versus isotype experiments were run in duplicate. Any miRNA with a Cq value > 37 for both antibody and isotype control within both replicates was excluded from analysis.
SEM and HIM of EVs immobilized on mTENPO and eluted EV-MNP complexes
Visualization of EVs immobilized on mTENPO was performed using SEM at the Cell & Developmental Biology Microscopy Core at the University of Pennsylvania. Pooled healthy human plasma (Zen-Bio) was preprocessed by thawing at 4 °C overnight, keeping the clear phase, and performing a triple spin (at 1600 x g, 3000 x g, and 3000 x g, for 10 min each), each time retaining the supernatant. For EV capture, the mTENPO protocol was run as described above until after the three washes with 700 μL PBS. Captured EVs were fixed overnight on-chip with 2% glutaraldehyde in 50 mM Na-cacodylate buffer (pH 7.3) at 4 °C and washed three times with 50 mM Na-cacodylate buffer using a syringe pump (Braintree Scientific). Membrane sections were cut from the mTENPO chip and dehydrated in a graded series of ethanol concentrations through 100% over a period of 1.5 hr.
For SEM/HIM of eluted EV-MNP complexes, EV-MNPs were washed off-chip by flushing devices off-magnet with 300 µL PBS at 50 mL/hr following the conclusion of the three 15 mL/hr 700 µL PBS washes. 30 µL of eluted EV-MNP complexes was then added to glass coverslips (Electron Microscopy Sciences, 72291-02) coated with 100 µg/mL of poly-L-ornithine (Sigma-Aldrich, P3655) and incubated overnight at 4 °C. Following removal of the supernatant from the glass coverslips, the coverslips were incubated with 30 µL of 2% glutaraldehyde in 50 mM Na-cacodylate buffer overnight at 4 °C. Samples were then washed three times with 200 µL of 50 mM Na-caodylate buffer, with buffer exchange taking place following a 10 min incubation each time. Dehydration in 100% ethanol was repeated three times.
Following the dehydration steps for both membrane sections and glass coverslips, samples were then incubated for 20 min in 50% hexamethyldisilazane (HMDS) in ethanol followed by three changes of 100% HMDS (Sigma-Aldrich). This was followed by overnight air-drying as described previously105. Samples were stub-mounted and sputter-coated with gold palladium before imaging using a Quanta 250 field emission gun scanning electron microscope (FEI, Hillsboro, OR, USA) at accelerating voltages between 10 and 30 kV. Additional eluted EV-MNP images were acquired using a Zeiss Orion NanoFab helium-ion microscope at an acceleration of approximately 25,000 kV and a 2 µm field of view.
Cryo-TEM of eluted EV-MNP complexes from mTENPO devices
For cryo-TEM, pre-concentration steps are often necessary to maximize the number of EVs visible within each acquired image48, as EVs may appear sparse due to binding to support films and loss during sample blotting106,107. To produce sufficient EVs for imaging, mTENPO was run as previously described but with an increased starting input volume of 2.5 mL of pooled healthy human plasma (Precision for Medicine). EV-MNPs were washed off-chip by flushing devices off-magnet with 300 µL PBS at 50 mL/hr following the conclusion of the three 15 mL/hr 700 µL PBS washes. EV suspensions were then concentrated to a final estimated volume of 15 µL (representing an approximate 167-fold increase in GluR2+ or GLAST + EV concentrations relative to their input sample concentrations) using Amicon Ultra-0.5 Centrifugal filters (100K and 30K, MilliporeSigma) before mounting on grids.
300 mesh EM grids (Quantifoil R 1.2/1.3 300 Copper Mesh) were glow discharged for 30 s. 3 µL of each EV suspension was deposited onto each grid and vitrified using a Vitrobot Mark IV (Thermo Scientific) set to 100% humidity and 4 °C, with a blot time of 4 s. Grids were stored in liquid nitrogen until cassette loading. Imaging was performed using a Glacios Cryo-TEM (Thermo Scientific) operated at 200 kV and visualized at ×79,000 magnification.
Western blotting of EVs isolated using mTENPO
For western blot analysis of EV protein markers, plasma was collected from subjects from the University of Pennsylvania’s Penn Presbyterian Medical Center under protocols approved by the University of Pennsylvania Institutional Review Board (IRB number 856959) with criteria as defined in Beard, K. et al., Brain Commun., 202132. mTENPO was run as described above using 0.5 µg of GluR2 antibody per sample or 1.5 µg of GLAST antibody per sample until after the three 700 μL PBS washes. Captured EVs were lysed in 1X radioimmunoprecipitation assay (RIPA) buffer (Abcam), 1X Halt Protease Inhibitor Cocktail (Thermo Fisher), 5 mM EDTA, (Thermo Fisher), and 1 mM phenylmethylsulfonyl fluoride (PMSF) (Cell Signaling Technologies) and stored at –80 °C until downstream analysis.
