
Abstract
The rapid evolution of molecular representation methods has significantly advanced the drug discovery process. Advances in language models, graph-based representations, and novel learning strategies have greatly improved the ability to characterize molecules. These AI-driven strategies extend beyond traditional structural data, facilitating exploration of broader chemical spaces and accelerating scaffold hopping. This review summarizes key advancements, discusses their advantages over conventional techniques, and highlights challenges in data quality and real-world applications.
Similar content being viewed by others
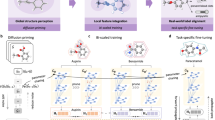

Introduction
Molecular representation
Drug discovery is a highly time-intensive and costly endeavor, driving researchers to continually develop new experimental and computational methods to accelerate drug development at all stages of drug discovery1. In recent years, advancements in Artificial Intelligence (AI) have positioned AI-assisted Drug Design as a prominent area of research. Cutting-edge methods have emerged for compound druggability evaluation, virtual screening for hit identification, and molecule generation for novel compound creation, etc1,2. These approaches play a crucial role in the early stages of drug development, enabling faster early screening and the identification of viable lead compounds3,4,5.
A key prerequisite for developing those methods is translating molecules into a computer-readable format, known as molecular representation, which serves as the foundation for training machine learning (ML) and deep learning (DL) models6. Molecular representation is a cornerstone of computational chemistry and drug design, bridging the gap between chemical structures and their biological, chemical, or physical properties7. It involves converting molecules into mathematical or computational formats that algorithms can process to model, analyze, and predict molecular behavior7,8. Effective molecular representation is essential for various drug discovery tasks, including virtual screening, activity prediction, and scaffold hopping, enabling efficient and precise navigation of chemical space9,10,11.
Advances in cheminformatics and AI have led to an increasing number of novel approaches to molecular representation (Fig. 1). Traditional representations rely on explicit, rule-based feature extraction methods, such as molecular descriptors that quantify the physical or chemical properties of molecules, and molecular fingerprints that typically encode substructural information as binary strings or numerical values. The most widely used method for molecular representation is the Simplified Molecular-Input Line-Entry System (SMILES)11,12, which provides a compact and efficient way to encode chemical structures as strings. However, despite its simplicity and convenience, SMILES has inherent limitations in capturing the full complexity of molecular interactions. As drug discovery tasks grow more sophisticated, traditional string-based representations often fall short in reflecting the intricate relationships between molecular structure and key drug-related characteristics such as biological activity and physicochemical properties8. Moreover, while traditional methods provide interpretable features, they often struggle to navigate the vast, nearly infinite chemical space in search of compounds with desired biological properties. Molecular representation must not only encode the chemical structure, but also enable efficient exploration of chemical space.

Key milestones illustrated with representative icons.
In recent years, AI-driven molecular representation methods employ DL techniques to learn continuous, high-dimensional feature embeddings directly from large and complex datasets. Models such as graph neural networks (GNNs), variational autoencoders (VAEs), and transformers enable these approaches to move beyond predefined rules, capturing both local and global molecular features13,14,15,16. These representations better reflect the subtle structural and functional relationships underlying molecular behavior, thereby providing powerful tools for molecular generation, scaffold hopping, lead compound optimization, and other key tasks in drug discovery17,18,19,20,21,22,23.
Scaffold hopping and its importance
In 1999, Schneider et al. introduced the concept of scaffold hopping as a key strategy in drug discovery and lead optimization, aimed at the discovery of new core structures (backbones) while retaining similar biological activity or target interaction as the original molecule24. Molecular representation and scaffold hopping are closely interconnected in drug design and medicinal chemistry, as the representation of molecules strongly influences the ability to identify structurally diverse yet functionally similar compounds. In 2012, Sun et al. classified scaffold hopping into four main categories (Fig. 2) of increasing degree of hopping, i.e., heterocyclic substitutions, open-or-closed rings, peptide mimicry, and topology-based hops25. Scaffold hopping plays a crucial role in drug discovery. On the one hand, existing lead compounds may have undesirable properties such as toxicity or metabolic instability, and new compounds discovered through scaffold hopping may have further enhancement in molecular activity and reduction of undesirable off-target effects, which may lead to improvement in pharmacokinetic and pharmacodynamic profiles26,27. On the other hand, by modifying the core structure of a molecule, it can help researchers discover novel compounds with similar biological effects but different structural features, thus breaking through the limitations of existing patents27,28,29. In conclusion, scaffold hopping is an important method to explore new chemical entities.

Representative strategies for scaffold hopping.
Scaffold hopping relies heavily on effective molecular representation, as the ability to identify new scaffolds that retain biological activity depends on accurately capturing and effectively representing the essential features of molecules. Traditional approaches to scaffold hopping typically utilize molecular fingerprinting and structure similarity searches to identify compounds with similar properties but different core structures28,29,30. These methods maintain key molecular interactions by substituting critical functional groups with alternatives that preserve binding contributions, such as hydrogen bonding patterns, hydrophobic interactions, and electrostatic forces, while incorporating new molecular fragment structures.
However, traditional methods are limited in their ability to explore diverse chemical spaces due to their reliance on predefined rules, fixed features, or expert knowledge. Modern methods, especially those utilizing DL, have greatly expanded the potential for scaffold hopping through more flexible and data-driven exploration of chemical diversity31,32,33. Researchers can identify novel scaffolds that were previously difficult to discover by leveraging advanced molecular representations, such as graph-based embedding or DL-generated features, which refer to latent embeddings (e.g., 128-dimensional vectors) learned through self-supervised tasks such as masked atom prediction, which capture non-linear relationships beyond manual descriptors31,32. These modern methods can capture nuances in molecular structure that may have been overlooked by traditional methods, allowing for a more comprehensive exploration of chemical space and the discovery of new scaffolds with unique properties33.
In recent years, AI-driven molecular generation methods have emerged as a transformative approach in scaffold hopping. Techniques such as VAEs and generative adversarial networks are increasingly utilized to design entirely new scaffolds absent from existing chemical libraries, while simultaneously tailoring molecules to possess desired properties32,34,35. This data-driven shift toward AI-enhanced scaffold generation equips researchers with advanced tools to explore the vast chemical space more efficiently, facilitating the discovery of novel bioactive compounds with enhanced efficacy and safety.
In this review, we chronologically examine traditional molecular representation methods and summarize modern mainstream AI-based molecular representation approaches from perspectives including language model-based, graph-based, as well as the recently popular multimodal learning and contrastive learning frameworks. Subsequently, we introduce how molecular representation methods are applied to scaffold hopping tasks and analyze the challenges that still exist.
Molecular representation: from classical rules to AI-driven innovation
Traditional approaches for molecular representation
Traditional molecular representation methods have laid a strong foundation for many computational approaches in drug discovery. These methods often rely on string-based formats to describe molecules. Alternatively, they encode molecular structures using predefined rules derived from chemical and physical properties, including molecular descriptors (e.g., molecular weight, hydrophobicity, or topological indices) and molecular fingerprints36,37,38,39,40.
The IUPAC name was first introduced by the International Chemical Congress in Geneva in 1892 and established by the International Union of Pure and Applied Chemistry (IUPAC). Over the following decades, methods such as Dyson cyphering41 and Wiswesser Line Notation (WLN)42 were proposed. The widely used Simplified Molecular Input Line Entry System (SMILES)12 was introduced in 1988 by Weininger et al. Subsequently, improved versions like ChemAxon Extended SMILES (CXSMILES), OpenSMILES, and SMILES Arbitrary Target Specification (SMARTS) were developed to extend the functionalities of the original SMILES43. In 2005, IUPAC introduced the InChI44. However, since InChI cannot guarantee the decoding back to their original molecular graphs and SMILES offers the advantage of being more human-readable, SMILES remains the mainstream molecular representation method. During this period, molecular fingerprints gained widespread application in Quantitative Structure-Activity Relationship (QSAR) analyses due to their effective representation of the physicochemical and structural properties of molecules.
For instance, extended-connectivity fingerprints36 are widely used to represent local atomic environments in a compact and efficient manner, making them invaluable for representing complex molecules. These traditional representations are particularly effective for tasks such as similarity search, clustering, and quantitative structure-activity relationship modeling45,46 due to their computational efficiency and concise format.
Traditional molecular representations have been widely applied to various drug design tasks. In early studies, for example, Bender et al. investigated molecular similarity searching and demonstrated that different molecular descriptors could yield distinct similarity evaluations, highlighting the impact of descriptor choice on virtual screening outcomes47. In addition, Chen et al. proposed combination rules for group fusion in similarity-based virtual screening, showing that integrating multiple molecular fingerprints could enhance screening performance48. More recently, Shen et al. proposed MolMapNet49, a model that transforms large-scale molecular descriptors and fingerprint features into two-dimensional feature maps. By capturing the intrinsic correlations of complex molecular properties, MolMapNet uses convolutional neural networks (CNNs) to predict molecular properties in an end-to-end manner. In FP-ADMET and MapLight45,46, the authors combined different molecular fingerprints with ML models to establish robust prediction frameworks for a wide range of ADMET-related properties. Similarly, BoostSweet represents a state-of-the-art (SOTA) ML framework for predicting molecular sweetness, leveraging a soft-vote ensemble model based on LightGBM and combining layered fingerprints with alvaDesc molecular descriptors50,51. The FP-BERT model employs a substructure masking pre-training strategy on extended-connectivity fingerprints (ECFP) to derive high-dimensional molecular representations. It then leverages CNNs to extract high-level features for classification or regression tasks52. Additionally, Li et al. proposed CrossFuse-XGBoost, a model that predicts the maximum recommended daily dose of compounds based on existing human study data. This approach provides valuable guidance for first-in-human dose selection53.