The protein concentration of EV protein lysates was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher) according to manufacturer’s instructions. 5-10 µg of protein was then separated on 10% sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline with Tween-20 (TBST) (20 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween-20) for 1 hr, followed by incubation with anti-TSG101 (1:1000, Santa Cruz Biotechnology, sc-7964), anti-GLAST antibodies (1:250, Miltenyi Biotec, 130-118-984), and anti-GluR2/3 (1:1000, MilliporeSigma, 07-598) in 5% BSA in TBST at 4 °C overnight. Membranes were then washed with TBST, incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Rockland Immunochemicals, Inc.), and then visualized using Immobilon Western Chemiluminescent HRP Substrate (MilliporeSigma) on the LI-COR Odyssey Fc Imager (LI-COR).
Isolation of EVs from neuron and astrocyte cell cultures
EVs derived from cell culture were obtained by taking the media from flasks cultured with either mixed cells (neuron and astrocyte) or astrocytes alone. Cells were provided by the Neurons R Us Core at the University of Pennsylvania and were derived from the neocortical tissue of day 18 embryos from Sprague-Dawley rats. Cultures were prepared by mesh-filtering (Crosswire Cloth) cells and resuspending in Minimum Essential Medium (MEM) with Earl’s salts, GlutaMAX (Gibco), 0.6% D-glucose (Sigma-Aldrich), 1% Pen-Strep (Gibco) and 10% Horse Serum (Gibco) for mixed cell culture; or Dulbecco’s Modified Eagle Medium (DMEM, Gibco) with 10% fetal bovine serum (FBS, Sigma-Aldrich) and 1% Pen-Strep for astrocyte cell culture. Cells were then added to T75 flasks (Corning) coated with both 0.08 mg/mL of poly-D-lysine (Sigma-Aldrich) and 0.001 mg/mL of laminin (BD Biosciences) for mixed cell culture, or only 0.08 mg/mL of poly-D-lysine (Sigma-Aldrich) for astrocyte cell culture, and allowed to adhere overnight in 37 °C at 5% CO2. Mixed cell cultures were maintained in B-27 neurobasal medium and 0.4 mM GlutaMAX, and media was collected after 21 days. Astrocyte cultures were maintained in DMEM with FBS and Pen-Strep and passaged at day 21, and media was collected at day 28. Media was frozen at –80 °C until mTENPO isolation. To prepare cell culture media for mTENPO, media was spun at 2500 x g for 15 min. The resulting supernatant was then filtered through a 0.22 µm PES filter (CELLTREAT Scientific Products) before proceeding with the abovementioned mTENPO, EV elution27, and NTA protocols using 500 µL of input sample volume.
Statistical analysis
Demographic, clinical, and neuropathologic data were summarized using measures of central tendency (mean or median) with associated dispersion [standard deviation or lower and upper quartiles, as determined by MATLAB R2024a’s (Mathworks) iqr function]. Differences in demographic and clinical data between disease cohorts were determined using Chi-square tests and one-way ANOVAs. PCA was performed using MATLAB R2024a’s pca function on log2-transformed CPM-normalized data. Pearson correlation and Mann–Whitney U analyses were performed using the corr and ranksum functions, respectively.
For the statistical analysis of RNA sequencing data, the Qiagen RNA-seq Analysis Portal was used to conduct tests of differential expression between disease cohorts using Wald tests based on negative binomial distributions, and multiple hypothesis testing correction using the Benjamini-Hochberg method with the Qiagen default FDR level of 0.1. The statistical significance of differences in protein measurements was assessed using the Mann–Whitney U test and implemented using MATLAB R2024a. FDR correction was again performed using the Benjamini-Hochberg method with an FDR level of 0.1. miRNA markers with zero expression across all samples were removed by the Qiagen RNA-seq Analysis Portal. Outliers were not removed from downstream analysis.
LASSO biomarker selection was implemented using Python. Data visualization, calculation of ROC and AUC, and evaluation of LASSO panels using an ensemble model with tenfold cross-validation repeated 5x (which included k-nearest neighbors, support vector machine, linear discriminate analysis, logistic regression, and Naive Bayes, based on our prior work)35 were conducted using the MATLAB R2024a/2025a Statistics and Machine Learning Toolbox. Error bars for AUC of individual markers were calculated by bootstrapping 10x. Bootstrapping for AUC analysis, 95% CI calculations, hierarchical clustering of subjects, biomarker correlation, and scrambled label experiments were performed using MATLAB’s randi, quantile, clustergram, corr, and randperm functions, respectively. Delong’s test for determining statistically significant differences between AUCs was applied using a framework from Sun, X. and Xu, W., IEEE Signal Process. Lett., 2014108, for which biomarkers were defined as significant relative to a non-predictive biomarker (AUC = 0.5) at P value < 0.1. P values for DIANA miRPath v4.0 GO and KEGG analyses were obtained using a one-sided Fisher’s exact test using FDR correction and the default P value threshold of 0.05104.
To develop a learning curve for our model, we used a methodology described by Breiman, L., Mach. Learn., 200164,109, in which a model is trained on bootstrapped samples, and out-of-bag data that was not used within the training set is used as a validation set, giving an estimate of model performance. AUC and accuracy were obtained by bootstrapping 100 times64, where using at least 100 bootstraps has been previously recommended for precise model performance66 and standard error65 estimates. For small sample sizes (n < 25), bootstrapping occasionally produced degenerate distributions for one or both classes. As this is a violation of the assumptions for Naïve Bayes, Naïve Bayes was conditionally excluded from the ensemble model for those training sets. All of the other ensemble classifiers remained unchanged.
Permutation feature importance was computed within the 5x repeated tenfold cross-validation framework by quantifying the decrease in AUC and accuracy64 following n = 50 permutations of each feature within each held-out fold and averaging these parameters across folds and repeats. 50 permutations per feature was selected to balance computational cost with stability of importance estimates. Prior work has shown that 50 permutations can provide robust feature rankings for a tenfold cross-validation model64. SHAP analyses67 were also conducted by taking the mean absolute SHAP values computed for each feature on held-out folds within repeated cross-validation. Permutation feature importance and SHAP analyses were implemented using MATLAB 2025a’s randperm and shapley functions, respectively.
To determine the necessary size of an independent validation cohort, we used powerROC70 (accessed January 8, 2026). Precision of the AUC of our model was defined as the total width of the 95% confidence interval.
Data availability
Data associated with this study are present in the paper or the Supplementary Materials. Sequencing and SIMOA expression data for the subjects within this study, as well as qPCR data for antibody versus isotype control experiments, have been deposited in Dryad (https://doi.org/10.5061/dryad.612jm64j4). Raw reads have been deposited at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under BioProject PRJNA1416371. Individual identifiers and demographic information have been removed to protect patient privacy.
Code availability
Specific MATLAB functions and toolboxes that were used for statistical analysis and data visualization are indicated where applicable; Python code developed for LASSO feature selection and MATLAB code for ensemble learning can be found in Dryad (https://doi.org/10.5061/dryad.612jm64j4).
References
Prasad, S. et al. Recent advances in Lewy body dementia: A comprehensive review. Dis. Mon. 69, 101441 (2023).
Boot, B. P., McDade, E. M., McGinnis, S. M. & Boeve, B. F. Treatment of dementia with lewy bodies. Curr. Treat. Options Neurol. 15, 738–764 (2013).
DeTure, M. A. & Dickson, D. W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14, 32 (2019).
Salvadó, G. et al. Specific associations between plasma biomarkers and postmortem amyloid plaque and tau tangle loads. EMBO Mol. Med. 15, e17123 (2023).
Barthélemy, N. R. et al. Highly accurate blood test for Alzheimer’s disease is similar or superior to clinical cerebrospinal fluid tests. Nat. Med. 30, 1085–1095 (2024).
Ashton, N. J. et al. Plasma p-tau231: A new biomarker for incipient Alzheimer’s disease pathology––. Acta Neuropathol. 141, 709–724 (2021).
Rajkumar, A. P. et al. Next-generation RNA-sequencing of serum small extracellular vesicles discovers potential diagnostic biomarkers for dementia with Lewy bodies. Am. J. Geriatr. Psychiatry 29, 573–584 (2021).
Chouliaras, L. et al. Differential levels of plasma biomarkers of neurodegeneration in Lewy body dementia, Alzheimer’s disease, frontotemporal dementia and progressive supranuclear palsy. J. Neurol. Neurosurg. Psychiatry 93, 651–658 (2022).
Vrillon, A. et al. Plasma biomarkers of amyloid, tau, axonal, and neuroinflammation pathologies in dementia with Lewy bodies. Alzheimers Res. Ther. 16, 146 (2024).
Tiraboschi, P. et al. What best differentiates Lewy body from Alzheimer’s disease in early-stage dementia? Brain 129, 729–735 (2006).
McKeith, I. G. et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89, 88–100 (2017).