However, as the complexity of drug discovery problems increases, these conventional methods often fall short in capturing the subtle and intricate relationships between molecular structure and function. This limitation has spurred the development of more advanced, data-driven molecular representation techniques that can better address the multifaceted challenges of modern drug discovery.
Modern approaches to molecular representation
Recent advancements in AI have ushered in a new era of molecular representation methods, shifting from predefined rules to data-driven learning paradigms6,11,43. These AI-driven approaches leverage DL models to directly extract and learn intricate features from molecular data, enabling a more sophisticated understanding of molecular structures and their properties. As illustrated in Fig. 3 and summarized in Table 1, these methods encompass a wide range of innovative strategies, including language model-based, graph-based, high-dimensional features-based, multimodal-based, and contrastive learning-based approaches, reflecting their diverse applications and transformative potential in drug discovery.

Illustration of the five main AI-based molecular representation models.
Language model-based molecular representation
Inspired by advances in natural language processing (NLP), models such as Transformers have been adapted for molecular representation by treating molecular sequences (e.g., SMILES or SELFIES) as a specialized chemical language54. Unlike traditional methods like ECFP fingerprints that encode predefined substructures, this approach tokenizes molecular strings at the atomic or substructure level (e.g., individual atom symbols such as “C” or “N” and bond characters like “=”). Each token is mapped into a continuous vector, and these vectors are then processed by architectures like Transformers or BERT using self-supervised pre-training strategies, such as random masking, to learn the deep semantic relationships within molecular structures. The learned latent embeddings encapsulate critical chemical and structural information that goes beyond what simpler, rule-based descriptors can offer. Consequently, these representations provide a robust foundation for downstream tasks, such as molecular property prediction, novel molecule generation, and scaffold hopping, thereby enabling enhanced performance and generalization in various drug design applications.
An example of NLP-inspired methods is the work by Jaeger et al., who introduced Mol2vec, an unsupervised ML approach inspired by NLP to represent molecular substructures as dense and information-rich vectors55. By treating molecular substructures derived from the Morgan algorithm as “words” and entire molecules as “sentences,” the model leverages the Word2vec algorithm56 to generate meaningful embeddings. Mol2vec overcomes limitations of traditional molecular representations, such as sparsity and bit collisions, and provides a pre-trained model capable of capturing chemically relevant substructure relationships, making it a valuable tool for cheminformatics and drug discovery.
BERT-based models have gained significant attention in molecular representation, with Mol-BERT, MTL-BERT, and MolRoPE-BERT standing out as notable examples57,58,59. These pre-trained models extend the BERT framework to capture both structural and contextual information of molecules, demonstrating remarkable utility in drug discovery and cheminformatics. Mol-BERT, proposed by Li et al. leverages the BERT architecture to encode molecular structure and context57. By pre-training on a masked language modeling task using SMILES sequences and fine-tuning on downstream molecular property prediction tasks, Mol-BERT demonstrated superior performance compared to traditional and SOTA graph-based models, showcasing its ability to learn intricate molecular relationships. MTL-BERT takes SMILES as input and applies the Transformer’s encoder with a multi-head self-attention mechanism to capture both long-term and short-term dependencies58. This design effectively addresses data scarcity issues by extracting rich and robust features from molecular sequences. MolRoPE-BERT incorporates rotational position embeddings alongside SMILES data to enhance molecular representation59,60. By pre-training on unlabeled molecular datasets, it focuses on capturing chemically relevant substructure information, providing a comprehensive and improved representation of molecular features. In addition to these models, DeepSA, proposed in 2023, exemplifies the application of BERT-based frameworks beyond molecular representation61. Designed for high-throughput prediction of compound synthesis accessibility, DeepSA intergrates pre-trained language models such as RoBERTa, DeBERTa, and ELECTRA18,62,63,64,65,66. It processes molecular data at token and position levels for embedding and uses a multi-layer perceptron (MLP) for feature decoding to output synthetic accessibility scores. These advancements highlight the adaptability and transformative potential of BERT-inspired methodologies in cheminformatics and drug discovery.
The Transformer-based MOLFORMER model further advances molecular representation by combining the chemical SMILES language with self-supervised learning, leading to significantly improvements in capturing molecular features67. T-SMILES builds on advanced NLP techniques to establish a hierarchical molecular representation framework centered on molecular fragments. This approach enhances the efficiency and rationality of molecular generation while improving adaptability and generalization, especially on low-resource datasets68. INTransformer employs a Transformer-style framework that integrates raw SMILES with noisy SMILES for data augmentation. This innovative design not only improves molecular representation but also addresses challenges in data diversity and robustness69.
In general, language model-based molecular representation methods leverage sequential data to capture the semantic relationships among atoms and substructures through pre-training and self-supervised learning. These approaches efficiently extract rich chemical features from large-scale datasets, enabling the identification of key structural motifs essential for tasks such as scaffold hopping. Moreover, their inherent flexibility also supports efficient fine-tuning across diverse downstream applications, including molecular property prediction and generation of novel scaffolds or molecules. However, a notable limitation lies in their reliance on linear representations, which may fail to fully capture the three-dimensional spatial and topological complexities of molecule structures. Consequently, while language model-based methods can generate structurally innovative scaffolds, incorporating complementary 3D structural information may be necessary to preserve critical bioactive features. In summary, language model-based methods offer a powerful and versatile framework for molecular representation in scaffold hopping, with further potential unlocked by addressing the challenges of multi-dimensional molecular encoding.
Graph-based molecular representation
Graph neural networks represent molecules as graphs, where atoms are treated as nodes and bonds as edges. This graph-based representation is highly effective for capturing the intricate structural details of molecules, enabling a more direct correlation with their physical and chemical properties. GNNs are well-suited for tasks such as molecular generation and molecular property prediction, as they can effectively learn from the relational structure of atoms within a molecule70. Numerous innovative approaches have been developed, each emphasizing different facets of molecular representation to enhance predictive performance.
Attentive FP, a widely recognized graph neural network architecture for molecular representation, propagates node information from nearby nodes to more distant ones, effectively capturing the local atomic environment. It leverages graph attention mechanisms to account for non-local effects within the molecule71. This architecture enables Attentive FP to efficiently identify hidden critical links between nodes while fully considering the molecule’s intrinsic structure. Another notable approach is GROVER, which constructs a multi-level molecular graph representation through self-supervised learning. By pre-training on large-scale unlabeled molecular data, GROVER enhances the structural expressiveness of molecular representations72. This model integrates a message-passing network with a Transformer architecture, allowing it to capture local structural information while simultaneously modeling global relationships within the molecule.
MPG, proposed by Li et al., utilizes a self-supervised pre-training strategy at both the node and graph levels, enabling the model to extract valuable chemical insights after pre-training on a dataset of 11 million unlabeled molecules73. This approach results in interpretable and chemically meaningful representations. Similarly, ReLMole enhances molecular representation through contrastive learning by analyzing similarities at both the atomic layer and functional group levels within molecular graphs74. GraphMVP emphasizes the alignment and consistency between 2D topological and 3D geometric views during self-supervised learning, achieving robust 2D molecular graph encoding without relying on explicit 3D structural information75. Complementing these methods, GEM employs a geometry-aware graph neural network combined with self-supervised learning strategies to incorporate molecular geometry knowledge into its representations76.
Further advancing graph-based methods, FunQG introduces the concept of quotient graphs from graph theory to condense molecular graphs into smaller, more informative representations77. Wang et al. proposed MolCAP, a chemically informed framework that leverages chemical reactivity knowledge through pre-training and prompted fine-tuning78. MolCAP incorporates self-supervised tasks at both the atom and bond levels, employing a balanced multi-task learning strategy to generate highly transferable representations. Similarly, SMPT utilizes a graph isomorphism network (GIN) architecture to aggregate molecular features while capturing molecular spatial geometry at multiple levels, thereby improving the performance of downstream prediction tasks79.
Several models employ hierarchical or multi-layered strategies to enhance molecular representation. HiMol utilizes a hierarchical molecular graph neural network paired with multi-layer self-supervised pre-training tasks for attribute prediction, effectively capturing complex molecular features80. The MMGX model incorporates multiple molecular graph representations, including Atom, Pharmacophore, Junction Tree, and Functional Group views, integrating these perspectives using a dynamic attention mechanism81. This approach captures complementary molecular features, enhances interpretability, and achieve SOTA performance across various prediction tasks. Similarly, PharmHGT targets pharmacophore-constrained molecular property prediction by encoding chemically rich features from heterogeneous molecular graphs, providing a tailored approach for chemically informed tasks82.