Beach, T. G. & Adler, C. H. Importance of low diagnostic accuracy for early Parkinson’s disease. Mov. Disord. 33, 1551–1554 (2018).
Turcano, P. et al. Clinicopathologic discrepancies in a population-based incidence study of parkinsonism in olmsted county: 1991-2010. Mov. Disord. 32, 1439–1446 (2017).
Fertan, E. et al. Lecanemab preferentially binds to smaller aggregates present at early Alzheimer’s disease. Alzheimers Dement 21, e70086 (2025).
Rajkumar, A. P. Progressing towards blood based diagnostic RNA biomarkers for dementia with lewy bodies. Am. J. Geriatr. Psychiatry 30, 976–978 (2022).
Quispialaya, K. M. et al. Comparison of Plasma p-tau217 and [18F] FDG-PET for identifying Alzheimer disease in people with early-onset or atypical dementia. Neurology 104, e210211 (2025).
Arnold, M. R. et al. α-synuclein seed amplification in CSF and brain from patients with different brain distributions of pathological α-synuclein in the context of co-pathology and non-LBD diagnoses. Ann. Neurol. 92, 650–662 (2022).
Hall, S. et al. Performance of αSynuclein RT-QuIC in relation to neuropathological staging of Lewy body disease. Acta Neuropathol. Commun. 10, 90 (2022).
Perez-Grijalba, V. et al. Plasma Aβ42/40 ratio detects early stages of Alzheimer’s disease and correlates with CSF and neuroimaging biomarkers in the AB255 study. J. Prev. Alzheimers Dis. 6, 34–41 (2019).
Brickman, A. M. et al. Plasma p-tau181, p-tau217, and other blood-based Alzheimer’s disease biomarkers in a multi-ethnic, community study. Alzheimers Dement 17, 1353–1364 (2021).
Donaghy, P. C. et al. Blood mRNA expression in Alzheimer’s disease and dementia with Lewy bodies. Am. J. Geriatr. Psychiatry 30, 964–975 (2022).
Gámez-Valero, A. et al. Platelet mirna biosignature discriminates between dementia with lewy bodies and alzheimer’s disease. Biomedicines 9, 1272 (2021).
Balogun, W. G., Zetterberg, H., Blennow, K. & Karikari, T. K. Plasma biomarkers for neurodegenerative disorders: ready for prime time? Curr. Opin. Psychiatry 36, 112–118 (2023).
Zhang, Y., Liu, Y., Liu, H. & Tang, W. H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 9, 19 (2019).
Holm, M. M., Kaiser, J. & Schwab, M. E. Extracellular vesicles: Multimodal envoys in neural maintenance and repair. Trends Neurosci. 41, 360–372 (2018).
Gámez-Valero, A., Beyer, K. & Borràs, F. E. Extracellular vesicles, new actors in the search for biomarkers of dementias. Neurobiol. Aging 74, 15–20 (2019).
Lin, A. A., Shen, H., Spychalski, G., Carpenter, E. L. & Issadore, D. Modeling and optimization of parallelized immunomagnetic nanopore sorting for surface marker specific isolation of extracellular vesicles from complex media. Sci. Rep. 13, 13292 (2023).
Gámez-Valero, A. et al. Exploratory study on microRNA profiles from plasma-derived extracellular vesicles in Alzheimer’s disease and dementia with Lewy bodies. Transl. Neurodegener. 8, 31 (2019).
Serpente, M. et al. MiRNA profiling in plasma neural-derived small extracellular vesicles from patients with Alzheimer’s disease. Cells 9, 1443 (2020).
Kumar, A. et al. MicroRNA expression in extracellular vesicles as a novel blood-based biomarker for Alzheimer’s disease. Alzheimers Dement 19, 4952–4966 (2023).
Ko, J. et al. Diagnosis of traumatic brain injury using miRNA signatures in nanomagnetically isolated brain-derived extracellular vesicles. Lab Chip 18, 3617–3630 (2018).
Beard, K. et al. Extracellular vesicles as distinct biomarker reservoirs for mild traumatic brain injury diagnosis. Brain Commun. 3, fcab151 (2021).
Ko, J. et al. Multi-dimensional mapping of brain-derived extracellular vesicle microRNA biomarker for traumatic brain injury diagnostics. J. Neurotrauma 37, 2424–2434 (2020).
Shen, H. et al. Brain-derived extracellular vesicles as serologic markers of brain injury following cardiac arrest: A pilot feasibility study. Resuscitation 191, 109937 (2023).
Yang, Z. et al. A multianalyte panel consisting of extracellular vesicle miRNAs and mRNAs, cfDNA, and CA19-9 shows utility for diagnosis and staging of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 26, 3248–3258 (2020).
Spychalski, G. B. et al. miRNA panel from HER2+ and CD24+ plasma extracellular vesicle subpopulations as biomarkers of early-stage breast cancer. Breast Cancer Res 27, 90 (2025).
Eitan, E. et al. Synaptic proteins in neuron-derived extracellular vesicles as biomarkers for Alzheimer’s disease: Novel methodology and clinical proof of concept. Extracell. Vesicles Circulating Nucleic Acids 4, 133 (2023).
Yousif, G. et al. Exosomes derived neuronal markers: Immunoaffinity isolation and characterization. Neuromol. Med. 24, 339–351 (2022).
Fauré, J. et al. Exosomes are released by cultured cortical neurones. Mol. Cell. Neurosci. 31, 642–648 (2006).
Lombino, F. L. et al. Extracellular vesicles released from cortical neurons influence spontaneous activity of recipient neurons. J. Neurochem. 169, e70231 (2025).
Lachenal, G. et al. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell. Neurosci. 46, 409–418 (2011).
Winston, C. N. et al. Evaluation of blood-based, extracellular vesicles as biomarkers for aging-related TDP-43 pathology. Alzheimer’s. Dement. Diagn. Assess. Dis. Monit. 14, e12365 (2022).
Wang, S., Zhang, H., Li, R., Liu, Z. & Xiang, D. Altered neuroplasticity in epilepsy is associated with neuroinflammation and oxidative stress: In vivo evidence of brain-derived extracellular vesicles. Int. J. Nanomed. 7185-7197 (2025).
Xie, X.-h. et al. Isolating astrocyte-derived extracellular vesicles from urine. Int. J. Nanomed. 2475-2484 (2025).
Forró, T., Manu, D. R., Barbu-Tudoran, L. & Bălașa, R. Astrocyte dysfunction reflected in ischemia-induced astrocyte-derived extracellular vesicles: A pilot study on acute ischemic stroke patients. Int. J. Mol. Sci. 25, 12471 (2024).
Ikezu, T., Yang, Y., Verderio, C. & Krämer-Albers, E.-M. Extracellular Vesicle-Mediated neuron–glia communications in the central nervous system. J. Neurosci. 44, e1170242024 (2024).
Savica, R. et al. Incidence of dementia with lewy bodies and Parkinson disease dementia. JAMA Neurol. 70, 1396–1402 (2013).
Chernyshev, V. S. et al. Size and shape characterization of hydrated and desiccated exosomes. Anal. Bioanal. Chem. 407, 3285–3301 (2015).
Tan, Y. J. et al. Altered cerebrospinal fluid exosomal microRNA levels in young-onset Alzheimer’s disease and frontotemporal dementia. J. Alzheimers Dis. Rep. 5, 805–813 (2021).
Doecke, J. D. et al. Total Aβ42/Aβ40 ratio in plasma predicts amyloid-PET status, independent of clinical AD diagnosis. Neurology 94, e1580–e1591 (2020).
Kinser, H. E. & Pincus, Z. MicroRNAs as modulators of longevity and the aging process. Hum. Genet. 139, 291–308 (2020).
Uchikado, H., Lin, W.-L., DeLucia, M. W. & Dickson, D. W. Alzheimer disease with amygdala Lewy bodies: A distinct form of α-synucleinopathy. J. Neuropathol. Exp. Neurol. 65, 685–697 (2006).
Lawrence, J. M., Schardien, K., Wigdahl, B. & Nonnemacher, M. R. Roles of neuropathology-associated reactive astrocytes: A systematic review. Acta Neuropathol. Commun. 11, 42 (2023).
Falcicchia, C., Tozzi, F., Arancio, O., Watterson, D. M. & Origlia, N. Involvement of p38 MAPK in synaptic function and dysfunction. Int. J. Mol. Sci. 21, 5624 (2020).
Sánchez-Alegría, K., Flores-León, M., Avila-Muñoz, E., Rodríguez-Corona, N. & Arias, C. PI3K signaling in neurons: A central node for the control of multiple functions. Int. J. Mol. Sci. 19, 3725 (2018).
Oliveira da Silva, M. I. et al. -Synuclein triggers cofilin pathology and dendritic spine impairment via a PrPC-CCR5 dependent pathway. Cell Death Dis. 15, 264 (2024).
Cardoso, S., López, I. P., Piñeiro-Hermida, S., Pichel, J. G. & Moreira, P. I. Igf1r deficiency modulates brain signaling pathways and disturbs mitochondria and redox homeostasis. Biomedicines 9, 158 (2021).