Other models focus on novel structural encoding mechanisms and visualization techniques to enhance interpretability. The IFGN model employs a multi-step focusing mechanism to pinpoint key atoms contributing significantly to predicted molecular properties83. Coupled with visualization techniques, this approach offers step-by-step insights into the prediction process. The GSL-MPP model updates node features via graph convolution to capture structural information within molecules, while introducing a molecular similarity graph to compute similarities and generate a similarity map84. This enables better relationship modeling between molecules, improving property prediction accuracy. The SME model provides a chemically intuitive interpretation framework for graph neural networks by analyzing combinations of substructures, addressing limitations of single-mask methods and offering a comprehensive exploration of structure-property relationships85. Xiang et al. proposed a Gram Matrix-based approach compresses 3D molecular spatial information into a 2D representation, facilitating more efficient downstream applications86. Additionally, KANO utilizes graph-based embedding to extract structural and functional cues from knowledge graphs, training word2vec models to enhance molecular representations87. Likewise, KPGT integrates a graph transformer specifically designed for molecular graphs with a knowledge-guided pre-training strategy, effectively capturing both structural and semantic information for improved molecular understanding88.
Innovative pre-training frameworks have emerged to address the limitations of traditional graph-based methods. R-MAT employs a graph transformer with a relative molecular self-attention module, enabling the model to generalize molecular information effectively through pre-training89. TOML-BERT introduces a two-layer pre-training strategy that combines node-level self-supervised learning with graph-level supervised learning, successfully mitigating challenges related to data scarcity90. In contrast, MolFormer departs from conventional graph-based representations by treating molecules as collections of atomic invariants, eliminating the need for explicit graph topology or molecular geometry91. This approach enhances flexibility and adaptability across diverse chemical contexts.
Collectively, these graph-based molecular representation models capture both local chemical environments and global molecular topology in a natural and interpretable manner, which is critical for identifying and preserving key scaffold features during scaffold hopping. Their strengths lie in the ability to directly model structural interactions and incorporate geometric and hierarchical information. However, these methods also face challenges, including high computational complexity, sensitivity to the design of graph construction and message-passing strategies, and sometimes limited scalability when applied to very large or complex molecular datasets. In summary, collectively, these graph-based molecular representation models showcase a wide range of innovations, addressing challenges such as data scarcity, interpretability, and the integration of geometric and hierarchical information. Their rapid development underscores the transformative potential of these methods in molecular property prediction and drug discovery applications.
High-dimensional features-based molecular representation
In addition to the methods mentioned above, incorporating high-dimensional features (e.g., molecular 3D structure, binding pocket, and cellular phenotype) has proven to significantly enhance a model’s ability to represent molecules. Uni-Mol, a general framework for molecular representation learning based on 3D molecular structures, leverages large-scale unlabeled data for pre-training20. The pre-training dataset includes two extensive 3D datasets: molecular structures and protein pocket structures. Specifically, the molecular dataset is constructed from several commercially available databases, comprising approximately 19 million molecules and 210 million 3D conformations. Molecular conformations are generated efficiently using RDKit in combination with molecular force field optimization. Pre-training is conducted on large-scale distributed clusters using a unified model framework and effective pre-training task strategies. Furthermore, GeminiMol introduces the concept of inter-molecular conformation space similarity2. During pre-training, pairs of drug-like molecules are independently encoded using the same molecular encoder to generate 2048-dimensional molecular representation vectors. These vectors are then projected into multiple molecular similarity metrics, using 2D maximum common substructure similarity and conformation space similarity as prediction heads. By capturing conformational space characteristics, GeminiMol demonstrates balanced and robust performance across various drug discovery tasks, including ligand-based virtual screening, target identification, and molecular property prediction. Recently, the authors further expanded the number of training samples and proposed the Ouroboros model, which significantly improved the performance of downstream tasks, indicating the effectiveness of this training strategy92. A more recent development, PhenoModel, builds on this by introducing information on cell morphological changes induced by chemical perturbations93. Using contrastive learning, PhenoModel aligns compound representations with perturbation-induced cell painting images in the feature space. This alignment allows the model to simultaneously capture both molecular conformation and potential activity information, offering a unique perspective that bridges molecular and phenotypic data.
In summary, incorporating high-dimensional features into molecular representation models has markedly improved the capture of complex spatial conformations and molecular interactions that are critical for biological activity. These models exemplify how leveraging extensive 3D structure data, interaction information between protein and ligand, potential molecular activity information, and innovative pre-training strategies can yield robust and predictive molecular embeddings. These advancements not only enhance performance in property prediction and virtual screening but also establish a critical link between molecular conformation and phenotypic response, thereby paving the way for more informed and effective drug discovery applications.
Multimodal-based molecular representation
Multimodal molecular representation learning has gained significant attention in recent years, leveraging complementary information from diverse molecular modalities to enhance the robustness and interpretability of molecular property predictions. These approaches integrate representations such as molecular graphs, SMILES sequences, fingerprints, images, and even molecular video data, enabling a more comprehensive understanding of molecular properties.
One widely adopted method in this domain is FP-GNN, which combines molecular graph information with molecular fingerprints through co-training for property prediction94. Similarly, MoleSG, introduced by Shen et al. in 2024, integrates SMILES and molecular graph representations using a unified transformer-based backbone95. By implementing a novel non-overlapping masking strategy, MoleSG ensures complementary yet independent interactions between these two modalities, achieving SOTA performance across 14 downstream tasks, thereby highlighting the potential of multimodal strategies. Lu et al. proposed MMFDL, which independently processes SMILES, ECFP, and molecular graphs using Transformer encoders, BiGRU, and GCN, respectively96. These features are then fused with calculated importance weights, effectively leveraging complementary information from different data sources. MMFDL underscores the value of integrating diverse modalities to improve prediction accuracy, enhance generalization, and increase resilience to noise in drug property prediction tasks.
Several approaches emphasize cross-modal contrastive learning to align and integrate molecular information. CLAMP utilizes a modular architecture comprising a chemical molecule encoder and a text encoder, pre-trained via cross-modal contrastive learning to enhance molecular understanding97. Similarly, COATI combines textual and 3D molecular representations within a contrastive learning framework to produce unbiased, general-purpose molecular embeddings that support downstream structural models98. Extending this concept, CGIP integrates molecular images with explicit graph information and implicit visual cues, leveraging both intra- and inter-modal contrastive learning to capture rich multimodal representations99.
Other methods employ multimodal fusion frameworks to achieve higher precision in molecular property predictions. DLF-MFF processes molecular fingerprints, 2D and 3D maps, and molecular images using specialized DL frameworks for each modality, subsequently fusing these representations for enhanced predictive performance100. UniMAP adopts a multi-layer Transformer model to decompose molecular graphs into fragments, generating SMILES-based multimodal inputs for deep cross-modal molecular feature fusion101. PremuNet introduces a dual-branch architecture, with PremuNet-L capturing low-dimensional features and PremuNet-H focusing on high-dimensional features, effectively integrating these representations to improve performance across diverse tasks102.
Molecular images have emerged as a crucial modality in molecular representation. ImageMol employs five pre-training strategies to integrate chemical knowledge and structural information into molecular image representations, enhancing their utility for predictive tasks103. ISMol combines molecular images and SMILES strings as bi-modal inputs, aligning and fusing them through a cross-modal attention mechanism using Vision Transformer and ChemBERTa-77M-MLM encoders104. Extending this idea, VideoMol leverages Vision Transformer to extract dynamic and physicochemical information from molecular video data, achieving highly precise molecular characterization105.
Multiscale and multi-view approaches further enrich multimodal representations. MvMRL integrates molecular fingerprints, SMILES sequences, and molecular maps using a multi-scale feature extractor and a dual cross-attention mechanism to capture both local and global information106. This design effectively models complex nonlinear relationships between molecular features, improving predictive performance.
By leveraging the strengths of each modality, these methods capture various aspects of molecular information, ranging from structural topology to spatial conformation and sequential patterns. This integration enhances the robustness and interpretability of predictive models, ultimately leading to improved molecular design and property prediction outcomes. However, effectively fusing these heterogeneous data types poses significant challenges, including the need for sophisticated alignment strategies to mitigate potential noise and redundancy. In summary, multimodal molecular representation methods harness diverse molecular information sources to develop robust, accurate, and interpretable models for property prediction. By integrating graph-based, sequence-based, image-based, and other molecular representations, these approaches pave a promising path for advancing drug discovery and materials science.
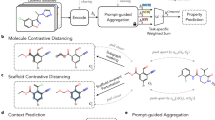
Contrastive learning-based molecular representation
Contrastive learning utilizes positive and negative pairs to learn rich, discriminative molecular embeddings. By contrasting similar and dissimilar molecules, these models effectively capture the key features that distinguish molecular activities. This approach is particularly valuable in scenarios with limited labeled data, as it enables learning from large, unlabeled datasets, making it a powerful tool for lead discovery and optimization. It has been widely adopted across various frameworks, each incorporating unique strategies for data augmentation, molecular featurization, and hierarchical learning.