Zhao, J. et al. Specific depletion of the motor protein KIF5B leads to deficits in dendritic transport, synaptic plasticity and memory. elife 9, e53456 (2020).
Chandrasekaran, S. & Bonchev, D. A network view on Parkinson’s disease. Comput. Struct. Biotechnol. J. 7, e201304004 (2013).
Zaitoun, I. S. et al. Bcl-2 expression in pericytes and astrocytes impacts vascular development and homeostasis. Sci. Rep. 9, 9700 (2019).
Annibali, D. et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat. Commun. 5, 4632 (2014).
Lee, S. et al. PTK2 regulates tau-induced neurotoxicity via phosphorylation of p62 at Ser403. J. Neurogenet. 37, 10–19 (2023).
Stone, M. Cross-validatory choice and assessment of statistical predictions. J. R. Stat. Soc.: Ser. B (Methodol.) 36, 111–133 (1974).
Altmann, A., Toloşi, L., Sander, O. & Lengauer, T. Permutation importance: A corrected feature importance measure. Bioinformatics 26, 1340–1347 (2010).
Efron, B. & Tibshirani, R. J. An introduction to the bootstrap. (Chapman and Hall/CRC, 1994).
Steyerberg, E. W., Bleeker, S. E., Moll, H. A., Grobbee, D. E. & Moons, K. G. Internal and external validation of predictive models: A simulation study of bias and precision in small samples. J. Clin. Epidemiol. 56, 441–447 (2003).
Ponce-Bobadilla, A. V., Schmitt, V., Maier, C. S., Mensing, S. & Stodtmann, S. Practical guide to SHAP analysis: Explaining supervised machine learning model predictions in drug development. Clin. Transl. Sci. 17, e70056 (2024).
Hajjar, M., Aldabbagh, G. & Albaradei, S. Interpretable multi-cancer early detection using SHAP-based machine learning on tumor-educated platelet RNA. Diagnostics 15, 2216 (2025).
Salman, R., Alzaatreh, A. & Sulieman, H. The stability of different aggregation techniques in ensemble feature selection. J. Big Data 9, 51 (2022).
Grolleau, F., Tibshirani, R. & Chen, J. H. powerROC: An Interactive Web Tool for Sample Size Calculation in Assessing Models’ Discriminative Abilities. AMIA Summits Transl. Sci. Proc. 2025, 196 (2025).
Gilbert, M. A. et al. CryoET of β-amyloid and tau within postmortem Alzheimer’s disease brain. Nature 631, 913–919 (2024).
Kuang, Y. et al. A skin-specific α-Synuclein seeding amplification assay for diagnosing Parkinson’s disease. NPJ Parkinsons Dis. 10, 129 (2024).
Okuzumi, A. et al. Propagative α-synuclein seeds as serum biomarkers for synucleinopathies. Nat. Med. 29, 1448–1455 (2023).
Rodriguez, J. D., Perez, A. & Lozano, J. A. Sensitivity analysis of k-fold cross validation in prediction error estimation. IEEE Trans. pattern Anal. Mach. Intell. 32, 569–575 (2009).
Robin, X. et al. PanelomiX: A threshold-based algorithm to create panels of biomarkers. Transl. Proteom. 1, 57–64 (2013).
King, R. D., Orhobor, O. I. & Taylor, C. C. Cross-validation is safe to use. Nat. Mach. Intell. 3, 276–276 (2021).
Coughlin, D. G. & Irwin, D. J. Fluid and biopsy based biomarkers in Parkinson’s disease. Neurotherapeutics 20, 932–954 (2023).
Silva-Rodríguez, J. et al. Differential effects of tau stage, Lewy body pathology, and substantia nigra degeneration on 18F-FDG PET patterns in clinical Alzheimer disease. J. Nucl. Med. 64, 274–280 (2023).
Liberini, P., Valerio, A., Memo, M. & Spano, P. Lewy-body dementia and responsiveness to cholinesterase inhibitors: A paradigm for heterogeneity of Alzheimer’s disease? Trends Pharmacol. Sci. 17, 155–160 (1996).
VandeVrede, L. et al. Co-pathology may impact outcomes of amyloid-targeting treatments: Clinicopathological results from two patients treated with aducanumab. Acta Neuropathol. 146, 777–781 (2023).
Seath, P., Macedo-Orrego, L. E. & Velayudhan, L. Clinical characteristics of early-onset versus late-onset Alzheimer’s disease: A systematic review and meta-analysis. Int. Psychogeriatr. 36, 1093–1109 (2024).
Riboldi, G. M., Frattini, E., Monfrini, E., Frucht, S. J. & Di Fonzo, A. A practical approach to early-onset parkinsonism. J. Parkinson’s. Dis. 12, 1–26 (2022).
Sim, J., Li, H., Hameed, S. & Ting, S. K. S. Clinical manifestations of early-onset dementia with Lewy bodies compared with late-onset dementia with Lewy bodies and early-onset Alzheimer disease. JAMA Neurol. 79, 702–709 (2022).
Choudhury, P. et al. The temporal onset of the core features in dementia with Lewy bodies. Alzheimers Dement 18, 591–601 (2022).
Graff-Radford, N. R., Besser, L. M., Crook, J. E., Kukull, W. A. & Dickson, D. W. Neuropathologic differences by race from the National Alzheimer’s Coordinating Center. Alzheimers Dement 12, 669–677 (2016).
Shami-Shah, A., Norman, M. & Walt, D. R. Ultrasensitive protein detection technologies for extracellular vesicle measurements. Mol. Cell. Proteom. 22, 100557 (2023).
Bravo-Miana, R. D. C. et al. Tetraspanins, GLAST and L1CAM quantification in single extracellular vesicles from cerebrospinal fluid and serum of people with multiple sclerosis. Biomedicines 12, 2245 (2024).
Wu, Z. Y. et al. AMPA receptors regulate exocytosis and insulin release in pancreatic β cells. Traffic 13, 1124–1139 (2012).
Usami, S. -i, Matsubara, A., Fujita, S., Shinkawa, H. & Hayashi, M. NMDA (NMDAR1) and AMPA-type (GluR2/3) receptor subunits are expressed in the inner ear. Neuroreport 6, 1161–1164 (1995).
Vivero, M., Doyle, L. A., Fletcher, C. D., Mertens, F. & Hornick, J. L. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology 65, 71–80 (2014).
Huggett, J., Mustafa, A., O’neal, L. & Mason, D. J. The glutamate transporter GLAST-I (EAAT-I) is expressed in the plasma membrane of osteocytes and is responsive to extracellular glutamate concentration. Biochem. Soc. Trans. 30, 890–893 (2002).
Carozzi, V. A. et al. Expression and distribution of ‘high affinity’glutamate transporters GLT1, GLAST, EAAC1 and of GCPII in the rat peripheral nervous system. J. Anat. 213, 539–546 (2008).
Berger, U. V. & Hediger, M. A. Distribution of the glutamate transporters GLT-1 (SLC1A2) and GLAST (SLC1A3) in peripheral organs. Anat. Embryol. (Berl.) 211, 595–606 (2006).
Newman, L. & Rowland, A. Detection and isolation of tissue-specific extracellular vesicles from the blood. J. Extracell. Biol. 4, e70059 (2025).
Shami-Shah, A., Travis, B. G. & Walt, D. R. Advances in extracellular vesicle isolation methods: a path towards cell-type specific EV isolation. Extracell. Vesicles Circulating Nucleic Acids 4, 447 (2023).
Mirra, S. S. et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–479 (1991).
Braak, H., Thal, D. R., Ghebremedhin, E. & Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969 (2011).
Halliday, G. M., Holton, J. L., Revesz, T. & Dickson, D. W. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 122, 187–204 (2011).
Hughes, C. P., Berg, L., Danziger, W., Coben, L. A. & Martin, R. L. A new clinical scale for the staging of dementia. Br. J. psychiatry 140, 566–572 (1982).
Reilly, J., Rodriguez, A. D., Lamy, M. & Neils-Strunjas, J. Cognition, language, and clinical pathological features of non-Alzheimer’s dementias: An overview. J. Commun. Disord. 43, 438–452 (2010).
Hale, J. M., Schneider, D. C., Mehta, N. K. & Myrskylä, M. Cognitive impairment in the US: Lifetime risk, age at onset, and years impaired. SSM-Popul. Health 11, 100577 (2020).
Lowe, V. J. et al. Amyloid PET detects the deposition of brain Aβ earlier than CSF fluid biomarkers. Alzheimers Dement. 20, 8097–8112 (2024).
Goetzl, E. J., Schwartz, J. B., Abner, E. L., Jicha, G. A. & Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 83, 544–552 (2018).
Tastsoglou, S. et al. DIANA-miRPath v4. 0: Expanding target-based miRNA functional analysis in cell-type and tissue contexts. Nucleic Acids Res. 51, W154–W159 (2023).
Braet, F., De Zanger, R. & Wisse, E. Drying cells for SEM, AFM and TEM by hexamethyldisilazane: A study on hepatic endothelial cells. J. Microsc. 186, 84–87 (1997).