MoCL, proposed by Sun et al., introduces a contrastive learning framework specifically tailored for molecular fingerprinting107. This method integrates chemical domain knowledge by employing substructure replacement for local-level augmentation and Tanimoto similarity for global-level guidance. MoCL optimizes molecular graph representations through hierarchical contrastive objectives, combining multi-view augmentation with domain-specific insights. This innovative approach delivers superior semantic understanding and robust performance across downstream tasks.
MolCLR, proposed by Wang et al., employs GNNs to contrast augmented molecular graphs, generating generalized molecular representations108. The model integrates diverse data augmentation strategies, a non-linear MLP projection head, and the NT-Xent contrastive loss. By pre-training on large chemical datasets, MolCLR demonstrates exceptional scalability, generalization, and transferability, particularly in low-data molecular tasks. Building upon MolCLR, iMolCLR incorporates a weighted contrastive loss to address false negatives and learns representations at both molecular and fragment levels109. Pretrained on approximately 10 million label-free molecules, iMolCLR exhibits robust performance across various molecular property prediction tasks. Similarly, MOCO employs multi-view molecular featurization by integrating 2D topology, 3D geometry, SMILES strings, and fingerprints110. Utilizing an attention mechanism for weighted aggregation and optimizing embeddings with InfoNCE loss, MOCO achieves superior generalization and transferability.
Several frameworks extend contrastive learning to address specific challenges in molecular representation. MolFeSCue combines few-shot learning with contrastive learning to tackle issues like data scarcity and class imbalance111. By integrating sequence-based (ChemBERTa), graph-based (HuGIN), and Jumping Knowledge Network models, MolFeSCue generates discriminative embeddings using a dynamic contrastive loss function, enhancing both efficiency and accuracy in molecular property prediction. Similarly, 3DGCL and 3D-MOL focus on leveraging 3D molecular structures, utilizing SchNet and hierarchical graph-based models, respectively, to capture spatial information while preserving molecular semantic consistency112,113.
Innovative designs in graph contrastive learning further strengthen model robustness and generalization. ATMOL employs attention-wise masked graph contrastive learning to enhance molecular attribute prediction through advanced graph enhancement and feature extraction techniques114. CasANGCL integrates Cascaded Attention Networks with graph contrastive tasks, effectively capturing both local and global molecular representations for improved robustness115. Meanwhile, SMICLR combines graph neural networks with Long Short-Term Memory, generating augmented positive and negative sample pairs from molecular maps and SMILES views for comprehensive representation learning116.
Multi-view and fragment-based strategies have also proven effective. FraSICL generates semantically invariant molecular views by decomposing molecular graphs into fragment pairs117. Leveraging multi-view fusion mechanisms and auxiliary similarity loss, FraSICL captures complementary information from different fragments. Similarly, UniCorn integrates multiple molecular views into a universal contrastive learning framework118, while GraphCL applies graph-specific augmentations to maximize consistency across graph views, yielding robust and transferable representations119.
Together, contrastive learning-based molecular representation methods enhance the discriminative power of molecular embeddings by comparing positive and negative sample pairs. By contrasting structurally similar and dissimilar molecules, these models can capture subtle features and differences that are critical for accurate property prediction and scaffold hopping. A key strength of this approach lies in its ability to leverage large volumes of unlabeled data, helping to mitigate challenges like data scarcity and class imbalance that often arise in chemical datasets. Nonetheless, the effectiveness of contrastive learning is highly dependent on the strategies used to construct positive and negative pairs, and training stability can be a concern. In summary, together, these models exemplify the transformative potential of contrastive learning in molecular representation. By addressing challenges such as data scarcity, class imbalance, and molecular complexity through innovative strategies and diverse molecular modalities, they lay a robust foundation for advancing molecular property prediction and drug discovery applications.
Scaffold hopping approaches using molecular representations
Early scaffold hopping primarily relied on molecular fingerprints, shape similarity, pharmacophore modeling, and fragment replacement. Over time, these methods have evolved into AI-driven approaches that enable data-driven scaffold exploration with enhanced efficiency and precision.
Traditional approaches for scaffold hopping
Traditional scaffold hopping encompasses a broad range of strategies designed to identify novel molecular scaffolds while maintaining biological activity. These methods have been foundational in drug discovery, leveraging approaches such as pharmacophore modeling, shape similarity methods, and molecular fingerprinting. Each technique offers distinct advantages in exploring chemical diversity and advancing lead discovery24,120,121,122.
Pharmacophore modeling, for instance, represents the spatial arrangement of molecular features critical for biological activity, such as hydrogen bond donors or acceptors, hydrophobic regions, and charged groups. This approach identifies molecules capable of fitting into a target binding site, facilitating the discovery of novel scaffolds with similar interaction profiles. A significant advancement in this area is NScaffold, which employs topological pharmacophore graphs (PhGs) to encode pharmacophoric features as graph nodes and their topological distances as edges123. NScaffold introduces a ranking method that prioritizes PhGs based on scaffold coverage, enabling the identification of scaffold-independent pharmacophoric features. Validated across six biological targets, NScaffold outperformed traditional scoring metrics like Coverage and Growth-rate, particularly when working with limited scaffold datasets. For example, in thrombin inhibitors, NScaffold successfully identified key hydrogen bonding interactions, underscoring its potential for interpretable scaffold hopping. By providing an explainable and efficient framework for exploring diverse chemical scaffolds, this approach represents a significant advancement in ligand-based virtual screening.
Shape similarity methods focus on comparing the three-dimensional shapes of molecules to identify scaffolds structurally similar to known bioactive compounds. Techniques like ROCS detect molecular volumes with similar binding properties by leveraging shape overlays124,125. Similarly, Phase Shape enables flexible ligand superposition and virtual screening, offering rapid and accurate 3D ligand alignments with high enrichment of active compounds126. Building on these principles, SHAFTS combines shape overlay scoring (ShapeScore) with pharmacophore feature matching (FeatureScore), employing a feature triplet hashing algorithm to enhance scaffold discovery efficiency127. Retrospective validation on datasets such as DUD and Jain’s benchmark demonstrated SHAFTS’s superior early enrichment and scaffold diversity compared to ROCS and ShaEP128. In prospective studies, SHAFTS identified 16 RSK2 inhibitors, including low micromolar hits with potent anti-migration activity. By bridging ligand-based virtual screening and scaffold hopping, SHAFTS exemplifies a robust tool for chemical space exploration and lead discovery129.
Fingerprints and similarity searches provide computationally efficient strategies for scaffold hopping, utilizing molecular descriptors like ECFP and pharmacophore-based approaches. The ErG method abstracts molecular graphs into reduced graphs with pharmacophore-type nodes, capturing biologically relevant features while preserving chemical diversity130. By incorporating fuzzy incrementation for inter-feature distances, ErG improves scaffold diversity and retrieval rates over traditional fingerprints such as DAYLIGHT. Validation across 11 activity classes in the MDDR database demonstrated that ErG outperformed traditional methods in 10 classes, offering a highly interpretable and computationally efficient alternative for ligand-based virtual screening. The WHALES descriptor provides a comprehensive 3D representation by integrating geometric, atomic distance, and molecular property information21. In retrospective screenings across 182 biological targets, WHALES achieved superior scaffold diversity compared to benchmark descriptors like MACCS and ECFP. Prospective validation further identified four novel RXR agonists, including a rare non-acidic chemotype with nanomolar activity and high selectivity. WHALES’s ability to navigate uncharted chemical space underscores its potential as a powerful scaffold-hopping tool.
Applications of modern AI-based molecular representations in scaffold hopping
Traditional scaffold hopping approaches, while effective, often rely on predefined libraries, limiting their ability to explore the vast chemical space. Recent advancements in AI, such as VAE-based, and diffusion-based generative models (Fig. 4), have revolutionized scaffold hopping by enabling efficient and diverse scaffold design while preserving biological relevance. These AI-driven methods (Table 2) offer innovative solutions for overcoming the limitations of traditional approaches, such as scalability and the ability to handle complex molecular modifications.

A VAE-based framework and B diffusion model-based framework.
Graph- and VAE-based generative models have established a strong foundation for advanced scaffold hopping by disentangling molecular components. GraphGMVAE, for instance, employs a Gaussian mixture variational autoencoder to encode scaffolds and side chains into separate distributions, facilitating precise scaffold modifications while preserving pharmacophoric features131. Validated on Janus kinase 1 (JAK1) inhibitors, it achieved a 97.9% success rate in generating novel scaffolds, with several synthesized compounds demonstrating strong bioactivity, including an IC50 of 5.0 nM. Building on this approach, ScaffoldGVAE incorporates multi-view graph neural networks to capture both scaffold-level and molecular dynamics features31. It achieves near-perfect success rates across various kinase targets and has successfully generated novel LRRK2 inhibitors with high activity.
Multimodal and pharmacophore-guided methods further enhance scaffold design by integrating diverse molecular data to ensure bioactivity. DeepHop, for example, combines 3D molecular structures with protein sequence embeddings, leveraging Transformer architectures to optimize scaffold generation33. Trained on over 57,000 scaffold-hopping pairs, DeepHop achieved a 65.2% success rate while maintaining high 3D similarity and generating bioactive scaffolds for previously unseen targets. Similarly, PGMG encodes pharmacophore features into complete graphs, addressing many-to-many relationships between pharmacophores and molecules to generate bioactive compounds132. This framework excels in scaffold hopping, producing novel EGFR inhibitors with enhanced bioactivity and drug-likeness.
Diffusion-based models have emerged as transformative tools in scaffold hopping, providing precise spatial control and enhanced scaffold diversity. DiffLinker utilizes E(3)-equivariant diffusion models for scaffold hopping and linker generation, significantly improving scaffold diversity and molecular connectivity133. Similarly, DiffHopp employs an E(3)-equivariant graph diffusion model tailored for scaffold hopping, leveraging conditional probability distributions to generate novel molecular scaffolds within protein pockets32. By integrating geometric vector perceptron (GVP)-based encoders and 3D molecular graph diffusion, DiffHopp enhances scaffold connectivity, diversity, and binding affinity. Validation on the PDBBind dataset highlights its superior performance across key metrics such as QED, SA, and Vina scores, positioning DiffHopp as a robust framework for exploring chemical space and advancing protein-ligand interaction studies.
DiffSBDD incorporates protein pocket information to enable context-aware scaffold hopping with optimized molecular properties like binding affinity and drug-likeness35. It excels at preserving critical substructures while efficiently generating diverse, chemically plausible scaffolds without retraining. Experimental validation on the Binding MOAD dataset demonstrates significant improvements in docking scores and synthetic accessibility compared to baseline methods like Pocket2Mol134 and ResGen135. DiffSBDD’s flexibility in scaffold hopping underscores its potential for exploring uncharted chemical spaces in drug discovery. Similarly, PMDM introduces dual diffusion strategies to model local and global molecular dynamics, achieving strong performance in challenging scaffold hopping tasks for targets such as SARS-CoV-2 Mpro and CDK2136. By accurately capturing protein pocket dynamics, PMDM provides a robust framework for structure-based drug design.
Addressing the computational inefficiencies of traditional diffusion models, TurboHopp introduces an efficient scaffold hopping algorithm using E(3)-equivariant consistency models and reinforcement learning to overcome the inference speed limitations of diffusion-based methods137. By reducing generation steps and incorporating task-specific reward functions, TurboHopp achieves up to 30× faster scaffold generation while improving molecular properties like connectivity, binding affinity, and synthesizability. Validated on the PDBBind dataset, TurboHopp outperforms models like DiffHopp across key metrics, setting a new benchmark for efficiency and quality in scaffold hopping and accelerating structure-based drug design.
In addition to these methods, REINVENT 4 is a comprehensive generative framework that integrates reinforcement learning, transfer learning, and curriculum learning to facilitate diverse scaffold modifications138. REINVENT 4 enables scaffold hopping by generating innovative linkers that bridge key fragments, and inducing novel core scaffold while optimizing for desired pharmacological properties. Similarly, Ouroboros adopts a directed chemical evolution strategy within the latent space of pre-trained molecular encoders92. By mapping molecules into a continuous latent space, Ouroboros employs iterative “mutation” and selection processes that guide molecular representations from one scaffold region to another, thereby achieving scaffold hopping. This method leverages the inherent continuity of the latent space to enable controlled and gradual scaffold transitions while preserving or enhancing target properties.
Collectively, these methods demonstrate the convergence of generative modeling and molecular representation in scaffold hopping. Each approach contributes distinct innovations, such as disentangled latent representations (GraphGMVAE, ScaffoldGVAE, Ouroboros), multimodal learning (DeepHop, PGMG), and 3D-equivariant diffusion (DiffLinker, DiffHopp, DiffSBDD, DiffPROTACs139). This progression reflects a growing emphasis on scalability, diversity, and biological relevance in scaffold hopping, establishing a robust foundation for exploring uncharted chemical spaces in drug discovery.
Current challenges and limitations
Despite the significant advances in both traditional and AI-driven molecular representation and scaffold hopping approaches, several challenges and limitations continue to hinder the full potential of these methods.
Overemphasis on benchmark performance
While numerous innovative molecular representation methods have been developed in recent years, with some extending into experimental applications, most are evaluated primarily on standard benchmark datasets for tasks such as molecular property prediction or virtual screening. This has led to a “leaderboard arms race,” where achieving SOTA performance on benchmarks is prioritized over addressing real scientific challenges. Although some methods introduce novel training paradigms or offer interpretability, they often lack logical consistency from an experimental perspective. Moreover, AI-driven approaches may sometimes exploit dataset-specific tricks to achieve high benchmark scores, potentially at the cost of generalization. In other words, while these methods appear to perform well on benchmark datasets, they may fail to extend their performance to new molecules that are not represented in those datasets. Interestingly, several studies have demonstrated that by effectively combining molecular graph information with molecular fingerprint features, and by carefully selecting model architectures along with appropriate parameter tuning, predictive models can also reach high levels of performance using traditional representation methods140,141,142. This suggests that the possible combination of AI-based and conventional molecular characterization approaches to develop a general, adaptive AI architecture, capable of self-adjusting to various benchmark scenarios, might partially overcome these limitations, much like recent advances in molecular interaction and property prediction have attempted to address similar challenges70.
Dependence on data quality and quantity
AI-driven approaches heavily rely on the quality and quantity of training data. Issues such as insufficient datasets, batch effects in experimental data, and biases in data labeling can severely affect model performance and generalization. Furthermore, acquiring high-quality labeled data for specific drug targets is expensive and time-consuming. Large-scale experimental datasets are often proprietary to commercial organizations, limiting accessibility for the broader research community and restricting the general applicability of AI-based models. To address these data challenges, several innovative strategies can be borrowed. For instance, federated learning enables multiple institutions or companies to collaboratively train AI models without directly sharing sensitive or proprietary data143. By aggregating model updates rather than raw data, federated learning not only alleviates privacy concerns but also leverages the collective strength of diverse datasets144. Moreover, recent advancements have combined federated learning with knowledge distillation, like the approach employed by advanced large language models such as DeepSeek (https://www.deepseek.com/), allowing complex local models (teachers) to transfer their learned knowledge into a compact global model (student). This hybrid strategy enhances the robustness and performance of AI models, thereby mitigating limitations related to data heterogeneity and limited generalizability.
Limitations in exploring chemical space
Traditional scaffold hopping methods often rely on predefined rules, which constrain their ability to explore diverse chemical spaces. Even with modern AI-based approaches, training dataset distributions can cause generated scaffolds to converge on specific chemotypes, reducing diversity and failing to explore novel or unconventional chemical structures. Scaffold hopping requires identifying structurally diverse compounds while retaining specific bioactivity, and the inability to strike this balance poses challenges for practical applications. A potentially viable solution is to adopt an attention-based multimodal fusion network that can adaptively learn the relationships among 2D, 3D, and deep learning-based representations. By designing dedicated fusion layers to integrate features from different modalities into a shared latent space, this approach captures a more comprehensive array of molecular information, thereby enhancing the efficiency of chemical space exploration and the success rate of scaffold hopping.
Synthetic accessibility and drug-likeness
Although AI-based generative models excel at creating novel scaffolds, ensuring their synthetic feasibility and drug-like properties remains a substantial challenge. Many generated molecules may be difficult or impractical to synthesize and could exhibit suboptimal pharmacokinetic or pharmacodynamic characteristics. This often necessitates additional filtering steps to exclude unsuitable candidates. Striking a balance between exploring novel chemical spaces, maintaining synthetic accessibility, and preserving key activity features is particularly demanding. To address these synthetic feasibility concerns, incorporating synthetic accessibility scores, functional reaction templates or retrosynthesis prediction algorithms directly into the reward function of generative models can help prioritize molecules that are not only novel but also synthetically tractable145,146.
Challenges in multimodal representation integration
Despite the rise of multimodal molecular representation models and increased focus on cross-modal integration, effectively combining 2D, 3D, and DL-based representations in the drug discovery workflow presents substantial challenges. A key concern is interpretability. While graph neural network models may provide some interpretability by visualizing node or edge weights, these insights are often system-specific and lack generalizability. Understanding how specific molecular features influence predicted activities is inherently difficult, and incorporating multimodal data further exacerbates this issue, hindering the rational optimization of molecules.
Scaffold hopping and bioactivity preservation
In scaffold hopping, modifying molecular scaffolds often risks compromising bioactivity. Neglecting target structural features during compound design can further diminish activity. Capturing complex interactions, such as protein-ligand binding, and explaining molecular interactions with protein 3D conformations and dynamics remain critical yet challenging tasks. While advancements like 3D graph-based models and diffusion models have made notable strides, they still struggle to effectively handle flexible and dynamic molecular systems.
Data availability
No datasets were generated or analysed during the current study.