Hartjes, T. A., Mytnyk, S., Jenster, G. W., van Steijn, V. & van Royen, M. E. Extracellular vesicle quantification and characterization: Common methods and emerging approaches. Bioengineering 6, 7 (2019).
Yuana, Y. et al. Cryo-electron microscopy of extracellular vesicles in fresh plasma. J. Extracell. Vesicles 2, 21494 (2013).
Sun, X. & Xu, W. Fast implementation of DeLong’s algorithm for comparing the areas under correlated receiver operating characteristic curves. IEEE Signal Process. Lett. 21, 1389–1393 (2014).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Acknowledgements
The authors thank Drs. Yangzhu Du and Honghong Sun of the Human Immunology Core at the Perelman School of Medicine at the University of Pennsylvania for assistance with digital ELISA assays. The authors also would like to thank Drs. Neha Srikumar, Yuri Veklich, and Jamie Ford for assistance with imaging of EVs with scanning electron microscopy and helium ion microscopy; Drs. Vera Moiseenkova-Bell, Prerana Gogoi, and Ruth Anne Pumroy for assistance with imaging EVs with transmission electron microscopy; Dr. Luca Musante for assistance with performing nanoparticle tracking analysis; Kyra Kovacic, Dr. Jonathan Schug, and Dr. C. Erik Nordgren for assistance with RNA sequencing; Drs. Kryshawna Beard and Bonnie L. Firestein for assistance in conducting cell culture and performing western blotting; Dr. Hiroaki Sekiya for taking representative pathology pictures for this manuscript; and Dr. Fabrice Lucien-Matteoni for sample storage. The authors particularly thank all participants, volunteers, and coordinators who contributed to this research. This work was supported by the American Academy of Neurology/American Brain Foundation Cure One Cure Many Award (D.I., P.J.M., O.A.R., B.F.B., D.T.J., S.O.B., R.S.); NIH grants P30 AG062677 (B.F.B., P.J.M., D.T.J., D.W.D., A.T.N., R.R.R.), U01 AG006786, U01 NS100620 (B.F.B.), U19 AG063911 (B.F.B., D.W.D., L.K.F.); the Little Family Foundation (B.F.B., O.A.R.); the Mangurian Foundation (B.F.B., D.W.D.); the LBD Center without Walls, NIH grant U54 NS110435 (P.J.M., O.A.R., D.W.D.); and the Ted Turner and Family Foundation (B.F.B., O.A.R., P.J.M., L.K.F.). The Human Immunology Core at the University of Pennsylvania is supported in part by NIH P30 AI045008 and P30 CA016520 (Human Immunology Core RRID: SCR_022380). Scanning electron microscopy, helium ion microscopy, and transmission electron microscopy of eluted EVs were carried out at the University of Pennsylvania’s Singh Center for Nanotechnology, which is supported by the NSF National Nanotechnology Coordinated Infrastructure Program under grant NNCI-2025608 and through facilities supported by the University of Pennsylvania Materials Research Science and Engineering Center (MRSEC) DMR-2309043.
Author information
Authors and Affiliations
Contributions
S.J.Y., A.A.L., B.F.B., O.A.R., P.J.M., and D.I. conceived, designed, and developed the methodology for the study; S.J.Y., A.A.L., H.S., L.W.P., and J.R. contributed to the acquisition of experimental data; and S.J.Y., A.A.L., H.S., G.B.S., and D.I. contributed to formal analyses and data visualization. Data curation was carried out by S.J.Y., A.A.L., L.K.F., K.M.G., B.F.B., O.A.R., and P.J.M., with provision of samples, reagents, and clinical and neuropathological data provided by L.K.F., K.M.G., D.W.D., R.R.R., A.T.N., D.F.M., B.F.B., O.A.R., P.J.M., and D.I. J.R.B. constructed the chips used in this study and developed the automated platform. B.F.B., O.A.R., P.J.M., and D.I. supervised the project. S.J.Y., A.A.L., and D.I. drafted the original manuscript. S.J.Y., A.A.L., H.S., G.B.S., R.S., S.O.B., D.T.J., D.F.M., B.F.B., O.A.R., P.J.M., and D.I. reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
D.I. is a founder and holds shares in Chip Diagnostics. J.R.B. holds ownership of Consonant Systems, LLC, which was paid to produce the mTENPO chips used in this study. The other authors do not have a competing interest. The mTENPO platform is protected under US patent US20240238802A1.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, S.J., Lin, A.A., Shen, H. et al. Microfluidic nanomagnetically isolated neuron- and astrocyte-derived extracellular vesicles to differentiate Lewy body and Alzheimer’s disease. npj Biosensing 3, 19 (2026). https://doi.org/10.1038/s44328-026-00086-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44328-026-00086-x