References
Bai, F., Li, S. L. & Li, H. L. AI enhances drug discovery and development. Natl. Sci. Rev. 11, nwad303 (2024).
Wang, L. et al. Conformational space profiling enhances generic molecular representation for AI-powered ligand-based drug discovery. Adv. Sci. 11, e2403998 (2024).
Jimenez-Luna, J., Grisoni, F., Weskamp, N. & Schneider, G. Artificial intelligence in drug discovery: recent advances and future perspectives. Expert Opin. Drug Discov. 16, 949–959 (2021).
Sabe, V. T. et al. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 224, 113705 (2021).
Wu, H. et al. A review of deep learning methods for ligand based drug virtual screening. Fundam. Res. 4, 715–737 (2024).
Nguyen-Vo, T. H., Teesdale-Spittle, P., Harvey, J. E. & Nguyen, B. P. Molecular representations in bio-cheminformatics. Memetic Comput. 16, 519–536 (2024).
Gallegos, L. C., Luchini, G., St John, P. C., Kim, S. & Paton, R. S. Importance of engineered and learned molecular representations in predicting organic reactivity, selectivity, and chemical properties. Acc. Chem. Res. 54, 827–836 (2021).
Li, Y., Liu, B. Y., Deng, J. Y., Guo, Y. & Du, H. B. Image-based molecular representation learning for drug development: a survey. Brief. Bioinform. 25, bbae294 (2024).
Geppert, H., Vogt, M. & Bajorath, J. Current trends in ligand-based virtual screening: molecular representations, data mining methods, new application areas, and performance evaluation. J. Chem. Inf. Model. 50, 205–216 (2010).
Atz, K., Grisoni, F. & Schneider, G. Geometric deep learning on molecular representations. Nat. Mach. Intell. 3, 1023–1032 (2021).
Wigh, D. S., Goodman, J. M. & Lapkin, A. A. A review of molecular representation in the age of machine learning. Wiley Interdiscip. Rev.-Comput. Mol. Sci. 12, 25010–25024 (2022).
Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988).
Kim, S. et al. PubChem substance and compound databases. Nucleic Acids Res. 44, D1202–D1213 (2016).
Mendez, D. et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 47, D930–D940 (2019).
Irwin, J. J. et al. ZINC20-A Free Ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model. 60, 6065–6073 (2020).
Liu, T. Q., Lin, Y. M., Wen, X., Jorissen, R. N. & Gilson, M. K. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 35, D198–D201 (2007).
Tong, X. C. et al. Generative models for De novo drug design. J. Med. Chem. 64, 14011–14027 (2021).
Ahmad, W., Simon, E., Chithrananda, S., Grand, G. & Ramsundar, B. ChemBERTa-2: Towards Chemical Foundation Models. arXiv:2209.01712, https://ui.adsabs.harvard.edu/abs/2022arXiv220901712A (2022).
Wang, S., Guo, Y., Wang, Y., Sun, H., Huang, J. Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics. Association for Computing Machinery: Niagara Falls, NY, USA, 2019). 429–436.
Zhou, G. et al. In International Conference on Learning Representations.
Grisoni, F., Merk, D., Byrne, R. & Schneider, G. Scaffold-Hopping from Synthetic Drugs by Holistic Molecular Representation. Sci. Rep. 8, 16469 (2018).
Kuz’min, V. et al. Simplex representation of molecular structure as universal QSAR/QSPR tool. Struct. Chem. 32, 1365–1392 (2021).
Li, Z., Jiang, M. J., Wang, S. & Zhang, S. G. Deep learning methods for molecular representation and property prediction. Drug Discov. Today 27, 103373 (2022).
Schneider, G., Neidhart, W., Giller, T. & Schmid, G. “Scaffold-hopping” by topological pharmacophore search: A contribution to virtual screening. Angew. Chem.-Int. Ed. 38, 2894–2896 (1999).
Sun, H. M., Tawa, G. & Wallqvist, A. Classification of scaffold-hopping approaches. Drug Discov. Today 17, 310–324 (2012).
Mishra, A. et al. Scaffold hopping approaches for dual-target antitumor drug discovery: opportunities and challenges. Expert Opin. Drug Discov. 19, 1355–1381 (2024).
Hu, Y., Stumpfe, D. & Bajorath, J. Recent advances in scaffold hopping. J. Med. Chem. 60, 1238–1246 (2017).
Schuffenhauer, A. Computational methods for scaffold hopping. Wiley Interdiscip. Rev.-Comput. Mol. Sci. 2, 842–867 (2012).
Hu, Y., Stumpfe, D. & Bajorath, J. Computational exploration of molecular scaffolds in medicinal chemistry. J. Med. Chem. 59, 4062–4076 (2016).
Callis, T. B., Garrett, T. R., Montgomery, A. P., Danon, J. J. & Kassiou, M. Recent scaffold hopping applications in central nervous system drug discovery. J. Med. Chem. 65, 13483–13504 (2022).
Hu, C. et al. ScaffoldGVAE: scaffold generation and hopping of drug molecules via a variational autoencoder based on multi-view graph neural networks. J. Cheminform. 15, 91 (2023).
Torge, J., Harris, C., Mathis, S. V. & Lio, P. DiffHopp: A Graph Diffusion Model for Novel Drug Design via Scaffold Hopping. arXiv:2308.07416, https://ui.adsabs.harvard.edu/abs/2023arXiv230807416T (2023).
Zheng, S. J. et al. Deep scaffold hopping with multimodal transformer neural networks. J. Cheminform. 13, 87 (2021).
Alakhdar, A., Poczos, B. & Washburn, N. Diffusion models in De Novo drug design. J. Chem. Inf. Model. 64, 7238–7256 (2024).
Schneuing, A. et al. Structure-based drug design with equivariant diffusion models. arXiv:2210.13695, https://ui.adsabs.harvard.edu/abs/2022arXiv221013695S (2022).
Rogers, D. & Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754 (2010).
Durant, J. L., Leland, B. A., Henry, D. R. & Nourse, J. G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 42, 1273–1280 (2002).
Landrum, G. “RDKit: Open-source cheminformatics. https://www.rdkit.org” (2022).
Carhart, R. E., Smith, D. H. & Venkataraghavan, R. Atom pairs as molecular features in structure-activity studies: definition and applications. J. Chem. Inf. Comput Sci. 25, 64–73 (1985).
Nilakantan, R., Bauman, N., Dixon, J. S. & Venkataraghavan, R. Topological torsion: a new molecular descriptor for SAR applications. Comparison with other descriptors. J. Chem. Inf. Comput. Sci. 27, 82–85 (1987).
Dyson, G. M., Lynch, M. F. & Morgan, H. L. A modified IUPAC-Dyson notation system for chemical structures. Inf. Storage Retr. 4, 27–83 (1968).
Wiswesser, W. J. How the WLN began in 1949 and how it might be in 1999. J. Chem. Inf. Comput. Sci. 22, 88–93 (1982).
David, L., Thakkar, A., Mercado, R. & Engkvist, O. Molecular representations in AI-driven drug discovery: a review and practical guide. J. Cheminform. 12, 56 (2020).
Heller, S. R., McNaught, A., Pletnev, I., Stein, S. & Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 7, 23 (2015).
Notwell, J. H. & Wood, M. W. ADMET property prediction through combinations of molecular fingerprints. arXiv:2310.00174, https://ui.adsabs.harvard.edu/abs/2023arXiv231000174N (2023).
Venkatraman, V. FP-ADMET: a compendium of fingerprint-based ADMET prediction models. J. Cheminform. 13, 75 (2021).
Bender, A. How similar are those molecules after all? Use two descriptors and you will have three different answers. Expert Opin. Drug Discov. 5, 1141–1151 (2010).
Chen, B. N., Mueller, C. & Willett, P. Combination rules for group fusion in similarity-based virtual screening. Mol. Inform. 29, 533–541 (2010).
Shen, W. X. et al. Out-of-the-box deep learning prediction of pharmaceutical properties by broadly learned knowledge-based molecular representations. Nat. Mach. Intell. 3, 334–343 (2021).
Lee, J., Song, S. B., Chung, Y. K., Jang, J. H. & Huh, J. BoostSweet: Learning molecular perceptual representations of sweeteners. Food Chem. 383, 132435 (2022).
Mauri, A. In Ecotoxicological QSARs (ed K., Roy) 801-820 (Springer US, 2020).
Wen, N. F. et al. A fingerprints based molecular property prediction method using the BERT model. J. Cheminform. 14, 71 (2022).
Li, Q., He, Y. & Pan, J. B. CrossFuse-XGBoost: accurate prediction of the maximum recommended daily dose through multi-feature fusion, cross-validation screening and extreme gradient boosting. Brief. Bioinform. 25, bbad511 (2024).
Shao, J. S., Jia, Q. F., Chen, X., Hao, Y. J. & Wang, L. Molecular fragmentation as a crucial step in the AI-based drug development pathway. Commun. Chem. 7, 20 (2024).
Jaeger, S., Fulle, S. & Turk, S. Mol2vec: Unsupervised machine learning approach with chemical intuition. J. Chem. Inf. Model. 58, 27–35 (2018).
Mikolov, T., Chen, K., Corrado, G. S. & Dean, J. in International Conference on Learning Representations.
Li, J. C. & Jiang, X. F. Mol-BERT: An Effective Molecular Representation with BERT for Molecular Property Prediction. Wireless Communications & Mobile Computing 2021 (2021).
Zhang, X. C. et al. Pushing the boundaries of molecular property prediction for drug discovery with multitask learning BERT enhanced by SMILES enumeration. Research 2022, 0004 (2022).
Liu, Y. W. et al. MolRoPE-BERT: An enhanced molecular representation with Rotary Position Embedding for molecular property prediction. J. Mol. Graph. Model. 118, 108344 (2023).
Su, J., Lu, Y., Pan, S., Wen, B. & Liu, Y. RoFormer: Enhanced Transformer with Rotary Position Embedding. ArXiv abs/2104.09864 (2021).
Wang, S. H., Wang, L., Li, F. L. & Bai, F. DeepSA: a deep-learning driven predictor of compound synthesis accessibility. J. Cheminform. 15, 103 (2023).
Liu, Y. et al. RoBERTa: A Robustly Optimized BERT Pretraining Approach. arXiv:1907.11692, https://ui.adsabs.harvard.edu/abs/2019arXiv190711692L (2019).
He, P., Liu, X., Gao, J. & Chen, W. DeBERTa: Decoding-enhanced. BERT with Disentangled Attention. arXiv:2006.03654, https://ui.adsabs.harvard.edu/abs/2020arXiv200603654H (2020).
Clark, K., Luong, M.-T., Le, Q. V. & Manning, C. D. ELECTRA: Pre-training Text Encoders as Discriminators Rather Than Generators. arXiv:2003.10555, https://ui.adsabs.harvard.edu/abs/2020arXiv200310555C (2020).
Bhargava, P., Drozd, A. & Rogers, A. Generalization in NLI: Ways (Not) To Go Beyond Simple Heuristics, arXiv:2110.01518, https://ui.adsabs.harvard.edu/abs/2021arXiv211001518B (2021).
Guo, D. et al. GraphCodeBERT: Pre-training Code Representations with Data Flow. arXiv:2009.08366, https://ui.adsabs.harvard.edu/abs/2020arXiv200908366G (2020).
Ross, J. et al. Large-scale chemical language representations capture molecular structure and properties. Nat. Mach. Intell. 4 (2022).
Wu, J. N. et al. t-SMILES: a fragment-based molecular representation framework for de novo ligand design. Nat. Commun. 15, 4993 (2024).
Jiang, J., Li, Y. C., Zhang, R. S. & Liu, Y. W. INTransformer: Data augmentation-based contrastive learning by injecting noise into transformer for molecular property prediction. J. Mol. Graph. Model. 128, 108703 (2024).
Li, Y. et al. An adaptive graph learning method for automated molecular interactions and properties predictions. Nat. Mach. Intell. 4, 645–651 (2022).
Xiong, Z. et al. Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism. J. Med. Chem. 63, 8749–8760 (2020).
Rong, Y. et al. In 34th Conference on Neural Information Processing Systems (NeurIPS). (2020).
Li, P. Y. et al. An effective self-supervised framework for learning expressive molecular global representations to drug discovery. Brief. Bioinform. 22 (2021).
Ji, Z. W., Shi, R. H., Lu, J. R., Li, F. & Yang, Y. ReLMole: Molecular Representation Learning Based on Two-Level Graph Similarities. J. Chem. Inf. Model. 62, 5361–5372 (2022).
Liu, S. et al. Pre-training Molecular Graph Representation with 3D Geometry. arXiv:2110.07728, https://ui.adsabs.harvard.edu/abs/2021arXiv211007728L (2021).
Fang, X. M. et al. Geometry-enhanced molecular representation learning for property prediction. Nat. Mach. Intell. 4, 127–134 (2022).
Hajiabolhassan, H., Taheri, Z., Hojatnia, A. & Yeganeh, Y. T. FunQG: Molecular Representation Learning via Quotient Graphs. J. Chem. Inf. Model. 63, 3275–3287 (2023).
Wang, Y., Zhang, J. J., Jin, J. R. & Wei, L. Y. MolCAP: Molecular Chemical reActivity Pretraining and prompted-finetuning enhanced molecular representation learning. Comput. Biol. Med. 167, 107666 (2023).
Li, Y. S., Wang, W., Liu, J. & Wu, C. K. Pre-training molecular representation model with spatial geometry for property prediction. Comput. Biol. Chem. 109, 108023 (2024).
Zang, X., Zhao, X. B. & Tang, B. Z. Hierarchical Molecular Graph Self-Supervised Learning for property prediction. Commun. Chem. 6, 34 (2023).
Kengkanna, A. & Ohue, M. Enhancing property and activity prediction and interpretation using multiple molecular graph representations with MMGX. Commun. Chem. 7, 74 (2024).
Jiang, Y. H. et al. Pharmacophoric-constrained heterogeneous graph transformer model for molecular property prediction. Commun. Chem. 6, 60 (2023).
Tian, Y. N., Wang, X. R., Yao, X. J., Liu, H. X. & Yang, Y. Predicting molecular properties based on the interpretable graph neural network with multistep focus mechanism. Brief. Bioinform. 24, bbac534 (2023).
Zhao, B. Y., Xu, W. X., Guan, J. H. & Zhou, S. G. Molecular property prediction based on graph structure learning. Bioinformatics 40 (2024).
Wu, Z. X. et al. Chemistry-intuitive explanation of graph neural networks for molecular property prediction with substructure masking. Nat. Commun. 14, 2585 (2023).
Xiang, W. K. et al. Gram matrix: an efficient representation of molecular conformation and learning objective for molecular pretraining. Brief. Bioinform. 25, bbae340 (2024).
Fang, Y. et al. Knowledge graph-enhanced molecular contrastive learning with functional prompt. Nat. Mach. Intell. 5, 542–553 (2023).
Li, H. et al. A knowledge-guided pre-training framework for improving molecular representation learning. Nat. Commun. 14, 7568 (2023).
Maziarka, L. et al. Relative molecule self-attention transformer. J. Cheminform. 16, 3 (2024).
Duan, Y. J. et al. Enhancing molecular property prediction through task-oriented transfer learning: integrating universal structural insights and domain-specific knowledge. J. Med. Chem. 67, 9575–9586 (2024).
Boulougouri, M., Vandergheynst, P. & Probst, D. Molecular set representation learning. Nat. Mach. Intell. 6 (2024).
Wang, L. et al. Directed Chemical Evolution via Navigating Molecular Encoding Space. bioRxiv, 2025.2003.2018.643899 (2025).
Wang, S. et al. PhenoScreen: a dual-space contrastive learning framework-based phenotypic screening method by linking chemical perturbations to cellular morphology. bioRxiv, 2024.2010.2023.619752 (2024).
Cai, H. X., Zhang, H. M., Zhao, D. C., Wu, J. X. & Wang, L. F. P.-G. N. N. a versatile deep learning architecture for enhanced molecular property prediction. Brief. Bioinform. 23, bbac408 (2022).
Shen, A., Yuan, M. Z., Ma, Y. F., Du, J. & Wang, M. N. Complementary multi-modality molecular self-supervised learning via non-overlapping masking for property prediction. Brief. Bioinform. 25 (2024).
Lu, X. H. et al. Multimodal fused deep learning for drug property prediction: Integrating chemical language and molecular graph. Comput. Struct. Biotechnol. J. 23, 1666–1679 (2024).
Seidl, P., Vall, A., Hochreiter, S. & Klambauer, G. Enhancing activity prediction models in drug discovery with the ability to understand human language. arXiv:2303.03363, https://ui.adsabs.harvard.edu/abs/2023arXiv230303363S (2023).
Kaufman, B. et al. COATI: Multimodal contrastive pretraining for representing and traversing chemical space. J. Chem. Inf. Model. 64, 1145–1157 (2024).
Xiang, H. X., Jin, S. T., Liu, X. R., Zeng, X. X. & Zeng, L. Chemical structure-aware molecular image representation learning. Brief. Bioinform. 24, bbad404 (2023).
Ma, M. & Lei, X. J. A deep learning framework for predicting molecular property based on multi-type features fusion. Comput. Biol. Med. 169, 107911 (2024).
Feng, S. et al. UniMAP: Universal SMILES-Graph Representation Learning. arXiv:2310.14216, https://ui.adsabs.harvard.edu/abs/2023arXiv231014216F (2023).
Zhang, H. H., Wu, J. T., Liu, S. C. & Han, S. A pre-trained multi-representation fusion network for molecular property prediction. Inf. Fusion 103 (2024).
Zeng, X. X. et al. Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework. Nat. Mach. Intell. 4, 1004–1016 (2022).
Zhang, X. et al. Dual-view learning based on images and sequences for molecular property prediction. IEEE J. Biomed. Health Inform. 28, 1564–1574 (2024).
Xiang, H. X. et al. A molecular video-derived foundation model for scientific drug discovery. Nat. Commun. 15, 9696 (2024).
Zhang, R. et al. MvMRL: a multi-view molecular representation learning method for molecular property prediction. Brief. Bioinform. 25, bbae298 (2024).
Sun, M. Y. et al. In 27th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD). 3585-3594 (2021).
Wang, Y. Y., Wang, J. R., Cao, Z. L. & Farimani, A. B. Molecular contrastive learning of representations via graph neural networks. Nat. Mach. Intell. 4, 279–287 (2022).
Wang, Y. Y., Magar, R., Liang, C. & Farimani, A. B. Improving molecular contrastive learning via faulty negative mitigation and decomposed fragment contrast. J. Chem. Inf. Model. 62, 2713–2725 (2022).
Zhu, Y. Q. et al. Molecular contrastive pretraining with collaborative featurizations. J. Chem. Inf. Model. 64, 1112–1122 (2024).
Zhang, R. C. et al. MolFeSCue: enhancing molecular property prediction in data-limited and imbalanced contexts using few-shot and contrastive learning. Bioinformatics 40, btae118 (2024).
Moon, K., Im, H. J. & Kwon, S. 3D graph contrastive learning for molecular property prediction. Bioinformatics 39, btad371 (2023).
Kuang, T. J., Ren, Y. M. & Ren, Z. X. 3D-Mol: a novel contrastive learning framework for molecular property prediction with 3D information. Pattern Anal. Appl. 27, 71 (2024).
Liu, H., Huang, Y. B. A., Liu, X. J. & Deng, L. Attention-wise masked graph contrastive learning for predicting molecular property. Brief. Bioinform. 23, bbac303 (2022).
Zheng, Z. X. et al. CasANGCL: pre -training and fine-tuning model based on cascaded attention network and graph contrastive learning for molecular property prediction. Brief. Bioinform. 24, bbac566 (2023).
Pinheiro, G. A., Silva, J. L. F. & Quiles, M. G. SMICLR: contrastive learning on multiple molecular representations for semisupervised and unsupervised representation learning. J. Chem. Inf. Model. 62, 3948–3960 (2022).
Zhang, Z. Q., Xie, A. L., Guan, J. H. & Zhou, S. G. Molecular property prediction by semantic-invariant contrastive learning. Bioinformatics 39 (2023).
Feng, S. et al. UniCorn: A Unified Contrastive Learning Approach for Multi-view Molecular Representation Learning. arXiv:2405.10343, https://ui.adsabs.harvard.edu/abs/2024arXiv240510343F (2024).
You, Y. et al. In 34th Conference on Neural Information Processing Systems (NeurIPS), (2020).
Kumar, A. & Zhang, K. Y. J. Advances in the development of shape similarity methods and their application in drug discovery. Front. Chem. 6, 315 (2018).
Maggiora, G., Vogt, M., Stumpfe, D. & Bajorath, J. Molecular similarity in medicinal chemistry. J. Med. Chem. 57, 3186–3204 (2014).
Brown, N. & Jacoby, E. On scaffolds and hopping in medicinal chemistry. Mini-Rev. Med. Chem. 6, 1217–1229 (2006).
Nakano, H., Miyao, T. & Funatsu, K. Exploring topological pharmacophore graphs for scaffold hopping. J. Chem. Inf. Model. 60, 2073–2081 (2020).
Rush, T. S., Grant, J. A., Mosyak, L. & Nicholls, A. A shape-based 3-D scaffold hopping method and its application to a bacterial protein−protein interaction. J. Med. Chem. 48, 1489–1495 (2005).
Hawkins, P. C. D., Skillman, A. G. & Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 50, 74–82 (2007).
Sastry, G. M., Dixon, S. L. & Sherman, W. Rapid shape-based ligand alignment and virtual screening method based on atom/feature-pair similarities and volume overlap scoring. J. Chem. Inf. Model. 51, 2455–2466 (2011).
Liu, X. F., Jiang, H. L. & Li, H. L. SHAFTS: A hybrid approach for 3D molecular similarity calculation. 1. Method and assessment of virtual screening. J. Chem. Inf. Model. 51, 2372–2385 (2011).
Vainio, M. J., Puranen, J. S. & Johnson, M. S. ShaEP: Molecular overlay based on shape and electrostatic potential. J. Chem. Inf. Model. 49, 492–502 (2009).
Lu, W. Q. et al. SHAFTS: A hybrid approach for 3D molecular similarity calculation. 2. prospective case study in the discovery of diverse p90 Ribosomal S6 Protein Kinase 2 inhibitors to suppress cell migration. J. Med. Chem. 54, 3564–3574 (2011).
Stiefl, N., Watson, T. A., Baumann, K. & Zaliani, A. ErG: 2D pharmacophore descriptions for scaffold hopping. J. Chem. Inf. Model. 46, 208–220 (2006).
Yu, Y. et al. A novel scalarized scaffold hopping algorithm with graph-based variational autoencoder for discovery of JAK1 inhibitors. Acs Omega 6, 22945–22954 (2021).
Zhu, H. M., Zhou, R. Y., Cao, D. S., Tang, J. & Li, M. A pharmacophore-guided deep learning approach for bioactive molecular generation. Nat. Commun. 14, 6234 (2023).
Igashov, I. et al. Equivariant 3D-conditional diffusion model for molecular linker design. Nat. Mach. Intell. 6, 417–427 (2024).
Peng, X. et al. Pocket2Mol: Efficient molecular sampling based on 3D protein pockets. arXiv:2205.07249, https://ui.adsabs.harvard.edu/abs/2022arXiv220507249P (2022).
Zhang, O. et al. ResGen is a pocket-aware 3D molecular generation model based on parallel multiscale modelling. Nat. Mach. Intell. 5, 1020–1030 (2023).
Huang, L. et al. A dual diffusion model enables 3D molecule generation and lead optimization based on target pockets. Nat. Commun. 15, 2657 (2024).
Yoo, K., Oertell, O., Lee, J., Lee, S. & Kang, J. TurboHopp: accelerated molecule scaffold hopping with consistency models. arXiv:2410.20660, https://ui.adsabs.harvard.edu/abs/2024arXiv241020660Y (2024).
Loeffler, H. H. et al. Reinvent 4: Modern AI–driven generative molecule design. J. Cheminform. 16, 20 (2024).
Li, F., Hu, Q., Zhou, Y., Yang, H. & Bai, F. DiffPROTACs is a deep learning-based generator for proteolysis targeting chimeras. Brief. Bioinform. 25, bbae358 (2024).
Liu, S., Chen, M., Yao, X. & Liu, H. Fingerprint-enhanced hierarchical molecular graph neural networks for property prediction. J. Pharm. Anal., 101242 (2025).
Han, H. et al. Employing Automated Machine Learning (AutoML) Methods to Facilitate the In Silico ADMET Properties Prediction. J. Chem. Inf. Model. (2025).
Myung, Y., de Sá, A. G. C. & Ascher, D. B. Deep-PK: deep learning for small molecule pharmacokinetic and toxicity prediction. Nucleic Acids Res. 52, W469–W475 (2024).
McMahan, H. B. et al. Communication-Efficient Learning of Deep Networks from Decentralized Data. arXiv:1602.05629, https://ui.adsabs.harvard.edu/abs/2016arXiv160205629M (2016).
Hanser, T. et al. Data-driven federated learning in drug discovery with knowledge distillation. Nat. Mach. Intell. 7, 423–436 (2025).
Yin, X. et al. Syn-MolOpt: a synthesis planning-driven molecular optimization method using data-derived functional reaction templates. J. Cheminform. 17, 27 (2025).
Dorna, V. et al. TAGMol: Target-Aware Gradient-guided Molecule Generation. arXiv:2406.01650, https://ui.adsabs.harvard.edu/abs/2024arXiv240601650D (2024).
Acknowledgements
This work was supported by Shanghai Science and Technology Development Funds (Grant IDs: 24JS2850200, 24YF2729200, 24JS2850100, and 22ZR1441400), ShanghaiTech AI4S Initiative SHTAI4S202404, National Key R&D Program of China (Grant IDs: 2022YFC3400501 & 2022YFC3400500), the National Natural Science Foundation of China (No 82341093), start-up package from ShanghaiTech University, and Shanghai Frontiers Science Center for Biomacromolecules and Precision Medicine at ShanghaiTech University. The authors appreciate the technical support provided by the high-performance computing cluster of ShanghaiTech University.
Author information
Authors and Affiliations
Contributions
S.W. and F.B. contributed to conceptualization. S.W., R.Z., X.L., F.C., X.M., Y.T., C.X., P.R., and L.L. contributed to data curation. S.W., L.W., and Q. Q. contributed to visualization. S.W., X.M., S. W., and F. B. contributed to original draft writing. All authors contributed to review and edit the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, S., Zhang, R., Li, X. et al. Recent advances in molecular representation methods and their applications in scaffold hopping. npj Drug Discov. 2, 14 (2025). https://doi.org/10.1038/s44386-025-00017-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44386-025-00017-2
